
Abstract
Overall lifespan and health span are dependent on maintaining neuromuscular function. Degeneration in neural and muscular etiological factors with advancing age can result in sarcopenia, dynapenia, and physical frailty. The relative contribution of these neuromuscular mechanisms to clinically meaningful skeletal muscle function with aging is poorly understood. Here, we posit that optimal neuromuscular function, which is critical for healthy aging, includes domains of motor function, contractile quality, muscle mass, and muscle metabolism. Thus, it is essential that research efforts identify mechanisms to attenuate neuromuscular dysfunction and skeletal muscle weakness so effective therapeutics can be provided in the clinical setting. This chapter highlights five major components of neuromuscular function that are potential targets for regenerative rehabilitation in relation to sarcopenia, dynapenia, and physical frailty. These are (1) neuromuscular excitation, (2) excitation–contraction (EC) coupling, (3) mitochondrial function, (4) protein homeostasis and (5) glucose metabolism.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
5.1 Introduction
5.1.1 The Aging Neuromuscular System
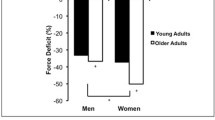
The neuromuscular system, consisting of the nervous and skeletal muscle tissues, experiences many deficits with advancing age. These deficits contribute to skeletal muscle weakness that manifests in pervasive chronic conditions and disability observed in many older adults (defined as 65 years and older by the National Institutes of Health) (Guidelines for the Review of Inclusion on the Basis of Sex/Gender, Race, Ethnicity, and Age in Clinical Research 2019). For instance, around 30% of women and 15% of men in the USA over 60 years self-report that they are unable to lift or carry 10 pounds (Louie and Ward 2010). Furthermore, over 40% of seniors have limitations in performing one or more daily tasks (e.g., walking two to three blocks, transferring from sitting to standing) that are essential for maintaining physical independence (Seeman et al. 2010). Scientific and medical communities agree that weakness (frequently referred to as dynapenia) is a major determinant of physical limitations and general poor health in older adults (Rantanen 2003; Rantanen et al. 1998, 1999, 2002; Visser et al. 2005; Newman et al. 2006; Manini et al. 2007; McGrath et al. 2018, 2019a, b). The neuromuscular mechanisms of dynapenia are known to be multi-factorial. One well-known contributor to dynapenia is the age-related loss of muscle mass (collectively referred to as sarcopeniaFootnote 1) (Clark and Manini 2008). Muscle wasting is not, however, the sole determinant of age-related weakness. In fact, when changes in quadriceps muscle size and leg extensor strength were assessed over a 5-year period (n = 1678, 70–79 years at baseline), it was observed that decreases in strength were two–five times greater than loss of muscle size in those who lost or maintained weight (Delmonico et al. 2009). Further, individuals that gained weight (n = 333) actually exhibited a small increase in muscle size, yet were not spared from dynapenia. These findings indicate that age-related loss of strength is not only attributed to muscle atrophy, but also complex neurologic and other skeletal muscle factors (Enoka et al. 2003; Tieland et al. 2018; Russ et al. 2012; Clark and Manini 2008; Narici and Maffulli 2010).
With continued and progressive loss of neuromuscular function, the risk of developing physical frailty increases, with physical frailty defined as “a medical syndrome with multiple causes and contributors that is characterized by diminished strength, endurance, and reduced physiologic function that increases an individual’s vulnerability for developing increased dependency and/or death” (Morley et al. 2013). Physical frailty is often considered to be on a continuum, in that frailty status can transition between non-frail, pre-frail (or intermediate) and frail, with status corresponding to mortality risk (Fried et al. 2001; Baumann et al. 2020). In a recent systematic review and meta-analysis, these transition states predicted mortality, with hazard ratios of 1.335 (95% CI: 1.260–1.414) and 2.000 (95% CI: 1.727–2.316) for pre-frail and frail individuals when compared to those that were non-frail, respectively (Chang and Lin 2015). Similar findings have also been observed in the C57BL/6 mouse (Baumann et al. 2018; Kwak et al. 2020; Baumann et al. 2020), a common strain used by laboratories to study aging (Mitchell et al. 2015). In both humans (Fried et al. 2001) and rodents (Liu et al. 2014; Baumann et al. 2020), skeletal muscle weakness and loss of function are salient components in the development of physical frailty. Therefore, if sarcopenia and dynapenia are left untreated, a vicious cycle of weakness and neuromuscular dysfunction will continue, leading to physical frailty, loss of dependence and ultimately, an early death.
5.1.2 Regenerative Rehabilitation in the Aging Neuromuscular System
Maintaining neuromuscular function is an important component to overall lifespan and health span. Sarcopenia, dynapenia and resultant physical frailty may result from primary neural or muscular etiological factors or a combination thereof (Fig. 5.1). The relative contribution of these neuromuscular mechanisms to clinically meaningful skeletal muscle deficits in advancing age is poorly understood. Moreover, muscle mass has broad health benefits that extend beyond just force generation capacity and locomotion, such as the critical role it plays in metabolism, specifically glucose regulation. Optimal neuromuscular function in the domains of motor function, contractile quality, muscle mass, and muscle metabolism are therefore critical to healthy aging. Thus, identifying and validating therapeutic approaches that focus on attenuating or delaying neuromuscular dysfunction and subsequently dynapenia are essential goals in laboratory and clinical settings. This chapter highlights five major components of neuromuscular function that are potential targets for regenerative rehabilitation in sarcopenia, dynapenia, and physical frailty. These are (1) neuromuscular excitation, (2) excitation-contraction (EC) coupling, (3) mitochondrial function, (4) protein homeostasis, and (5) glucose metabolism. Each component or section within this chapter is further divided into the following subsections: Introduction, Age-induced Pathophysiology, and Regenerative Rehabilitation.

Domains that contribute to neuromuscular deficits with age. Loss of neuromuscular excitation, EC coupling failure, sarcopenia (via loss of protein homeostasis) and metabolic dysfunction (via mitochondrial dysfunction and impaired glucose metabolism) contribute to physical frailty, as measured by dynapenia and muscle dysfunction
5.2 Neuromuscular Excitation
5.2.1 Introduction to Neuromuscular Excitation
There are two important constructs to muscular strength, the ability to generate sufficient force and the ability to generate force rapidly. The capacity of the central nervous system to excite motor units (i.e., muscle fibers innervated by a motor neuron) is critical to both. It is also important to recognize that the ability to selectively engage muscles in a coordinated, context sensitive manner is critical to motor control of skeletal muscle. For instance, if a perturbation occurs during gait, the adaptive response that is observed extends across the whole body, involving coordinated lower limb muscle activity, but also goal-directed engagement of torso and upper limb muscles (Marigold and Misiaszek 2009). The pattern of muscle activation under such circumstances is often described with the term “synergy.” Synergy is defined as mechanisms used by the central nervous system to coordinate groups of motor units into functional assemblies (Windhort et al. 1991). While it is perhaps obvious that measures of muscle strength depend upon the ability to recruit motor units, it is less widely appreciated that all voluntary contractions and movements reflect the organization of muscle synergies. Thus, strength is the accumulation of both skeletal muscle and the motor system (Enoka 1988). Voluntary engagement occurs through activity in brain networks (e.g., in the primary motor cortex), which results in elevated firing of (descending) corticospinal neurons and the consequential recruitment of spinal motor neurons and muscle fibers. As descending neural drive increases, a greater number of spinal motor neurons are recruited, discharge more rapidly, and increase contractile force (Ashe 1997). When a motor neuron fires sufficiently fast, the muscle fibers it innervates produce a fused contraction. In this context, the state of the spinal motor neurons (in addition to descending drive from the motor cortex and other supraspinal centers) are influenced by many factors, such as those mediated by excitatory and inhibitory afferent projections and alterations in motor neuron properties that may make them more or less responsive to synaptic input (Berardelli et al. 2001).
5.2.2 Age-Induced Pathophysiology of Neuromuscular Excitation
5.2.2.1 Age-Induced Loss of Voluntary (Central) Muscle Activation
Numerous studies have compared electrically-stimulated force production vs. voluntary force production to examine whether aging results in an impaired ability of the nervous system to fully activate skeletal muscle volitionally (Clark and Taylor 2011). These reports have yielded discrepant findings, likely due to variations in the muscle groups investigated, as well as the inherent heterogeneity of aging (Lowsky et al. 2014). In a recent report, older adults with clinically meaningful leg extensor weakness, exhibited deficits in the ability of their nervous system to fully activate their leg extensor muscles, while stronger older adults did not (Clark et al. 2019). Data such as this support the notion that the nervous system is a key culprit in older adults with clinically meaningful age-related weakness.
In addition to the leg extensor musculature, there is also evidence that grip strength measures are largely reflective of the integrity of the nervous system (Carson 2018). For instance, there is a well-described decline in the flexion force that can be generated by an individual finger, as the number of other fingers that contribute to the grip is increased (Ohtsuki 1981). The observation that the magnitude of this deficit is greater in older adults than in the young (Shinohara et al. 2003b) is consistent with evidence that deficiencies in muscle synergy formation contribute to the difficulties experienced by older adults in many movement tasks (Shinohara et al. 2003a; Barry et al. 2005). This finding is particularly intriguing when one considers that low hand grip strength is associated with a wide range of negative health outcomes in older adults, including cardiovascular disease (Celis-Morales et al. 2018), diabetes (McGrath et al. 2017), dementia (Buchman et al. 2007; Carson 2018), functional disability (McGrath et al. 2018; Al Snih et al. 2004), depression (Fukumori et al. 2015), mobility limitations (Hicks et al. 2012; Bhasin et al. 2020), and early all-cause mortality (Duchowny 2019; McGrath et al. 2019b).
5.2.2.2 Age-Induced Loss of Neural Excitability
Neural excitability can broadly be defined as the readiness of a nerve cell or a neural circuit to respond to a stimulus (Konstantinovic and Fliipovic 2019; Kandel et al. 2013; Schulz et al. 2006). The response is typically in the form of an action potential, a transient change of electrical charge (polarization) of the neuronal membrane. The action potential can be measured either individually, at the level of an individual nerve cell, or as the sum of action potentials in the form of a compound action potential or an evoked potential, at the level of groups of neurons or neural circuits (Kandel et al. 2013). There is a strong theoretical basis for the contention that neural hypoexcitability serves as a key contributor to weakness. That is, a neuron with low excitability will, conceptually, have a lower maximal steady-state firing frequency (Schulz et al. 2006).
Dynapenia, as well as a myriad of other disorders and conditions (e.g., disuse, injury, and sepsis), may be due, in part, to neural hypoexcitability (Clark et al. 2020; Clark et al. 2014; Nardelli et al. 2013; Stefanelli et al. 2019). In the context of aging, older adults with clinically meaningful leg extensor weakness exhibit indexes indicative of corticospinal hypoexcitability (e.g., magnetic brain stimulation derived motor evoked potentials about half that of the strong older adults) (Clark et al. 2020). Moreover, these indexes could explain ~33% of the between-subject variability in older adult’s leg extensor strength, which was slightly more than that explained by thigh lean mass (Clark et al. 2020). These findings suggest that weakness is mechanistically, partially mediated by neural hypoexcitability. Whether dysfunction is at the level of the cortical or spinal motor neurons has yet to be determined; though it has been hypothesized that age-induced hypoexcitability occurs in both upper and lower motor neurons. For instance, paired-pulse brain stimulation paradigms that permit inferences in relation to intracortical excitability demonstrate that older adults have greater indices of cortical hypoexcitability in comparison to young adults (McGinley et al. 2010; Clark et al. 2015). Human and animal studies also suggest that aging results in a reduction in α-motor neuron excitability (e.g., greater and longer hyperpolarization potentials and lower minimal and maximal steady-state firing frequencies) (Kalmar et al. 2009; Christie and Kamen 2006, 2010; Kamen et al. 1995).
5.2.2.3 Age-Induced Loss of Dopaminergic Function
Within the central nervous system, the basal ganglia may theoretically be linked to age-related reductions in mobility capacity via its associated dopaminergic function. Progressive degeneration of mid-brain dopaminergic neurons has been associated with deficits in the initiation, speed, and fluidity of voluntary movement (Berardelli et al. 2001; Buhusi and Meck 2005; Turner and Desmurget 2010). With regards to aging, slower rates of voluntary force development have been linked to risk of falls (Kamo et al. 2019), and ability of the nervous system to rapidly drive muscle force production is associated with overall mobility (Moskowitz et al. 2020). Moreover, peak horizontal saccade velocity, which theoretically should not be impacted by musculoskeletal mechanisms and processes, has been reported to decrease with age (Irving et al. 2006). In fact, older adults have a saccade velocity about half that of young adults (Irving et al. 2006). Studies of the aging human brain have also shown that regulation of dopamine action is significantly reduced in older age via structural degradation, including neuronal loss, fewer neuroreceptor sites, and loss of transporter molecules (Kaasinen and Rinne 2002). For instance, age-dependent declines of brain dopamine agonist (DA) levels have been reported in the basal ganglia, specifically the dorsal striatum post-mortem (Carlsson and Winblad 1976). In vivo imaging studies have since confirmed these findings (Kaasinen and Rinne 2002).
After the age of 20, the availability of dopamine D1-like receptors also declines in the human striatum at a rate of ~7% per decade (Suhara et al. 1991; Wang et al. 1998). The D2-like family demonstrates a similar decrease in receptor density (~5–10% per decade) (Rinne et al. 1993; Wong et al. 1997) and receptor binding potential (~6–8% per decade) (Rinne et al. 1993; Antonini and Leenders 1993; Volkow et al. 1996). There are several studies that have directly examined the relationship between striatal DA and age-related changes in gait and other parameters of motor function. Volkow et al. (1998) reported that age-related decreases in brain dopamine activity in non-Parkinsonian older adults, coincided with reductions in finger tapping speed. Similar studies have reported that lower striatal dopamine transporter activity explained ~23% and 35% of the between-subject variance in “comfortable pace” gait speed and cadence, respectively (Cham et al. 2008). Moreover, several investigations have reported a relationship between dopaminergic genotype (catechol-O-methyltransferase (COMT) genotype) and mobility (Moskowitz et al. 2020; Metti et al. 2017; Holtzer et al. 2010). These studies suggest that the genotype resulting in intermediate levels of tonic DA (i.e., the Val158Met polymorphism) is associated with faster gait and movement speeds, which is likely due to it balancing the roles of tonic and phasic DA (i.e., tonic–phasic regulation of DA transmission) (Bilder et al. 2004; Grace 1991; Schacht 2016).
5.2.3 Regenerative Rehabilitation and Neuromuscular Excitation
Physical exercise is known to enhance motor control and function via neural adaptations in older adults, and therefore cannot be overemphasized (Watson 2017). However, general physical exercise is beyond the scope of this chapter. Here, we discuss goal-directed motor training as well as other novel neurotherapeutic approaches that may be leveraged to improve neural excitation of muscle. We should note at the outset that, to our knowledge, there are no FDA-approved neuro-based therapies that have been approved for use in situations of muscle weakness or mobility limitations. All approaches that we will discuss should be considered experimental and only used in the context of research studies (as opposed to clinical practice per se). There are several potential approaches for enhancing neural excitability; herein, we discuss; goal-based motor training, non-invasive brain stimulation and pharmacological compounds.
5.2.3.1 Goal-Based Motor Training
The attachments between neurons are incredibly dynamic; they change and grow (or shrink) constantly. Working together in a network, neurons organize themselves into specialized groups to form different kinds of information processing. When one neuron sends a signal to another, the synapse between the two strengthens (hence the adage “neurons that fire together, wire together”). The more often a particular signal is sent between them, the stronger the connection grows. Novel experiences and learning cause new dendrites to form, whereas repeated behavior and learning cause existing dendrites to become more entrenched. These basic principles are fundamental to the development and rationale of exercise training strategies for enhancing motor function.
Motor representations (i.e., movement memories) are formed and stored in the brain, just like our memories of people and events. Motor representations are created by a series of remarkably complex coordinated processes dispersed throughout the brain that involve multiple neural networks that interact to help individuals perform learned movements. Thus, memory is the cornerstone of all learning. Motor adaptation refers to learning a new movement skill. When someone practices a movement over and over again, they perform better, partly because they develop new motor memories.
Progressive resistance exercise is largely considered to be the first-line therapy to manage sarcopenia (Dent et al. 2018). We believe this is rational in that it has been shown to have innumerable benefits (Churchward-Venne et al. 2015; Law et al. 2016). However, advancing age has been suggested to result in increased cortical processing for mobility tasks (i.e., less automaticity) (Sorond et al. 2015). Thus, we encourage increased attention to be given to interventional strategies that incorporate goal-based motor skill training (note: “goal-based” refers to the practice of certain activities that lead to improved performance). There are many exercise modalities that can incorporate aspects of goal-based motor skill training, but the more classic approaches are exercise modalities such as Tai Chi, dancing, boxing, and mixed martial arts. The central premise of goal-based motor skill training is that it facilitates learning through feedback (reinforcement learning), involves engagement of the prefrontal cognitive circuits that are involved in early phases of motor learning, and incorporates parameters important for experience-dependent neuroplasticity (e.g., intensity, repetition, difficulty, specificity, complexity of practice, etc.). Feedback (verbal cueing, proprioceptive, etc.) has numerous purposes, such as challenging individuals beyond their self-selected levels of perceived capability, maintaining motivation, and increasing cognitive awareness of movements that were previously automatic and unconscious. Figure 5.2 provides an illustration of the neural structures and connections involved in the cognitive and automatic aspects of motor control in relation to goal-directed learning. Goal-based exercise strategies have become a mainstay in neurorehabilitation for the improvement or recovery of impaired or lost motor function in overt neurological disease (e.g., Petzinger et al. 2013), and for the reasons stated in this chapter, we believe these strategies hold promise for dynapenia.

Goal directed and habitual control circuits of behavior. Motor control incorporates numerous cortical and subcortical structures with the most critical connections being those between the basal ganglia and cortex as these deeply involved in the cognitive (a) and automatic (b) aspects of motor control. In a, arrows represent the cognitive (or volitional) circuits. In b, the arrows represent the automatic (or the unconscious/habitual) circuits. Aging has been suggested to result in impaired automaticity. As such, we advocate for increased investigations on whether goal-based motor training programs can be used to mitigate and treat dynapenia. Figure recreated based on that of Petzinger et al. (2013). This image was created using BioRender (https://BioRender.com)
5.2.3.2 Non-invasive Brain Stimulation
Non-invasive brain stimulation consists of techniques including transcranial magnetic stimulation and transcranial direct current stimulation (tDCS). High-frequency (e.g., 10–20 Hz) magnetic brain stimulation and anodal transcranial direct current stimulation both have been demonstrated to transiently increase excitability of the motor cortex (Reis et al. 2008; Kobayashi and Pascual-Leone 2003; Lefebvre and Liew 2017). Whether they have the potential to modify strength and physical function, for use during rehabilitation or as a stand-alone therapy is not clear. In relatively small studies (based on sample size) consecutive sessions of anodal transcranial direct current stimulation enhanced hand dexterity (i.e., ~20–25% improvement on the Purdue Pegboard Test performance) (Rostami et al. 2020) and improved elbow flexor fatigue-resistance (i.e., ~15% increase the time to task failure of a sustained, submaximal contraction) (Oki et al. 2016) (Fig 5.3a). Thus, while this is far from conclusive data, it does suggest that non-invasive brain stimulation has the ability to modify indexes of motoric and muscle function in older adults.

Mean effects of various approaches that increase neural excitability on enhancing measures of physical function in older adults. (a) Single session of anodal transcranial direct current stimulation (tDCS), which increases cortical excitability, increased the time to task failure of a sustained, submaximal elbow flexion contraction (Oki et al. 2016), and five consecutive days of anodal tDCS improved the Purdue Pegboard Task performance (a measure of hand dexterity) (Rostami et al. 2020). (b) Acute ingestion of caffeine (3 mg/kg, which is the equivalence of 2–3 cups of coffee) increased manual dexterity, timed up and go time, six-minute walk gait speed (Duncan et al. 2014), as well as the rate of force development during a sit to stand task (Tallis et al. 2020). (c) A single dose of methylphenidate improved timed up and go performance (Ben-Itzhak et al. 2008). (d) Three weeks of L-DOPA improves single (blue) and dual task (violet) usual gait speed in older adults with depression (Rutherford et al. 2020). Graphs recreated based on data published in the respective articles
5.2.3.3 Pharmacological Compounds
Compounds that increase neural excitability can also enhance motor function. One such potential compound is caffeine. Caffeine is a well-established performance enhancing nutritional compound in young, healthy humans. There are limited data on the effects of caffeine on measures of human performance in older adults. The few studies that do exist are small-scale trials (e.g., 10–30 subjects) and have reported mixed effects. Specifically, some studies suggest that caffeine supplementation (typically around 3 mg/kg, or the equivalence of two–three cups of coffee) does not alter leg extensor muscle strength, static balance, as well as various measures of physical performance (Tallis et al. 2020). Conversely, some studies have reported that caffeine, when compared to a placebo, modestly enhances (~6–8%) manual dexterity, timed up and go time, and six-minute walk gait speed (Duncan et al. 2014), and more robustly increases the rate of force development during a sit to stand task around 13% (Tallis et al. 2020) (Fig. 5.3b). It should be noted that all of the prior work has been conducted in relatively young older adults (e.g., mean age of late 60s to early 70s) who were extremely high functioning. Thus, it is possible that a ceiling effect may exist as it relates to any enhancement in function amongst older adults with such high levels of physical function and mobility. Thus, more work is needed, and the existing results must be interpreted cautiously and critically.
Another potential compound of interest is the serotonin 5-HT2c receptor agonist, although the effects of serotonin 5-HT2c receptor agonist on motor function in aged rodents or humans have yet to be tested. However, motor neuron hypoexcitability has been reported to contribute to sepsis-induced weakness in rat muscle (Nardelli et al. 2013, 2016), and subthreshold voltage-activated currents are a key mechanism underlying defective repetitive firing observed in this model (Nardelli et al. 2017). More recently, it was demonstrated that treatment of septic rats with injection of a selective serotonin 5-HT2c agonist (lorcaserin; 3 mg/kg) significantly improved repetitive motor neuronal firing and dramatically increased motor unit force production. To our knowledge, only one prior study has examined the effects of lorcaserin on any behavioral measure related to motor function. In that study, young rats (n = 8) received single, varying 5-HT2c agonist doses, and a 21% increase in Rotarod performance (i.e., walking speed, motor performance) was observed at a dose of 0.6 mg/kg (Higgins et al. 2012). However, worsened performance was observed at higher doses, consistent with reports that high doses result in malaise (Higgins et al. 2020). Thus, additional research is needed.
With regards to the dopaminergic system, there are several studies that support the potential modulation of this system to enhance physical function and mobility outcomes in older adults. The earliest of these evaluated the potential of a single dose of methylphenidate (20 mg) on measures of physical function (Ben-Itzhak et al. 2008). Methylphenidate has multiple mechanisms of action, yielding its stimulant effect, with one of the mechanisms of action being inhibition of dopamine uptake (PubChem 2021a). This study reported that methylphenidate improved timed up and go performance and gait (stride time variability) in 26 community dwelling older adults when compared to placebo (Ben-Itzhak et al. 2008) (Fig. 5.3c). The second of these studies also examined the effects of a single dose of methylphenidate (short-acting 10 mg) on gait in thirty healthy older adults (Shorer et al. 2013). Here, it was also observed that methylphenidate improved mobility by reducing step errors during a standard gait task as well as when the gait task was overlayed with a cognitive load component. The findings that the effect was most robust in a dual task requiring higher executive control suggests the effects could be due to improvements in sustained attention as well as potential direct effects on the motor system. While beyond the scope of this chapter, it should be noted that there is growing scientific interest in the role of cognitive and motor system interactions and their interrelationship with age-related declines in both systems (Cohen and Verghese 2019). Lastly, there is one study that has examined the potential utility of levodopa (L-DOPA), an amino acid precursor of dopamine (PubChem 2021b), for enhancing physical function in older adult. This recent pilot study treated sixteen older adults who suffered from depression with L-DOPA for 3-weeks, and reported a 16% and 28% significant increase in single task and dual task usual gait speed, respectively (Rutherford et al. 2020) (Fig. 5.3d). These above-mentioned findings clearly indicate the need for further work examining the role of dopaminergic function in age-related mobility capacity.
With the above-mentioned said, a pharmaceutical approach to enhancing physical function in older adults could, in and of itself, be problematic as polypharmacy, defined as being prescribed five or more medications, has been shown to be associated with a decline in mental and physical functioning in elderly patients (Williams et al. 2019). In particular, anticholinergics, benzodiazepines, antipsychotics, and opioids were all found to have significant adverse effects in the elderly population (Williams et al. 2019). It should be noted that, in general, these classes of medications have sedative properties that reducing neural excitability. Thus, it is very possible that a “less is more” pharmaceutical approach may be the most beneficial approach to enhancing physical function in older adults.
5.3 Excitation–Contraction (EC) Coupling
5.3.1 Introduction to EC Coupling
In addition to the loss of neuromuscular excitation, sites and structures within the EC coupling pathway have also been linked to age-induced weakness (Delbono et al. 1995; Renganathan et al. 1997a; Ryan and Ohlendieck 2004; Delbono 2011; Baumann et al. 2016). EC coupling can broadly be defined as the sequence of events linking plasmalemma depolarization to the release of Ca2+ from the sarcoplasmic reticulum (SR) (Sandow 1952; Hernández-Ochoa and Schneider 2018; Calderón et al. 2014). Briefly, after a threshold potential is initiated at the motor endplate, a membrane action potential propagates along the plasmalemma down the transverse (T)-tubules. Action potential conduction is dependent on a coordinated response of various plasmalemmal ion channels and pumps. Depolarization of the plasma membrane stimulates the voltage-sensitive dihydropyridine receptors (DHPRs) located in the T-tubules, which in turn, activate the ryanodine receptors (RyRs). In skeletal muscle, the DHPRs and RyRs are in close proximity as to permit physical contact, thereby allowing direct interaction and communication (Fill and Copello 2002; Grabner and Dayal 2010; Calderón et al. 2014). The RyR is a Ca2+ release channel composed of four monomers embedded in the SR membrane. Each RyR monomer interacts with numerous ancillary proteins (e.g., calstabin, calmodulin) and enzymes (e.g., phosphatase; PP1, phosphodiesterase; PDE4D3) known to alter its gating (Bellinger et al. 2008a; Santulli et al. 2018). Upon RyR activation, Ca2+ is released into the cytosol and cross-bridge formation subsequently occurs. Theoretically, if any step in the EC coupling pathway is disrupted (termed EC coupling failure), voltage-induced SR Ca2+ release will be impaired, and less cytosolic Ca2+ will be available for cross-bridge formation and force generation (Baumann et al. 2016). With increasing age, peak intracellular Ca2+ transients evoked by plasmalemmal depolarization have been shown to decrease (Wang et al. 2000; Gonzalez et al. 2003; Delbono et al. 1995; Russ et al. 2011; Umanskaya et al. 2014), indicative of impaired SR Ca2+ release. Age-induced EC coupling failure can be observed as dynapenia without a concomitant change in muscle mass, as measured by reductions in skeletal muscle-specific force (i.e., force normalized to muscle mass or cross-sectional area; CSA) (Lynch et al. 1999; Moran et al. 2005; Hill et al. 2020; Goodpaster et al. 2006; Russ et al. 2011). Potential mechanisms for EC coupling failure in aged muscle include reduced content of EC coupling proteins, loss of EC coupling protein-protein interactions and/or modifications to EC coupling proteins (Fig. 5.4).

Possible sites and structures within the excitation-contraction (EC) coupling pathway implicated in age-induced skeletal muscle weakness (i.e., dynapenia). EC coupling failure with advanced age this thought to stem from DHPR-RyR uncoupling via the loss of DHPR, RyR and/or the triad structure. (A) Loss of triad structure with (B) corresponding reductions in triadic proteins, such as JP45 and MG29. (C) Loss of the DHPR and/or RyR causing a decrease in the DHPR/RyR ratio (in figure, DHPR content is reduced). (D) Loss or depletion of calstabin from RyR causing SR Ca2+ leak and (E) increased intracellular Ca2+ concentrations. This image was created using BioRender (https://BioRender.com)
5.3.2 Age-Induced Pathophysiology of EC Coupling Failure
5.3.2.1 Age-Induced Loss of EC Coupling Proteins and Protein-Protein Interactions
Any reduction in EC coupling proteins or the ability of these proteins to interact could result in EC coupling failure. Although many sites of age-induced EC coupling failure have yet to be established, several research groups have reported reduced expression, lowered content, and altered protein-protein interactions among the DHPR and RyR proteins (Fig. 5.4). Reductions in either DHPR (O’Connell et al. 2008; Renganathan et al. 1997a; Ryan et al. 2000), specifically the CaV1.1 subunit of DHPR (DHPRα1s) (Catterall et al. 2005), or RyR (Fodor et al. 2020) expression or protein content have been reported by many, but not by all (Lamboley et al. 2016; Ryan et al. 2003, 2011). With loss of DHPR and/or RyR, the DHPR-RyR interaction is uncoupled, as measured by decreases in the DHPR/RyR ratio (Renganathan et al. 1997a; Ryan et al. 2000). Delbono and colleagues (Renganathan et al. 1997a) reported that reduced DHPR expression in rat skeletal muscle is the molecular basis for age-induced EC coupling failure. Decreased expression of DHPR was suggested to cause significant impairment in action potential transduction into a DHPR signal, consequently reducing RyR activation, SR Ca2+ release and cross-bridge formation.
Besides loss of DHPR and/or RyR, age-induced uncoupling between DHPR and RyR may stem from diminished communication between these two proteins (Fig. 5.4). This is supported by fragmented SR (Weisleder et al. 2006) and disarranged triads (Boncompagni et al. 2006; Zampieri et al. 2015) observed using electron microscopy. Essentially, with triad disarrangement, the proximity of the DHPRs to the RyRs will be lost, reducing the number of proteins that can physically interact. Disorganized alignment of SR and T-tubule membranes coincide with reductions in the triadic proteins JP-45 (Anderson et al. 2003) and MG29 (Weisleder et al. 2006), proteins thought to interact with DHPR and RyR or maintain the structure of triad junction. Loss of these triadic proteins would inevitably increase the distance between the T-tubule and SR, and subsequently the DHPR-RyR interaction. Aligning with these aforementioned changes, Ca2+ handling proteins (e.g., sarcalumenin) (O’Connell et al. 2008), plasmalemmal ion channels (e.g., chloride channels) (Pierno et al. 1999) and plasmalemmal pumps (Ca2+ ATPase and the Na+-Ca2+ exchanger) (O’Connell et al. 2008) have also been reported to exhibit age-related reductions in content. Further research will be needed to elucidate whether all these age-related changes occur in synchronization or if there is an initiating event. Taken together, many EC coupling structures and proteins appear to be influenced by age, but most data suggest EC coupling failure stems from DHPR-RyR uncoupling via the loss of DHPR, RyR and/or the triad structure.
5.3.2.2 Age-Induced Modifications to EC Coupling Proteins
Modifications that occur to EC coupling proteins will also result in EC coupling failure. The most documented proteins are at the triad junction, specifically the RyRs. The RyR is a macromolecule complex that interacts with numerous proteins and is prone to various modifications (Bellinger et al. 2008a; Santulli et al. 2018). With advancing age, the RyR becomes increasingly oxidized and nitrosylated, promoting RyR dysfunction (Andersson et al. 2011; Umanskaya et al. 2014; Lamboley et al. 2016). Interestingly, these redox modifications do not appear to directly alter RyR function per se, but rather through their effects on the ancillary proteins associated with the channel’s activity. One in particular is calstabin (FK506 binding protein, FKBP12) (Andersson et al. 2011), a 12-kDa protein that normally binds to each RyR monomer (Fig. 5.4). Calstabin is thought to stabilize the channel, preventing it from opening to subconductance states (Ahern et al. 1997; Brillantes et al. 1994). Age-associated oxidation and nitrosylation of RyR depletes the channel of calstabin, diminishing the calstabin-RyR interaction (Andersson et al. 2011; Umanskaya et al. 2014). These changes are thought to result in “leaky” RyRs that manifest in increased single-channel open probability, increased Ca2+ spark frequency (Andersson et al. 2011; Umanskaya et al. 2014) and reduced SR Ca2+ content (Lamboley et al. 2015, 2016). Under these conditions, voltage-mediated SR Ca2+ release appears to be impaired through loss of RyR function rather than RyR content (Andersson et al. 2011; Lamboley et al. 2016; Russ et al. 2011). In support of the loss in the calstabin-RyR interaction causing SR Ca2+ leak, others have reported resting Ca2+ concentrations are elevated in aged muscle (Mijares et al. 2020). Importantly, RyR dysfunction mediated by depletion of calstabin is not only observed with age (Russ et al. 2011; Andersson et al. 2011; Umanskaya et al. 2014), but also heart failure (Shan et al. 2010), muscular dystrophy (Bellinger et al. 2009), chronic muscle fatigue (Bellinger et al. 2008b), and contraction-induced injury (Baumann et al. 2014). These results suggest that age-related EC coupling failure predominantly stems from redox-induced RyR dysfunction.
5.3.3 Regenerative Rehabilitation and EC Coupling
Data suggests age-induced weakness due to EC coupling failure stems from (1) a reduction in the DHPR-RyR interaction via the loss of the DHPR, RyR and/or triad structure and (2) redox-induced RyR dysfunction. Although the initiating events that induce EC coupling failure are likely many, loss of mitochondria homeostasis and oxidative stress appear to be central catalysts (Andersson et al. 2011; Qaisar et al. 2018; Umanskaya et al. 2014). With advancing age, oxidative stress is thought to occur due to an overproduction of reactive oxygen and nitrogen species (ROS/RNS) and an impaired ability to neutralize them (Mijares et al. 2020; Vasilaki et al. 2006). ROS/RNS can increase with age due to mitochondrial dysfunction caused by age-related mitochondrial DNA mutations, deletions and/or damage (Boengler et al. 2017) (see Sect. 5.4.2.1). With the accumulation of ROS/RNS, the RyRs are subject to oxidative stress, which leads to SR Ca2+ leak and elevated cytosolic Ca2+ concentrations (see Sect. 5.3.2.2) (Andersson et al. 2011; Umanskaya et al. 2014). These events can lead to a vicious cycle that exacerbates mitochondrial dysfunction by causing mitochondrial Ca2+ overload, stimulation of additional ROS/RNS and greater SR Ca2+ leak (i.e., further depleting RyR of its stabilizing subunit calstabin). These data indicate that overproduction of ROS/RNS occurring in aged skeletal muscle may alter the expression and function of key EC coupling proteins, thereby causing dynapenia.
When considering regenerative rehabilitation to combat age-associated EC coupling failure, it is essential to reduce oxidative stress via lowering ROS/RNS production, increasing ROS/RNS clearance or by attenuating the negative effects of ROS/RNS. Likely one of the most robust and potent strategies is modifying lifestyle with physical exercise (Distefano and Goodpaster 2018; Lanza et al. 2008). For instance, muscle from older adults who exercised regularly had lower expression of genes related to oxygen species detoxification, better preserved fiber morphology, better preserved ultrastructure of intracellular organelles involved in Ca2+ handling and produced greater maximal isometric knee extensor force when compared to age-matched sedentary participants (Zampieri et al. 2015). Moreover, long-term training (i.e., access to a voluntary running wheel) promoted maintenance of the triad junction (Boncompagni et al. 2020), prevented age-induced elevations in resting intracellular Ca2+, loss of RyR content and reductions in ex vivo specific force in C57BL/6 mouse muscle (Fodor et al. 2020). Although physical exercise is a robust lifestyle modifier that improves function, it is difficult to pinpoint its precise mechanisms of action, especially if it is lifelong. Moreover, physical exercise may not be practical or feasible for all older adults. Methods to mimic physical activity have therefore become attractive therapies to attenuate age-induced oxidative stress and its downstream effects. Here, we highlight the use of antioxidants and the pharmaceutical S107 as potential therapeutic approaches, and briefly discuss the role of insulin-like growth factor-1 (IGF-1) in EC coupling maintenance with age.
5.3.3.1 Antioxidants
Preventing or restoring the redox environment of skeletal muscle has been reported to improve EC coupling and muscular strength in aged muscle. Recently, in vivo supplementation of selenium for 2 months, a trace element with antioxidant properties, was found to increase the maximal rate of Ca2+ efflux and specific force in aged C57BL/6 mouse muscle (Fodor et al. 2020). Furthermore, antioxidant treatment of dithiothreitol (DTT; a strong reducing agent) reduced RyR channel oxidation, SR Ca2+ leak and RyR Ca2+ sparks in aged C57BL/6 mouse fibers (Umanskaya et al. 2014) and increased maximal SR Ca2+ accumulation in human fibers of old subjects (Lamboley et al. 2016). To determine if increasing lifelong mitochondrial antioxidant activity would prevent age-related EC coupling failure and dynapenia, Umanskaya et al. (Umanskaya et al. 2014) overexpressed the human catalase gene in mitochondria. Catalase is an antioxidant enzyme that catalyzes the decomposition of hydrogen peroxide into water and oxygen. When compared to aged-matched controls, mice overexpressing catalase had less mitochondrial ROS (Lee et al. 2010), increased SR Ca2+ load, increased Ca2+ transients and reduced SR Ca2+ leak, all of which translated into greater skeletal muscle-specific force (Umanskaya et al. 2014). Taken together, these studies indicate mitochondrial ROS production is a molecular mechanism for age-related EC coupling failure and that reducing ROS through antioxidants (particularly those that target the mitochondrial or RyRs) may improve Ca2+ homeostasis and attenuate or delay the onset of dynapenia.
5.3.3.2 S107
Pharmacological agents can also be used to improve age-induced EC coupling failure caused by excessive oxidative stress. Marks and colleagues (Andersson et al. 2011) eloquently accomplished this by treating aged mice with S107, a drug that preserves the calstabin-RyR interaction. Impressively, 4 weeks of S107 treatment in old C57BL/6 mice was able to stabilize calstabin to RyR, prevent SR Ca2+ leak, restore tetanic Ca2+ release and increase muscle-specific force when compared to muscle of age-matched, untreated mice. These beneficial effects were observed despite persistent RyR oxidation and nitrosylation, indicating treatment was not able to reduce RyR modifications, but was able to ameliorate the detrimental effects of oxidative stress. These reports align with data demonstrating that when catalase is overexpressed in aged C57BL/6 mice, less RyR is oxidized and depleted of calstabin (Umanskaya et al. 2014). Reports such as these indicate EC coupling failure due to RyR dysfunction mediated by depletion of calstabin can be improved through preventing the loss of calstabin (via the increased capacity to neutralize oxidative stress) or by restoring calstabin to RyR (via S107). From a practical perspective, pharmacological agents like S107 yield significant promise for individuals that may have already experienced dynapenia or passed the timepoint when age-related RyR oxidation or nitrosylation is reversible.
5.3.3.3 Insulin-Like Growth Factor-1 (IGF-1)
An alternative strategy, not directly associated with the mitochondria or oxidative stress, is the use of IGF-1. Insulin-like growth factor (IGF-1) is a peptide primarily known for its canonical role in promoting skeletal muscle differentiation and growth (Florini et al. 1996). Another, less recognized function of IGF-1 is expression of DHPR. Data from S1S2 mice overexpressing human IGF-1 (20–30-fold increase in concentration) in skeletal muscle resulted in a significant increase (over 50%) in the number of DHPRs, specifically the CaV1.1 subunit (Renganathan et al. 1997b). Moreover, with advancing age, muscle of these IGF-1 overexpressing mice maintained a high DHPR number, DHPR/RyR ratio and muscle specific force when compared to the reductions observed in muscle of control mice (i.e., wildtype) (Renganathan et al. 1998). However, more recent reports in human skeletal muscle did not observe age-related reductions in DHPR expression (Ryan et al. 2003) or content (Lamboley et al. 2016), contrary to what has been measured in rodent (O’Connell et al. 2008; Renganathan et al. 1997a) and rabbit muscle (Ryan et al. 2000). These equivocal findings question the utility of IGF-1 as a beneficial therapy for improving EC coupling in aging skeletal muscle, however, additional research is needed before definitive conclusions can be drawn.
The use of antioxidants, the pharmaceutical S107 and IGF-1 all appear, to some extent, to improve EC coupling and strength in aged skeletal muscle. However, some caution should be considered when looking at the breadth of literature as several caveats will be noted. Some include the model used (human vs. animal models) and muscle/fibers analyzed (fast twitch vs., slow twitch)—as differences are likely to be observed. Despite these, there remains great potential for regenerative rehabilitation to improve EC coupling failure, loss of Ca2+ homeostasis and strength (i.e., attenuate dynapenia) in aging skeletal muscle.
5.4 Mitochondrial Function
5.4.1 Introduction to Mitochondrial Function
Skeletal muscles require energy to carry out their functions. Amongst other things, energy is required for the myofilament contractions that enable movement, for the interconversion and storage of nutrients that contribute to maintaining whole-body homeostasis, and for the continual maintenance of muscle itself (for example, repair and replacement of old and damaged contractile proteins). As with most cells, the main source of energy employed by muscle is adenosine triphosphate (ATP) which is produced by oxidative phosphorylation in the mitochondria and glycolysis in the cytosol (Baker et al. 2010). The number of mitochondria varies from muscle to muscle in accordance with its function. For example, cells with constant high energy demand such as oxidative, slow twitch, type I fibers are rich in mitochondria, whereas cells with low, sporadic energy demand such as glycolytic, fast twitch, type II fibers contain fewer mitochondria (Carter et al. 2015).
Unlike other cells, skeletal muscle contains two pools of mitochondria, one in the cytoplasm and one in the sarcomeric contractile fibers (Carter et al. 2015). The cytoplasmic mitochondria, or subplasmalemmal, are believed to be more responsive to the energy needs of the cell for growth and metabolism (Crescenzo et al. 2006; Koves et al. 2005; Ritov et al. 2005), whereas the intrasarcomeric or intermyofibrillar mitochondria are believed to provide energy specifically to the contractile fibers (Ferreira et al. 2010). Interestingly, in addition to producing ATP, mitochondria also provide cells with an ability to buffer Ca2+ gradients and are often found localized in areas of Ca2+ influx (Parekh 2003a). Thus, another possible function of having both cytoplasmic and intrasarcomeric mitochondria is to provide an additional level of regulation of Ca2+ levels during EC coupling (see example in Sect. 5.3.3) via the interplay of the plasmalemma, SR and mitochondria (Parekh 2003b).
Mitochondrial content is established and increased via mitochondrial biogenesis (Gureev et al. 2019) and is decreased by degradation via a specialized form of autophagy termed mitophagy (Chen et al. 2020b) (Fig. 5.5). Mitochondrial biogenesis is controlled by the activity of several transcription factors, including PGC-1α, PGC-1β, NRF1, and NRF2 (Gureev et al. 2019). These, and other, transcription factors regulate the transcription of roughly 1500 nuclear encoded genes (Hendrickson et al. 2010; Hill 2014) that are translated and translocated into the mitochondria, largely through the mitochondrial TOM20/TOM40 import complex (Boengler et al. 2011). Slightly more than a dozen mitochondrial encoded proteins also contribute to maintaining mitochondrial structure and function, which are regulated by TFAM (Ngo et al. 2014). Removal of mitochondrial via autophagy requires regulation/induction of autophagy and the ability of autophagic vesicles to recognize mitochondrial via surface proteins such as the PINK1-PARKIN complex, NIX, and BNIP3 and thus target them for degradation (Chen et al. 2020b).

Conceptual diagram of control of mitochondrial structure and function. Cellular energy demand (A) drives rates of mitochondrial biogenesis with increased demand increasing the mitochondrial pool and decreased demand leading to the mitochondrial pool to tend towards mitophagy (B). Increases in energy demand can lead to mitochondrial fusion (C) which increases the mitochondrial surface area to enable increased proton motive force and derived functions (D). Mitochondrial function can also be affected by post-translational modifications. Impaired mitochondria function or needs to remodel mitochondrial location can lead to mitochondrial fission (E) enabling mitochondria to renter the pool where they can undergo further fusion, mitophagy, or rupture with smaller consequences that rupture of fussed mitochondria
Mitochondrial structure is dynamic with mitochondria undergoing fission to separate into smaller structures and fusion to establish larger ones (Hood et al. 2019). Fission is controlled by DRP1 and associated binding proteins. Fusion is controlled by OPA1, MFN1 and MFN2 and associated regulator proteins. Fission produces smaller structures that can be more easily targeted for degradation by mitophagy and/or act independently (Fig. 5.5). Fusion produces larger structures with increased surface area which allows for more coordinated action and more efficient establishment, maintenance, and utilization of membrane gradients for ATP production, heat production, and Ca2+ buffering. Mitochondria function then is a function of cellular environment/demands (e.g., fiber type, recruitment pattern, etc.), content (regulated by biogenesis and mitophagy), structure (regulated by fission and fusion), and also post-translational modifications (Stram and Payne 2016); for example, oxidation, persulfidation, and Ca2+ binding.
5.4.2 Age-Induced Alterations in Mitochondria
Loss of physiologic functions and increased risk of death are observed with age. Molecularly, these losses of molecular physiologic function have been characterized into nine hallmarks of aging (Lopez-Otin et al. 2013); reductions in mitochondria being one of the hallmarks. These reductions in mitochondria appear to take place across species and tissues. Below we discuss alterations in human skeletal muscle mitochondria with advancing age that have been shown to contribute to reduced physical function, increased fatigability, sarcopenia, dynapenia, and cardiorespiratory fitness (Coen et al. 2018).
5.4.2.1 Age-Associated Loss in Mitochondrial Number and Volume
With age there is a reduction of mitochondria in human muscle as assessed by mitochondrial DNA content, mitochondrial enzyme activity assays, and the gold standard method of counting mitochondria in electron micrographs (Seo et al. 2016). However, more research is needed to understand when, chronologically, mitochondria may be lost and at what rate. Human muscle appears to retain the ability to invoke mitochondrial biogenesis with age (Konopka et al. 2014), however, this response may be blunted when compared to younger human muscle (Deane et al. 2019). Thus, it may be the case that muscle loses the ability to rejuvenate/maintain mitochondria with age and that this also impacts the ability to rejuvenate/maintain muscle as a whole (Chen et al. 2020c); some speculate that this may be related to reductions in stem cell function with age (Lopez-Otin et al. 2013). While more research remains to be done to understand why mitochondrial response to exercise may be blunted with age, it is clear that mitochondria reman responsive to exercise interventions. Therefore, there is reason to suspect that reductions in mitochondrial number and volume can be overcome by further understanding how exercise influences mitochondrial structure in old human muscle.
5.4.2.2 Age-Associated Loss in Mitochondrial Function
Loss of mitochondrial function with age can be observed as reductions in ATP production, ability to buffer intramuscular Ca2+, ability to process energetic substrates, and in efficiency of oxygen use (Short et al. 2005; Seo et al. 2016; Hood et al. 2019). These declines in function are seen in both humans and animal models and occur prior to age-associated functional decline and the onset of frailty (Andreux et al. 2018). Mitochondrial dysfunction has been tied to cellular senescence (Chapman et al. 2019), another hallmark of aging (Lopez-Otin et al. 2013). While alterations in mitochondrial structure and function are both observed in human muscle with age, it is not clear if one causes the other and/or if individual specific (genetic and/or epigenetic) differences occur. For example, single nucleotide polymorphisms in nuclear encoded mitochondrial genes have been linked with disease progression (Hendrickson et al. 2010). As structural defects can cause functional defects and vice versa, it is entirely possible that one causing the other may differ between individuals. Similarly, as different individuals may have different causes of mitochondrial dysfunctions, they may show similar changes in mitochondrial structure but as the result of different dysfunctions. As discussed above, the ability of exercise to invoke similar improvements in mitochondrial function, in addition to structure, suggests that mitochondrial function can be restored in aged human muscle.
5.4.3 Regenerative Rehabilitation and Mitochondrial Function
5.4.3.1 Exercise Mimetics
It is clear that exercise promotes mitochondrial biogenesis and mitochondrial function, and that some of these improvements via the action of the transcription factor PGC-1α (Konopka et al. 2014; Seo et al. 2016; Gureev et al. 2019; Hood et al. 2019). Thus, pharmaceutical or nutraceutical activation of PGC-1α could be a potential mimetic for exercise. PGC-1α stimulation has also been suggested as a potential therapeutic for mitochondrial disease and diabetes (Wenz 2009; Yuan et al. 2019). To date, drugs in preclinical trials have targeted PPAR (bezafibrate, rosiglitazone), AMPK (AICAR, metformin), and Sirt1 (resveratrol) (Hofer et al. 2014). See Sect. 5.6.3 for more details on some of these treatments. Additionally, there are novel small molecular activators for PGC-1α under development (for example, Zhang et al. 2013). Another interesting mimetic is carbon monoxide (CO). As a gasotransmitter, CO has recently been shown to have mitochondrial biogenesis effects and enhance the effects of exercise alone (Rhodes et al. 2009).
5.4.3.2 Hormone and Diet Supplementation
A number of hormones regulate mitochondrial content and/or function (Ritz et al. 2005). Some of these, like the androgens change with age and are present at different levels in across biological sex and gender. Correction of clinical deficiencies in hormones has clear benefits in promoting mitochondrial health. For instance, a recent study supplementing testosterone found improved efficacy of the response to the exercise training when the supplement was provided as an adjunct (Gharahdaghi et al. 2019). Similarly, clinical deficiencies in most nutrients (protein, fatty acids, vitamins) lead to impaired metabolism and correction of these had benefits in promoting mitochondrial health (Du et al. 2016). For example, in relation to Vitamin D, intramuscular Vitamin D receptor concentration correlates with the extent of muscle improvement in response to exercise training (Bass et al. 2020). As another example, supplementation of NAD, a pyridine-nucleoside similar to vitamin B3, has been found to have beneficial effects across species (Romani et al. 2021). The reason for this beneficial effect has been suggested to be due to an age-associated decline in NAD levels (Schultz and Sinclair 2016), which may or may not be associated with mitochondrial deficits.
5.5 Protein Homeostasis
5.5.1 Introduction to Protein Homeostasis
Muscle is the largest store of nitrogen, in the form of protein, in the human body. In a young, healthy individual muscle comprises roughly 40% of all the protein in the body (Brook et al. 2016b). Accordingly, protein turnover accounts for roughly 35% of daily whole-body resting energy expenditure (Carbone et al. 2019). Within muscle, more than 40% of protein can be found in the contractile apparatus (i.e., sarcomeres) (Ojima 2019). Thus, there is a strong link between muscle protein content and sarcomere content (Sweeney and Hammers 2018). This means that the human body needs to balance regulation of protein content for maintenance of whole-body energy homeostasis. Muscle lengths tend to be dictated by the skeletal attachment points, whereas muscle volume can change based upon hydration/inflammatory status and growth, termed hypertrophy (Haun et al. 2019). When muscle does grow in length, sarcomeres are added in series, and this results in alterations in the strength and contraction velocity of the muscles (Wisdom et al. 2015). Thus, muscle growth for increased strength requires energy at the whole-body level to maintain whole-body energy balance. Conversely, when muscle atrophies (Atherton et al. 2016), strength will generally be lost as the result of breakdown of sarcomeres in parallel. As muscle size is largely dictated by balancing energy and physical performance needs, muscle hypertrophy and atrophy are largely dictated by alterations in muscle protein synthesis, with or without increased protein degradation (Brook et al. 2016b). Rates of muscle protein synthesis and/or degradation are controlled by a wide variety of anabolic (growth promoting) and catabolic (atrophy promoting) stimuli (McCarthy and Esser 2010).
A number of signaling pathways acting downstream of growth factor receptors have been demonstrated to regulate protein synthesis in animal models and cell culture. In human adult muscle, it is clear that the mechanistic target of rapamycin (mTOR) is a central regulator of muscle protein synthesis, serving to integrate anabolic signals such as amino acid content and energy status, as signaled by 5′ AMP-activated protein kinase (AMPK) and likely other growth and mechanical signals (Brook et al. 2016b). mTOR regulates protein synthesis via 4EBP1, S6K and downstream signals.
The control of protein degradation in human muscle is less well defined, in part due to greater technical challenges in studying degradation in human muscle. While the signals regulating the enzymes that carry out protein degradation, termed proteases, may not be well defined, the proteases themselves are (Szewczyk and Jacobson 2005). Lysosomes and proteasomes are the major proteases within muscle. Both lysosomes and proteasomes are normally “on” but only degrade proteins that are sent to them. Thus, regulation of lysosomal and proteasomal degradation is largely at the level of trafficking proteins to the proteases rather that at the level of turning these proteases “on” or “off” (Szewczyk and Jacobson 2005). In the case of lysosomes, this occurs largely via autophagy and increased autophagy appears to be a basal state in the absence of positive growth factors in yeast, C. elegans, and rodents, so it may similarly be the case for humans (Klionsky et al. 2021). For proteasomes, proteins destined for degradation are typically targeted to the proteasome by tagging the protein with ubiquitin via ubiquitin conjugating enzymes (Lecker et al. 2006). Importantly, at least 30% of all newly synthesized proteins do not properly mature and are degraded by the proteasome, meaning that the proteasome plays a significant role in the degradation of newly synthesized improperly folded proteins (Schubert et al. 2000). Calpains and caspases are the other two main proteolytic systems in muscle and both are constitutively inactive, or “off” (Szewczyk and Jacobson 2005), meaning that their activation results in the degradation of proteins in the immediate vicinity of the proteases once activated. The majority of proteins degraded by calpains (Goll et al. 2003) and caspases are cytoskeletal proteins, implying that a major role of each protease is in the structural remodeling of muscle (Crawford and Wells 2011). In the case of calpains, these tend to be membrane associated and activated by Ca2+, suggesting they may have a role in maintaining the structures necessary for proper EC coupling (Verburg et al. 2009) (see Sect. 5.3.1 for EC coupling) and other Ca2+ signaling. For caspases, these are associated with mitochondria and appear to be activated in response to mitochondrial Ca2+ overload and/or other toxic stressors to mitochondria (Vringer and Tait 2019), suggesting that caspases may have a role in appropriately localizing mitochondria within muscle.
5.5.2 Age-Induced Loss of Protein Homeostasis
Declines in maintaining protein homeostasis, termed proteostasis, is one of nine hallmarks of aging (Lopez-Otin et al. 2013). Like other hallmarks of aging, declines in proteostasis appear to take place across species and tissues. Loss of proteostasis results in accumulation of protein aggregates, disruption of cellular structures, and leads to general declines in the ability to maintain cellular homeostasis (Santra et al. 2019). Below we discuss two aspects of altered protein homeostasis in muscle with advancing age: (1) loss of muscle size and (2) increased anabolic resistance. As previously mentioned, sarcopenia (Evans et al. 2019) and dynapenia are associated with increased physical frailty, increased risk of morbidity, and increased risk of mortality (Wilkinson et al. 2018).
5.5.2.1 Age-Loss of Muscle Size (Sarcopenia)
Human muscle grows in size from birth and reaches a peak in the mid-20s to early-30s depending on the individual. After this point muscle size declines for reasons not still fully understood (Nair 2005). It has been suggested that this may be due to lack of evolutionary pressure for prolonged muscle function with age based upon shorter human life expectancy as little as 2000 years ago (Goldspink 2012). Some of the loss of muscle mass with age may be due to biological effects of aging such as alterations in hormonal signaling and/or loss of mitochondrial or protein homeostasis, and some of this may be determined by lifestyle changes such as nutritional excesses or deficits and/or level of activity (Andreux et al. 2018; Brook et al. 2016b; Carter et al. 2015; Coen et al. 2018; Gharahdaghi et al. 2019; Wilkinson et al. 2018). Once individuals reach the age of 60, there is an exponential decline in muscle size which when associated with declines in muscle function is termed sarcopenia (Nair 2005; Wilkinson et al. 2018). For reasons currently unknown (Papadopoulou 2020), this decline differs between males and females. Males usually display changes earlier than females, yet females usually displaying greater rates of decline than males. Recent studies examining the effect of exercise on human muscle growth have found that older human muscle does not respond the same as younger muscle in terms of signaling growth of the extracellular matrix that surrounds muscle, possibly signifying that alterations in the extramuscular scaffold underlie decreased growth with age (Deane et al. 2019; Wessner et al. 2019). This suggestion is yet to be tested with an intervention to restore growth.
5.5.2.2 Age-Associated Anabolic Resistance
Despite proteostasis being altered with age and across species (Lopez-Otin et al. 2013), it is not immediately clear that the causes and consequences are identical across species. For example, wide spread protein aggregation is not reported for human muscle with age (Wilkinson et al. 2018), unlike in some human neurodegenerative diseases (Currais et al. 2017). Further, baseline rates of muscle protein synthesis and degradation are not widely reported to be altered in older versus younger human muscle (Wilkinson et al. 2018). Rather, the responsiveness of muscle to anabolic stimuli such as feeding (Mitchell et al. 2016) and exercise (Durham et al. 2010) seems to be blunted (Wilkinson et al. 2018). That is, the increase in protein synthesis in response to anabolic stimuli is less in old muscle than in young. Thus, with similar rates of basal protein synthesis and degradation in young vs. old muscle but smaller post-meal increases in synthesis, muscle protein content gradually declines over time (Fig. 5.6). Accordingly, this gradual decline is sufficient to account for loss of muscle size which contributes to decreased function. It remains to be determined what causes the blunted anabolic responses in older muscle, presumably this is at the level of translation or later as anabolic signaling pathways via mTOR seem normally activated in older human muscle (Brook et al. 2016b). A recent study has suggested that impaired ribosomal biogenesis with advancing age may underly anabolic resistance (Brook et al. 2016a), which could lead to novel therapies for combating age-associated anabolic resistance.

Conceptual diagram of how changes in human muscle protein homeostasis drive changes in muscle size. Rates of Muscle Protein Synthesis (MPS) and Muscle Protein Breakdown (MPB) fluctuate during the day with rates of MPS increasing and rates of MPB decreasing immediately after a meal or ingestion of Essential Amino Acids (EAA). Decreases (a) or increases (b) in physical activity can decrease or increase the rate at which MPS is increased in response to eating a meal or ingesting EAA. Net losses (a, red curve) in the responsiveness of MPS (termed anabolic blunting) lead to muscle atrophy, whereas net increases (b, green curve) in the responsiveness of MPS lead to muscle hypertrophy. Note that in all cases, there is a net loss of muscle protein between meals (bottom, red) and a net increase in muscle protein post-meal (bottom, green). The anabolic blunting displayed in response to inactivity is also observed with age and disease, raising the issue of how much anabolic blunting with age or disease is due to decreased physical activity
5.5.3 Regenerative Rehabilitation and Protein Homeostasis
5.5.3.1 Hormone and Dietary Strategies
As discussed in Sect. 5.4.3.2, correction of clinical deficiencies in hormone levels or dietary intake have clear implications for muscle health, including size and protein content. It is possible that in the absence of clinical deficiencies supplementation combined with exercise may have positive effects, with both testosterone (Gharahdaghi et al. 2019) and vitamin D (McKendry et al. 2020) supplementation enhancing skeletal muscle responses to exercise. In addition, protein intake or absorption is often deficient in older individuals (Mitchell et al. 2016), especially in frail individuals. The amino acid leucine and its metabolite HMB have both been shown to be sufficient to produce the same anabolic muscle protein synthesis response as eating a protein-rich meal (Mitchell et al. 2016). Thus, targeted strategies using these nutrients and others, particularly as adjuncts to activity level, have been proposed and are under study, with a focus on particular factors such as concentration and timing (McKendry et al. 2020).
5.5.3.2 Ribosomal Biogenesis
The full cause of anabolic blunting currently remains unknown, but is associated with decreased levels of ribosomes that cause further declines in ribosome biogenesis (Brook et al. 2016a). As rRNA comprises the bulk of cellular RNA, which is a major component of ribosomes (Figueiredo and McCarthy 2019), it is possible that age-related declines in nucleic acids and/or RNA (Tahoe et al. 2004) manifest most notably as declines in rRNA. This would be entirely in keeping with the recent findings of cross-species NAD declines with advancing age (Schultz and Sinclair 2016). The idea that declines in ribosomal biogenesis underly age-related muscular deficits has been proposed as a general feature of aging (Steffen and Dillin 2016). If this proves true, then supplementation of nucleic acids rather than proteins, lipids, carbohydrates, or vitamins may be a viable strategy for increasing anabolic responses to exercise with age.
5.6 Glucose Metabolism
5.6.1 Introduction to Glucose Metabolism
Glucose homeostasis is critical to human health and survival due to the important role that glucose has in providing cellular energy. Any disturbances in glucose homeostasis can result in disease, most notably type 2 diabetes, but can also contribute to other life-altering conditions, including sarcopenia (Sugimoto et al. 2019) and dynapenia (Nebuloni et al. 2020; Kalyani et al. 2015). In healthy individuals, systemic glucose homeostasis is intricately regulated between tissue (i.e., muscle, liver, adipose, pancreas). Liver plays a primary role in regulating glucose homeostasis (glycogenolysis and gluconeogenesis) in the fasted state, while peripheral tissue plays a critical role in promoting glucose uptake and restoring normoglycemia after eating. As the largest organ in the body, skeletal muscle is considered a major regulator of whole-body glucose homeostasis, with 70–80% of insulin-stimulated glucose uptake occurring in this tissue (DeFronzo et al. 1981). Therefore, it is not surprising that the muscle atrophy that occurs with aging, plays a contributing role in the disruption of glucose homeostasis and pathogenesis of insulin resistance in these individuals (Sugimoto et al. 2019; Nebuloni et al. 2020; Kalyani et al. 2015).
In healthy adults, insulin-mediated glucose uptake occurs when insulin binds to the insulin receptor, initiating a signaling cascade including phosphorylation of the insulin receptor, insulin receptor substrate-1 (IRS-1) association with p85 subunit of phosphoinositide 3-kinase (PI3K), Akt2 phosphorylation on threonine 308 and serine 473 sites, and phosphorylation of the TBC1 domain family member 4 (TBC1D4) on multiple sites, subsequently allowing the translocation of glucose transporter type 4 (GLUT4) to the plasma membrane (Thorell et al. 1999; Kramer et al. 2006) (Fig. 5.7). In resting muscle, the majority of insulin-stimulated glucose undergoes nonoxidative metabolism (primarily converted to glycogen), while the remaining is oxidized (DeFronzo and Tripathy 2009). Impairments in any of these signaling pathways can cause skeletal muscle insulin resistance, which is believed to be the primary defect leading to the development of type 2 diabetes (DeFronzo and Tripathy 2009).

Potential cellular mechanisms for therapeutic enhancement of glucose metabolism and prevention of age-induced skeletal muscle dysfunction. (A) Decreased myostatin protein expression (i.e., via Ex-4), inhibition of myostatin (i.e., via anti-myostatin antibody landogrozumab), or inhibition of the myostatin receptor (i.e., via anti-ActRII antibody bimagrumab) results in (B) reductions in SMAD2/SMAD3 phosphorylation leading to (C) inhibition of the SMAD2/SMAD3/SMAD4 complex entering the nucleus, preventing the upregulation of atrophy genes involved in sarcopenia. SMAD2/SMAD3 dephosphorylation also (D) suppresses myostatin’s inhibition of insulin-stimulated Akt phosphorylation leading to (E) TBC1D4 phosphorylation, GLUT4 translocation to the plasma membrane, and increased glucose uptake, as well as, (F) increased protein synthesis (via mTOR signaling) and suppression of protein degradation (phosphorylation of FoxO inhibits its translocation to the nucleus where it would upregulate genes involved in autophagy and protein degradation). (G) Metformin increases the phosphorylation of AMPK, which increases glucose uptake (via TBC1D4) and protein synthesis (via mTOR signaling). (H) DPP-4 inhibitors increase GLUT4 content and insulin-stimulated glucose uptake. This image was created using BioRender (https://BioRender.com)
It is well known that the aging process is associated with whole-body insulin resistance (Consitt et al. 2013; Petersen et al. 2003; Fink et al. 1983, 1986; Rowe et al. 1983; Petersen et al. 2015). The factors contributing to age-related insulin resistance are likely multifaceted and include chronological age (Consitt et al. 2013), reduced physical activity (Amati et al. 2009), inflammation (Greiwe et al. 2001), and/or increased body fat (Kohrt et al. 1993; Amati et al. 2009). The obesity epidemic combined with the growing prevalence of sarcopenia in older adults has resulted in the concept of “sarcopenic obesity” (i.e., coexistence of obesity and sarcopenia conditions) (Stenholm et al. 2008; Cauley 2015). These individuals experience the negative metabolic effects of obesity, plus they have reduced skeletal muscle mass available for insulin-stimulated glucose uptake (Reaven 1988), putting them at even greater risk for developing insulin resistance and type 2 diabetes. While the prevalence of sarcopenic obesity varies depending on the operational definition, the National Health and Nutrition Examination Survey (NHANES) III study reported that 19% of women and 28% of men over the age of 60 suffered from this condition (Batsis et al. 2015), further highlighting the need to understand the interaction between metabolic dysfunction and sarcopenia.
A discussion on the effects of glucose metabolism and age-related muscle dysfunction would be remiss without mentioning that over 70% of Americans over the age of 65 are classified as either type 2 diabetic or prediabetic (Cowie et al. 2009). While the detrimental effects of insulin resistance and type 2 diabetes have been well documented for the general population, the consequences are even more dire for the elderly. Poor glycemic control and insulin resistance are well-known risk factors for sarcopenia (Sugimoto et al. 2019) and dynapenia (Nebuloni et al. 2020; Kalyani et al. 2015) and have been shown to accelerate age-related losses in both skeletal muscle mass (Park et al. 2009) and strength (Park et al. 2007). Therefore, the aging adult is often caught in a vicious cycle of glucose dysregulation and muscle dysfunction, which may be further exacerbated by conditions of obesity. The mechanism(s) responsible for age-related related impairments in glucose metabolism and muscle function remain unclear and are likely complex. Here, age-related impairments in insulin-stimulated glucose metabolism, accumulation of advanced glycation end-products (AGEs) and increases in myostatin are discussed.
5.6.2 Age-Induced Pathophysiology of Impaired Glucose Metabolism
5.6.2.1 Age-Related Impairments in Insulin-Stimulated Glucose Metabolism
Age-related impairments in the skeletal muscle insulin signaling cascade have been reported in both human (Consitt et al. 2013; Petersen et al. 2015) and animal models (Gupte et al. 2008; Sharma et al. 2010; Consitt et al. 2018). In aging humans, several impairments in distal insulin signaling have been reported. Peterson et al. (Petersen et al. 2015) reported diminished Akt phosphorylation after 20 min of hyperinsulinemia in aged individuals compared to their younger counterparts when body mass index (BMI), fat mass, and habitual physical activity were matched (Petersen et al. 2015). Additionally, in a cross-sectional study including individuals across a wide range of the adult life span (18–84 years of age), insulin-stimulated phosphorylation of AS160 on multiple sites (serine-588, threonine-642, and serine-666) were impaired in conjunction with age-related whole-body insulin resistance (Consitt et al. 2013). Skeletal muscle GLUT4 has also been reported to decrease with age in some (Consitt et al. 2013; Gaster et al. 2000; Houmard et al. 1995; Xu et al. 2017) but not all (Houmard et al. 1995; Cox et al. 1999; Dela et al. 1994) studies, and at least one study has suggested that age-related changes may be fiber-type specific with older adults having reduced GLUT4 in type II, but not type I fibers (Gaster et al. 2000).
Age-related impairments have been reported in both the insulin-stimulated oxidative and nonoxidative pathways (Bryhni et al. 2005; Poulsen et al. 2005; Franssila-Kallunki et al. 1992; Petersen et al. 2015; Consitt et al. 2016). Early findings that older adults had ~60% less skeletal muscle glycogen stores compared to their younger counterparts (Meredith et al. 1989), led to subsequent findings of diminished insulin-stimulated glycogen synthase activity in aged (Poulsen et al. 2005; Bienso et al. 2015) compared to younger adults (Poulsen et al. 2005; Pehleman et al. 2005). Reduced skeletal muscle pyruvate dehydrogenase (PDH) flux during insulin stimulation has also been reported in the elderly compared to young individuals (Petersen et al. 2015; Consitt et al. 2016), suggesting that under hyperinsulinemic conditions, PDH regulation may be compromised in older adults putting them at increased risk for metabolic inflexibility and the development of insulin resistance (Constantin-Teodosiu et al. 2015; Hansen et al. 2019).
Deletion of the insulin receptor in the skeletal muscle of mice has been reported to reduce skeletal muscle mass and grip strength by approximately 20% (O’Neill et al. 2016), supporting a direct role for insulin signaling on muscle mass and function. Besides stimulating glucose uptake, insulin-mediated Akt phosphorylation can promote protein synthesis through the Akt/mTOR/p70S6K signaling pathway (Wang and Proud 2006) and inhibit protein atrophy through the Akt/FoxO pathway (O’Neill et al. 2016). Given the crosstalk between these pathways, it is conceivable that age-related impairments in insulin signaling are responsible for both reduced glucose uptake and sarcopenia. Animal research suggests insulin acts to maintain muscle mass by suppressing Akt/FoxO1/3/4-mediated autophagy and protein degradation pathways (O’Neill et al. 2016). In humans, there remains considerable debate regarding which insulin-stimulated pathway is responsible for age-related sarcopenia and may be related additionally to insulin’s effect on blood flow, as well as amino acid availability and uptake (Rasmussen et al. 2006; Fujita et al. 2009; Wilkes et al. 2009).
5.6.2.2 Age-Related Accumulation of Advanced Glycation End-Products (AGEs)
With age-related insulin resistance, postprandial blood glucose levels become elevated for pronged periods of time. Poor glycemic control and chronic hyperglycemia (often measured as hemoglobin A1c, HbA1c) lead to the accumulation of advanced glycation end-products (AGEs). Elevated serum AGEs have been associated with decreased muscular performance, including reduced grip strength (Dalal et al. 2009), and slower walking speed (Semba et al. 2010) in adults aged 65 or older. Incubation of C2C12 myotubes (muscle cells) with AGE has been reported to reduce cell diameter (muscle atrophy) and increase atrogin-1 (protein involved in muscle atrophy) (Chiu et al. 2016), suggesting AGEs have a direct role in skeletal muscle wasting. While the mechanism remains to be fully elucidated, Chiu et al. (2016) proposed AGEs act through their receptor to activate AMPK and downregulate Akt signaling, resulting in atrophy and impairments in myogenesis. Recently, it was suggested that AGEs may promote mitochondrial dysfunction in skeletal muscle (Daussin et al. 2021), which theoretically would contribute further to loss of muscle mass and function. It should also be noted that chronic hyperglycemia and AGEs are believed to play an essential role in the development of diabetic peripheral neuropathy, a condition reported to be an independent risk factor for sarcopenia (Yang et al. 2020; Erbas et al. 2011; Addison et al. 2018; Sugimoto et al. 2008).
5.6.2.3 Age-Related Increases in Myostatin
The myokine myostatin, also referred to as growth and differentiation factor-8 (GDF-8), is a member of the transforming growth factor β (TGF-β) superfamily and is well-known negative regulator of skeletal muscle growth (McPherron et al. 1997). A number of studies have reported a link between increased myostatin levels, age (Yarasheski et al. 2002; Leger et al. 2008), declining muscle mass (Yarasheski et al. 2002; Leger et al. 2008), decreased muscular strength (Han et al. 2011; Patel et al. 2014), as well as, obesity (Hittel et al. 2009) and insulin resistance (Hjorth et al. 2016; Hittel et al. 2009; Ryan et al. 2013; Guo et al. 2009), suggesting this protein contributes to both muscle atrophy and insulin resistance in the elderly, especially those suffering from sarcopenic obesity.
At the cellular level, muscle growth is dependent on myogenesis which includes the differentiation and fusion of single-nucleated muscle cells (myoblasts) into multi-nucleated, mature myotubes. Myostatin is believed to promote muscle atrophy by suppressing the expression of genes involved in myogenesis through its canonical signaling pathway. Myostatin binds to the activin type IIB receptor (ActRIIB), leading to phosphorylation of Small Mothers Against Decapentaplegic (Smad) 2 and Smad3, which form a complex with Smad4 and translocate to the nucleus to inhibit the transcription of myogenic genes, including myogenic differentiation factor (MyoD) and myogenin (Langley et al. 2002; Trendelenburg et al. 2009) (Fig. 5.7). Myostatin-induced SMAD3 phosphorylation can also interact and inhibit the IGF-1/Akt/mTOR pathway, via inhibition of Akt phosphorylation (Trendelenburg et al. 2009; Goodman et al. 2013; Morissette et al. 2009). Since signaling pathways for protein synthesis and glucose uptake converge on Akt, it is not surprising that several studies have also provided convincing evidence that myostatin can inhibit insulin signaling by reducing Akt phosphorylation (Hittel et al. 2010; Zhang et al. 2011; Guo et al. 2009). Taken together, these findings support myostatin-induced inhibition of Akt as a mechanism for age-related muscle wasting and insulin resistance, especially in conditions of sarcopenic obesity.
5.6.3 Regenerative Medicine and Age-Impaired Glucose Metabolism
Therapeutic interventions to combat muscle wasting and insulin resistance have been historically studied independent of each other. For example, the discovery of myostatin as a negative regulator of skeletal muscle growth (McPherron et al. 1997) led to therapeutic strategies to target the myostatin signaling pathway, which focused on muscle mass/strength outcomes but neglected potential metabolic effects. The lack of translation of promising preclinical data to muscular dystrophy models (Wagner et al. 2008; St Andre et al. 2017; Wagner et al. 2020) combined with more recent findings that myostatin acts as a negative regulator of insulin signaling (Guo et al. 2009; Zhang et al. 2011) led to speculation that these therapeutics could be more beneficial in older adults, especially those suffering from sarcopenic obesity. In contrast to the lack of FDA-approved drugs to treat sarcopenia, several pharmacological treatments are available for older adults with impaired glucose metabolism. Whereas the metabolic outcomes from these treatments have been well documented (Musi et al. 2002; Kim et al. 2002; Kulkarni et al. 2018; Hirst et al. 2012; Sasaki et al. 2019; Huthmacher et al. 2020; Gedulin et al. 2005; Henry et al. 2018), research continues to examine if these therapeutics could also prevent sarcopenia in older adults. The following provides examples of each of these types of therapeutics with special attention towards how each can regulate glucose metabolism, as well as skeletal muscle mass and function.
5.6.3.1 Myostatin/ActIIB Inhibitors
There has been considerable interest in developing therapeutics to block myostatin’s inhibitory action on glucose uptake and muscle growth. Promising preclinical data demonstrated that mice treated with an anti-myostatin polyclonal antibody had a reversal of diet-induced whole-body insulin resistance, in conjunction with skeletal muscle changes including increased muscle mass, enhanced PI3K activity, Akt phosphorylation, GLUT4 protein expression, and increased phosphorylation of mTOR (Tang et al. 2014). Unfortunately, no known clinical studies have examined the effectiveness of these therapeutics in older sarcopenic patients with accompanying insulin resistance; however, early studies examining each condition separately have provided beneficial insight.
Phase II clinical studies blocking myostatin or its receptor (ActRIIB) suggest that despite increases in muscle mass, improvements in muscular function in non-obese, older adults, may be dependent on mobility/activity levels preceding or during treatment. For example, old (75 years or older), frail adults that had experienced at least one fall in the previous year, treated with the human monoclonal anti-myostatin antibody landogrozumab (LY2495655, Eli Lilly) demonstrated increased appendicular lean mass (~2.5%) and improvements in muscular performance on 12-step stair climb time (~ –1.3 s) (Becker et al. 2015). In contrast, when older non-obese, sarcopenic patients added bimagrumab (BYM338; Novartis), a human monoclonal anti-ActRII antibody, to nutritional counseling and a home-based exercise program, no additional improvements in muscular strength or mobility were observed, despite increases in lean mass (Rooks et al. 2020). This latter finding is of interest given an earlier proof-of-concept study that reported bimagrumab treatment improved muscle function in older sarcopenic patients when combined with nutritional counselling alone (Rooks et al. 2017). The effectiveness of myostatin inhibition on specific muscle contractile properties has been less clear with preclinical data producing concern that myostatin deletion could reduce specific force (Mendias et al. 2006; Mendias et al. 2011). Reduced SR Ca2+ release, possibly due to a decreased ability to refill the SR, has been suggested as a mechanism to explain the reduced specific force observed in hypermuscular (“compact”) mice with a mutation in the myostatin gene (Sztretye et al. 2017; Bodnar et al. 2014). It is possible that postnatal therapeutic blockade may be less detrimental on specific force, at least in mice (Bogdanovich et al. 2002, 2005; Mendias et al. 2015).
Early clinical studies suggest that therapeutics that block myostatin’s receptor can improve insulin sensitivity and glycemic control in individuals with impaired glucose metabolism. For example, a single intravenous dose of bimagrubmab increased insulin sensitivity by ~20% (measured by hyperinsulinemic-euglycemic clamp) and reduced HbA1C in middle-aged (mean age: ~45 years), insulin-resistant individuals (Garito et al. 2018). These metabolic improvements were recently extended to overweight and obese type 2 diabetics when bimagrubmab treatment was added to a nutritional and exercise counseling (Heymsfield et al. 2021). Given recent findings that increased intramuscular lipids play an inhibitory role (Choi et al. 2016) in the age-related reductions in specific force (Ochala et al. 2007; Larsson et al. 1997; Choi et al. 2016) and that myostatin inhibition consistently demonstrates reductions in fat mass (Garito et al. 2018; Heymsfield et al. 2021), future research in obese, sarcopenic adults is warranted.
While purely speculative, these clinical findings suggest that older adults without co-existing conditions of obesity or uncontrolled glycemia can achieve increases in muscle mass with therapeutics that block myostatin or its receptor; however, improvements in muscle performance appear to be specific to frail or inactive older adults where these therapeutics could compensate for the lack of metabolic adaptations that regular muscle contractions induce. In contrast, it is possible that these therapeutics can improve insulin sensitivity and glycemic control above the benefits achieved by lifestyle modifications in insulin-resistant individuals; however, the potential effects on muscle mass, contractile properties, and muscle function in these individuals remain to be elucidated.
5.6.3.2 Pharmacological Agents
Metformin, an AMPK agonist, is often the first-line of treatment for insulin resistance, including those of advancing age due to its ability to enhance insulin sensitivity (Musi et al. 2002; Kim et al. 2002), improve glucose tolerance (Kulkarni et al. 2018), lower HbA1c levels (Hirst et al. 2012) and reduce type 2 diabetes incidence rates (Diabetes Prevention Program Research Group 2015). The mechanism(s) through which metformin improves glucose homeostasis are still under debate but likely involves the drug’s ability to target numerous tissues, including the liver, skeletal muscle, and adipose tissue (Konopka and Miller 2019). Metformin is commonly cross-referenced as a calorie restrictive or exercise mimetic since it increases AMPK activity and has been shown to extend lifespan (Novelle et al. 2016; Martin-Montalvo et al. 2013; Sharoff et al. 2010). Additionally, metformin treatment has improved glucose intolerance in older adults (Kulkarni et al. 2018) and whole-body insulin sensitivity in type 2 diabetics (Musi et al. 2002; Kim et al. 2002). Of interest, these improvements in glucose metabolism do not appear to be related to changes in proximal insulin signaling (Kim et al. 2002). Instead, metformin incubation of muscle cells has demonstrated increases in AS160 phosphorylation (Lee et al. 2011b), suggesting improvements may occur distally in the insulin signaling cascade where older adults appear to be most susceptible (Consitt et al. 2013). The effects of metformin on sarcopenia and dynapenia are less clear with reports that it may prevent sarcopenia in elderly adults with diabetes (Chen et al. 2020a; Lee et al. 2011a), but may also prevent gains in muscle mass (Walton et al. 2019; Long et al. 2021) and strength (Long et al. 2021) when prescribed in combination with resistance training, potentially due to the lack of increase in type II fiber frequency in older adults (Long et al. 2021) and/or hyperphosphorylation of AMPK and dampening of mTOR signaling (Walton et al. 2019).
Sodium-glucose cotransporter 2 (SGLT2) inhibitors are effective in lowering blood glucose and HbA1c levels (Sasaki et al. 2019) by preventing glucose absorption in the kidneys, but their impact on sarcopenia and dynapenia remains unclear. Despite studies in middle to older aged type 2 diabetics reporting increased grip strength (Sano et al. 2016) and no loss in muscle mass (Sugiyama et al. 2018) with SGLT2 treatment, others have reported these drugs may accelerate diabetes-associated sarcopenia, especially in the elderly (Yabe et al. 2015; Sasaki et al. 2019). While the mechanisms responsible for reductions in muscle mass remain unknown, Yabe et al. (2015) speculated it may be related hypoglycemia events causing low circulating insulin levels to reduce glucose and amino acid entry into the skeletal muscle.
Treatment with GLP-1 receptor (GLP-1R) agonists has proven to be useful in managing hyperglycemia and decreasing HbA1c levels (Huthmacher et al. 2020; Gedulin et al. 2005; Henry et al. 2018). Recent in vitro research suggests these agonists may have a direct role on muscle. Treatment of C2C12 myotubes with the GLP1R agonist, exendin-4 (Ex-4), resulted in the suppression of myostatin and muscle atrophic genes (atrogin-1 and Murf-1) along with increasing protein synthesis (Hong et al. 2019). In addition, it has also been suggested Ex-4 may have neuroprotective properties based on findings that treatment of a mouse model of Parkinson’s disease protected dopaminergic neurons against degeneration and improved motor function (Li et al. 2009). DPP-4 inhibitors block the DPP-4 degradation of circulating incretins, including GLP-1 (Herman et al. 2005; Berg et al. 2011) and have proven effective in reducing HbA1c levels (Dicker 2011), increasing skeletal muscle insulin-stimulated glucose uptake (Sato et al. 2016) and increasing GLUT4 expression (Giannocco et al. 2013). In addition, DPP-4 inhibitors have been reported to reduce the progression of sarcopenia in elderly type 2 diabetics (Rizzo et al. 2016). Although it remains unclear if DPP-4 inhibitors act directly on the muscle to provide this protective effect on muscle mass, these findings are encouraging and warrant future research.
Taken together, there is promising data suggesting inhibitors that target the myostatin pathway and some pharmacologic agents traditionally used to regulate glycemic control could provide improvements in both glucose metabolism, as well as muscle mass and strength in older adults. However, it is evident that despite a clear link between glucose dysregulation, sarcopenia and dynapenia, the cellular mechanisms contributing to this relationship and the type of therapeutics required may differ among individuals. Factors including, but not limited to, obesity, degree of glucose dysregulation, previous and current physical activity can all impact responses depending on the therapeutic used and should be considered.
5.7 Conclusions of the Chapter
Deficits in neuromuscular physiological factors contribute to skeletal muscle dysfunction that manifests in pervasive chronic conditions and disability are observed in many older adults. Moreover, scientific and medical communities agree that dynapenia is a major determinant of physical limitations and general poor health in older adults. The neuromuscular mechanisms of dynapenia and more broadly, muscle dysfunction, are multi-factorial and likely include several domains that are affected by the aging process. We propose some of these domains are loss of neuromuscular excitation, EC coupling failure, sarcopenia (via loss of protein homeostasis) and metabolic dysfunction (via mitochondrial dysfunction and impaired glucose metabolism) (Fig. 5.1). However, we acknowledge these domains are likely a few of many, and are all interconnected. It appears that if dynapenia and sarcopenia continue to worsen with age that a vicious cycle of continued neuromuscular deficits ensues, which can ultimately lead to physical frailty. Thus, maintenance of neuromuscular function is a critical component to healthy aging. Within this chapter, we have listed several approaches that yield promise in delaying or preventing many domains of age-related neuromuscular deficits. However, we urge caution in that many of our discussed approaches are speculative and in the early stages of investigations. With that stated, regenerative rehabilitation in sarcopenia, dynapenia, and frailty in both basic laboratory and clinical settings that utilizes a team science approach is essential to advancing the aging field forward.
Notes
- 1.
In this article we use terms that have been more commonly used over the past decade and longer to specifically refer to age-related muscle weakness (i.e., dynapenia) and wasting (i.e., sarcopenia). We should note, however, that the operational definition of “sarcopenia” is still fluid and that recent consensus statements now emphasizing low muscle strength as the primary characteristic of sarcopenia (Cruz-Jentoft et al. 2019).
References
Addison O, Prior SJ, Kundi R, Serra MC, Katzel LI, Gardner AW, Ryan AS (2018) Sarcopenia in peripheral arterial disease: prevalence and effect on functional status. Arch Phys Med Rehabil 99(4):623–628. https://doi.org/10.1016/j.apmr.2017.10.017
Ahern GP, Junankar PR, Dulhunty AF (1997) Subconductance states in single-channel activity of skeletal muscle ryanodine receptors after removal of FKBP12. Biophys J 72(1):146–162. https://doi.org/10.1016/s0006-3495(97)78654-5
Al Snih S, Markides KS, Ottenbacher KJ, Raji MA (2004) Hand grip strength and incident ADL disability in elderly Mexican Americans over a seven-year period. Aging Clin Exp Res 16(6):481–486. https://doi.org/10.1007/BF03327406
Amati F, Dube JJ, Coen PM, Stefanovic-Racic M, Toledo FG, Goodpaster BH (2009) Physical inactivity and obesity underlie the insulin resistance of aging. Diabetes Care 32(8):1547–1549. https://doi.org/10.2337/dc09-0267
Anderson AA, Treves S, Biral D, Betto R, Sandonà D, Ronjat M, Zorzato F (2003) The novel skeletal muscle sarcoplasmic reticulum JP-45 protein. Molecular cloning, tissue distribution, developmental expression, and interaction with alpha 1.1 subunit of the voltage-gated calcium channel. J Biol Chem 278(41):39987–39992. https://doi.org/10.1074/jbc.M305016200
Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR (2011) Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging. Cell Metab 14(2):196–207. https://doi.org/10.1016/j.cmet.2011.05.014
Andreux PA, van Diemen MPJ, Heezen MR, Auwerx J, Rinsch C, Groeneveld GJ, Singh A (2018) Mitochondrial function is impaired in the skeletal muscle of pre-frail elderly. Sci Rep 8(1):8548. https://doi.org/10.1038/s41598-018-26944-x
Antonini A, Leenders KL (1993) Dopamine D2 receptors in normal human brain: effect of age measured by positron emission tomography (PET) and [11C]-raclopride. Ann N Y Acad Sci 695:81–85. https://doi.org/10.1111/j.1749-6632.1993.tb23033.x
Ashe J (1997) Force and the motor cortex. Behav Brain Res 87(2):255–269. https://doi.org/10.1016/s0166-4328(97)00752-3
Atherton PJ, Greenhaff PL, Phillips SM, Bodine SC, Adams CM, Lang CH (2016) Control of skeletal muscle atrophy in response to disuse: clinical/preclinical contentions and fallacies of evidence. Am J Physiol Endocrinol Metab 311(3):E594–E604. https://doi.org/10.1152/ajpendo.00257.2016
Baker JS, McCormick MC, Robergs RA (2010) Interaction among skeletal muscle metabolic energy systems during intense exercise. J Nutr Metab 2010:905612. https://doi.org/10.1155/2010/905612
Barry BK, Riek S, Carson RG (2005) Muscle coordination during rapid force production by young and older adults. J Gerontol A Biol Sci Med Sci 60(2):232–240. https://doi.org/10.1093/gerona/60.2.232
Bass JJ, Nakhuda A, Deane CS, Brook MS, Wilkinson DJ, Phillips BE, Philp A, Tarum J, Kadi F, Andersen D, Garcia AM, Smith K, Gallagher IJ, Szewczyk NJ, Cleasby ME, Atherton PJ (2020) Overexpression of the vitamin D receptor (VDR) induces skeletal muscle hypertrophy. Mol Metab 42:101059. https://doi.org/10.1016/j.molmet.2020.101059
Batsis JA, Mackenzie TA, Lopez-Jimenez F, Bartels SJ (2015) Sarcopenia, sarcopenic obesity, and functional impairments in older adults: National Health and Nutrition Examination Surveys 1999-2004. Nutr Res 35(12):1031–1039. https://doi.org/10.1016/j.nutres.2015.09.003
Baumann CW, Rogers RG, Gahlot N, Ingalls CP (2014) Eccentric contractions disrupt FKBP12 content in mouse skeletal muscle. Physiol Rep 2(7). https://doi.org/10.14814/phy2.12081
Baumann CW, Kwak D, Liu HM, Thompson LV (2016) Age-induced oxidative stress: how does it influence skeletal muscle quantity and quality? J Appl Physiol (1985) 121(5):1047–1052. https://doi.org/10.1152/japplphysiol.00321.2016
Baumann CW, Kwak D, Thompson LV (2018) Assessing onset, prevalence and survival in mice using a frailty phenotype. Aging (Albany NY) 10(12):4042–4053. https://doi.org/10.18632/aging.101692
Baumann CW, Kwak D, Thompson LV (2020) Phenotypic frailty assessment in mice: development, discoveries, and experimental considerations. Physiology (Bethesda) 35(6):405–414. https://doi.org/10.1152/physiol.00016.2020
Becker C, Lord SR, Studenski SA, Warden SJ, Fielding RA, Recknor CP, Hochberg MC, Ferrari SL, Blain H, Binder EF, Rolland Y, Poiraudeau S, Benson CT, Myers SL, Hu L, Ahmad QI, Pacuch KR, Gomez EV, Benichou O, STEADY Group (2015) Myostatin antibody (LY2495655) in older weak fallers: a proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol 3(12):948–957. https://doi.org/10.1016/S2213-8587(15)00298-3
Bellinger AM, Mongillo M, Marks AR (2008a) Stressed out: the skeletal muscle ryanodine receptor as a target of stress. J Clin Invest 118(2):445–453. https://doi.org/10.1172/jci34006
Bellinger AM, Reiken S, Dura M, Murphy PW, Deng SX, Landry DW, Nieman D, Lehnart SE, Samaru M, LaCampagne A, Marks AR (2008b) Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci U S A 105(6):2198–2202. https://doi.org/10.1073/pnas.0711074105
Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, Matecki S, Lacampagne A, Marks AR (2009) Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med 15(3):325–330. https://doi.org/10.1038/nm.1916
Ben-Itzhak R, Giladi N, Gruendlinger L, Hausdorff JM (2008) Can methylphenidate reduce fall risk in community-living older adults? A double-blind, single-dose cross-over study. J Am Geriatr Soc 56(4):695–700. https://doi.org/10.1111/j.1532-5415.2007.01623.x
Berardelli A, Rothwell JC, Thompson PD, Hallett M (2001) Pathophysiology of bradykinesia in Parkinson’s disease. Brain 124(Pt 11):2131–2146. https://doi.org/10.1093/brain/124.11.2131
Berg JK, Shenouda SK, Heilmann CR, Gray AL, Holcombe JH (2011) Effects of exenatide twice daily versus sitagliptin on 24-h glucose, glucoregulatory and hormonal measures: a randomized, double-blind, crossover study. Diabetes Obes Metab 13(11):982–989. https://doi.org/10.1111/j.1463-1326.2011.01428.x
Bhasin S, Travison TG, Manini TM, Patel S, Pencina KM, Fielding RA, Magaziner JM, Newman AB, Kiel DP, Cooper C, Guralnik JM, Cauley JA, Arai H, Clark BC, Landi F, Schaap LA, Pereira SL, Rooks D, Woo J, Woodhouse LJ, Binder E, Brown T, Shardell M, Xue QL, D’Agostino RB Sr, Orwig D, Gorsicki G, Correa-De-Araujo R, Cawthon PM (2020) Sarcopenia definition: the position statements of the sarcopenia definition and outcomes consortium. J Am Geriatr Soc. https://doi.org/10.1111/jgs.16372
Bienso RS, Olesen J, Gliemann L, Schmidt JF, Matzen MS, Wojtaszewski JF, Hellsten Y, Pilegaard H (2015) Effects of exercise training on regulation of skeletal muscle glucose metabolism in elderly men. J Gerontol A Biol Sci Med Sci 70(7):866–872. https://doi.org/10.1093/gerona/glv012
Bilder RM, Volavka J, Lachman HM, Grace AA (2004) The catechol-O-methyltransferase polymorphism: relations to the tonic-phasic dopamine hypothesis and neuropsychiatric phenotypes. Neuropsychopharmacology 29(11):1943–1961. https://doi.org/10.1038/sj.npp.1300542
Bodnar D, Geyer N, Ruzsnavszky O, Olah T, Hegyi B, Sztretye M, Fodor J, Dienes B, Balogh A, Papp Z, Szabo L, Muller G, Csernoch L, Szentesi P (2014) Hypermuscular mice with mutation in the myostatin gene display altered calcium signalling. J Physiol 592(6):1353–1365. https://doi.org/10.1113/jphysiol.2013.261958
Boengler K, Heusch G, Schulz R (2011) Nuclear-encoded mitochondrial proteins and their role in cardioprotection. Biochim Biophys Acta 1813(7):1286–1294. https://doi.org/10.1016/j.bbamcr.2011.01.009
Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S (2017) Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle 8(3):349–369. https://doi.org/10.1002/jcsm.12178
Bogdanovich S, Krag TO, Barton ER, Morris LD, Whittemore LA, Ahima RS, Khurana TS (2002) Functional improvement of dystrophic muscle by myostatin blockade. Nature 420(6914):418–421. https://doi.org/10.1038/nature01154
Bogdanovich S, Perkins KJ, Krag TO, Whittemore LA, Khurana TS (2005) Myostatin propeptide-mediated amelioration of dystrophic pathophysiology. FASEB J 19(6):543–549. https://doi.org/10.1096/fj.04-2796com
Boncompagni S, d’Amelio L, Fulle S, Fanò G, Protasi F (2006) Progressive disorganization of the excitation-contraction coupling apparatus in aging human skeletal muscle as revealed by electron microscopy: a possible role in the decline of muscle performance. J Gerontol A Biol Sci Med Sci 61(10):995–1008. https://doi.org/10.1093/gerona/61.10.995
Boncompagni S, Pecorai C, Michelucci A, Pietrangelo L, Protasi F (2020) Long-term exercise reduces formation of tubular aggregates and promotes maintenance of Ca(2+) entry units in aged muscle. Front Physiol 11:601057. https://doi.org/10.3389/fphys.2020.601057
Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasová E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR (1994) Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell 77(4):513–523. https://doi.org/10.1016/0092-8674(94)90214-3
Brook MS, Wilkinson DJ, Mitchell WK, Lund JN, Phillips BE, Szewczyk NJ, Greenhaff PL, Smith K, Atherton PJ (2016a) Synchronous deficits in cumulative muscle protein synthesis and ribosomal biogenesis underlie age-related anabolic resistance to exercise in humans. J Physiol 594(24):7399–7417. https://doi.org/10.1113/JP272857
Brook MS, Wilkinson DJ, Phillips BE, Perez-Schindler J, Philp A, Smith K, Atherton PJ (2016b) Skeletal muscle homeostasis and plasticity in youth and ageing: impact of nutrition and exercise. Acta Physiol (Oxf) 216(1):15–41. https://doi.org/10.1111/apha.12532
Bryhni B, Jenssen TG, Olafsen K, Bendikssen A (2005) Oxidative and nonoxidative glucose disposal in elderly vs younger men with similar and smaller body mass indices and waist circumferences. Metabolism 54(6):748–755. https://doi.org/10.1016/j.metabol.2005.01.016
Buchman AS, Wilson RS, Boyle PA, Bienias JL, Bennett DA (2007) Grip strength and the risk of incident Alzheimer’s disease. Neuroepidemiology 29(1–2):66–73. https://doi.org/10.1159/000109498
Buhusi CV, Meck WH (2005) What makes us tick? Functional and neural mechanisms of interval timing. Nat Rev Neurosci 6(10):755–765. https://doi.org/10.1038/nrn1764
Calderón JC, Bolaños P, Caputo C (2014) The excitation-contraction coupling mechanism in skeletal muscle. Biophys Rev 6(1):133–160. https://doi.org/10.1007/s12551-013-0135-x
Carbone JW, McClung JP, Pasiakos SM (2019) Recent advances in the characterization of skeletal muscle and whole-body protein responses to dietary protein and exercise during negative energy balance. Adv Nutr 10(1):70–79. https://doi.org/10.1093/advances/nmy087
Carlsson A, Winblad B (1976) Influence of age and time interval between death and autopsy on dopamine and 3-methoxytyramine levels in human basal ganglia. J Neural Transm 38(3–4):271–276. https://doi.org/10.1007/BF01249444
Carson RG (2018) Get a grip: individual variations in grip strength are a marker of brain health. Neurobiol Aging 71:189–222. https://doi.org/10.1016/j.neurobiolaging.2018.07.023
Carter HN, Chen CC, Hood DA (2015) Mitochondria, muscle health, and exercise with advancing age. Physiology (Bethesda) 30(3):208–223. https://doi.org/10.1152/physiol.00039.2014
Catterall WA, Goldin AL, Waxman SG (2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 57(4):397–409. https://doi.org/10.1124/pr.57.4.4
Cauley JA (2015) An overview of sarcopenic obesity. J Clin Densitom 18(4):499–505. https://doi.org/10.1016/j.jocd.2015.04.013
Celis-Morales CA, Welsh P, Lyall DM, Steell L, Petermann F, Anderson J, Iliodromiti S, Sillars A, Graham N, Mackay DF, Pell JP, Gill JMR, Sattar N, Gray SR (2018) Associations of grip strength with cardiovascular, respiratory, and cancer outcomes and all cause mortality: prospective cohort study of half a million UK Biobank participants. BMJ 361:k1651. https://doi.org/10.1136/bmj.k1651
Cham R, Studenski SA, Perera S, Bohnen NI (2008) Striatal dopaminergic denervation and gait in healthy adults. Exp Brain Res 185(3):391–398. https://doi.org/10.1007/s00221-007-1161-3
Chang SF, Lin PL (2015) Frail phenotype and mortality prediction: a systematic review and meta-analysis of prospective cohort studies. Int J Nurs Stud 52(8):1362–1374. https://doi.org/10.1016/j.ijnurstu.2015.04.005
Chapman J, Fielder E, Passos JF (2019) Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 593(13):1566–1579. https://doi.org/10.1002/1873-3468.13498
Chen F, Xu S, Wang Y, Chen F, Cao L, Liu T, Huang T, Wei Q, Ma G, Zhao Y, Wang D (2020a) Risk factors for sarcopenia in the elderly with type 2 diabetes mellitus and the effect of metformin. J Diabetes Res 2020:3950404. https://doi.org/10.1155/2020/3950404
Chen G, Kroemer G, Kepp O (2020b) Mitophagy: an emerging role in aging and age-associated diseases. Front Cell Dev Biol 8:200. https://doi.org/10.3389/fcell.2020.00200
Chen W, Datzkiw D, Rudnicki MA (2020c) Satellite cells in ageing: use it or lose it. Open Biol 10(5):200048. https://doi.org/10.1098/rsob.200048
Chiu CY, Yang RS, Sheu ML, Chan DC, Yang TH, Tsai KS, Chiang CK, Liu SH (2016) Advanced glycation end-products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE-mediated, AMPK-down-regulated, Akt pathway. J Pathol 238(3):470–482. https://doi.org/10.1002/path.4674
Choi SJ, Files DC, Zhang T, Wang ZM, Messi ML, Gregory H, Stone J, Lyles MF, Dhar S, Marsh AP, Nicklas BJ, Delbono O (2016) Intramyocellular lipid and impaired myofiber contraction in normal weight and obese older adults. J Gerontol A Biol Sci Med Sci 71(4):557–564. https://doi.org/10.1093/gerona/glv169
Christie A, Kamen G (2006) Doublet discharges in motoneurons of young and older adults. J Neurophysiol 95(5):2787–2795. https://doi.org/10.1152/jn.00685.2005. 00685.2005 [pii]
Christie A, Kamen G (2010) Short-term training adaptations in maximal motor unit firing rates and afterhyperpolarization duration. Muscle Nerve 41(5):651–660. https://doi.org/10.1002/mus.21539
Churchward-Venne TA, Tieland M, Verdijk LB, Leenders M, Dirks ML, de Groot LC, van Loon LJ (2015) There are no nonresponders to resistance-type exercise training in older men and women. J Am Med Dir Assoc 16(5):400–411. https://doi.org/10.1016/j.jamda.2015.01.071
Clark BC, Manini TM (2008) Sarcopenia =/= dynapenia. J Gerontol A Biol Sci Med Sci 63(8):829–834. 63/8/829 [pii]
Clark BC, Taylor JL (2011) Age-related changes in motor cortical properties and voluntary activation of skeletal muscle. Curr Aging Sci. https://doi.org/10.2174/1874609811104030192
Clark BC, Mahato NK, Nakazawa M, Law TD, Thomas JS (2014) The power of the mind: the cortex as a critical determinant of muscle strength/weakness. J Neurophysiol 112(12):3219–3226. https://doi.org/10.1152/jn.00386.2014
Clark BC, Taylor JL, Hong SL, Law TD, Russ DW (2015) Weaker seniors exhibit motor cortex hypoexcitability and impairments in voluntary activation. J Gerontol A Biol Sci Med Sci 70(9):1112–1119. https://doi.org/10.1093/gerona/glv030
Clark BC, Manini TM, Wages NP, Simon JE, Clark LA (2019) Voluntary vs electrically stimulated activation in age-related muscle weakness. JAMA Netw Open 2(9):e1912052. https://doi.org/10.1001/jamanetworkopen.2019.12052
Clark LA, Manini TM, Wages NP, Simon JE, Russ DW, Clark BC (2020) Reduced neural excitability and activation contribute to clinically-meaningful weakness in older adults. J Gerontol A Biol Sci Med Sci. https://doi.org/10.1093/gerona/glaa157
Coen PM, Musci RV, Hinkley JM, Miller BF (2018) Mitochondria as a target for mitigating sarcopenia. Front Physiol 9:1883. https://doi.org/10.3389/fphys.2018.01883
Cohen JA, Verghese J (2019) Gait and dementia. Handb Clin Neurol 167:419–427. https://doi.org/10.1016/B978-0-12-804766-8.00022-4
Consitt LA, Van Meter J, Newton CA, Collier DN, Dar MS, Wojtaszewski JF, Treebak JT, Tanner CJ, Houmard JA (2013) Impairments in site-specific AS160 phosphorylation and effects of exercise training. Diabetes 62(10):3437–3447. https://doi.org/10.2337/db13-0229
Consitt LA, Saxena G, Saneda A, Houmard JA (2016) Age-related impairments in skeletal muscle PDH phosphorylation and plasma lactate are indicative of metabolic inflexibility and the effects of exercise training. Am J Physiol Endocrinol Metab 311(1):E145–E156. https://doi.org/10.1152/ajpendo.00452.2015
Consitt LA, Saxena G, Slyvka Y, Clark BC, Friedlander M, Zhang Y, Nowak FV (2018) Paternal high-fat diet enhances offspring whole-body insulin sensitivity and skeletal muscle insulin signaling early in life. Physiol Rep 6(5):doi:10.14814/phy2.13583
Constantin-Teodosiu D, Stephens FB, Greenhaff PL (2015) Perpetual muscle PDH activation in PDH kinase knockout mice protects against high-fat feeding-induced muscle insulin resistance. Proc Natl Acad Sci U S A 112(8):E824. https://doi.org/10.1073/pnas.1422929112
Cowie CC, Rust KF, Ford ES, Eberhardt MS, Byrd-Holt DD, Li C, Williams DE, Gregg EW, Bainbridge KE, Saydah SH, Geiss LS (2009) Full accounting of diabetes and pre-diabetes in the U.S. population in 1988-1994 and 2005-2006. Diabetes Care 32(2):287–294. https://doi.org/10.2337/dc08-1296
Cox JH, Cortright RN, Dohm GL, Houmard JA (1999) Effect of aging on response to exercise training in humans: skeletal muscle GLUT-4 and insulin sensitivity. J Appl Physiol (1985) 86(6):2019–2025. https://doi.org/10.1152/jappl.1999.86.6.2019
Crawford ED, Wells JA (2011) Caspase substrates and cellular remodeling. Annu Rev Biochem 80:1055–1087. https://doi.org/10.1146/annurev-biochem-061809-121639
Crescenzo R, Lionetti L, Mollica MP, Ferraro M, D’Andrea E, Mainieri D, Dulloo AG, Liverini G, Iossa S (2006) Altered skeletal muscle subsarcolemmal mitochondrial compartment during catch-up fat after caloric restriction. Diabetes 55(8):2286–2293. https://doi.org/10.2337/db06-0312
Cruz-Jentoft AJ, Bahat G, Bauer J, Boirie Y, Bruyère O, Cederholm T, Cooper C, Landi F, Rolland Y, Sayer AA (2019) Sarcopenia: revised European consensus on definition and diagnosis. Age Ageing 48(1):16–31
Currais A, Fischer W, Maher P, Schubert D (2017) Intraneuronal protein aggregation as a trigger for inflammation and neurodegeneration in the aging brain. FASEB J 31(1):5–10. https://doi.org/10.1096/fj.201601184
Dalal M, Ferrucci L, Sun K, Beck J, Fried LP, Semba RD (2009) Elevated serum advanced glycation end products and poor grip strength in older community-dwelling women. J Gerontol A Biol Sci Med Sci 64(1):132–137. https://doi.org/10.1093/gerona/gln018
Daussin FN, Boulanger E, Lancel S (2021) From mitochondria to sarcopenia: role of inflammaging and RAGE-ligand axis implication. Exp Gerontol 146:111247. https://doi.org/10.1016/j.exger.2021.111247
Deane CS, Ames RM, Phillips BE, Weedon MN, Willis CRG, Boereboom C, Abdulla H, Bukhari SSI, Lund JN, Williams JP, Wilkinson DJ, Smith K, Gallagher IJ, Kadi F, Szewczyk NJ, Atherton PJ, Etheridge T (2019) The acute transcriptional response to resistance exercise: impact of age and contraction mode. Aging (Albany NY) 11(7):2111–2126. https://doi.org/10.18632/aging.101904
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163. https://doi.org/10.2337/dc09-S302
DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP (1981) The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 30(12):1000–1007
Dela F, Ploug T, Handberg A, Petersen LN, Larsen JJ, Mikines KJ, Galbo H (1994) Physical training increases muscle GLUT4 protein and mRNA in patients with NIDDM. Diabetes 43(7):862–865. https://doi.org/10.2337/diab.43.7.862
Delbono O (2011) Expression and regulation of excitation-contraction coupling proteins in aging skeletal muscle. Curr Aging Sci 4(3):248–259. https://doi.org/10.2174/1874609811104030248
Delbono O, O’Rourke KS, Ettinger WH (1995) Excitation-calcium release uncoupling in aged single human skeletal muscle fibers. J Membr Biol 148(3):211–222. https://doi.org/10.1007/bf00235039
Delmonico MJ, Harris TB, Visser M, Park SW, Conroy MB, Velasquez-Mieyer P, Boudreau R, Manini TM, Nevitt M, Newman AB, Goodpaster BH (2009) Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am J Clin Nutr 90(6):1579–1585. https://doi.org/10.3945/ajcn.2009.28047. ajcn.2009.28047 [pii]
Dent E, Morley JE, Cruz-Jentoft AJ, Arai H, Kritchevsky SB, Guralnik J, Bauer JM, Pahor M, Clark BC, Cesari M, Ruiz J, Sieber CC, Aubertin-Leheudre M, Waters DL, Visvanathan R, Landi F, Villareal DT, Fielding R, Won CW, Theou O, Martin FC, Dong B, Woo J, Flicker L, Ferrucci L, Merchant RA, Cao L, Cederholm T, Ribeiro SML, Rodriguez-Manas L, Anker SD, Lundy J, Gutierrez Robledo LM, Bautmans I, Aprahamian I, Schols J, Izquierdo M, Vellas B (2018) International Clinical Practice Guidelines for Sarcopenia (ICFSR): screening, diagnosis and management. J Nutr Health Aging 22(10):1148–1161. https://doi.org/10.1007/s12603-018-1139-9
Diabetes Prevention Program Research Group (2015) Long-term effects of lifestyle intervention or metformin on diabetes development and microvascular complications over 15-year follow-up: the Diabetes Prevention Program Outcomes Study. Lancet Diabetes Endocrinol 3(11):866–875. https://doi.org/10.1016/S2213-8587(15)00291-0
Dicker D (2011) DPP-4 inhibitors: impact on glycemic control and cardiovascular risk factors. Diabetes Care 34(Suppl 2):S276–S278. https://doi.org/10.2337/dc11-s229
Distefano G, Goodpaster BH (2018) Effects of exercise and aging on skeletal muscle. Cold Spring Harb Perspect Med 8(3). https://doi.org/10.1101/cshperspect.a029785
Du J, Zhu M, Bao H, Li B, Dong Y, Xiao C, Zhang GY, Henter I, Rudorfer M, Vitiello B (2016) The role of nutrients in protecting mitochondrial function and neurotransmitter signaling: implications for the treatment of depression, PTSD, and suicidal behaviors. Crit Rev Food Sci Nutr 56(15):2560–2578. https://doi.org/10.1080/10408398.2013.876960
Duchowny K (2019) Do nationally representative cutpoints for clinical muscle weakness predict mortality? Results from 9 years of follow-up in the health and retirement study. J Gerontol A Biol Sci Med Sci 74(7):1070–1075. https://doi.org/10.1093/gerona/gly169
Duncan MJ, Clarke ND, Tallis J, Guimaraes-Ferreira L, Leddington Wright S (2014) The effect of caffeine ingestion on functional performance in older adults. J Nutr Health Aging 18(10):883–887. https://doi.org/10.1007/s12603-014-0474-8
Durham WJ, Casperson SL, Dillon EL, Keske MA, Paddon-Jones D, Sanford AP, Hickner RC, Grady JJ, Sheffield-Moore M (2010) Age-related anabolic resistance after endurance-type exercise in healthy humans. FASEB J 24(10):4117–4127. https://doi.org/10.1096/fj.09-150177
Enoka RM (1988) Muscle strength and its development. New perspectives. Sports Med 6(3):146–168
Enoka RM, Christou EA, Hunter SK, Kornatz KW, Semmler JG, Taylor AM, Tracy BL (2003) Mechanisms that contribute to differences in motor performance between young and old adults. J Electromyogr Kinesiol 13(1):1–12. S1050641102000846 [pii]
Erbas T, Ertas M, Yucel A, Keskinaslan A, Senocak M, TURNEP Study Group (2011) Prevalence of peripheral neuropathy and painful peripheral neuropathy in Turkish diabetic patients. J Clin Neurophysiol 28(1):51–55. https://doi.org/10.1097/WNP.0b013e3182051334
Evans WJ, Hellerstein M, Orwoll E, Cummings S, Cawthon PM (2019) D3 -Creatine dilution and the importance of accuracy in the assessment of skeletal muscle mass. J Cachexia Sarcopenia Muscle 10(1):14–21. https://doi.org/10.1002/jcsm.12390
Ferreira R, Vitorino R, Alves RM, Appell HJ, Powers SK, Duarte JA, Amado F (2010) Subsarcolemmal and intermyofibrillar mitochondria proteome differences disclose functional specializations in skeletal muscle. Proteomics 10(17):3142–3154. https://doi.org/10.1002/pmic.201000173
Figueiredo VC, McCarthy JJ (2019) Regulation of ribosome biogenesis in skeletal muscle hypertrophy. Physiology (Bethesda) 34(1):30–42. https://doi.org/10.1152/physiol.00034.2018
Fill M, Copello JA (2002) Ryanodine receptor calcium release channels. Physiol Rev 82(4):893–922. https://doi.org/10.1152/physrev.00013.2002
Fink RI, Kolterman OG, Griffin J, Olefsky JM (1983) Mechanisms of insulin resistance in aging. J Clin Invest 71(6):1523–1535. https://doi.org/10.1172/jci110908
Fink RI, Wallace P, Olefsky JM (1986) Effects of aging on glucose-mediated glucose disposal and glucose transport. J Clin Invest 77(6):2034–2041. https://doi.org/10.1172/JCI112533
Florini JR, Ewton DZ, Coolican SA (1996) Growth hormone and the insulin-like growth factor system in myogenesis. Endocr Rev 17(5):481–517. https://doi.org/10.1210/edrv-17-5-481
Fodor J, Al-Gaadi D, Czirják T, Oláh T, Dienes B, Csernoch L, Szentesi P (2020) Improved calcium homeostasis and force by selenium treatment and training in aged mouse skeletal muscle. Sci Rep 10(1):1707. https://doi.org/10.1038/s41598-020-58500-x
Franssila-Kallunki A, Schalin-Jantti C, Groop L (1992) Effect of gender on insulin resistance associated with aging. Am J Physiol 263(4 Pt 1):E780–E785. https://doi.org/10.1152/ajpendo.1992.263.4.E780
Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, Seeman T, Tracy R, Kop WJ, Burke G, McBurnie MA (2001) Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci 56(3):M146–M156. https://doi.org/10.1093/gerona/56.3.m146
Fujita S, Glynn EL, Timmerman KL, Rasmussen BB, Volpi E (2009) Supraphysiological hyperinsulinaemia is necessary to stimulate skeletal muscle protein anabolism in older adults: evidence of a true age-related insulin resistance of muscle protein metabolism. Diabetologia 52(9):1889–1898. https://doi.org/10.1007/s00125-009-1430-8
Fukumori N, Yamamoto Y, Takegami M, Yamazaki S, Onishi Y, Sekiguchi M, Otani K, Konno S, Kikuchi S, Fukuhara S (2015) Association between hand-grip strength and depressive symptoms: locomotive syndrome and health outcomes in Aizu Cohort Study (LOHAS). Age Ageing 44(4):592–598. https://doi.org/10.1093/ageing/afv013
Garito T, Roubenoff R, Hompesch M, Morrow L, Gomez K, Rooks D, Meyers C, Buchsbaum MS, Neelakantham S, Swan T, Filosa LA, Laurent D, Petricoul O, Zakaria M (2018) Bimagrumab improves body composition and insulin sensitivity in insulin-resistant individuals. Diabetes Obes Metab 20(1):94–102. https://doi.org/10.1111/dom.13042
Gaster M, Poulsen P, Handberg A, Schroder HD, Beck-Nielsen H (2000) Direct evidence of fiber type-dependent GLUT-4 expression in human skeletal muscle. Am J Physiol Endocrinol Metab 278(5):E910–E916. https://doi.org/10.1152/ajpendo.2000.278.5.E910
Gedulin BR, Nikoulina SE, Smith PA, Gedulin G, Nielsen LL, Baron AD, Parkes DG, Young AA (2005) Exenatide (exendin-4) improves insulin sensitivity and {beta}-cell mass in insulin-resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology 146(4):2069–2076. https://doi.org/10.1210/en.2004-1349
Gharahdaghi N, Rudrappa S, Brook MS, Idris I, Crossland H, Hamrock C, Abdul Aziz MH, Kadi F, Tarum J, Greenhaff PL, Constantin-Teodosiu D, Cegielski J, Phillips BE, Wilkinson DJ, Szewczyk NJ, Smith K, Atherton PJ (2019) Testosterone therapy induces molecular programming augmenting physiological adaptations to resistance exercise in older men. J Cachexia Sarcopenia Muscle 10(6):1276–1294. https://doi.org/10.1002/jcsm.12472
Giannocco G, Oliveira KC, Crajoinas RO, Venturini G, Salles TA, Fonseca-Alaniz MH, Maciel RM, Girardi AC (2013) Dipeptidyl peptidase IV inhibition upregulates GLUT4 translocation and expression in heart and skeletal muscle of spontaneously hypertensive rats. Eur J Pharmacol 698(1–3):74–86. https://doi.org/10.1016/j.ejphar.2012.09.043
Goldspink G (2012) Age-related loss of muscle mass and strength. J Aging Res 2012:158279. https://doi.org/10.1155/2012/158279
Goll DE, Thompson VF, Li H, Wei W, Cong J (2003) The calpain system. Physiol Rev 83(3):731–801. https://doi.org/10.1152/physrev.00029.2002
Gonzalez E, Messi ML, Zheng Z, Delbono O (2003) Insulin-like growth factor-1 prevents age-related decrease in specific force and intracellular Ca2+ in single intact muscle fibres from transgenic mice. J Physiol 552(Pt 3):833–844. https://doi.org/10.1113/jphysiol.2003.048165
Goodman CA, McNally RM, Hoffmann FM, Hornberger TA (2013) Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol Endocrinol 27(11):1946–1957. https://doi.org/10.1210/me.2013-1194
Goodpaster BH, Park SW, Harris TB, Kritchevsky SB, Nevitt M, Schwartz AV, Simonsick EM, Tylavsky FA, Visser M, Newman AB (2006) The loss of skeletal muscle strength, mass, and quality in older adults: the health, aging and body composition study. J Gerontol A Biol Sci Med Sci 61(10):1059–1064. https://doi.org/10.1093/gerona/61.10.1059
Grabner M, Dayal A (2010) Crosstalk via the sarcoplasmic gap: the DHPR-RyR interaction. Curr Top Membr 66:115–138. https://doi.org/10.1016/s1063-5823(10)66006-1
Grace AA (1991) Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: a hypothesis for the etiology of schizophrenia. Neuroscience 41(1):1–24. https://doi.org/10.1016/0306-4522(91)90196-u
Greiwe JS, Cheng B, Rubin DC, Yarasheski KE, Semenkovich CF (2001) Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J 15(2):475–482. https://doi.org/10.1096/fj.00-0274com
Guidelines for the Review of Inclusion on the Basis of Sex/Gender, Race, Ethnicity, and Age in Clinical Research (2019) Older adults. National Institutes of Health (NIH)
Guo T, Jou W, Chanturiya T, Portas J, Gavrilova O, McPherron AC (2009) Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS One 4(3):e4937. https://doi.org/10.1371/journal.pone.0004937
Gupte AA, Bomhoff GL, Geiger PC (2008) Age-related differences in skeletal muscle insulin signaling: the role of stress kinases and heat shock proteins. J Appl Physiol (1985) 105(3):839–848. https://doi.org/10.1152/japplphysiol.00148.2008
Gureev AP, Shaforostova EA, Popov VN (2019) Regulation of mitochondrial biogenesis as a way for active longevity: interaction between the Nrf2 and PGC-1alpha signaling pathways. Front Genet 10:435. https://doi.org/10.3389/fgene.2019.00435
Han DS, Chen YM, Lin SY, Chang HH, Huang TM, Chi YC, Yang WS (2011) Serum myostatin levels and grip strength in normal subjects and patients on maintenance haemodialysis. Clin Endocrinol (Oxf) 75(6):857–863. https://doi.org/10.1111/j.1365-2265.2011.04120.x
Hansen SL, Svendsen PF, Jeppesen JF, Hoeg LD, Andersen NR, Kristensen JM, Nilas L, Lundsgaard AM, Wojtaszewski JFP, Madsbad S, Kiens B (2019) Molecular mechanisms in skeletal muscle underlying insulin resistance in women who are lean with polycystic ovary syndrome. J Clin Endocrinol Metab 104(5):1841–1854. https://doi.org/10.1210/jc.2018-01771
Haun CT, Vann CG, Roberts BM, Vigotsky AD, Schoenfeld BJ, Roberts MD (2019) A critical evaluation of the biological construct skeletal muscle hypertrophy: size matters but so does the measurement. Front Physiol 10:247. https://doi.org/10.3389/fphys.2019.00247
Hendrickson SL, Lautenberger JA, Chinn LW, Malasky M, Sezgin E, Kingsley LA, Goedert JJ, Kirk GD, Gomperts ED, Buchbinder SP, Troyer JL, O’Brien SJ (2010) Genetic variants in nuclear-encoded mitochondrial genes influence AIDS progression. PLoS One 5(9):e12862. https://doi.org/10.1371/journal.pone.0012862
Henry RR, Rosenstock J, Denham DS, Prabhakar P, Kjems L, Baron MA (2018) Clinical impact of ITCA 650, a novel drug-device GLP-1 Receptor agonist, in uncontrolled type 2 diabetes and very high baseline HbA1c: the FREEDOM-1 HBL (high baseline) study. Diabetes Care 41(3):613–619. https://doi.org/10.2337/dc17-1519
Herman GA, Stevens C, Van Dyck K, Bergman A, Yi B, De Smet M, Snyder K, Hilliard D, Tanen M, Tanaka W, Wang AQ, Zeng W, Musson D, Winchell G, Davies MJ, Ramael S, Gottesdiener KM, Wagner JA (2005) Pharmacokinetics and pharmacodynamics of sitagliptin, an inhibitor of dipeptidyl peptidase IV, in healthy subjects: results from two randomized, double-blind, placebo-controlled studies with single oral doses. Clin Pharmacol Ther 78(6):675–688. https://doi.org/10.1016/j.clpt.2005.09.002
Hernández-Ochoa EO, Schneider MF (2018) Voltage sensing mechanism in skeletal muscle excitation-contraction coupling: coming of age or midlife crisis? Skelet Muscle 8(1):22. https://doi.org/10.1186/s13395-018-0167-9
Heymsfield SB, Coleman LA, Miller R, Rooks DS, Laurent D, Petricoul O, Praestgaard J, Swan T, Wade T, Perry RG, Goodpaster BH, Roubenoff R (2021) Effect of bimagrumab vs placebo on body fat mass among adults with type 2 diabetes and obesity: a phase 2 randomized clinical trial. JAMA Netw Open 4(1):e2033457. https://doi.org/10.1001/jamanetworkopen.2020.33457
Hicks GE, Shardell M, Alley DE, Miller RR, Bandinelli S, Guralnik J, Lauretani F, Simonsick EM, Ferrucci L (2012) Absolute strength and loss of strength as predictors of mobility decline in older adults: the InCHIANTI study. J Gerontol A Biol Sci Med Sci 67(1):66–73. https://doi.org/10.1093/gerona/glr055
Higgins GA, Silenieks LB, Rossmann A, Rizos Z, Noble K, Soko AD, Fletcher PJ (2012) The 5-HT2C receptor agonist lorcaserin reduces nicotine self-administration, discrimination, and reinstatement: relationship to feeding behavior and impulse control. Neuropsychopharmacology 37(5):1177–1191. https://doi.org/10.1038/npp.2011.303
Higgins GA, Fletcher PJ, Shanahan WR (2020) Lorcaserin: a review of its preclinical and clinical pharmacology and therapeutic potential. Pharmacol Ther 205:107417. https://doi.org/10.1016/j.pharmthera.2019.107417
Hill GE (2014) Sex linkage of nuclear-encoded mitochondrial genes. Heredity (Edinb) 112(5):469–470. https://doi.org/10.1038/hdy.2013.125
Hill C, James RS, Cox VM, Seebacher F, Tallis J (2020) Age-related changes in isolated mouse skeletal muscle function are dependent on sex, muscle, and contractility mode. Am J Physiol Regul Integr Comp Physiol 319(3):R296–r314. https://doi.org/10.1152/ajpregu.00073.2020
Hirst JA, Farmer AJ, Ali R, Roberts NW, Stevens RJ (2012) Quantifying the effect of metformin treatment and dose on glycemic control. Diabetes Care 35(2):446–454. https://doi.org/10.2337/dc11-1465
Hittel DS, Berggren JR, Shearer J, Boyle K, Houmard JA (2009) Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes 58(1):30–38. https://doi.org/10.2337/db08-0943
Hittel DS, Axelson M, Sarna N, Shearer J, Huffman KM, Kraus WE (2010) Myostatin decreases with aerobic exercise and associates with insulin resistance. Med Sci Sports Exerc 42(11):2023–2029. https://doi.org/10.1249/MSS.0b013e3181e0b9a8
Hjorth M, Pourteymour S, Gorgens SW, Langleite TM, Lee S, Holen T, Gulseth HL, Birkeland KI, Jensen J, Drevon CA, Norheim F (2016) Myostatin in relation to physical activity and dysglycaemia and its effect on energy metabolism in human skeletal muscle cells. Acta Physiol (Oxf) 217(1):45–60. https://doi.org/10.1111/apha.12631
Hofer A, Noe N, Tischner C, Kladt N, Lellek V, Schauss A, Wenz T (2014) Defining the action spectrum of potential PGC-1alpha activators on a mitochondrial and cellular level in vivo. Hum Mol Genet 23(9):2400–2415. https://doi.org/10.1093/hmg/ddt631
Holtzer R, Ozelius L, Xue X, Wang T, Lipton RB, Verghese J (2010) Differential effects of COMT on gait and executive control in aging. Neurobiol Aging 31(3):523–531. https://doi.org/10.1016/j.neurobiolaging.2008.05.011
Hong Y, Lee JH, Jeong KW, Choi CS, Jun HS (2019) Amelioration of muscle wasting by glucagon-like peptide-1 receptor agonist in muscle atrophy. J Cachexia Sarcopenia Muscle 10(4):903–918. https://doi.org/10.1002/jcsm.12434
Hood DA, Memme JM, Oliveira AN, Triolo M (2019) Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu Rev Physiol 81:19–41. https://doi.org/10.1146/annurev-physiol-020518-114310
Houmard JA, Weidner MD, Dolan PL, Leggett-Frazier N, Gavigan KE, Hickey MS, Tyndall GL, Zheng D, Alshami A, Dohm GL (1995) Skeletal muscle GLUT4 protein concentration and aging in humans. Diabetes 44(5):555–560. https://doi.org/10.2337/diab.44.5.555
Huthmacher JA, Meier JJ, Nauck MA (2020) Efficacy and safety of short- and long-acting glucagon-like peptide 1 receptor agonists on a background of basal insulin in type 2 diabetes: a meta-analysis. Diabetes Care 43(9):2303–2312. https://doi.org/10.2337/dc20-0498
Irving EL, Steinbach MJ, Lillakas L, Babu RJ, Hutchings N (2006) Horizontal saccade dynamics across the human life span. Invest Ophthalmol Vis Sci 47(6):2478–2484. https://doi.org/10.1167/iovs.05-1311
Kaasinen V, Rinne JO (2002) Functional imaging studies of dopamine system and cognition in normal aging and Parkinson’s disease. Neurosci Biobehav Rev 26(7):785–793. https://doi.org/10.1016/s0149-7634(02)00065-9
Kalmar JM, Button DC, Gardiner K, Cahill F, Gardiner PF (2009) Caloric restriction does not offset age-associated changes in the biophysical properties of motoneurons. J Neurophysiol 101(2):548–557. https://doi.org/10.1152/jn.90617.2008
Kalyani RR, Metter EJ, Egan J, Golden SH, Ferrucci L (2015) Hyperglycemia predicts persistently lower muscle strength with aging. Diabetes Care 38(1):82–90. https://doi.org/10.2337/dc14-1166
Kamen G, Sison SV, Du CC, Patten C (1995) Motor unit discharge behavior in older adults during maximal-effort contractions. J Appl Physiol 79(6):1908–1913. https://doi.org/10.1152/jappl.1995.79.6.1908
Kamo T, Asahi R, Azami M, Ogihara H, Ikeda T, Suzuki K, Nishida Y (2019) Rate of torque development and the risk of falls among community dwelling older adults in Japan. Gait Posture 72:28–33. https://doi.org/10.1016/j.gaitpost.2019.05.019
Kandel ER, Schwartz JH, Jessel TM, Siegelbaum SA, Hudspeth AJ (2013) Principles of neural science, 5th edn. Mc-Graw Hill, New York
Kim YB, Ciaraldi TP, Kong A, Kim D, Chu N, Mohideen P, Mudaliar S, Henry RR, Kahn BB (2002) Troglitazone but not metformin restores insulin-stimulated phosphoinositide 3-kinase activity and increases p110beta protein levels in skeletal muscle of type 2 diabetic subjects. Diabetes 51(2):443–448. https://doi.org/10.2337/diabetes.51.2.443
Klionsky DJ et al (2021) Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 17(1):1–382. https://doi.org/10.1080/15548627.2020.1797280
Kobayashi M, Pascual-Leone A (2003) Transcranial magnetic stimulation in neurology. Lancet Neurol 2(3):145–156. S1474442203003211 [pii]
Kohrt WM, Kirwan JP, Staten MA, Bourey RE, King DS, Holloszy JO (1993) Insulin resistance in aging is related to abdominal obesity. Diabetes 42(2):273–281
Konopka AR, Miller BF (2019) Taming expectations of metformin as a treatment to extend healthspan. Geroscience 41(2):101–108. https://doi.org/10.1007/s11357-019-00057-3
Konopka AR, Suer MK, Wolff CA, Harber MP (2014) Markers of human skeletal muscle mitochondrial biogenesis and quality control: effects of age and aerobic exercise training. J Gerontol A Biol Sci Med Sci 69(4):371–378. https://doi.org/10.1093/gerona/glt107
Konstantinovic LM, Fliipovic SR (2019) Effects of near-infrared low-level laser stimulation on neuronal excitability. In: Photobiomodulation in the brain. Elsevier, San Diego
Koves TR, Noland RC, Bates AL, Henes ST, Muoio DM, Cortright RN (2005) Subsarcolemmal and intermyofibrillar mitochondria play distinct roles in regulating skeletal muscle fatty acid metabolism. Am J Physiol Cell Physiol 288(5):C1074–C1082. https://doi.org/10.1152/ajpcell.00391.2004
Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ (2006) Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 55(7):2067–2076. https://doi.org/10.2337/db06-0150
Kulkarni AS, Brutsaert EF, Anghel V, Zhang K, Bloomgarden N, Pollak M, Mar JC, Hawkins M, Crandall JP, Barzilai N (2018) Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 17(2). https://doi.org/10.1111/acel.12723
Kwak D, Baumann CW, Thompson LV (2020) Identifying characteristics of frailty in female mice using a phenotype assessment tool. J Gerontol A Biol Sci Med Sci 75(4):640–646. https://doi.org/10.1093/gerona/glz092
Lamboley CR, Wyckelsma VL, Dutka TL, McKenna MJ, Murphy RM, Lamb GD (2015) Contractile properties and sarcoplasmic reticulum calcium content in type I and type II skeletal muscle fibres in active aged humans. J Physiol 593(11):2499–2514. https://doi.org/10.1113/jp270179
Lamboley CR, Wyckelsma VL, McKenna MJ, Murphy RM, Lamb GD (2016) Ca(2+) leakage out of the sarcoplasmic reticulum is increased in type I skeletal muscle fibres in aged humans. J Physiol 594(2):469–481. https://doi.org/10.1113/jp271382
Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R (2002) Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J Biol Chem 277(51):49831–49840. https://doi.org/10.1074/jbc.M204291200
Lanza IR, Short DK, Short KR, Raghavakaimal S, Basu R, Joyner MJ, McConnell JP, Nair KS (2008) Endurance exercise as a countermeasure for aging. Diabetes 57(11):2933–2942. https://doi.org/10.2337/db08-0349
Larsson L, Li X, Frontera WR (1997) Effects of aging on shortening velocity and myosin isoform composition in single human skeletal muscle cells. Am J Physiol 272(2 Pt 1):C638–C649. https://doi.org/10.1152/ajpcell.1997.272.2.C638
Law TD, Clark LA, Clark BC (2016) Resistance exercise to prevent and manage sarcopenia and dynapenia. Annu Rev Gerontol Geriatr 36(1):205–228. https://doi.org/10.1891/0198-8794.36.205
Lecker SH, Goldberg AL, Mitch WE (2006) Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17(7):1807–1819. https://doi.org/10.1681/ASN.2006010083
Lee HY, Choi CS, Birkenfeld AL, Alves TC, Jornayvaz FR, Jurczak MJ, Zhang D, Woo DK, Shadel GS, Ladiges W, Rabinovitch PS, Santos JH, Petersen KF, Samuel VT, Shulman GI (2010) Targeted expression of catalase to mitochondria prevents age-associated reductions in mitochondrial function and insulin resistance. Cell Metab 12(6):668–674. https://doi.org/10.1016/j.cmet.2010.11.004
Lee CG, Boyko EJ, Barrett-Connor E, Miljkovic I, Hoffman AR, Everson-Rose SA, Lewis CE, Cawthon PM, Strotmeyer ES, Orwoll ES, Osteoporotic Fractures in Men Study Research Group (2011a) Insulin sensitizers may attenuate lean mass loss in older men with diabetes. Diabetes Care 34(11):2381–2386. https://doi.org/10.2337/dc11-1032
Lee JO, Lee SK, Jung JH, Kim JH, You GY, Kim SJ, Park SH, Uhm KO, Kim HS (2011b) Metformin induces Rab4 through AMPK and modulates GLUT4 translocation in skeletal muscle cells. J Cell Physiol 226(4):974–981. https://doi.org/10.1002/jcp.22410
Lefebvre S, Liew SL (2017) Anatomical parameters of tDCS to modulate the motor system after stroke: a review. Front Neurol 8:29. https://doi.org/10.3389/fneur.2017.00029
Leger B, Derave W, De Bock K, Hespel P, Russell AP (2008) Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res 11(1):163–175B. https://doi.org/10.1089/rej.2007.0588
Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, Brossi A, Lahiri DK, Mattson MP, Hoffer BJ, Wang Y, Greig NH (2009) GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A 106(4):1285–1290. https://doi.org/10.1073/pnas.0806720106
Liu H, Graber TG, Ferguson-Stegall L, Thompson LV (2014) Clinically relevant frailty index for mice. J Gerontol A Biol Sci Med Sci 69(12):1485–1491. https://doi.org/10.1093/gerona/glt188
Long DE, Peck BD, Tuggle SC, Villasante Tezanos AG, Windham ST, Bamman MM, Kern PA, Peterson CA, Walton RG (2021) Associations of muscle lipid content with physical function and resistance training outcomes in older adults: altered responses with metformin. Geroscience. https://doi.org/10.1007/s11357-020-00315-9
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217. https://doi.org/10.1016/j.cell.2013.05.039
Louie GH, Ward MM (2010) Sex disparities in self-reported physical functioning: true differences, reporting bias, or incomplete adjustment for confounding? J Am Geriatr Soc 58(6):1117–1122. https://doi.org/10.1111/j.1532-5415.2010.02858.x
Lowsky DJ, Olshansky SJ, Bhattacharya J, Goldman DP (2014) Heterogeneity in healthy aging. J Gerontol A Biol Sci Med Sci 69(6):640–649. https://doi.org/10.1093/gerona/glt162
Lynch NA, Metter EJ, Lindle RS, Fozard JL, Tobin JD, Roy TA, Fleg JL, Hurley BF (1999) Muscle quality. I. Age-associated differences between arm and leg muscle groups. J Appl Physiol (1985) 86(1):188–194. https://doi.org/10.1152/jappl.1999.86.1.188
Manini TM, Visser M, Won-Park S, Patel KV, Strotmeyer ES, Chen H, Goodpaster B, De Rekeneire N, Newman AB, Simonsick EM, Kritchevsky SB, Ryder K, Schwartz AV, Harris TB (2007) Knee extension strength cutpoints for maintaining mobility. J Am Geriatr Soc 55(3):451–457. https://doi.org/10.1111/j.1532-5415.2007.01087.x. JGS1087 [pii]
Marigold DS, Misiaszek JE (2009) Whole-body responses: neural control and implications for rehabilitation and fall prevention. Neuroscientist 15(1):36–46. https://doi.org/10.1177/1073858408322674
Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, de Cabo R (2013) Metformin improves healthspan and lifespan in mice. Nat Commun 4:2192. https://doi.org/10.1038/ncomms3192
McCarthy JJ, Esser KA (2010) Anabolic and catabolic pathways regulating skeletal muscle mass. Curr Opin Clin Nutr Metab Care 13(3):230–235. https://doi.org/10.1097/MCO.0b013e32833781b5
McGinley M, Hoffman RL, Russ DW, Thomas JS, Clark BC (2010) Older adults exhibit more intracortical inhibition and less intracortical facilitation than young adults. Exp Gerontol 45(9):671–678. https://doi.org/10.1016/j.exger.2010.04.005
McGrath R, Vincent BM, Al Snih S, Markides KS, Peterson MD (2017) The association between muscle weakness and incident diabetes in older Mexican Americans. J Am Med Dir Assoc 18(5):452 e457–452 e412. https://doi.org/10.1016/j.jamda.2017.01.017
McGrath RP, Vincent BM, Lee IM, Kraemer WJ, Peterson MD (2018) Handgrip strength, function, and mortality in older adults: a time-varying approach. Med Sci Sports Exerc 50(11):2259–2266. https://doi.org/10.1249/MSS.0000000000001683
McGrath R, Erlandson KM, Vincent BM, Hackney KJ, Herrmann SD, Clark BC (2019a) Decreased handgrip strength is associated with impairments in each autonomous living task for aging adults in the United States. J Frailty Aging 8(3):141–145. https://doi.org/10.14283/jfa.2018.47
McGrath R, Vincent BM, Peterson MD, Jurivich DA, Dahl LJ, Hackney KJ, Clark BC (2019b) Weakness may have a causal association with early mortality in older Americans: a matched cohort analysis. J Am Med Dir Assoc. https://doi.org/10.1016/j.jamda.2019.10.016
McKendry J, Currier BS, Lim C, McLeod JC, Thomas ACQ, Phillips SM (2020) Nutritional supplements to support resistance exercise in countering the sarcopenia of aging. Nutrients 12(7). https://doi.org/10.3390/nu12072057
McPherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387(6628):83–90. https://doi.org/10.1038/387083a0
Mendias CL, Marcin JE, Calerdon DR, Faulkner JA (2006) Contractile properties of EDL and soleus muscles of myostatin-deficient mice. J Appl Physiol (1985) 101(3):898–905. https://doi.org/10.1152/japplphysiol.00126.2006
Mendias CL, Kayupov E, Bradley JR, Brooks SV, Claflin DR (2011) Decreased specific force and power production of muscle fibers from myostatin-deficient mice are associated with a suppression of protein degradation. J Appl Physiol (1985) 111(1):185–191. https://doi.org/10.1152/japplphysiol.00126.2011
Mendias CL, Bakhurin KI, Gumucio JP, Shallal-Ayzin MV, Davis CS, Faulkner JA (2015) Haploinsufficiency of myostatin protects against aging-related declines in muscle function and enhances the longevity of mice. Aging Cell 14(4):704–706. https://doi.org/10.1111/acel.12339
Meredith CN, Frontera WR, Fisher EC, Hughes VA, Herland JC, Edwards J, Evans WJ (1989) Peripheral effects of endurance training in young and old subjects. J Appl Physiol (1985) 66(6):2844–2849. https://doi.org/10.1152/jappl.1989.66.6.2844
Metti AL, Rosano C, Boudreau R, Massa R, Yaffe K, Satterfield S, Harris T, Rosso AL (2017) Catechol-O-methyltransferase genotype and gait speed changes over 10 years in older adults. J Am Geriatr Soc 65(9):2016–2022. https://doi.org/10.1111/jgs.14980
Mijares A, Allen PD, Lopez JR (2020) Senescence is associated with elevated intracellular resting [Ca(2 +)] in mice skeletal muscle fibers. An in vivo study. Front Physiol 11:601189. https://doi.org/10.3389/fphys.2020.601189
Mitchell SJ, Scheibye-Knudsen M, Longo DL, de Cabo R (2015) Animal models of aging research: implications for human aging and age-related diseases. Annu Rev Anim Biosci 3:283–303. https://doi.org/10.1146/annurev-animal-022114-110829
Mitchell WK, Wilkinson DJ, Phillips BE, Lund JN, Smith K, Atherton PJ (2016) Human skeletal muscle protein metabolism responses to amino acid nutrition. Adv Nutr 7(4):828S–838S. https://doi.org/10.3945/an.115.011650
Moran AL, Warren GL, Lowe DA (2005) Soleus and EDL muscle contractility across the lifespan of female C57BL/6 mice. Exp Gerontol 40(12):966–975. https://doi.org/10.1016/j.exger.2005.09.005
Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A (2009) Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol 297(5):C1124–C1132. https://doi.org/10.1152/ajpcell.00043.2009
Morley JE, Vellas B, van Kan GA, Anker SD, Bauer JM, Bernabei R, Cesari M, Chumlea WC, Doehner W, Evans J, Fried LP, Guralnik JM, Katz PR, Malmstrom TK, McCarter RJ, Gutierrez Robledo LM, Rockwood K, von Haehling S, Vandewoude MF, Walston J (2013) Frailty consensus: a call to action. J Am Med Dir Assoc 14(6):392–397. https://doi.org/10.1016/j.jamda.2013.03.022
Moskowitz S, Russ DW, Clark LA, Wages NP, Grooms DR, Woods AJ, Suhr J, Simon JE, O’Shea A, Criss CR, Fadda P, Clark BC (2020) Is impaired dopaminergic function associated with mobility capacity in older adults? GeroScience. https://doi.org/10.1007/s11357-020-00303-z
Musi N, Hirshman MF, Nygren J, Svanfeldt M, Bavenholm P, Rooyackers O, Zhou G, Williamson JM, Ljunqvist O, Efendic S, Moller DE, Thorell A, Goodyear LJ (2002) Metformin increases AMP-activated protein kinase activity in skeletal muscle of subjects with type 2 diabetes. Diabetes 51(7):2074–2081. https://doi.org/10.2337/diabetes.51.7.2074
Nair KS (2005) Aging muscle. Am J Clin Nutr 81(5):953–963. https://doi.org/10.1093/ajcn/81.5.953
Nardelli P, Khan J, Powers R, Cope TC, Rich MM (2013) Reduced motoneuron excitability in a rat model of sepsis. J Neurophysiol 109(7):1775–1781. https://doi.org/10.1152/jn.00936.2012
Nardelli P, Vincent JA, Powers R, Cope TC, Rich MM (2016) Reduced motor neuron excitability is an important contributor to weakness in a rat model of sepsis. Exp Neurol 282:1–8. https://doi.org/10.1016/j.expneurol.2016.04.020
Nardelli P, Powers R, Cope TC, Rich MM (2017) Increasing motor neuron excitability to treat weakness in sepsis. Ann Neurol 82(6):961–971. https://doi.org/10.1002/ana.25105
Narici MV, Maffulli N (2010) Sarcopenia: characteristics, mechanisms and functional significance. Br Med Bull 95:139–159. https://doi.org/10.1093/bmb/ldq008
Nebuloni CC, Maximo RO, de Oliveira C, Alexandre TDS (2020) Uncontrolled diabetes as an associated factor with dynapenia in adults aged 50 years or older: sex differences. J Gerontol A Biol Sci Med Sci 75(6):1191–1197. https://doi.org/10.1093/gerona/glz257
Newman AB, Simonsick EM, Naydeck BL, Boudreau RM, Kritchevsky SB, Nevitt MC, Pahor M, Satterfield S, Brach JS, Studenski SA, Harris TB (2006) Association of long-distance corridor walk performance with mortality, cardiovascular disease, mobility limitation, and disability. JAMA 295(17):2018–2026. https://doi.org/10.1001/jama.295.17.2018
Ngo HB, Lovely GA, Phillips R, Chan DC (2014) Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat Commun 5:3077. https://doi.org/10.1038/ncomms4077
Novelle MG, Ali A, Dieguez C, Bernier M, de Cabo R (2016) Metformin: a hopeful promise in aging research. Cold Spring Harb Perspect Med 6(3):a025932. https://doi.org/10.1101/cshperspect.a025932
Ochala J, Frontera WR, Dorer DJ, Van Hoecke J, Krivickas LS (2007) Single skeletal muscle fiber elastic and contractile characteristics in young and older men. J Gerontol A Biol Sci Med Sci 62(4):375–381. https://doi.org/10.1093/gerona/62.4.375
O’Connell K, Gannon J, Doran P, Ohlendieck K (2008) Reduced expression of sarcalumenin and related Ca2+ -regulatory proteins in aged rat skeletal muscle. Exp Gerontol 43(10):958–961. https://doi.org/10.1016/j.exger.2008.07.006
Ohtsuki T (1981) Inhibition of individual fingers during grip strength exertion. Ergonomics 24(1):21–36. https://doi.org/10.1080/00140138108924827
Ojima K (2019) Myosin: formation and maintenance of thick filaments. Anim Sci J 90(7):801–807. https://doi.org/10.1111/asj.13226
Oki K, Mahato NK, Nakazawa M, Amano S, France CR, Russ DW, Clark BC (2016) Preliminary evidence that excitatory transcranial direct current stimulation extends time to task failure of a sustained, submaximal muscular contraction in older adults. J Gerontol A Biol Sci Med Sci 71(8):1109–1112. https://doi.org/10.1093/gerona/glw011
O’Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, Stanford KI, Robinson MM, Cai W, Kleinridders A, Pereira RO, Hirshman MF, Abel ED, Accili D, Goodyear LJ, Nair KS, Kahn CR (2016) Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest 126(9):3433–3446. https://doi.org/10.1172/JCI86522
Papadopoulou SK (2020) Sarcopenia: a contemporary health problem among older adult populations. Nutrients 12(5). https://doi.org/10.3390/nu12051293
Parekh AB (2003a) Mitochondrial regulation of intracellular Ca2+ signaling: more than just simple Ca2+ buffers. News Physiol Sci 18:252–256. https://doi.org/10.1152/nips.01458.2003
Parekh AB (2003b) Store-operated Ca2+ entry: dynamic interplay between endoplasmic reticulum, mitochondria and plasma membrane. J Physiol 547(Pt 2):333–348. https://doi.org/10.1113/jphysiol.2002.034140
Park SW, Goodpaster BH, Strotmeyer ES, Kuller LH, Broudeau R, Kammerer C, de Rekeneire N, Harris TB, Schwartz AV, Tylavsky FA, Cho YW, Newman AB, Health A, Body Composition S (2007) Accelerated loss of skeletal muscle strength in older adults with type 2 diabetes: the health, aging, and body composition study. Diabetes Care 30(6):1507–1512. https://doi.org/10.2337/dc06-2537
Park SW, Goodpaster BH, Lee JS, Kuller LH, Boudreau R, de Rekeneire N, Harris TB, Kritchevsky S, Tylavsky FA, Nevitt M, Cho YW, Newman AB, Health A, Body Composition S (2009) Excessive loss of skeletal muscle mass in older adults with type 2 diabetes. Diabetes Care 32(11):1993–1997. https://doi.org/10.2337/dc09-0264
Patel HP, Al-Shanti N, Davies LC, Barton SJ, Grounds MD, Tellam RL, Stewart CE, Cooper C, Sayer AA (2014) Lean mass, muscle strength and gene expression in community dwelling older men: findings from the Hertfordshire Sarcopenia Study (HSS). Calcif Tissue Int 95(4):308–316. https://doi.org/10.1007/s00223-014-9894-z
Pehleman TL, Peters SJ, Heigenhauser GJ, Spriet LL (2005) Enzymatic regulation of glucose disposal in human skeletal muscle after a high-fat, low-carbohydrate diet. J Appl Physiol (1985) 98(1):100–107. https://doi.org/10.1152/japplphysiol.00686.2004
Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300(5622):1140–1142. https://doi.org/10.1126/science.1082889
Petersen KF, Morino K, Alves TC, Kibbey RG, Dufour S, Sono S, Yoo PS, Cline GW, Shulman GI (2015) Effect of aging on muscle mitochondrial substrate utilization in humans. Proc Natl Acad Sci U S A 112(36):11330–11334. https://doi.org/10.1073/pnas.1514844112
Petzinger GM, Fisher BE, McEwen S, Beeler JA, Walsh JP, Jakowec MW (2013) Exercise-enhanced neuroplasticity targeting motor and cognitive circuitry in Parkinson’s disease. Lancet Neurol 12(7):716–726. https://doi.org/10.1016/S1474-4422(13)70123-6
Pierno S, De Luca A, Beck CL, George AL Jr, Conte Camerino D (1999) Aging-associated down-regulation of ClC-1 expression in skeletal muscle: phenotypic-independent relation to the decrease of chloride conductance. FEBS Lett 449(1):12–16. https://doi.org/10.1016/s0014-5793(99)00202-1
Poulsen P, Wojtaszewski JF, Petersen I, Christensen K, Richter EA, Beck-Nielsen H, Vaag A (2005) Impact of genetic versus environmental factors on the control of muscle glycogen synthase activation in twins. Diabetes 54(5):1289–1296. https://doi.org/10.2337/diabetes.54.5.1289
PubChem (2021a) PubChem Compound Summary for CID 4158, Methylphenidate. National Library of Medicine (US), National Center for Biotechnology Information. https://pubchem.ncbi.nlm.nih.gov/compound/Methylphenidate. Accessed 29 Jan 2021
PubChem (2021b) PubChem Compound Summary for CID 6047, Levodopa. National Library of Medicine (US), National Center for Biotechnology Information. https://pubchem.ncbi.nlm.nih.gov/compound/Levodopa. Accessed 29 Jan 2021
Qaisar R, Bhaskaran S, Premkumar P, Ranjit R, Natarajan KS, Ahn B, Riddle K, Claflin DR, Richardson A, Brooks SV, Van Remmen H (2018) Oxidative stress-induced dysregulation of excitation-contraction coupling contributes to muscle weakness. J Cachexia Sarcopenia Muscle 9(5):1003–1017. https://doi.org/10.1002/jcsm.12339
Rantanen T (2003) Muscle strength, disability and mortality. Scand J Med Sci Sports 13(1):3–8
Rantanen T, Guralnik JM, Izmirlian G, Williamson JD, Simonsick EM, Ferrucci L, Fried LP (1998) Association of muscle strength with maximum walking speed in disabled older women. Am J Phys Med Rehabil 77(4):299–305. https://doi.org/10.1097/00002060-199807000-00008
Rantanen T, Guralnik JM, Foley D, Masaki K, Leveille S, Curb JD, White L (1999) Midlife hand grip strength as a predictor of old age disability. JAMA 281(6):558–560. jbr80447 [pii]
Rantanen T, Avlund K, Suominen H, Schroll M, Frandin K, Pertti E (2002) Muscle strength as a predictor of onset of ADL dependence in people aged 75 years. Aging Clin Exp Res 14(3 Suppl):10–15
Rasmussen BB, Fujita S, Wolfe RR, Mittendorfer B, Roy M, Rowe VL, Volpi E (2006) Insulin resistance of muscle protein metabolism in aging. FASEB J 20(6):768–769. https://doi.org/10.1096/fj.05-4607fje
Reaven GM (1988) Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37(12):1595–1607
Reis J, Swayne OB, Vandermeeren Y, Camus M, Dimyan MA, Harris-Love M, Perez MA, Ragert P, Rothwell JC, Cohen LG (2008) Contribution of transcranial magnetic stimulation to the understanding of cortical mechanisms involved in motor control. J Physiol 586(2):325–351. https://doi.org/10.1113/jphysiol.2007.144824. jphysiol.2007.144824 [pii]
Renganathan M, Messi ML, Delbono O (1997a) Dihydropyridine receptor-ryanodine receptor uncoupling in aged skeletal muscle. J Membr Biol 157(3):247–253. https://doi.org/10.1007/s002329900233
Renganathan M, Messi ML, Schwartz R, Delbono O (1997b) Overexpression of hIGF-1 exclusively in skeletal muscle increases the number of dihydropyridine receptors in adult transgenic mice. FEBS Lett 417(1):13–16. https://doi.org/10.1016/s0014-5793(97)01225-8
Renganathan M, Messi ML, Delbono O (1998) Overexpression of IGF-1 exclusively in skeletal muscle prevents age-related decline in the number of dihydropyridine receptors. J Biol Chem 273(44):28845–28851. https://doi.org/10.1074/jbc.273.44.28845
Rhodes MA, Carraway MS, Piantadosi CA, Reynolds CM, Cherry AD, Wester TE, Natoli MJ, Massey EW, Moon RE, Suliman HB (2009) Carbon monoxide, skeletal muscle oxidative stress, and mitochondrial biogenesis in humans. Am J Physiol Heart Circ Physiol 297(1):H392–H399. https://doi.org/10.1152/ajpheart.00164.2009
Rinne JO, Hietala J, Ruotsalainen U, Sako E, Laihinen A, Nagren K, Lehikoinen P, Oikonen V, Syvalahti E (1993) Decrease in human striatal dopamine D2 receptor density with age: a PET study with [11C]raclopride. J Cereb Blood Flow Metab 13(2):310–314. https://doi.org/10.1038/jcbfm.1993.39
Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE (2005) Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes 54(1):8–14. https://doi.org/10.2337/diabetes.54.1.8
Ritz P, Dumas JF, Ducluzeau PH, Simard G (2005) Hormonal regulation of mitochondrial energy production. Curr Opin Clin Nutr Metab Care 8(4):415–418. https://doi.org/10.1097/01.mco.0000172582.86890.19
Rizzo MR, Barbieri M, Fava I, Desiderio M, Coppola C, Marfella R, Paolisso G (2016) Sarcopenia in elderly diabetic patients: role of dipeptidyl peptidase 4 inhibitors. J Am Med Dir Assoc 17(10):896–901. https://doi.org/10.1016/j.jamda.2016.04.016
Romani M, Sorrentino V, Oh CM, Li H, de Lima TI, Zhang H, Shong M, Auwerx J (2021) NAD(+) boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep 34(3):108660. https://doi.org/10.1016/j.celrep.2020.108660
Rooks D, Praestgaard J, Hariry S, Laurent D, Petricoul O, Perry RG, Lach-Trifilieff E, Roubenoff R (2017) Treatment of sarcopenia with bimagrumab: results from a phase II, randomized, controlled, proof-of-concept study. J Am Geriatr Soc 65(9):1988–1995. https://doi.org/10.1111/jgs.14927
Rooks D, Swan T, Goswami B, Filosa LA, Bunte O, Panchaud N, Coleman LA, Miller RR, Garcia Garayoa E, Praestgaard J, Perry RG, Recknor C, Fogarty CM, Arai H, Chen LK, Hashimoto J, Chung YS, Vissing J, Laurent D, Petricoul O, Hemsley S, Lach-Trifilieff E, Papanicolaou DA, Roubenoff R (2020) Bimagrumab vs optimized standard of care for treatment of sarcopenia in community-dwelling older adults: a randomized clinical trial. JAMA Netw Open 3(10):e2020836. https://doi.org/10.1001/jamanetworkopen.2020.20836
Rostami M, Mosallanezhad Z, Ansari S, Kidgell D, Rezaeian T, Bakhshi E, Ghodrati M, Jaberzadeh S (2020) The effects of consecutive sessions of anodal transcranial direct current stimulation over the primary motor cortex on hand function in healthy older adults. Arch Gerontol Geriatr 89:104063. https://doi.org/10.1016/j.archger.2020.104063
Rowe JW, Minaker KL, Pallotta JA, Flier JS (1983) Characterization of the insulin resistance of aging. J Clin Invest 71(6):1581–1587. https://doi.org/10.1172/jci110914
Russ DW, Grandy JS, Toma K, Ward CW (2011) Ageing, but not yet senescent, rats exhibit reduced muscle quality and sarcoplasmic reticulum function. Acta Physiol (Oxf) 201(3):391–403. https://doi.org/10.1111/j.1748-1716.2010.02191.x
Russ DW, Gregg-Cornell K, Conaway MJ, Clark BC (2012) Evolving concepts on the age-related changes in “muscle quality”. J Cachexia Sarcopenia Muscle. https://doi.org/10.1007/s13539-011-0054-2
Rutherford BR, Choi J, Slifstein M, O’Boyle K, Abi-Dargham A, Brown PJ, Wall MW, Vanegas-Arroyave N, Sakhardande J, Stern Y, Roose SP (2020) Neuroanatomical predictors of L-DOPA response in older adults with psychomotor slowing and depression: a pilot study. J Affect Disord 265:439–444. https://doi.org/10.1016/j.jad.2020.01.066
Ryan M, Ohlendieck K (2004) Excitation-contraction uncoupling and sarcopenia. Basic Appl Myol 14(3):141–154
Ryan M, Carlson BM, Ohlendieck K (2000) Oligomeric status of the dihydropyridine receptor in aged skeletal muscle. Mol Cell Biol Res Commun 4(4):224–229. https://doi.org/10.1006/mcbr.2001.0282
Ryan M, Butler-Browne G, Erzen I, Mouly V, Thornell LE, Wernig A, Ohlendieck K (2003) Persistent expression of the alpha1S-dihydropyridine receptor in aged human skeletal muscle: implications for the excitation-contraction uncoupling hypothesis of sarcopenia. Int J Mol Med 11(4):425–434
Ryan AS, Li G, Blumenthal JB, Ortmeyer HK (2013) Aerobic exercise + weight loss decreases skeletal muscle myostatin expression and improves insulin sensitivity in older adults. Obesity (Silver Spring) 21(7):1350–1356. https://doi.org/10.1002/oby.20216
Sandow A (1952) Excitation-contraction coupling in muscular response. Yale J Biol Med 25(3):176–201
Sano M, Meguro S, Kawai T, Suzuki Y (2016) Increased grip strength with sodium-glucose cotransporter 2. J Diabetes 8(5):736–737. https://doi.org/10.1111/1753-0407.12402
Santra M, Dill KA, de Graff AMR (2019) Proteostasis collapse is a driver of cell aging and death. Proc Natl Acad Sci U S A 116(44):22173–22178. https://doi.org/10.1073/pnas.1906592116
Santulli G, Lewis D, des Georges A, Marks AR, Frank J (2018) Ryanodine receptor structure and function in health and disease. Subcell Biochem 87:329–352. https://doi.org/10.1007/978-981-10-7757-9_11
Sasaki T, Sugawara M, Fukuda M (2019) Sodium-glucose cotransporter 2 inhibitor-induced changes in body composition and simultaneous changes in metabolic profile: 52-week prospective LIGHT (Luseogliflozin: the Components of Weight Loss in Japanese Patients with Type 2 Diabetes Mellitus) Study. J Diabetes Invest 10(1):108–117. https://doi.org/10.1111/jdi.12851
Sato H, Kubota N, Kubota T, Takamoto I, Iwayama K, Tokuyama K, Moroi M, Sugi K, Nakaya K, Goto M, Jomori T, Kadowaki T (2016) Anagliptin increases insulin-induced skeletal muscle glucose uptake via an NO-dependent mechanism in mice. Diabetologia 59(11):2426–2434. https://doi.org/10.1007/s00125-016-4071-8
Schacht JP (2016) COMT val158met moderation of dopaminergic drug effects on cognitive function: a critical review. Pharmacogenomics J 16(5):430–438. https://doi.org/10.1038/tpj.2016.43
Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR (2000) Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404(6779):770–774. https://doi.org/10.1038/35008096
Schultz MB, Sinclair DA (2016) Why NAD(+) declines during aging: it’s destroyed. Cell Metab 23(6):965–966. https://doi.org/10.1016/j.cmet.2016.05.022
Schulz DJ, Baines RA, Hempel CM, Li L, Liss B, Misonou H (2006) Cellular excitability and the regulation of functional neuronal identity: from gene expression to neuromodulation. J Neurosci 26(41):10362–10367. https://doi.org/10.1523/JNEUROSCI.3194-06.2006
Seeman TE, Merkin SS, Crimmins EM, Karlamangla AS (2010) Disability trends among older Americans: National Health And Nutrition Examination Surveys, 1988-1994 and 1999-2004. Am J Public Health 100(1):100–107. https://doi.org/10.2105/AJPH.2008.157388
Semba RD, Bandinelli S, Sun K, Guralnik JM, Ferrucci L (2010) Relationship of an advanced glycation end product, plasma carboxymethyl-lysine, with slow walking speed in older adults: the InCHIANTI study. Eur J Appl Physiol 108(1):191–195. https://doi.org/10.1007/s00421-009-1192-5
Seo DY, Lee SR, Kim N, Ko KS, Rhee BD, Han J (2016) Age-related changes in skeletal muscle mitochondria: the role of exercise. Integr Med Res 5(3):182–186. https://doi.org/10.1016/j.imr.2016.07.003
Shan J, Betzenhauser MJ, Kushnir A, Reiken S, Meli AC, Wronska A, Dura M, Chen BX, Marks AR (2010) Role of chronic ryanodine receptor phosphorylation in heart failure and β-adrenergic receptor blockade in mice. J Clin Invest 120(12):4375–4387. https://doi.org/10.1172/jci37649
Sharma N, Arias EB, Sajan MP, MacKrell JG, Bhat AD, Farese RV, Cartee GD (2010) Insulin resistance for glucose uptake and Akt2 phosphorylation in the soleus, but not epitrochlearis, muscles of old vs. adult rats. J Appl Physiol (1985) 108(6):1631–1640. https://doi.org/10.1152/japplphysiol.01412.2009
Sharoff CG, Hagobian TA, Malin SK, Chipkin SR, Yu H, Hirshman MF, Goodyear LJ, Braun B (2010) Combining short-term metformin treatment and one bout of exercise does not increase insulin action in insulin-resistant individuals. Am J Physiol Endocrinol Metab 298(4):E815–E823. https://doi.org/10.1152/ajpendo.00517.2009
Shinohara M, Latash ML, Zatsiorsky VM (2003a) Age effects on force produced by intrinsic and extrinsic hand muscles and finger interaction during MVC tasks. J Appl Physiol (1985) 95(4):1361–1369. https://doi.org/10.1152/japplphysiol.00070.2003
Shinohara M, Li S, Kang N, Zatsiorsky VM, Latash ML (2003b) Effects of age and gender on finger coordination in MVC and submaximal force-matching tasks. J Appl Physiol (1985) 94(1):259–270. https://doi.org/10.1152/japplphysiol.00643.2002
Shorer Z, Bachner Y, Guy T, Melzer I (2013) Effect of single dose methylphenidate on walking and postural stability under single- and dual-task conditions in older adults—a double-blind randomized control trial. J Gerontol A Biol Sci Med Sci 68(10):1271–1280. https://doi.org/10.1093/gerona/glt035
Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS (2005) Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A 102(15):5618–5623. https://doi.org/10.1073/pnas.0501559102
Sorond FA, Cruz-Almeida Y, Clark DJ, Viswanathan A, Scherzer CR, De Jager P, Csiszar A, Laurienti PJ, Hausdorff JM, Chen WG, Ferrucci L, Rosano C, Studenski SA, Black SE, Lipsitz LA (2015) Aging, the central nervous system, and mobility in older adults: neural mechanisms of mobility impairment. J Gerontol A Biol Sci Med Sci 70(12):1526–1532. https://doi.org/10.1093/gerona/glv130
St Andre M, Johnson M, Bansal PN, Wellen J, Robertson A, Opsahl A, Burch PM, Bialek P, Morris C, Owens J (2017) A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys. Skelet Muscle 7(1):25. https://doi.org/10.1186/s13395-017-0141-y
Stefanelli L, Lockyer EJ, Collins BW, Snow NJ, Crocker J, Kent C, Power KE, Button DC (2019) Delayed-onset muscle soreness and topical analgesic alter corticospinal excitability of the biceps brachii. Med Sci Sports Exerc 51(11):2344–2356. https://doi.org/10.1249/MSS.0000000000002055
Steffen KK, Dillin A (2016) A ribosomal perspective on proteostasis and aging. Cell Metab 23(6):1004–1012. https://doi.org/10.1016/j.cmet.2016.05.013
Stenholm S, Harris TB, Rantanen T, Visser M, Kritchevsky SB, Ferrucci L (2008) Sarcopenic obesity: definition, cause and consequences. Curr Opin Clin Nutr Metab Care 11(6):693–700. https://doi.org/10.1097/MCO.0b013e328312c37d
Stram AR, Payne RM (2016) Post-translational modifications in mitochondria: protein signaling in the powerhouse. Cell Mol Life Sci 73(21):4063–4073. https://doi.org/10.1007/s00018-016-2280-4
Sugimoto K, Yasujima M, Yagihashi S (2008) Role of advanced glycation end products in diabetic neuropathy. Curr Pharm Des 14(10):953–961. https://doi.org/10.2174/138161208784139774
Sugimoto K, Tabara Y, Ikegami H, Takata Y, Kamide K, Ikezoe T, Kiyoshige E, Makutani Y, Onuma H, Gondo Y, Ikebe K, Ichihashi N, Tsuboyama T, Matsuda F, Kohara K, Kabayama M, Fukuda M, Katsuya T, Osawa H, Hiromine Y, Rakugi H (2019) Hyperglycemia in non-obese patients with type 2 diabetes is associated with low muscle mass: the multicenter study for clarifying evidence for sarcopenia in patients with diabetes mellitus. J Diabetes Invest 10(6):1471–1479. https://doi.org/10.1111/jdi.13070
Sugiyama S, Jinnouchi H, Kurinami N, Hieshima K, Yoshida A, Jinnouchi K, Nishimura H, Suzuki T, Miyamoto F, Kajiwara K, Jinnouchi T (2018) Dapagliflozin reduces fat mass without affecting muscle mass in type 2 diabetes. J Atheroscler Thromb 25(6):467–476. https://doi.org/10.5551/jat.40873
Suhara T, Fukuda H, Inoue O, Itoh T, Suzuki K, Yamasaki T, Tateno Y (1991) Age-related changes in human D1 dopamine receptors measured by positron emission tomography. Psychopharmacology (Berl) 103(1):41–45. https://doi.org/10.1007/BF02244071
Sweeney HL, Hammers DW (2018) Muscle contraction. Cold Spring Harb Perspect Biol 10(2). https://doi.org/10.1101/cshperspect.a023200
Szewczyk NJ, Jacobson LA (2005) Signal-transduction networks and the regulation of muscle protein degradation. Int J Biochem Cell Biol 37(10):1997–2011. https://doi.org/10.1016/j.biocel.2005.02.020
Sztretye M, Geyer N, Vincze J, Al-Gaadi D, Olah T, Szentesi P, Kis G, Antal M, Balatoni I, Csernoch L, Dienes B (2017) SOCE is important for maintaining sarcoplasmic calcium content and release in skeletal muscle fibers. Biophys J 113(11):2496–2507. https://doi.org/10.1016/j.bpj.2017.09.023
Tahoe NM, Mokhtarzadeh A, Curtsinger JW (2004) Age-related RNA decline in adult Drosophila melanogaster. J Gerontol A Biol Sci Med Sci 59(9):B896–B901. https://doi.org/10.1093/gerona/59.9.b896
Tallis J, Bradford C, Duncan MJ, Leddington-Wright S, Higgins MF, Hill M (2020) The effect of acute caffeine ingestion on cognitive dual task performance during assessment of static and dynamic balance in older adults. Nutrients 12(12). https://doi.org/10.3390/nu12123653
Tang L, Liu CT, Wang XD, Luo K, Zhang DD, Chi AP, Zhang J, Sun LJ (2014) A prepared anti-MSTN polyclonal antibody reverses insulin resistance of diet-induced obese rats via regulation of PI3K/Akt/mTOR&FoxO1 signal pathways. Biotechnol Lett 36(12):2417–2423. https://doi.org/10.1007/s10529-014-1617-z
Thorell A, Hirshman MF, Nygren J, Jorfeldt L, Wojtaszewski JF, Dufresne SD, Horton ES, Ljungqvist O, Goodyear LJ (1999) Exercise and insulin cause GLUT-4 translocation in human skeletal muscle. Am J Physiol 277(4):E733–E741. https://doi.org/10.1152/ajpendo.1999.277.4.E733
Tieland M, Trouwborst I, Clark BC (2018) Skeletal muscle performance and ageing. J Cachexia Sarcopenia Muscle 9(1):3–19. https://doi.org/10.1002/jcsm.12238
Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ (2009) Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol 296(6):C1258–C1270. https://doi.org/10.1152/ajpcell.00105.2009
Turner RS, Desmurget M (2010) Basal ganglia contributions to motor control: a vigorous tutor. Curr Opin Neurobiol 20(6):704–716. https://doi.org/10.1016/j.conb.2010.08.022
Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR (2014) Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci U S A 111(42):15250–15255. https://doi.org/10.1073/pnas.1412754111
Vasilaki A, McArdle F, Iwanejko LM, McArdle A (2006) Adaptive responses of mouse skeletal muscle to contractile activity: the effect of age. Mech Ageing Dev 127(11):830–839. https://doi.org/10.1016/j.mad.2006.08.004
Verburg E, Murphy RM, Richard I, Lamb GD (2009) Involvement of calpains in Ca2+-induced disruption of excitation-contraction coupling in mammalian skeletal muscle fibers. Am J Physiol Cell Physiol 296(5):C1115–C1122. https://doi.org/10.1152/ajpcell.00008.2009
Visser M, Goodpaster BH, Kritchevsky SB, Newman AB, Nevitt M, Rubin SM, Simonsick EM, Harris TB (2005) Muscle mass, muscle strength, and muscle fat infiltration as predictors of incident mobility limitations in well-functioning older persons. J Gerontol A Biol Sci Med Sci 60(3):324–333. 60/3/324 [pii]
Volkow ND, Wang GJ, Fowler JS, Logan J, Gatley SJ, MacGregor RR, Schlyer DJ, Hitzemann R, Wolf AP (1996) Measuring age-related changes in dopamine D2 receptors with 11C-raclopride and 18F-N-methylspiroperidol. Psychiatry Res 67(1):11–16. https://doi.org/10.1016/0925-4927(96)02809-0
Volkow ND, Gur RC, Wang GJ, Fowler JS, Moberg PJ, Ding YS, Hitzemann R, Smith G, Logan J (1998) Association between decline in brain dopamine activity with age and cognitive and motor impairment in healthy individuals. Am J Psychiatry 155(3):344–349. https://doi.org/10.1176/ajp.155.3.344
Vringer E, Tait SWG (2019) Mitochondria and inflammation: cell death heats up. Front Cell Dev Biol 7:100. https://doi.org/10.3389/fcell.2019.00100
Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, Eagle M, Florence JM, King WM, Pandya S, Straub V, Juneau P, Meyers K, Csimma C, Araujo T, Allen R, Parsons SA, Wozney JM, Lavallie ER, Mendell JR (2008) A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann Neurol 63(5):561–571. https://doi.org/10.1002/ana.21338
Wagner KR, Abdel-Hamid HZ, Mah JK, Campbell C, Guglieri M, Muntoni F, Takeshima Y, McDonald CM, Kostera-Pruszczyk A, Karachunski P, Butterfield RJ, Mercuri E, Fiorillo C, Bertini ES, Tian C, Statland J, Sadosky AB, Purohit VS, Sherlock SP, Palmer JP, Binks M, Charnas L, Marraffino S, Wong BL (2020) Randomized phase 2 trial and open-label extension of domagrozumab in Duchenne muscular dystrophy. Neuromuscul Disord 30(6):492–502. https://doi.org/10.1016/j.nmd.2020.05.002
Walton RG, Dungan CM, Long DE, Tuggle SC, Kosmac K, Peck BD, Bush HM, Villasante Tezanos AG, McGwin G, Windham ST, Ovalle F, Bamman MM, Kern PA, Peterson CA (2019) Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: a randomized, double-blind, placebo-controlled, multicenter trial: the MASTERS trial. Aging Cell 18(6):e13039. https://doi.org/10.1111/acel.13039
Wang X, Proud CG (2006) The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 21:362–369. https://doi.org/10.1152/physiol.00024.2006
Wang Y, Chan GL, Holden JE, Dobko T, Mak E, Schulzer M, Huser JM, Snow BJ, Ruth TJ, Calne DB, Stoessl AJ (1998) Age-dependent decline of dopamine D1 receptors in human brain: a PET study. Synapse 30(1):56–61. https://doi.org/10.1002/(SICI)1098-2396(199809)30:1<56::AID-SYN7>3.0.CO;2-J
Wang ZM, Messi ML, Delbono O (2000) L-type Ca(2+) channel charge movement and intracellular Ca(2+) in skeletal muscle fibers from aging mice. Biophys J 78(4):1947–1954. https://doi.org/10.1016/s0006-3495(00)76742-7
Watson R (2017) Physical activity and the aging brain: effects of exercise on neurological function. Elsevier, London
Weisleder N, Brotto M, Komazaki S, Pan Z, Zhao X, Nosek T, Parness J, Takeshima H, Ma J (2006) Muscle aging is associated with compromised Ca2+ spark signaling and segregated intracellular Ca2+ release. J Cell Biol 174(5):639–645. https://doi.org/10.1083/jcb.200604166
Wenz T (2009) PGC-1alpha activation as a therapeutic approach in mitochondrial disease. IUBMB Life 61(11):1051–1062. https://doi.org/10.1002/iub.261
Wessner B, Liebensteiner M, Nachbauer W, Csapo R (2019) Age-specific response of skeletal muscle extracellular matrix to acute resistance exercise: a pilot study. Eur J Sport Sci 19(3):354–364. https://doi.org/10.1080/17461391.2018.1526974
Wilkes EA, Selby AL, Atherton PJ, Patel R, Rankin D, Smith K, Rennie MJ (2009) Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age-related sarcopenia. Am J Clin Nutr 90(5):1343–1350. https://doi.org/10.3945/ajcn.2009.27543
Wilkinson DJ, Piasecki M, Atherton PJ (2018) The age-related loss of skeletal muscle mass and function: measurement and physiology of muscle fibre atrophy and muscle fibre loss in humans. Ageing Res Rev 47:123–132. https://doi.org/10.1016/j.arr.2018.07.005
Williams S, Miller G, Khoury R, Grossberg GT (2019) Rational deprescribing in the elderly. Ann Clin Psychiatry 31(2):144–152
Windhort U, Burke R, Dieringer N, Evinger C, Feldman A, Hasan Z, Humphrey DR, Freund H-J (1991) What are the outputs of motor behavior and how are they controlled? In: Humphrey DR, Freund H-J (eds) Motor control: concepts and issues. Wiley, New York, pp 101–119
Wisdom KM, Delp SL, Kuhl E (2015) Use it or lose it: multiscale skeletal muscle adaptation to mechanical stimuli. Biomech Model Mechanobiol 14(2):195–215. https://doi.org/10.1007/s10237-014-0607-3
Wong DF, Young D, Wilson PD, Meltzer CC, Gjedde A (1997) Quantification of neuroreceptors in the living human brain: III. D2-like dopamine receptors: theory, validation, and changes during normal aging. J Cereb Blood Flow Metab 17(3):316–330. https://doi.org/10.1097/00004647-199703000-00009
Xu H, Lamb GD, Murphy RM (2017) Changes in contractile and metabolic parameters of skeletal muscle as rats age from 3 to 12 months. J Muscle Res Cell Motil 38(5–6):405–420. https://doi.org/10.1007/s10974-017-9484-6
Yabe D, Nishikino R, Kaneko M, Iwasaki M, Seino Y (2015) Short-term impacts of sodium/glucose co-transporter 2 inhibitors in Japanese clinical practice: considerations for their appropriate use to avoid serious adverse events. Expert Opin Drug Saf 14(6):795–800. https://doi.org/10.1517/14740338.2015.1034105
Yang Q, Zhang Y, Zeng Q, Yang C, Shi J, Zhang C, Ni X, Du Z, Tang Z, Hu J, Li X, Cai J, Li Q, Cheng Q (2020) Correlation between diabetic peripheral neuropathy and sarcopenia in patients with type 2 diabetes mellitus and diabetic foot disease: a cross-sectional study. Diabetes Metab Syndr Obes 13:377–386. https://doi.org/10.2147/DMSO.S237362
Yarasheski KE, Bhasin S, Sinha-Hikim I, Pak-Loduca J, Gonzalez-Cadavid NF (2002) Serum myostatin-immunoreactive protein is increased in 60-92 year old women and men with muscle wasting. J Nutr Health Aging 6(5):343–348
Yuan D, Xiao D, Gao Q, Zeng L (2019) PGC-1alpha activation: a therapeutic target for type 2 diabetes? Eat Weight Disord 24(3):385–395. https://doi.org/10.1007/s40519-018-0622-y
Zampieri S, Pietrangelo L, Loefler S, Fruhmann H, Vogelauer M, Burggraf S, Pond A, Grim-Stieger M, Cvecka J, Sedliak M, Tirpáková V, Mayr W, Sarabon N, Rossini K, Barberi L, De Rossi M, Romanello V, Boncompagni S, Musarò A, Sandri M, Protasi F, Carraro U, Kern H (2015) Lifelong physical exercise delays age-associated skeletal muscle decline. J Gerontol A Biol Sci Med Sci 70(2):163–173. https://doi.org/10.1093/gerona/glu006
Zhang C, McFarlane C, Lokireddy S, Bonala S, Ge X, Masuda S, Gluckman PD, Sharma M, Kambadur R (2011) Myostatin-deficient mice exhibit reduced insulin resistance through activating the AMP-activated protein kinase signalling pathway. Diabetologia 54(6):1491–1501. https://doi.org/10.1007/s00125-011-2079-7
Zhang LN, Zhou HY, Fu YY, Li YY, Wu F, Gu M, Wu LY, Xia CM, Dong TC, Li JY, Shen JK, Li J (2013) Novel small-molecule PGC-1alpha transcriptional regulator with beneficial effects on diabetic db/db mice. Diabetes 62(4):1297–1307. https://doi.org/10.2337/db12-0703
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Baumann, C.W., Clark, B.C., Phillips, B.E., Szewczyk, N.J., Consitt, L.A. (2022). Regenerative Rehabilitation in Sarcopenia, Dynapenia, and Frailty. In: Greising, S.M., Call, J.A. (eds) Regenerative Rehabilitation. Physiology in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-95884-8_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-95884-8_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-95883-1
Online ISBN: 978-3-030-95884-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)