
Abstract
It is established that persistent inflammation promotes tumor formation and disease progression. The inflammatory microenvironment within solid tumors is composed of soluble and cellular components, including several cytokines and chemokines, growth factors, enzymes, as well as host cells: endothelial cells, fibroblasts, and leukocytes. Tumor-associated myeloid cells (TAMC) represent the preponderant part of tumor-infiltrating leukocytes; they are a very heterogeneous entity of cells regulating diverse aspects of cancer biology. TAMC can directly support cancer growth and survival and regulate angiogenesis, metastasis, as well as immunosuppression. Accordingly, TAMC have recently become targets of new and innovative anticancer therapy approaches. This chapter will review the main characteristics of tumor-associated myeloid cells and their role in promoting tumor growth and dissemination. In addition, we will briefly address new therapeutic possibilities based on targeting of TAMC.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Vascular Endothelial Growth Factor
- Hepatocyte Growth Factor
- Tumor Microenvironment
- Myeloid Cell
- Circulate Tumor Cell
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
Inflammation is a consistent feature of the tumor microenvironment and has been considered the seventh hallmark of cancer [1–6]. As suggested by current estimates, 25 % of cancers are associated with chronic inflammation sustained by infections (e.g., hepatitis) or inflammatory conditions of diverse origin (e.g., prostatitis) [6]. In addition, even tumors not directly connected to inflammation are characterized by the presence of cells and mediators of the inflammatory response [7].
Apart from malignant cells, host cells infiltrate tumors, including leukocytes, fibroblasts, and endothelial cells. Leukocytes, and in particular myeloid cells, are the most consistent cellular component of solid tumors. Tumor-associated myeloid cells (TAMC) mainly support tumor growth and progression, thereby contrasting the T-cell infiltrate, which mainly has antitumoral activity. TAMC all arise from hematopoietic stem cells (HSC) within the bone marrow (Fig. 2.1) and further differentiate into macrophage/granulocyte progenitors. The tumor infiltrate comprising the myeloid populations skews tumor-mediated immunosuppression, tissue remodeling, tumor progression and metastasis [8, 9]. TAMC demonstrated high plasticity, resulting in two extreme polarized macrophage (M1 and M2) and neutrophil (N1 and N2) phenotypes [10, 11]. Cross talk between the different cellular components was demonstrated, resulting in tuning of the adaptive immune response, promotion of angiogenesis, and tissue remodeling [8].

Differentiation pathways of tumor-associated myeloid cells. Myeloid cells originate from hematopoietic stem cells (HSC) in the bone marrow. Here the networks that give rise to the various myeloid cell lineages in diverse compartments (bone marrow, blood/spleen, and tumor) and their precursors are illustrated. In the tumor tissue, macrophages and neutrophils display a gradient of differently polarized phenotypes whose extreme are M1–M2 for TAM and N1–N2 for neutrophils. CMP common myeloid progenitors, IMC immature myeloid cells, TEM Tie2-expressing monocytes, MDSC myeloid-derived suppressor cells, M-MDSC myeloid MDSC, G-MDSC granulocytic MDSC, TAM tumor-associated macrophages, TAN tumor-associated neutrophils, iDC immature dendritic cells, TADC tumor-associated dendritic cells
Results obtained so far clearly indicate that TAMC are major players in the connection between inflammation and cancer. Ongoing efforts, which led to a better understanding of their biological properties, indicated that myeloid cell-infiltrating growing tumor could have a prognostic value, thus representing an attractive target for novel biological therapies of tumors.
In this chapter, we will mainly focus on myeloid cells infiltrating tumors and mention soluble mediators involved in their recruitment or released by TAMC, which affect tumor progression and dissemination (cytokines, chemokines, and proteases). Furthermore, new therapeutic approaches based on targeting of tumor-infiltrating myeloid cells and/or soluble mediators will be discussed.
2.2 Heterogeneity of Myeloid Cells in the Tumor Microenvironment
2.2.1 Myeloid Subsets in the Tumor Microenvironment
Solid tumors are characterized by the presence of a leukocyte infiltrate including lymphocytes and myeloid cells from early stages. Growing evidence indicated that the leukocyte infiltrate has a prognostic value. For instance, it has been described that infiltrating T lymphocytes are associated with a favorable prognosis in colorectal cancer, melanoma, ovarian cancer, and breast cancer [12, 13]. In contrast, myeloid cells are most frequently associated with a poor prognosis [14]. TAMC (Fig. 2.1) comprise five distinct myeloid populations, namely, tumor-associated macrophages (TAM), monocytes expressing the angiopoietin-2 (Ang-2) receptor Tie2 (known as Tie2-expressing monocytes or TEM), myeloid-derived suppressor cells (MDSC), tumor-associated neutrophils (TAN), and tumor-associated dendritic cells (TADC).
Tumor-associated macrophages belong to the early infiltrating leukocyte populations within tumors, thus preceding lymphocytes, and are usually the most abundant immune population in the tumor microenvironment [6, 15]. They derive from blood monocytes actively recruited from the circulation into tumor tissues. Early studies demonstrated that appropriately stimulated macrophages are able to kill tumor cells in vitro; however, TAM, conditioned by the tumor microenvironment, loose the cytotoxic capability and rather exert several pro-tumoral functions, mediating cancer-related inflammation, angiogenesis, immunosuppression, tissue remodeling, and metastasis [16, 17, 6].
The heterogeneous behavior of TAM is a hallmark of myeloid cells and is oversimplified in a polarization concept with two extreme M1 and M2 phenotypes [18–20] with distinct and somehow opposite functions. M1 macrophages are classically activated by bacterial products and Th1 cytokines (e.g., LPS/interferon-γ). They are potent producers of inflammatory and immunostimulating cytokines, trigger adaptive responses, secrete reactive oxygen species (ROS) and nitrogen intermediates, and have cytotoxic effect towards transformed cells. On the other hand, M2 macrophages or alternatively activated macrophages differentiate in response to Th2 cytokines (e.g., interleukin (IL)-4, IL-13) [21]. In contrast to their M1 counterpart, M2 macrophages produce growth factors, leading to tissue repair and angiogenesis activation, have high scavenging activity, and inhibit adaptive immune responses [22, 14, 23, 11, 24]. Thus, macrophages are a very heterogeneous cell population, able to display different functions depending on the context. Macrophages can be either immunostimulatory at the beginning of the inflammatory response or immunosuppressive which dampen inflammation [25, 18, 14, 26, 27].
A similar dichotomy with polarization towards two extreme phenotypes (N1 and N2) has been also described for neutrophils [28]. Besides exerting antibacterial functions, neutrophils can infiltrate tumors playing a major role as key mediators in malignant transformation, tumor progression, and regulation of antitumor activity [29]. Tumor-associated neutrophils (TAN) have received interest only recently, mainly due to their short life span and the observation that tumor microenvironment can sustain and prolong the survival of polymorphonuclear leukocytes (PMN) [30, 31].
A particular small subset of TAMC is represented by Tie2-expressing monocytes (TEM): they express several monocyte/macrophage markers, along with the angiopoetin-2 receptor, Tie2, and are endowed with proangiogenic properties [32–35]. Tie2-expressing monocytes can be distinguished from the majority of TAM by their surface marker profile (Tie2+, CD11b+) and their preferential localization to areas of angiogenesis [33], while they are largely missing in nonneoplastic area adjacent to tumors [35]. Indeed, Tie2 is constitutively expressed at low levels by a substantial fraction (20 %) of circulating monocytes and is overexpressed upon monocyte homing into growing tumors or regenerating tissues [33, 36]. Following Ang-2 stimulation, Tie2+ monocytes acquire an M2-like phenotype, with increased expression of IL-10, CCL17, arginase 1 (Arg-1), and scavenger and mannose receptors and low expression of proinflammatory molecules such as IL-12 and TNF-α [37, 38].
Myeloid-derived suppressor cells (MDSC) are a heterogeneous population of immature myeloid cells, having the ability to suppress T-cell functions [39, 40]. They are derived from myeloid progenitors in bone marrows which do not differentiate into mature granulocytes, macrophages, or dendritic cells. MDSC have been isolated from blood, spleen, and bone marrow of tumor-bearing mice and infiltrate the tumor tissue, where local tumor-associated factors promote their activation [41]. In tumor-bearing mice, two main subsets of MDSC were identified: monocytic MDSC (M-MDSC), characterized by CD11b+, Ly6G−, and Ly6Chigh, and granulocytic MDSC (G-MDSC), characterized by CD11b+, Ly6Ghigh, and Ly6C− [42]. M-MDSC were shown to govern the ability of differentiating into monocytes (macrophages) and (DC), whereas G-MDSC do not possess this potential [43]. These subsets are functionally different: M-MDSC-mediated immunosuppression is based on upregulation of inducible nitric oxide synthase (iNOS), expression of Arg-1, and production of suppressive cytokines, whereas G-MDSC-mediated immunosuppression is characterized by antigen-specific responses (including ROS release requiring prolonged MDSC and T-cell contacts) [44]. Tumor-associated MDSC generally exhibit an M2-like phenotype, while M1 and M2 phenotypes could coexist in some mouse tumor models [45, 46].
Human MDSC are still poorly defined [47], even if they have been isolated from blood of patients with glioblastoma, colon cancer, breast cancer, lung cancer, or kidney cancer [48–52]. Recent studies have proposed that human MDSC have a characteristic CD34+, CD33+, CD11b+, and HLA-DR− profile [42]. Similarly to the murine counterpart, human MDSC are divided into two main subsets: monocytic MDSC (M-MDSC), characterized by the expression of CD14, and granulocytic MDSC (G-MDSC), identified by positivity for CD15.
A small number of dendritic (DC) are found in most human and murine neoplasms. Similarly to macrophages and neutrophils, plasticity is a main feature of these cells. DC are differentially localized in tumors; for example, in breast cancer immature langerin+ DC are interspersed within the tumor mass, whereas more mature CD83+, DC-LAMP+ DC are confined to the peritumoral area [53]. In contrast to TAM, tumor-associated dendritic cells (TADC) were found in the invasive front of papillary thyroid carcinoma [54]. Growing evidences demonstrate that the majority of TADC found within the tumor microenvironment have an immature phenotype (iDC) [55–57]. The immature stage of TADC is responsible for the tolerogenic response of adaptive immunity against tumors and strongly contributes to tumor immune evasion [58].
2.2.2 Recruitment of Myeloid Cells in Tumors
TAMC derive from monocytes and granulocytes, extravasated from the circulation and infiltrating the tumor mass. Recruitment of blood cells into tumors is mediated by chemoattractants released by tumor and stromal cells. CC chemokine 2 (CCL2), originally known as monocyte chemotactic protein 1 (MCP1), was the first relevant tumor-derived chemotactic factor described [59, 60]. Several other chemokines attracting myeloid cells have been identified, including CCL5, CCL7, CCL8, and CXC chemokine 1 (CXCL1) and CXCL12 [61–63]. Furthermore, urokinase plasminogen activator (uPA); growth factors such as colony-stimulating factor (CSF)-1, transforming growth factor β (TGF-β), basic fibroblast growth factor (bFGF, also known as FGF-2), and vascular endothelial growth factor (VEGF); and antimicrobial peptides (e.g., human beta-defensin-3) were shown to be involved in myeloid recruitment into neoplastic tissues [64, 9, 65–67].
The prototypic chemoattractant for neutrophils, CXCL8, is mainly responsible for the recruitment of TAN; other related chemokines of the CXC subfamily are also involved, including CXCL1, CXCL2, and CXCL6 [68, 69]. Moreover, tumor-derived TGF-β can promote neutrophil migration [70].
CC chemokine receptor 2 (CCR2), CCL2 receptor, CXCL12, CXCL5, and stem cell factor (SCF, also known as KIT ligand) play a pivotal role in the recruitment of MDSC into tumors [71–73]; in addition, Bv8, also known as prokineticine 2 (PROK2), might be essential for MDSC recruitment [74, 75]. Finally, the proinflammatory proteins S-100A9 and S-100A8, produced by MDSC, are implicated in an autocrine loop promoting accumulation of suppressor cells into tumors [76, 77].
TEM do not express CCR2 and are therefore recruited towards tumors by different mechanisms [35, 78, 79]. Other CC chemokines, such as CCL3, CCL5, and CCL8, are produced by tumor cells and could play a role in TEM recruitment [80]. Ang-2, overexpressed by tumor cells and inflamed tissues, has been shown to exert a chemotactic effect on Tie2-expressing blood monocytes in vitro, suggesting that the Ang-2/Tie2 axis might be involved in recruiting TEM into tumors [81, 32, 35, 34, 82]. In addition, recent data suggest the involvement of the CXCL12-CXCR4 homing axis for TEM infiltration [82].
In recent years, it has been shown that tumor-derived factors such as VEGF, CXCL12, CXCL8, β-defensins, and hepatocyte growth factor (HGF) are secreted into the bloodstream and are believed to attract iDC into the tumor bed [83–86]. Moreover, CCL20, CCL7, as well as the receptors CCR5 and CCR6 were demonstrated to be important for TADC recruitment towards the tumor [87].
Proliferation can also contribute to sustaining TAMC levels in solid tumors. A paracrine loop has been evidenced for TAM, with production of colony-stimulating factor 1 (CSF-1) by murine fibrosarcoma cells acting on TAM-expressing CSF-1 receptor (CSF-1R) [88]. A finding confirmed more recently by Condeelis and Pollard [89] showed the effect of epidermal growth factor (EGF) produced by TAM and tumor-derived CSF-1 on recruitment and survival of macrophages during tumor growth. Indeed, macrophage proliferation has been demonstrated to occur during type II inflammation [90].
2.2.3 Tumor-Derived Factors Affecting Myeloid Differentiation and Polarized Functions
Upon arrival in the tumor, monocytes differentiate to macrophages primarily in response to CSF-1 produced by tumor cells. Although coexistence of diverse TAM subpopulations with distinct functions depending on tumor stage and geographical localization within the same tumor has been proposed, they mostly have an M2-like phenotype [91]. Many different studies demonstrated that M2 (pro-tumoral) TAM polarization is driven by cytokines and other signals released in the tumor microenvironment [92]. Among these IL-10, IL-6, CCL2, CSF-1, and prostaglandin E2 (PGE2) were reported to promote M2-like polarization [93, 94]. TGF-β is overexpressed by tumor cells and plays a crucial role in promoting an immunosuppressive phenotype, in addition to driving N2 polarization of TAN [31].
Many tumor-derived factors were implicated in MDSC expansion such as GM-CSF, M-CSF, IL-6, IL-1β, VEGF, and PGE2 [44, 95]. In addition, Bronte and coworkers recently found that cytokine-mediated induction of MDSC was completely dependent on the transcription factor CCAT/enhancer-binding protein b (C/EBPb), shown to function as a master regulator in this process [96]. Further it was proposed that a combination of at least two signals is necessary for MDSC functionality and expansion, for example, GM-CSF, inhibiting maturation of myeloid cells, and a proinflammatory molecule such as interferon-γ (INF-γ) [41].
Soluble factors released by tumor cells (i.e., IL-10, VEGF, TGF-β, etc.) contribute to keep DC in an immature pro-tumorigenic phenotype. Furthermore, in preclinical studies of breast cancer, it was shown that tumor-derived factors altered DC maturation by secretion of thymic stromal lymphopoietin (TSLP), which in turn induces the expression and secretion of the OX40 ligand, a molecule that contributes to sustain the M2-like phenotype of TAM.
2.3 Pro-tumoral Functions of Tumor-Associated Myeloid Cells
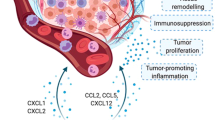
Myeloid cells exposed to the tumor microenvironment most frequently promote tumor progression. They can secrete soluble factors which support proliferation and invasion of tumor cells, activate angiogenesis, and promote resistance to therapies (Fig. 2.2). High TAM or TAN infiltration generally correlates with poor patient outcome [97, 6, 16, 11, 98–101], but few exceptions to this finding are also reported. For instance, in colorectal cancer (CRC) contrasting results reported that TAM density is associated with positive or negative patient outcome [102, 103–105]. On the same line, TAN infiltrate is associated with a favorable prognosis in patients with gastric carcinomas [106], but also with more aggressive pancreatic tumors [107]. Macrophage subsets might have distinct roles, as observed in lung adenocarcinoma were the number of CD204+ TAM showed a strong association with poor patient outcome, while the CD68+ TAM population did not [108]. The concept that not only the number and the presence of specific cell subsets but also the localization of infiltrating cells might have specific functions and predictive values is increasingly emerging. Accordingly, peritumoral TAM density with high expression of co-stimulatory molecules (CD80 and CD86) was associated with better patient survival in CRC, whereas the same cell population within the tumors did not have any predictive value [109, 110]. Thus, TAMC exert complex roles on growing tumors affecting different aspects of tumor progression, i.e. tumor cell proliferation and survival, angiogenesis, tumor dissemination, and resistance to therapies.

Pro-tumoral functions of tumor-associated myeloid cells. TAMC exposed to the tumor microenvironment exert several pro-tumoral functions, including promotion of angiogenesis, matrix degradation, and suppression of adaptive immunity. These effects are mediated through the release of soluble factors (i.e., cytokines, growth and proangiogenic factors, proteolytic enzymes, etc.) and result in higher tumor survival and proliferation, local invasion and dissemination, resistance to therapies
2.3.1 Tumor Proliferation and Survival
TAM were shown to have the ability to promote tumor growth directly through the production of trophic and activating factors for stromal and cancer cells (EGF, bFGF, VEGF, platelet-derived growth factor β [PDGF], TGF-β) [111, 112, 6, 113] in response to stimuli from the tumor microenvironment. For example, IL-13 and IL-4 produced by CD4+ T-cell-infiltrating tumors, such as breast cancer, led to the production and secretion of EGF by TAM [114]. Moreover, production of proinflammatory cytokines, including TNF-α and IL-6, by TAM and other cells of the tumor microenvironment (e.g., epithelial cells), sustains tumor growth and inhibits apoptosis [115–119].
Several lines of evidence suggest that TAN are required for the rapid growth of tumor cells and their depletion inhibits tumor development [120, 28]. Proteins stored within neutrophil granules (e.g., elastase) may have a role in tumor initiation [121]. In addition, neutrophil-derived ROS have been associated with DNA damage [122]. TAN were shown to be able to produce soluble factors (cytokines and chemokines, HGF, oncostatin M), driving processes like angiogenesis, wound healing, and hematopoiesis and thus exerting a role in tumor promotion and growth [123–125, 121, 101]. For instance, HGF released by neutrophils enhances the invasiveness of human cholangiocellular and hepatocellular carcinoma cells in vitro, and HGF levels in bronchoalveolar lavage fluids were found to correlate with neutrophil number in patients with bronchoalveolar carcinomas, which further correlates with poor patient prognosis [101].
2.3.2 Angiogenesis
To sustain the increased metabolic demand of growing tumors, the development of a tumor vasculature is required. VEGF is the primary, but not the only, angiogenic factor released by tumor cells and is involved in the “angiogenic switch” that can occur at various stages of tumor progression, depending on the tumor type and the microenvironment. Other factors are involved, including PDGF-β, bFGF, angiopoietins, and CXCL12 (SDF-1) [126]. Tumor-associated myeloid cells were shown to contribute to tumor angiogenesis by production of growth factors, cytokines, and proteases [80] such as VEGFA, Bv8, and metalloproteases (MMP) [10, 127, 65, 128].
The prototypic myeloid cell with angiogenic properties is the Tie2 monocyte [32, 35]. TEM can be found in close proximity to nascent blood vessels within solid tumors. In addition, TEM depletion completely prevented neovascularization in preclinical models (spontaneous pancreatic adenocarcinoma, human glioma grown orthotopically in the mouse) [33]. Interestingly, TEM ablation did not affect the number of infiltrating TAM or TAN, suggesting that TEM are an entity on their own and not just precursors of TAM [35]. How TEM stimulate angiogenesis has not been clarified yet, but preliminary indications in murine tumor models point to the fact that perivascular TEM secrete bFGF. It is believed that release of such factors in close proximity to vessels could directly stimulate angiogenesis or MMP9 secretion, which in turn would release growth factors entrapped within the extracellular matrix (ECM).
TAM have also a profound influence on the regulation of tumor angiogenesis [129]. It was demonstrated in several preclinical studies that TAM positively correlated with microvascular density (MVD) [130–133]. Lin and coworkers were the first to describe the direct role of TAM in driving the “angiogenic switch” in a spontaneous mammary carcinoma mouse model [134]. Likewise, depletion of monocytes by clodronate treatment in a preclinical model with Lewis lung carcinoma led to lower TAM infiltration and angiogenesis, further underlining the importance and the involvement of macrophages in tumor angiogenesis [135].
TAM express various molecules modulating angiogenesis, such as VEGF, bFGF, TNF-α, IL-1β, CXCL8, cyclooxygenase 2 (COX2, also known as PTGS2), plasminogen activator, uPA, PDGF-β, MMP7, MMP9, and MMP12 [136]. Hypoxia exerts a crucial role in the upregulation of gene transcription in TAM, promoting VEGF expression [137–141]. Other recent studies showed a direct involvement of TAM in tumor angiogenesis and neovascularization via transdifferentiation into endothelial cells when stimulated by angiogenic factors [142, 143].
More recent studies have shown that MDSC can contribute to tumor angiogenesis. In a preclinical model for colon cancer, MDSC positively correlated with tumor growth rate and blood vessel density [144]. Moreover, tumor angiogenesis was significantly lowered by blocking Bv8 with a neutralizing antibody, a treatment that significantly reduced the number of MDSC [74]. Metalloproteases, particularly MMP9, MMP2, MMP13, and MMP14, produced by MDSC, were shown to enhance VEGF bioavailability by mobilization from the ECM [144, 145]. Increased recruitment of MDSC has also been demonstrated in the presence of hypoxia, possibly stimulating tumor angiogenesis [126, 74]. Parallel to TEM, MDSC were also observed to be localized in the vicinity of blood vessels. Under certain conditions, some MDSC acquire endothelial cell shape, start to express endothelial markers including CD31 and VEGFR2, and are eventually incorporated into the tumor endothelium [144].
TAN were shown to rapidly release VEGF from internal storage compartments, leading to endothelial proliferation and tubule formation [146, 147]. In addition, TNF-α and GM-CSF secreted by tumor cells were shown to trigger the release of proangiogenic chemokines by TAN. The number of TAN in myxofibrosarcoma positively correlated with tumor MVD [148]. Furthermore, in a xenograft mouse model of human melanoma where cancer cells were engineered to constitutively produce CXCL6, it was found that the number of TAN as well as angiogenesis was markedly increased [149]. Studies in the RIP1-TAG2 mouse model for pancreatic carcinogenesis revealed formation of dysplastic, neutrophil-bearing, angiogenic islets upon malignant transformation. In the abovementioned model, neutrophil depletion of the islets led to dramatically lowered angiogenesis [150].
In recent years, it has become more and more apparent that iDC make a profound contribution to tumor angiogenesis [85]. TNF-α and CXCL8 produced by iDC from ovarian cancer ascites triggered the release of various growth factors from EC [85, 151]. Moreover, iDC were shown to release osteopontin which promotes monocyte secretion of the proangiogenic IL-1β [152]. Finally, it was recently observed that iDC produced high levels of VEGF and CXCL8 under hypoxic conditions, which, in turn, might inhibit DC maturation and further promote angiogenesis via this autocrine loop [153, 151].
2.3.3 Cancer Cell Dissemination
The major cause of death in cancer results from therapy-resistant metastases. Stephen Paget’s conclusion in the late nineteenth century that the metastatic process depends on cross talk between selected cancer cells (the “seeds”) and a specific organ microenvironment (“the soil”) is still valid and is experimentally confirmed [154, 155]. Tumor metastasis is a complex multistep process, during which malignant cells spread from the primary tumor site to secondary distant organs. The different steps of cancer cell dissemination can be subdivided into local invasion, entry into the bloodstream (intravasation), survival in the bloodstream, extravasation, and colonization [156]. Mesenchymal, endothelial, and immune cells are required to form an appropriate microenvironment for tumor progression [157]. Immune cells, particularly macrophages, neutrophils, T lymphocytes, and natural killer (NK) cells, are major sources of proteases that degrade the host tissue, allowing cancer cells to disseminate.
The set of proteolytic enzymes found in tumor microenvironment comprises matrix metalloproteases, serine proteases, and cysteine proteases (i.e., cathepsin) [158–162]. Matrix proteases exert essential functions in physiological conditions as active regulators of postnatal tissue development and remodeling. In addition, they are important for tissue repair in response to injury and regulate cancer progression modulating the tumor microenvironment, particularly the leukocyte infiltrate [163]. MMP were shown to activate TGF-β, which is an important regulator of T-cell and TAN functions [164]. Proteases also produce specific cleavage fragments of target chemokines with independent biological activity, ranging from anergic products (CXCL7, CXCL4, CXCL1), antagonists (CCL7), or more potent chemoattractants (CXCL8), thereby modulating the leukocyte composition within a tumor [165–167].
Besides their influence on the tumor infiltrate, proteases were shown to promote cancer cell invasion and intravasation. The cleavage of cell-adhesion molecules like E-cadherin induces the disruption of cell-cell junctions leading to loosening of cell-cell contacts which, together with ECM protein turnover, facilitated cancer cell migration and invasion into the surrounding tissue and vasculature. Tight regulation of the single proteases within the tumor microenvironment allows the control of tumor cell invasion [168].
After invasion to the surrounding tissues, cancer cells enter the blood circulatory system directly or indirectly via the lymphatic system. Since the majority of circulating tumor cells (CTC) are eliminated by NK cells [169], only about 0.01 % of CTC survive in the bloodstream [157]. Platelets play a key role in hematogenous metastasis and contribute to the survival of CTC in the bloodstream by both thrombin-dependent and thrombin-independent mechanisms [170]. After a passage into the bloodstream, CTC adhere to vessel walls for extravasation when they are in the vicinity of secondary metastatic organs. Circulating tumor cells take advantage of the capability of neutrophils and platelets to produce and secrete adhesion molecules, such as integrins and selectins which all aid the nearby CTC to adhere and ultimately extravasate [170, 171].
The arrest of cancer cells to specific organs seems to be primarily “mechanical” [172]. However, chemokines and chemokine receptors are also involved in organ-specific colonization, which finally drive cells along tissue-specific chemokine gradients. Furthermore, a non-chemokine pathway also exists, in which immune cells support organ-specific cancer cell dissemination. One example is represented by the two inflammatory mediators S100-A8 and S100-A9, which were shown to promote metastasis through serum amyloid A 3 (SAA-3) [173].
The subsequent growth of arrested tumor cells will depend on the molecular interactions between cancer cells and the microenvironment of the new organ. Although cancer cells are sometimes said to “home” to specific organs (e.g., breast tumors metastasizing to bone), it is more likely that this organ specificity is due to efficient organ-specific growth rather than preferential “homing” of cells to a particular organ.
It has been suggested that tumor cells can influence the microenvironment of secondary organs promoting the formation of a pre-metastatic niche [174, 175]. Tumor-derived factors and HSC are crucial components of the pre-metastatic niche. VEGF derived from tumor cells promote recruitment to the secondary organs of VEGFR1-expressing HSC that induce fibronectin and MMP9 expression by resident fibroblasts, creating favorable conditions for settlement of future metastases [176]. Other soluble factors released by tumor cells can promote the formation of pre-metastatic niche. In a murine model of breast cancer, tumor cells were found to induce production of CCL17 and CCL22 in the lung; both attracting CCR4+ tumor and immune cells which establish a microenvironment for metastases settlement at secondary organs [177]. Moreover, it was demonstrated that the prototypic hypoxia-induced protein lysyl oxidase (LOX), often found in tumors, leads to cross-linking of collagen IV in basement membranes, in addition to recruitment of CD11b+ myeloid cells which adhere to the abovementioned collagen meshwork. The captured CD11b+ myeloid cells were shown to secrete MMP2, which facilitated invasion and recruitment of metastasizing tumor cells [178].
TAMC, TAM and MDSC in particular, are important players of tumor progression and metastatic colonization through the cross talk with tumor cells. For instance, macrophages play a crucial role in conferring an invasive phenotype to epidermal keratinocytes from Snail transgenic mice [179]. TAM contribute to cancer cell dissemination by releasing enzymes involved in degradation of the ECM (i.e., MMP and cathepsin) [168, 161, 180, 76], or motility factors. Recently we found that tumor-derived soluble factors, particularly CSF-1, activate a transcription program in macrophages resulting in upregulation of a series of genes, especially migration-stimulating factor (MSF). MSF is a truncated isoform of human fibronectin 1, physiologically expressed during fetal life and upregulated in M2-like macrophages [181, 182]. MSF exerts a chemotactic effect on tumor cells, indicating that macrophage products released in the tumor microenvironment can support the pro-invasive phenotype of tumor cells [181]. An example of the cross talk between TAM and tumor cells involved in metastatic colonization is shown in breast cancer, where EGF secreted by TAM increases migration and invasion of neighboring breast cancer cells which express high levels of EGF receptor (EGFR). On the other hand, cancer cells secrete high levels of CSF-1, a main chemoattractant for TAM which expresses the cognate receptor CSF-R1. Therapies aiming at inhibiting this cross talk by blocking CSF-R1 and/or EGFR were shown to be successful [183, 184]. Macrophages and their reciprocal cross talk with tumor cells are mandatory for tumor cell migration, regardless of the factor inducing cell invasion (i.e., SDF-1).
A myeloid cell population involved in tumor progression, including invasion, is represented by MDSC. A direct role for MDSC in tumor metastasis has not been demonstrated; however, a connection was suggested by the study on mice deficient for the TGF-β receptor type 2 (TGF-β-R2), in which MDSC were concentrated on the invasive margin. In addition, it is possible to reduce lung metastases by antagonizing CXCR2 and CXCR4, two receptors involved in homing of MDSC [145]. As previously mentioned, PGE2 and the proinflammatory molecule S100A9 have been identified as main effectors of MDSC accumulation and function. Accordingly, S100A9 deficient mice rejected implantation of colorectal cancer, while administration of wild-type MDSC reverted the phenotype and colorectal cancer cells could successfully engraft [76]. In addition, TGF-β was demonstrated to be instrumental in MDSC homing, mediated via CXCL12-CXCR4 and CXCL5-CXCR2 axis in a preclinical mammary cancer model [145].
2.3.4 Suppression of Adaptive Immunity
Besides the effect on tumor growth and dissemination, TAMC have also the potential to suppress the adaptive immune response, leading to cancer immune evasion [185].
M2-like polarized tumor-infiltrating macrophages are characterized by an immunosuppressive phenotype, with production of high levels of the immunosuppressive cytokines IL-10 and TGF-β and reduced expressions of IL-12 [19, 186, 92, 187, 188]. In addition, they have reduced tumoricidal activity and are poor in antigen presentation [189]. Furthermore, TAM secret chemokines, such as CCL17 or CCL22, that preferentially attract Th1, Th2, and T regulatory (Treg) lymphocytes with defective cytotoxic functions, or such as CCL18, that recruit naïve T cells which become anergic in contact with M2 macrophages and iDC [8, 190–192].
MDSC play a prominent role in the inhibition of tumor-specific immune responses. MDSC localized within the tumor microenvironment has an M2-like phenotype and mediate immunosuppression through multiple pathways, that is, production of Arg-1 [193], iNOS [194, 195], ROI, and suppressive cytokines including IL-10 and TGF-β [196], or via the activation and recruitment of Treg [196, 197]. MDSC inhibit homing to lymph nodes of CD4+ and CD8+ T cells and suppress their activation [198, 199]. It was found that cysteine uptake by MDSC limited its availability for uptake by T cells, which in turn disables their activation and renders them nonfunctional. Furthermore, it was shown that posttranslational T-cell receptor modifications mediated via generation of peroxynitrite species led to anergy of effector CD8+ T cells [196]. MDSC can also impair innate immunity through cross talk with macrophages which led to decreased production of IL-12 by macrophages and increased production of IL-10 by MDSC, thus driving a polarization towards an M2-like phenotype [200].
In addition to the above described mechanisms in TAM and MDSC, TADC were found to be involved in suppression of adaptive immunity. One mechanism leading to the induction of tumor-specific T-cell tolerance was via upregulation of inhibitory molecules such as B7-H1 [201] or by inducing the expression of Arg-1 [202]. Moreover, it was shown that the induction of oxygen-dependent pathways led to the downregulation of CD3 epsilon and T-cell apoptosis [203]. Furthermore, Muller and coworkers demonstrated that upregulation of indoleamine 2,3-dioxygenase (IDO) in TADC contributed to immunosuppression [204].
2.4 Selected Aspects of Therapeutic Targeting of TAMC
The above summarized data describing the pro-tumoral role of the myeloid infiltrate of tumors make clear that TAMC are reasonable targets for novel therapeutic approaches. As illustrated above, TAMC can directly promote tumor cell growth releasing growth factors and proangiogenic molecules, in addition to suppression of tumor-specific immune responses. Strategies explored in the last years are focused on the stoppage of the mechanisms leading to suppression of lymphocyte activity and, on the other side, on the reduction of recruitment of myeloid cells and repolarization of M2-like pro-tumoral cells to proinflammatory M1 macrophages. There is a wide range of preclinical and clinical research aimed at eliminating or reprogramming TAMC [39]: here we only mention some examples of the results obtained so far in this growing field of anticancer research.
Many studies have shown that targeting TAM might be a successful strategy to limit tumor growth and metastasization and to achieve better therapeutic responses [32, 44, 59, 82, 189, 205, 206, 207]. One example is represented by bisphosphonates [208] traditionally used in the clinic to treat osteoporosis, which were shown to be very effective in depleting TAM and inhibiting angiogenesis as well as metastatic spread in preclinical animal models for breast cancer [209, 210]. Furthermore, Germano and coworkers recently showed that specific targeting of macrophages with the marine antitumor agent trabectedin was very successful in four different preclinical tumor animal models [211].
An alternative strategy is to target circulating monocytes known as precursors of TAM. Two candidate molecules are the M-CSF receptor (solely expressed by monocyte-macrophages) and the chemokine CCL2, involved in monocyte recruitment within tumors. Since preclinical studies on prostate and colon cancer [212–215] identified CCR2+Ly6C+ cells as targets involved in cancer progression and metastasis, CCL2 antibodies are currently investigated for therapeutic applications in human cancer treatment. Another approach to affect TAM specifically is to try to reeducate them to become tumoricidal or, in terms of polarization, to try to repolarize them towards an M1 phenotype. Several successful trials using CpG-oligodeoxynucleotide (TLR9 agonists) were performed in combination with anti-IL-10 receptor or anti-CD40 antibodies, which reverted pro-tumoral M2-like TAM to M1 macrophages displaying antitumor activity [216–218]. Rolny et al. recently demonstrated that skewing of M2 TAM towards M1 leads to effective antitumoral activity of host histidine-rich glycoprotein (HRG), which in consequence leads to inhibition of angiogenesis and promoted antitumor immune responses [219]. Gazzaniga and coworkers reported promising results using the molecule legumain, which targets M2 polarized TAM specifically, and was able to induce a robust CD8+ T-cell answer leading to reduced tumor growth and inhibition of tumor angiogenesis [220]. Furthermore, it was shown that zoledronic acid was able to revert M2 towards M1 TAM and inhibit breast carcinogenesis by targeting the mevalonate pathway [221]. Moreover, it was demonstrated that direct reeducation of TAM using the prototypical M1 polarizing cytokine INF-γ [222] is successful in promoting antitumor activity in minimal residual disease [8]. In line with the abovementioned results are the findings that inhibition of M2 polarization led to restoration of M1 proinflammatory phenotype and inhibition of tumor growth in several preclinical animal models [92, 223, 224].
To counteract the pro-tumoral activities of MDSC, two general strategies can be envisaged; the first consists of transforming these immature cells into mature cells devoid of suppressive activity, and the second is focused on blocking MDSC suppressive functions. Depletion of MDSC producing high levels of TGF-β (in an IL-13-dependent manner) led to the restoration of T-cell-mediated immunosurveillance in a preclinical mouse model for fibrosarcoma [225]. Several studies have shown that metabolites of all-trans-retinoic acid are able to differentiate MDSC into DC and macrophages, reducing MDSC accumulation [226, 227]. This effect was demonstrated to be beneficial for patients suffering from metastasizing renal cancer, since in these patient less circulating MDSC were detected in the bloodstream [228]. Furthermore, one of the beneficial effects of the anticancer drug gemcitabine is its potential to eliminate MDSC without affecting T, B, NK cells, or macrophages [229].
The second possibility to counteract MDSC function is to block their inhibitory function, for example, by using COX2 inhibitors, phosphodiesterase (PDE5), and nonsteroidal anti-inflammatory drugs releasing NO [44]. Blocking of IL-1β inhibits cancer progression and metastasis [230] and decreases MDSC accumulation and suppressive activity [42]. Moreover, the proangiogenic chemokine Bv8 was shown to be important for mobilization and homing of MDSC to tumor sites and therefore qualifies as an interesting therapeutic target [74].
Complete neutrophil depletion in already immunocompromised patients is not desirable; therefore, the strategy of choice concerning TAN might be to disturb their tumor homing ability, in other words to interfere with their ability to migrate. To this purpose, preclinical experiments using anti-CXCR2 antibodies were performed and were shown to be successful [231]. Furthermore, considering the well-documented key role of TGF-β in skewing TAN towards a N2 phenotype, this cytokine keeps promising potential for treatment [70, 31].
Some studies indicate that blocking IL-10 together with the administration of CpG oligonucleotides are able to unblock the functionally paralyzed TADCs and to reactivate antitumor responses [232]. Another strategy enhancing immunotherapy might be targeting of soluble factors like VEGF, IL-10, TGF-β, gangliosides, and others, which are all tumor secreted factors leading to abnormal differentiation of DC, often leaving them in an immature state [233]. Other and more recent strategies make use of siRNA nano-complexes which lead to reprogramming of TADC from an immunosuppressive to an activated anticancer phenotype [234]. Furthermore, it was shown that in situ stimulated CD40 and toll-like receptor 3 (TLR3) TADC were successfully transformed from immunosuppressive to immunostimulatory cells [235]. More recently it was demonstrated that delivery of regulatory miRNA, particularly miRNA 155 in a nanoparticle formulation, leads to reprogramming of immunosuppressive TADC to highly active antitumoral TADC which provoked regression of established ovarian tumors [236].
In light of the recent results, tumor therapy with drugs targeting the inflammatory tumor microenvironment in combination with treatment aimed at defeating TAM, TAN, and other myeloid cells holds promise for the future.
2.5 Concluding Remarks
In recent years, it has become clear that inflammation has an essential role in tumor promotion [1–6]. The inflammatory tumor microenvironment, mainly consisting of soluble factors and host cells, has a predominant role in all aspects of the disease (progression, angiogenesis, immune surveillance). In particular, a heterogeneous group of myeloid cells is the most consistent host cell component of solid tumor [8, 9]. TAM, TEM, MDSC, TAN, and TADC display distinct specialized functions, as well as overlapping activities (e.g. angiogenesis). Tumor and stromal cells release different chemoattractants involved in the recruitment of myeloid cells from the blood into the growing tumor. Cytokines and other soluble factors released in the tumor microenvironment can contribute to induce a protumoral phenotype, promoting M2 polarization of TAM [92], N2 polarization of TAN [31], MDSC expansion [41], or preventing maturation of DCs. Thus the different TAMC populations potentially represent a target for new therapeutic approaches aimed at breaking the protumoral networks established by cancer-associated myeloid cells.
References
Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339(6117):286–91.
Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40.
DiDonato JA, Mercurio F, Karin M. NF-kappaB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400.
Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073–81.
Mantovani A. Cancer: inflaming metastasis. Nature. 2009;457(7225):36–7.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–7.
Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117(5):1155–66.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Laghi L, Bianchi P, Miranda E, Balladore E, Pacetti V, Grizzi F, et al. CD3+ cells at the invasive margin of deeply invading (pT3-T4) colorectal cancer and risk of post-surgical metastasis: a longitudinal study. Lancet Oncol. 2009;10(9):877–84.
Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–66.
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95.
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–27.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8.
Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med. 1996;183(4):1323–9.
Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55.
Goerdt S, Politz O, Schledzewski K, Birk R, Gratchev A, Guillot P, et al. Alternative versus classical activation of macrophages. Pathobiology. 1999;67(5–6):222–6.
Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–83.
Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604.
Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23(4):344–6.
Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–85.
Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–92.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86.
Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–44.
Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33(5):949–55.
Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. 2011;11(8):519–31.
Sica A, Melillo G, Varesio L. Hypoxia: a double-edged sword of immunity. J Mol Med (Berl). 2011;89(7):657–65.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94.
De Palma M, Murdoch C, Venneri MA, Naldini L, Lewis CE. Tie2-expressing monocytes: regulation of tumor angiogenesis and therapeutic implications. Trends Immunol. 2007;28(12):519–24.
De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8(3):211–26.
Murdoch C, Tazzyman S, Webster S, Lewis CE. Expression of Tie-2 by human monocytes and their responses to angiopoietin-2. J Immunol. 2007;178(11):7405–11.
Venneri MA, De Palma M, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood. 2007;109(12):5276–85.
De Palma M, Mazzieri R, Politi LS, Pucci F, Zonari E, Sitia G, et al. Tumor-targeted interferon-alpha delivery by Tie2-expressing monocytes inhibits tumor growth and metastasis. Cancer Cell. 2008;14(4):299–311.
Coffelt SB, Tal AO, Scholz A, De Palma M, Patel S, Urbich C, et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010;70(13):5270–80.
Pucci F, Venneri MA, Biziato D, Nonis A, Moi D, Sica A, et al. A distinguishing gene signature shared by tumor-infiltrating Tie2-expressing monocytes, blood “resident” monocytes, and embryonic macrophages suggests common functions and developmental relationships. Blood. 2009;114(4):901–14.
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–68.
Nagaraj S, Gabrilovich DI. Regulation of suppressive function of myeloid-derived suppressor cells by CD4+ T cells. Semin Cancer Biol. 2012;22(4):282–8.
Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011;32(1):19–25.
Ostrand-Rosenberg S, Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182(8):4499–506.
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. 2008;181(8):5791–802.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74.
Ochando JC, Chen SH. Myeloid-derived suppressor cells in transplantation and cancer. Immunol Res. 2012;54(1–3):275–85.
Umemura N, Saio M, Suwa T, Kitoh Y, Bai J, Nonaka K, et al. Tumor-infiltrating myeloid-derived suppressor cells are pleiotropic-inflamed monocytes/macrophages that bear M1- and M2-type characteristics. J Leukoc Biol. 2008;83(5):1136–44.
Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S. Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother. 2012;61(8):1155–67.
Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11(7):802–7.
Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kubler H, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2008;14(24):8270–8.
Diaz-Montero CM, Salem ML, Nishimura MI, Garrett-Mayer E, Cole DJ, Montero AJ. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol Immunother. 2009;58(1):49–59.
Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood. 2011;118(8):2254–65.
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. 2009;182(9):5693–701.
Bell D, Chomarat P, Broyles D, Netto G, Harb GM, Lebecque S, et al. In breast carcinoma tissue, immature dendritic cells reside within the tumor, whereas mature dendritic cells are located in peritumoral areas. J Exp Med. 1999;190(10):1417–26.
Scarpino S, Stoppacciaro A, Ballerini F, Marchesi M, Prat M, Stella MC, et al. Papillary carcinoma of the thyroid: hepatocyte growth factor (HGF) stimulates tumor cells to release chemokines active in recruiting dendritic cells. Am J Pathol. 2000;156(3):831–7.
Troy AJ, Summers KL, Davidson PJ, Atkinson CH, Hart DN. Minimal recruitment and activation of dendritic cells within renal cell carcinoma. Clin Cancer Res. 1998;4(3):585–93.
Fricke I, Gabrilovich DI. Dendritic cells and tumor microenvironment: a dangerous liaison. Immunol Invest. 2006;35(3–4):459–83.
Dhodapkar MV, Dhodapkar KM, Palucka AK. Interactions of tumor cells with dendritic cells: balancing immunity and tolerance. Cell Death Differ. 2008;15(1):39–50.
Kusmartsev S, Gabrilovich DI. Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol Immunother. 2006;55(3):237–45.
Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunol Today. 1992;13(7):265–70.
Zachariae CO, Anderson AO, Thompson HL, Appella E, Mantovani A, Oppenheim JJ, et al. Properties of monocyte chemotactic and activating factor (MCAF) purified from a human fibrosarcoma cell line. J Exp Med. 1990;171(6):2177–82.
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4(7):540–50.
Allavena P, Sica A, Vecchi A, Locati M, Sozzani S, Mantovani A. The chemokine receptor switch paradigm and dendritic cell migration: its significance in tumor tissues. Immunol Rev. 2000;177:141–9.
Vicari AP, Caux C. Chemokines in cancer. Cytokine Growth Factor Rev. 2002;13(2):143–54.
Zhang J, Sud S, Mizutani K, Gyetko MR, Pienta KJ. Activation of urokinase plasminogen activator and its receptor axis is essential for macrophage infiltration in a prostate cancer mouse model. Neoplasia. 2011;13(1):23–30.
Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–70.
Lin EY, Gouon-Evans V, Nguyen AV, Pollard JW. The macrophage growth factor CSF-1 in mammary gland development and tumor progression. J Mammary Gland Biol Neoplasia. 2002;7(2):147–62.
Jin G, Kawsar HI, Hirsch SA, Zeng C, Jia X, Feng Z, et al. An antimicrobial peptide regulates tumor-associated macrophage trafficking via the chemokine receptor CCR2, a model for tumorigenesis. PLoS One. 2010;5(6):e10993.
Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol. 1998;152(1):83–92.
Kobayashi Y. The role of chemokines in neutrophil biology. Front Biosci. 2008;13:2400–7.
Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10(8):554–67.
Sawanobori Y, Ueha S, Kurachi M, Shimaoka T, Talmadge JE, Abe J, et al. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111(12):5457–66.
Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, et al. Modulation of the antitumor immune response by complement. Nat Immunol. 2008;9(11):1225–35.
Pan PY, Wang GX, Yin B, Ozao J, Ku T, Divino CM, et al. Reversion of immune tolerance in advanced malignancy: modulation of myeloid-derived suppressor cell development by blockade of stem-cell factor function. Blood. 2008;111(1):219–28.
Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature. 2007;450(7171):825–31.
LeCouter J, Zlot C, Tejada M, Peale F, Ferrara N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc Natl Acad Sci U S A. 2004;101(48):16813–8.
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–49.
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181(7):4666–75.
Nowak G, Karrar A, Holmen C, Nava S, Uzunel M, Hultenby K, et al. Expression of vascular endothelial growth factor receptor-2 or Tie-2 on peripheral blood cells defines functionally competent cell populations capable of reendothelialization. Circulation. 2004;110(24):3699–707.
Stratmann A, Risau W, Plate KH. Cell type-specific expression of angiopoietin-1 and angiopoietin-2 suggests a role in glioblastoma angiogenesis. Am J Pathol. 1998;153(5):1459–66.
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–31.
Coffelt SB, Chen YY, Muthana M, Welford AF, Tal AO, Scholz A, et al. Angiopoietin 2 stimulates TIE2-expressing monocytes to suppress T cell activation and to promote regulatory T cell expansion. J Immunol. 2011;186(7):4183–90.
Welford AF, Biziato D, Coffelt SB, Nucera S, Fisher M, Pucci F, et al. TIE2-expressing macrophages limit the therapeutic efficacy of the vascular-disrupting agent combretastatin A4 phosphate in mice. J Clin Invest. 2011;121(5):1969–73.
Gabrilovich DI, Chen HL, Girgis KR, Cunningham HT, Meny GM, Nadaf S, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103.
Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ, et al. Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A. Nat Med. 2004;10(9):950–8.
Curiel TJ, Cheng P, Mottram P, Alvarez X, Moons L, Evdemon-Hogan M, et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004;64(16):5535–8.
Okunishi K, Dohi M, Nakagome K, Tanaka R, Mizuno S, Matsumoto K, et al. A novel role of hepatocyte growth factor as an immune regulator through suppressing dendritic cell function. J Immunol. 2005;175(7):4745–53.
Viola A, Sarukhan A, Bronte V, Molon B. The pros and cons of chemokines in tumor immunology. Trends Immunol. 2012;33(10):496–504.
Bottazzi B, Erba E, Nobili N, Fazioli F, Rambaldi A, Mantovani A. A paracrine circuit in the regulation of the proliferation of macrophages infiltrating murine sarcomas. J Immunol. 1990;144(6):2409–12.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–6.
Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332(6035):1284–8.
Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66(2):605–12.
Biswas SK, Gangi L, Paul S, Schioppa T, Saccani A, Sironi M, et al. A distinct and unique transcriptional program expressed by tumor-associated macrophages (defective NF-kappaB and enhanced IRF-3/STAT1 activation). Blood. 2006;107(5):2112–22.
Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, et al. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006;176(8):5023–32.
Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b + peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 2009;284(49):34342–54.
Narita Y, Wakita D, Ohkur T, Chamoto K, Nishimura T. Potential differentiation of tumor bearing mouse CD11b + Gr-1+ immature myeloid cells into both suppressor macrophages and immunostimulatory dendritic cells. Biomed Res. 2009;30(1):7–15.
Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32(6):790–802.
Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–65.
Yang P, Bamlet WR, Sun Z, Ebbert JO, Aubry MC, Krowka MJ, et al. Alpha1-antitrypsin and neutrophil elastase imbalance and lung cancer risk. Chest. 2005;128(1):445–52.
Jensen HK, Donskov F, Marcussen N, Nordsmark M, Lundbeck F, von der Maase H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J Clin Oncol. 2009;27(28):4709–17.
Trellakis S, Farjah H, Bruderek K, Dumitru CA, Hoffmann TK, Lang S, et al. Peripheral blood neutrophil granulocytes from patients with head and neck squamous cell carcinoma functionally differ from their counterparts in healthy donors. Int J Immunopathol Pharmacol. 2011;24(3):683–93.
Wislez M, Rabbe N, Marchal J, Milleron B, Crestani B, Mayaud C, et al. Hepatocyte growth factor production by neutrophils infiltrating bronchioloalveolar subtype pulmonary adenocarcinoma: role in tumor progression and death. Cancer Res. 2003;63(6):1405–12.
Erreni M, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) and Inflammation in colorectal cancer. Cancer Microenviron. 2011;4(2):141–54.
Forssell J, Oberg A, Henriksson ML, Stenling R, Jung A, Palmqvist R. High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res. 2007;13(5):1472–9.
Ohno S, Inagawa H, Dhar DK, Fujii T, Ueda S, Tachibana M, et al. The degree of macrophage infiltration into the cancer cell nest is a significant predictor of survival in gastric cancer patients. Anticancer Res. 2003;23(6D):5015–22.
Sconocchia G, Zlobec I, Lugli A, Calabrese D, Iezzi G, Karamitopoulou E, et al. Tumor infiltration by FcgammaRIII (CD16) + myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer. 2011;128(11):2663–72.
Caruso RA, Bellocco R, Pagano M, Bertoli G, Rigoli L, Inferrera C. Prognostic value of intratumoral neutrophils in advanced gastric carcinoma in a high-risk area in northern Italy. Mod Pathol. 2002;15(8):831–7.
Reid MD, Basturk O, Thirabanjasak D, Hruban RH, Klimstra DS, Bagci P, et al. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod Pathol. 2011;24(12):1612–9.
Ohtaki Y, Ishii G, Nagai K, Ashimine S, Kuwata T, Hishida T, et al. Stromal macrophage expressing CD204 is associated with tumor aggressiveness in lung adenocarcinoma. J Thorac Oncol. 2010;5(10):1507–15.
Ohtani H, Naito Y, Saito K, Nagura H. Expression of costimulatory molecules B7-1 and B7-2 by macrophages along invasive margin of colon cancer: a possible antitumor immunity? Lab Invest. 1997;77(3):231–41.
Sugita J, Ohtani H, Mizoi T, Saito K, Shiiba K, Sasaki I, et al. Close association between Fas ligand (FasL; CD95L)-positive tumor-associated macrophages and apoptotic cancer cells along invasive margin of colorectal carcinoma: a proposal on tumor-host interactions. Jpn J Cancer Res Gann. 2002;93(3):320–8.
Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev Dyn. 2006;235(12):3222–9.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401.
Moussai D, Mitsui H, Pettersen JS, Pierson KC, Shah KR, Suarez-Farinas M, et al. The human cutaneous squamous cell carcinoma microenvironment is characterized by increased lymphatic density and enhanced expression of macrophage-derived VEGF-C. J Invest Dermatol. 2011;131(1):229–36.
Aspord C, Pedroza-Gonzalez A, Gallegos M, Tindle S, Burton EC, Su D, et al. Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. J Exp Med. 2007;204(5):1037–47.
Bollrath J, Greten FR. IKK/NF-kappaB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. EMBO Rep. 2009;10(12):1314–9.
Fukuda A, Wang SC, Morris JP, Folias AE, Liou A, Kim GE, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer cell. 2011;19(4):441–55.
Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–13.
Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19(4):456–69.
Ribatti D, Vacca A. The role of monocytes-macrophages in vasculogenesis in multiple myeloma. Leukemia. 2009;23(9):1535–6.
Pekarek LA, Starr BA, Toledano AY, Schreiber H. Inhibition of tumor growth by elimination of granulocytes. J Exp Med. 1995;181(1):435–40.
Houghton AM. The paradox of tumor-associated neutrophils: fueling tumor growth with cytotoxic substances. Cell Cycle. 2010;9(9):1732–7.
Gungor N, Knaapen AM, Munnia A, Peluso M, Haenen GR, Chiu RK, et al. Genotoxic effects of neutrophils and hypochlorous acid. Mutagenesis. 2010;25(2):149–54.
Cassatella MA, Locati M, Mantovani A. Never underestimate the power of a neutrophil. Immunity. 2009;31(5):698–700.
Mantovani A. The yin-yang of tumor-associated neutrophils. Cancer Cell. 2009;16(3):173–4.
Piccard H, Muschel RJ, Opdenakker G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit Rev Oncol Hematol. 2012;82(3):296–309.
Du R, Lu KV, Petritsch C, Liu P, Ganss R, Passegue E, et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell. 2008;13(3):206–20.
Lin EY, Li JF, Gnatovskiy L, Deng Y, Zhu L, Grzesik DA, et al. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006;66(23):11238–46.
Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Jung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124(1):175–89.
Knowles H, Leek R, Harris AL. Macrophage infiltration and angiogenesis in human malignancy. Novartis Found Symp. 2004;256:189–200; discussion -4, 59–69.
Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56(20):4625–9.
Onita T, Ji PG, Xuan JW, Sakai H, Kanetake H, Maxwell PH, et al. Hypoxia-induced, perinecrotic expression of endothelial Per-ARNT-Sim domain protein-1/hypoxia-inducible factor-2alpha correlates with tumor progression, vascularization, and focal macrophage infiltration in bladder cancer. Clin Cancer Res. 2002;8(2):471–80.
Takanami I, Takeuchi K, Kodaira S. Tumor-associated macrophage infiltration in pulmonary adenocarcinoma: association with angiogenesis and poor prognosis. Oncology. 1999;57(2):138–42.
Valkovic T, Dobrila F, Melato M, Sasso F, Rizzardi C, Jonjic N. Correlation between vascular endothelial growth factor, angiogenesis, and tumor-associated macrophages in invasive ductal breast carcinoma. Virchows Arch. 2002;440(6):583–8.
Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67(11):5064–6.
Kimura YN, Watari K, Fotovati A, Hosoi F, Yasumoto K, Izumi H, et al. Inflammatory stimuli from macrophages and cancer cells synergistically promote tumor growth and angiogenesis. Cancer Sci. 2007;98(12):2009–18.
Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. J Leukoc Biol. 2006;80(6):1183–96.
Murdoch C, Muthana M, Lewis CE. Hypoxia regulates macrophage functions in inflammation. J Immunol. 2005;175(10):6257–63.
Elbarghati L, Murdoch C, Lewis CE. Effects of hypoxia on transcription factor expression in human monocytes and macrophages. Immunobiology. 2008;213(9–10):899–908.
White JR, Harris RA, Lee SR, Craigon MH, Binley K, Price T, et al. Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics. 2004;83(1):1–8.
Burke B, Giannoudis A, Corke KP, Gill D, Wells M, Ziegler-Heitbrock L, et al. Hypoxia-induced gene expression in human macrophages: implications for ischemic tissues and hypoxia-regulated gene therapy. Am J Pathol. 2003;163(4):1233–43.
Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000;192(2):150–8.
Fernandez Pujol B, Lucibello FC, Gehling UM, Lindemann K, Weidner N, Zuzarte ML, et al. Endothelial-like cells derived from human CD14 positive monocytes. Differ Res Biol Divers. 2000;65(5):287–300.
Kuwana M, Okazaki Y, Kodama H, Satoh T, Kawakami Y, Ikeda Y. Endothelial differentiation potential of human monocyte-derived multipotential cells. Stem Cells. 2006;24(12):2733–43.
Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr + CD11b + cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell. 2004;6(4):409–21.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1 + CD11b + myeloid cells that promote metastasis. Cancer Cell. 2008;13(1):23–35.
McCourt M, Wang JH, Sookhai S, Redmond HP. Proinflammatory mediators stimulate neutrophil-directed angiogenesis. Arch Surg. 1999;134(12):1325–31; discussion 31–2.
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–44.
Mentzel T, Brown LF, Dvorak HF, Kuhnen C, Stiller KJ, Katenkamp D, et al. The association between tumour progression and vascularity in myxofibrosarcoma and myxoid/round cell liposarcoma. Virchows Archiv. 2001;438(1):13–22.
Van Coillie E, Van Aelst I, Wuyts A, Vercauteren R, Devos R, De Wolf-Peeters C, et al. Tumor angiogenesis induced by granulocyte chemotactic protein-2 as a countercurrent principle. Am J Pathol. 2001;159(4):1405–14.
Nozawa H, Chiu C, Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A. 2006;103(33):12493–8.
Feijoo E, Alfaro C, Mazzolini G, Serra P, Penuelas I, Arina A, et al. Dendritic cells delivered inside human carcinomas are sequestered by interleukin-8. Int J Cancer. 2005;116(2):275–81.
Naldini A, Leali D, Pucci A, Morena E, Carraro F, Nico B, et al. Cutting edge: IL-1beta mediates the proangiogenic activity of osteopontin-activated human monocytes. J Immunol. 2006;177(7):4267–70.
Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood. 1998;92(11):4150–66.
Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–8.
Talmadge JE, Fidler IJ. AACR centennial series: the biology of cancer metastasis: historical perspective. Cancer Res. 2010;70(14):5649–69.
Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–84.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74.
Lynch CC, Matrisian LM. Matrix metalloproteinases in tumor-host cell communication. Differentiation. 2002;70(9–10):561–73.
Mohamed MM, Sloane BF. Cysteine cathepsins: multifunctional enzymes in cancer. Nat Rev Cancer. 2006;6(10):764–75.
Gocheva V, Joyce JA. Cysteine cathepsins and the cutting edge of cancer invasion. Cell Cycle. 2007;6(1):60–4.
Laufs S, Schumacher J, Allgayer H. Urokinase-receptor (u-PAR): an essential player in multiple games of cancer: a review on its role in tumor progression, invasion, metastasis, proliferation/dormancy, clinical outcome and minimal residual disease. Cell Cycle. 2006;5(16):1760–71.
Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–33.
Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14(2):163–76.
McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM. Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science. 2000;289(5482):1202–6.
Boulay A, Masson R, Chenard MP, El Fahime M, Cassard L, Bellocq JP, et al. High cancer cell death in syngeneic tumors developed in host mice deficient for the stromelysin-3 matrix metalloproteinase. Cancer Res. 2001;61(5):2189–93.
Kataoka H, Uchino H, Iwamura T, Seiki M, Nabeshima K, Koono M. Enhanced tumor growth and invasiveness in vivo by a carboxyl-terminal fragment of alpha1-proteinase inhibitor generated by matrix metalloproteinases: a possible modulatory role in natural killer cytotoxicity. Am J Pathol. 1999;154(2):457–68.
Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21(4):228–37.
Gassmann P, Haier J. The tumor cell-host organ interface in the early onset of metastatic organ colonisation. Clin Exp Metastasis. 2008;25(2):171–81.
Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104(2):397–401.
McDonald B, Spicer J, Giannais B, Fallavollita L, Brodt P, Ferri LE. Systemic inflammation increases cancer cell adhesion to hepatic sinusoids by neutrophil mediated mechanisms. Int J Cancer. 2009;125(6):1298–305.
Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2(8):563–72.
Hiratsuka S, Watanabe A, Sakurai Y, Akashi-Takamura S, Ishibashi S, Miyake K, et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat Cell Biol. 2008;10(11):1349–55.
Kaplan RN, Psaila B, Lyden D. Bone marrow cells in the ‘pre-metastatic niche’: within bone and beyond. Cancer Metastasis Rev. 2006;25(4):521–9.
Sceneay J, Smyth MJ, Moller A. The pre-metastatic niche: finding common ground. Cancer Metastasis Rev. 2013;32(3–4):449–64.
Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438(7069):820–7.
Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69(14):5996–6004.
Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44.
Du F, Nakamura Y, Tan TL, Lee P, Lee R, Yu B, et al. Expression of snail in epidermal keratinocytes promotes cutaneous inflammation and hyperplasia conducive to tumor formation. Cancer Res. 2010;70(24):10080–9.
Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241–55.
Solinas G, Schiarea S, Liguori M, Fabbri M, Pesce S, Zammataro L, et al. Tumor-conditioned macrophages secrete migration-stimulating factor: a new marker for m2-polarization, influencing tumor cell motility. J Immunol. 2010;185(1):642–52.
Schor SL, Ellis IR, Jones SJ, Baillie R, Seneviratne K, Clausen J, et al. Migration-stimulating factor: a genetically truncated onco-fetal fibronectin isoform expressed by carcinoma and tumor-associated stromal cells. Cancer Res. 2003;63(24):8827–36.
Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64(19):7022–9.
Goswami S, Sahai E, Wyckoff JB, Cammer M, Cox D, Pixley FJ, et al. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005;65(12):5278–83.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60.
Biswas SK, Sica A, Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008;180(4):2011–7.
Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205(6):1261–8.
Sica A, Saccani A, Bottazzi B, Polentarutti N, Vecchi A, van Damme J, et al. Autocrine production of IL-10 mediates defective IL-12 production and NF-kappa B activation in tumor-associated macrophages. J Immunol. 2000;164(2):762–7.
Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–79.
Schutyser E, Struyf S, Proost P, Opdenakker G, Laureys G, Verhasselt B, et al. Identification of biologically active chemokine isoforms from ascitic fluid and elevated levels of CCL18/pulmonary and activation-regulated chemokine in ovarian carcinoma. J Biol Chem. 2002;277(27):24584–93.
Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711.
Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306.
Ochoa AC, Zea AH, Hernandez C, Rodriguez PC. Arginase, prostaglandins, and myeloid-derived suppressor cells in renal cell carcinoma. Clin Cancer Res. 2007;13(2 Pt 2):721s–6.
Bronte V, Kasic T, Gri G, Gallana K, Borsellino G, Marigo I, et al. Boosting antitumor responses of T lymphocytes infiltrating human prostate cancers. J Exp Med. 2005;201(8):1257–68.
Capuano G, Rigamonti N, Grioni M, Freschi M, Bellone M. Modulators of arginine metabolism support cancer immunosurveillance. BMC Immunol. 2009;10:1.
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, et al. Gr-1 + CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–31.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Kruger C, Manns MP, et al. A new population of myeloid-derived suppressor cells in hepatocellular carcinoma patients induces CD4(+)CD25(+)Foxp3(+) T cells. Gastroenterology. 2008;135(1):234–43.
Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111(8):4233–44.
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13(7):828–35.
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–83.
Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–7.
Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J Immunol. 2009;182(10):6207–16.
Kuang DM, Zhao Q, Xu J, Yun JP, Wu C, Zheng L. Tumor-educated tolerogenic dendritic cells induce CD3epsilon down-regulation and apoptosis of T cells through oxygen-dependent pathways. J Immunol. 2008;181(5):3089–98.
Muller AJ, Sharma MD, Chandler PR, Duhadaway JB, Everhart ME, Johnson 3rd BA, et al. Chronic inflammation that facilitates tumor progression creates local immune suppression by inducing indoleamine 2,3 dioxygenase. Proc Natl Acad Sci U S A. 2008;105(44):17073–8.
Aharinejad S, Sioud M, Lucas T, Abraham D. Targeting stromal-cancer cell interactions with siRNAs. Methods Mol Biol. 2009;487:243–66.
Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–40.
Ferrara N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr Opin Hematol. 2010;17(3):219–24.
Morgan G, Lipton A. Antitumor effects and anticancer applications of bisphosphonates. Semin Oncol. 2010;37 Suppl 2:S30–40.
Brown HK, Holen I. Anti-tumour effects of bisphosphonates–what have we learned from in vivo models? Curr Cancer Drug Targets. 2009;9(7):807–23.
Zeisberger SM, Odermatt B, Marty C, Zehnder-Fjallman AH, Ballmer-Hofer K, Schwendener RA. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. Br J Cancer. 2006;95(3):272–81.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62.
Li X, Loberg R, Liao J, Ying C, Snyder LA, Pienta KJ, et al. A destructive cascade mediated by CCL2 facilitates prostate cancer growth in bone. Cancer Res. 2009;69(4):1685–92.
Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67(19):9417–24.
Popivanova BK, Kostadinova FI, Furuichi K, Shamekh MM, Kondo T, Wada T, et al. Blockade of a chemokine, CCL2, reduces chronic colitis-associated carcinogenesis in mice. Cancer Res. 2009;69(19):7884–92.
Hoos A, Wolf MJ, Bauer J, Borsig L, Heikenwalder M. Endothelial chemokine receptors as facilitators of tumor cell extravasation? Oncotarget. 2012;3(9):919–20.
Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65(8):3437–46.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6.
Buhtoiarov IN, Sondel PM, Wigginton JM, Buhtoiarova TN, Yanke EM, Mahvi DA, et al. Anti-tumour synergy of cytotoxic chemotherapy and anti-CD40 plus CpG-ODN immunotherapy through repolarization of tumour-associated macrophages. Immunology. 2011;132(2):226–39.
Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44.
Gazzaniga S, Bravo AI, Guglielmotti A, van Rooijen N, Maschi F, Vecchi A, et al. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J Invest Dermatol. 2007;127(8):2031–41.
Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. 2010;14(12):2803–15.
Duluc D, Corvaisier M, Blanchard S, Catala L, Descamps P, Gamelin E, et al. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. 2009;125(2):367–73.
Rauh MJ, Ho V, Pereira C, Sham A, Sly LM, Lam V, et al. SHIP represses the generation of alternatively activated macrophages. Immunity. 2005;23(4):361–74.
Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A. 2009;106(35):14978–83.
Terabe M, Matsui S, Park JM, Mamura M, Noben-Trauth N, Donaldson DD, et al. Transforming growth factor-beta production and myeloid cells are an effector mechanism through which CD1d-restricted T cells block cytotoxic T lymphocyte-mediated tumor immunosurveillance: abrogation prevents tumor recurrence. J Exp Med. 2003;198(11):1741–52.
Gabrilovich DI, Velders MP, Sotomayor EM, Kast WM. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166(9):5398–406.
Kusmartsev S, Cheng F, Yu B, Nefedova Y, Sotomayor E, Lush R, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63(15):4441–9.
Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006;66(18):9299–307.
Ko HJ, Kim YJ, Kim YS, Chang WS, Ko SY, Chang SY, et al. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 2007;67(15):7477–86.
Dinarello CA. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010;29(2):317–29.
Gregory AD, Houghton AM. Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 2011;71(7):2411–6.
Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, et al. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196(4):541–9.
Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4(12):941–52.
Cubillos-Ruiz JR, Engle X, Scarlett UK, Martinez D, Barber A, Elgueta R, et al. Polyethylenimine-based siRNA nanocomplexes reprogram tumor-associated dendritic cells via TLR5 to elicit therapeutic antitumor immunity. J Clin Invest. 2009;119(8):2231–44.
Scarlett UK, Cubillos-Ruiz JR, Nesbeth YC, Martinez DG, Engle X, Gewirtz AT, et al. In situ stimulation of CD40 and Toll-like receptor 3 transforms ovarian cancer-infiltrating dendritic cells from immunosuppressive to immunostimulatory cells. Cancer Res. 2009;69(18):7329–37.
Cubillos-Ruiz JR, Baird JR, Tesone AJ, Rutkowski MR, Scarlett UK, Camposeco-Jacobs AL, et al. Reprogramming tumor-associated dendritic cells in vivo using miRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res. 2012;72(7):1683–93.
Acknowledgments
The authors would like to gratefully acknowledge the financial supports of the European Research Council (ERC project HIIS), the European Commission (FP7-HEALTH-2011-ADITEC-280873), the Italian Association for Cancer Research, the Italian Ministry of Health and University, Fondazione CARIPLO (project 2009–2582), and Regione Lombardia (project Metadistretti – SEPSIS).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Brennecke, P., Allavena, P., Laface, I., Mantovani, A., Bottazzi, B. (2015). Inflammatory and Innate Immune Cells in Cancer Microenvironment and Progression. In: Rezaei, N. (eds) Cancer Immunology. Springer, Berlin, Heidelberg. https://doi.org/10.1007/978-3-662-44006-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-662-44006-3_2
Published:
Publisher Name: Springer, Berlin, Heidelberg
Print ISBN: 978-3-662-44005-6
Online ISBN: 978-3-662-44006-3
eBook Packages: MedicineMedicine (R0)