
Abstract
Traditionally, interventions to treat skeletal muscle aging have largely targeted sarcopenia—the age-related loss of skeletal muscle mass. Dynapenia refers to the age-related loss in skeletal muscle function due to factors outside of muscle mass, which helps to inform treatment strategies for aging skeletal muscle. There is evidence that mechanisms to maintain protein homeostasis and proteostasis, deteriorate with age. One key mechanism to maintain proteostasis is protein turnover, which is an energetically costly process. When there is a mismatch between cellular energy demands and energy provision, inelastic processes related to metabolism are maintained, but there is competition for the remaining energy between the elastic processes of somatic maintenance and growth. With aging, mitochondrial dysfunction reduces ATP generation capacity, constraining the instantaneous supply of energy, thus compromising growth and somatic maintenance processes. Further, with age the need for somatic maintenance increases because of the accumulation of protein damage. In this review, we highlight the significant role mitochondria have in maintaining skeletal muscle proteostasis through increased energy provision, protein turnover, and substrate flux. In addition, we provide evidence that improving mitochondrial function could promote a cellular environment that is conducive to somatic maintenance, and consequently for mitigating dynapenia. Finally, we highlight interventions, such as aerobic exercise, that could be used to improve mitochondrial function and improve outcomes related to dynapenia.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sarcopenia, the age-related loss of muscle mass, affects 28% of men and 19% of women over 60 years of age (Batsis et al. 2015), and over half of individuals over the age of 80 (Baumgartner et al. 1998). Over the past decade, the definition of sarcopenia has evolved to incorporate the age-related loss of muscle function as well (Cruz-Jentoft et al. 2010; Fielding et al. 2011). Few studies have measured the economic burden of sarcopenia, but one study in 2000 estimated that it cost $18.5 billion in the United States (Janssen et al. 2004), with similar burdens likely in European communities (European Commission Directorate-General for Economic and Financial Affairs 2015). A number of excellent review articles summarize interventions to minimize sarcopenia (Doherty 2003; Paddon-Jones and Rasmussen 2009; Cruz-Jentoft et al. 2014; Murton 2015). In general, the collective data support the use of resistance exercise and amino acid supplementation, with some consideration to timing of supplementation, to minimize muscle loss with age. Manini introduced the concept of dynapenia (Clark and Manini 2008, 2012) to account for factors outside of muscle mass that contribute to the failure to maintain force-generating capacity with age. The distinction between sarcopenia and dynapenia is important because dynapenia defines the actual functional deficit, and it frames potential strategies to treat it. To treat dynapenia, maintaining muscle mass might not be the only, or the most effective, way to slow age-related decline in muscle function. In this brief review, we build a case for targeting mitochondria and the consequent change in cellular energetics as a potential treatment for dynapenia. To do so, we highlight how cellular energetics determine protein-synthetic responses, how protein-synthetic responses in turn affect cellular energetics, and how we might, therefore, target mitochondrial protein synthesis to minimize dynapenia.
The role of proteostasis in dynapenia
Proteostasis refers to the maintenance of protein homeostasis through mechanisms that involve the location, concentration, conformation, and turnover of individual proteins (Balch et al. 2008). Of particular importance to the current review is the role of protein turnover in maintaining proteostasis. In skeletal muscle, impaired protein turnover with age leads to the loss of contractile protein quality and quantity because of the accumulation of protein damage (Fig. 1) (Haus et al. 2007; Toyama and Hetzer 2013). For example, with age there is an increase in non-enzymatic modifications of proteins such as advanced glycation end-products (AGEs) (Dalal et al. 2009; Semba et al. 2010). Through cross-bridge formation, AGEs and the modified proteins they are attached to, are resistant to breakdown leading to further accumulation (Barreiro and Hussain 2010; Drenth et al. 2016). In addition to AGEs, oxidatively modified proteins also accumulate in muscle with age leading to enzymatic dysfunction (Stadtman et al. 1988; Levine and Stadtman 2001; Gavrilov and Gavrilova 2002; Toyama and Hetzer 2013; Ayyadevara et al. 2016). Since there is a limited capacity to enzymatically repair proteins (Mortimore and Pösö 1987; Poppek and Grune 2005), protein turnover is the primary mechanism to prevent or reverse age-related accumulation of modified or damaged proteins. Age-related increases in skeletal muscle oxidatively modified proteins and AGEs contribute to declines in strength, independent of actin and myosin concentration, emphasizing the importance of protein quality and its effect on skeletal muscle function independent of protein quantity (Brocca et al. 2017). Therefore, degradation of damaged proteins and synthesis of new functional proteins (protein turnover) represents an important mechanism to maintain skeletal muscle protein quality with age.

Conceptual role of mitochondrial dysfunction in the development of dynapenia. A Aging skeletal muscle is characterized by a mitochondrial dysfunction that includes both decreased ATP availability (B) and a chronic mismatch between reactive oxygen species (ROS) produced and scavenged by endogenous antioxidants (C). A decline in metabolic flux also contributes to accumulation of lipotoxic intermediates which cause greater oxidative stress (D). Damaged proteins, including those modified by advanced glycation end-products (AGEs) and ROS, accumulate; this accumulation is exacerbated by a decline in protein turnover (E), a primary component of skeletal muscle dyshomeostasis. The age-related decline in the rate of ATP production constrains energy available for cellular function. F This energetic constraint forces cells to increase the proportion of energy allocated toward metabolism “M”, while sacrificing processes related to growth “G” and somatic maintenance “S”. Collectively, these inter-related aspects of mitochondrial dysfunction contribute to dynapenia
Protein turnover and cellular energetics
Protein turnover is an energetically costly process. The energy required to synthesize new proteins represents approximately 20% of basal metabolism, (Waterlow 1984; Dennis and Bier 1999), while protein breakdown accounts for another 5–15% of basal metabolism (Rolfe and Brown 1997). Protein synthesis is an energy intensive series of processes that involves translating mRNA into amino acids and assembling the amino acids into peptide chains. Amino acid synthesis is energetically costly, requiring 12–72 ATP per amino acid (Buttgereit and Brand 1995; Lynch and Marinov 2015). The synthesis of peptide bonds between amino acids requires four high-energy phosphates from ATP or GTP hydrolysis per peptide bond (Buttgereit and Brand 1995; Lynch and Marinov 2015). The energetic cost of protein breakdown is more difficult to ascertain. Breakdown occurs through protein ubiquitination or direct lysosomal degradation. Ubiquitination for subsequent proteasome-mediated breakdown requires 2 ATP per ubiquitin tag (Lynch and Marinov 2015). Subsequent proteasome activity requires the hydrolysis of between 100 and 200 ATP per protein depending on the length and other characteristics of the protein (Lynch and Marinov 2015). Lysosomal degradation costs approximately 1 ATP for every 3–4 amino acids in a given protein (Lynch and Marinov 2015). The cost of protein turnover explains a substantial (30–50%) portion of the basal metabolism not accounted for by mitochondrial proton leak (Brand 1990; Buttgereit and Brand 1995). Because the energy demands of protein turnover are substantial, protein synthesis and breakdown are tightly regulated processes.
The Dynamic Energy Budget theory posits that organisms have a finite amount (or budget) of energy to sustain cellular processes (van Leeuwen et al. 2010; Nisbet et al. 2012). Even though the supply of energy in the form of lipid and carbohydrate stores is enough to last a human for weeks, there are energetic constraints due to the fact that the rate of oxidizing stored energy to usable ATP on demand is limited by mitochondrial function (Drew et al. 2003; Figueiredo et al. 2009). Therefore, limited energetic resources must be allocated based on cellular priorities (Martin et al. 2012). While energetic accounting is challenging, there appears to be a hierarchy of cellular energetic processes broadly categorized as metabolism, growth, or somatic maintenance (Hou et al. 2008). The category of metabolism encompasses processes that sustain life including energy production, locomotion, feeding, and ion channel and pump maintenance. The growth processes involve synthesis of new biomass, and are usually accompanied by DNA replication to form new cells or to maintain a DNA to cytoplasm ratio (Gregory 2001). Finally, somatic maintenance processes preserve existing biomass and include protein turnover (Shanley and Kirkwood 2000; Kapahi 2010). The latter two categories, growth and somatic maintenance, often compete for energetic resources (Hou 2013).
To understand supply and demand in cellular energetics, using terms from the field of economics is helpful. Elasticity refers to the degree to which demand for a good or service is sensitive to changes in its supply. Demand for inelastic commodities are constant or inflexible regardless of supply because inelastic commodities are essential or indispensable. For example, commodities such as water and petrol are inelastic whereas brand-name clothes and specialty cheeses are elastic. In cells, metabolic processes such as maintaining proton pumps are indispensable, or inelastic, whereas repair and growth are elastic. When cellular demand for energy exceeds the rate of energy production, cells will allocate energy toward inelastic cellular processes. Under energetic constraints, somatic maintenance and growth are elastic processes that can be sacrificed for the inelastic metabolic processes (Buttgereit and Brand 1995; Brand 1997; Martin et al. 2012). In addition, there can be tradeoff between the elastic processes so that as growth increases, for example, less energy can be allocated to somatic maintenance and vice versa. Therefore, energy provision for metabolic processes are maintained, while growth and somatic maintenance compete for the remaining energetic budget (Hou 2013).
Mitochondrial dysfunction contributes to a loss of proteostasis
While it is controversial whether mitochondrial dysfunction is a cause or a consequence of aging and age-related chronic diseases, it is clear that it is a characteristic (Dai et al. 2014; Gonzalez-Freire et al. 2015; Bhatti et al. 2016). When there is dysfunction within a mitochondrial unit, increasing the size of the mitochondrial reticulum can compensate for the lack of energy-producing capacity. By this mechanism, total electron transport capacity is increased rather than the relative capacity of individual components (Picard et al. 2010; Larsen et al. 2012; Porter and Wall 2012; Morrow et al. 2017). However, the ability to expand the reticulum is limited implying that this compensatory mechanism is constrained (Suarez 1998; Suarez and Suarez 2012). The inability of mitochondrial reticulum expansion to fully compensate may further compound mitochondrial dysfunction and its consequences.
Age-related declines in mitochondrial bioenergetics contribute to a decline in skeletal muscle proteostatic processes (Figueiredo et al. 2009). When mitochondrial function is decreased, provision of reducing equivalents such as NADH in excess of electron transport system capacity can lead to an imbalance that contributes to reactive oxygen species (ROS) formation (Anderson et al. 2009). This dysfunction is associated with the aging process and exacerbates ROS production and oxidative damage (Wanagat et al. 2001; Moghaddas et al. 2003). In addition to ROS-induced damage, mitochondrial dysfunction leads to the accumulation of metabolic byproducts that also cause cellular damage. For example, decreases in free fatty acid flux from impaired mitochondrial function, leads to the accumulation of lipotoxic intermediates such as diacylglycerides (DAGs) and ceramides (Coen et al. 2010). DAG and ceramide accumulation leads to inflammation and oxidative stress that damage myofibrillar and mitochondrial proteins (Dumitru et al. 2007; Salminen et al. 2012; Rivas et al. 2016), membrane lipids, and DNA (Wiley et al. 2016).
An additional problem associated with mitochondrial dysfunction is the shortage of readily available energy. As mentioned, protein quality control is an energetically costly process. Therefore, dysfunctional mitochondria may not be able to provide enough ATP to meet the energetic demands for both metabolism and cellular repair (Conley et al. 2000; Marcinek et al. 2005; Nair 2005; Amara et al. 2007). When faced with this problem, cells compromise elastic energetically costly proteostatic processes, such as protein turnover, in favor of inelastic metabolic processes (Buttgereit and Brand 1995; Brand 1997). Accordingly, impaired mitochondrial function contributes to the loss of skeletal muscle proteostasis and dynapenia in two ways: by increasing the accumulation of damaged proteins and by decreasing the ability of the mitochondria to generate energy for somatic maintenance.
Whereas mitochondrial dysfunction contributes to protein damage, maintaining mitochondrial function facilitates proteostatic mechanisms. First, the maintenance of mitochondrial proteins facilitates the coupling of electron transport system to ATP production (Zangarelli et al. 2006; Greggio et al. 2017), thus decreasing ROS production. Further, functional mitochondria readily adapt to fluctuating energy demands with efficient energy production (Madeira 2012). Efficient aerobic energy production facilitates the maintenance of proteostatic processes by not having to compromise elastic processes such as somatic maintenance. Key to this somatic maintenance is the ability to maintain protein turnover, which minimizes accumulation of protein damage and further improves mitochondrial function (Jensen and Jasper 2014; Ryu et al. 2016; Hamilton and Miller 2017).
It is worth making the point that mitochondrial function can improve without a change in mitochondrial content. Increasing mitochondrial content is just one mechanism to improve electron transport capacity. However, increasing mitochondrial protein turnover, even in the absence of an increase in content, can also improve mitochondrial function (Menshikova et al. 2006; Siegel et al. 2013; Romanello and Sandri 2015). It is even possible that with aging, improving the turnover of existing mitochondrial proteins is a better strategy to improve muscle function, since increasing mitochondrial content without improving function, may just lead to greater ROS production (Stern 2017). Therefore, when examining strategies to mitigate dynapenia, assessments of mitochondrial function are equally, if not more important than mitochondrial content.
To summarize, damaged proteins accumulate in skeletal muscle with age and there is an impairment in cellular energetics. The increased protein damage and decreased energy availability constrains somatic maintenance and mechanisms of proteostasis (Drew et al. 2003; Figueiredo et al. 2009), which exacerbates the decline in protein quality (Wiley et al. 2016). To counter this downward spiral, improving the rate of energy production in skeletal muscle by increasing mitochondrial function could relieve energetic constraints (Siegel et al. 2013; Ryu et al. 2016), improving proteostatic mechanisms and thus somatic maintenance (Stern 2017). Therefore, interventions that improve mitochondrial function could be useful for mitigating dynapenia.
Targeting mitochondria to mitigate dynapenia
Improving mitochondrial function is a promising target to prevent dynapenia by increasing the efficiency of energy production to match energy demand, minimizing oxidative damage and improving capacity for proteostatic processes (Kruse et al. 2016; Ryu et al. 2016; Heeman et al. 2011; Gonzalez-Freire et al. 2015). Restoring mitochondrial function, even at older ages, improves energetic production and improves skeletal muscle function (Siegel et al. 2013). The ability to efficiently produce energy directly facilitates somatic maintenance by increasing the energy budget. Allocation of energetic resources to somatic maintenance may, in turn, facilitate an environment that is conducive to growth. Although it is true that treatments that improve mitochondrial function do not always also increase muscle size, they likely have the potential to do so. Below, we discuss how aerobic exercise and mechanistic target of rapamycin (mTOR) inhibition allow for improved proteostatic mechanisms and thus somatic maintenance (Fig. 2).

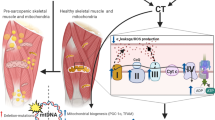
Improving mitochondrial dysfunction mitigates dynapenia. A Interventions such as aerobic exercise and mTOR inhibition, via caloric restriction (CR) or rapamycin treatment (RAP), induce adaptations that improve mitochondrial function (B), which results in improved rates of ATP production (C) and a better balance between ROS production and antioxidant scavenging (D). Aerobic exercise stimulates metabolic flux of free fatty acids, decreasing the accumulation of lipotoxic products (E). Because of decreases in chronic oxidative stress and improved antioxidant scavenging, oxidative modifications to proteins decrease (F), leading to improved proteostatic maintenance. The improvement in ATP production, in addition to inhibition of mTOR and AMPK, causes cells to allocate energy more toward somatic maintenance “S” than toward growth “G” (G). The relief of energetic constraint in addition to the maintenance of the rate of energy production facilitates the maintenance of the skeletal muscle proteome. Altogether, interventions that improve mitochondrial function maintain skeletal muscle function and mitigate dynapenia (H)
Aerobic exercise has well-known benefits on mitochondria. These benefits occur primarily via an increase in mitochondrial content (Holloszy 1967) and mitochondrial function (Menshikova et al. 2006; Jacobs et al. 2013; Greggio et al. 2017). Increases in mitochondrial content and function are mediated through increases in mitochondrial protein synthesis (Scalzo et al. 2014; Robinson et al. 2017), and mitochondrial-specific autophagy (mitophagy) (Drake et al. 2016). These changes increase mitochondrial ATP production and \(V{{\text{O}}_{{\text{2max}}}}\). The increased capacity to produce energy on demand improves the cellular energetic budget allowing cells to allocate energetic resources to elastic processes (Wibom et al. 1992; Berthon et al. 1995). In older adults, aerobic exercise training improves single muscle fiber size and function (Harber et al. 2009), whole muscle size and strength (Harber et al. 2012; Konopka and Harber 2014), and whole muscle power (Konopka et al. 2011). In healthy young adults, aerobic exercise training improved myofibrillar protein synthesis at rest (Pikosky et al. 2006). In addition, lifelong, predominantly aerobic, physical activity can delay the loss of skeletal muscle (Zampieri et al. 2015). Although the referenced studies did not assess temporal responses, it is possible that muscle growth was secondary to mitochondrial adaptations that improved bioenergetics to facilitate growth.
In addition to improving energy production, mitochondrial adaptations from aerobic exercise training facilitate important metabolic improvements. First, the increased energetic demands of aerobic exercise increase the flux of fatty acid substrates. As demonstrated in previous studies, increased flux of fatty acid substrates diminishes the accumulation of lipotoxic intermediates (Goodpaster et al. 2001; Corcoran et al. 2007; Befroy et al. 2008; Rivas et al. 2016). Improvements in mitochondrial electron flux also decrease formation of AGEs and oxidatively modified proteins (Snow et al. 2007; Rivas et al. 2012; Distefano et al. 2016; Kent and Fitzgerald 2016; Brocca et al. 2017). Finally, aerobic exercise stimulates endogenous antioxidant production, protecting myofibers from oxidative damage (Gomez-Cabrera et al. 2008). Therefore, in addition to increasing the capacity for maintaining proteostasis, increased mitochondrial function and metabolic flux decreases the demand for somatic maintenance.
Inhibition of mTOR by rapamycin treatment and caloric restriction also mitigates dynapenia, but by different mechanisms than aerobic exercise training. The chronic activation of 5′ adenosine monophosphate-activated protein kinase (AMPK) and inhibition of mTOR, provide a cellular signal that energy is restricted. For many years, whether calorie restriction increases mitochondrial biogenesis was controversial (Nisoli et al. 2005; Civitarese et al. 2007; Hancock et al. 2011). However, recent work by our group confirmed that mitochondrial biogenesis increases in a variety of energy-restricted states (Miller et al. 2012a, b; Drake et al. 2014; Hamilton and Miller 2017). Compared to aerobic exercise, which imposes transient energy deficits, the prolonged signaling of energy shortage during calorie restriction causes cells to allocate energy toward somatic maintenance at the expense of growth. Therefore, under these conditions, growth is restricted but proteostatic processes, and consequently somatic maintenance, improve thus improving overall function.
The positive effects of activating energetic signaling on muscle function seem somewhat underappreciated. For example rapamycin treatment, which inhibits mTOR, attenuates the age-related losses of strength, lean body mass, and endurance capacity in mice (Fischer et al. 2015; Xue and Leng 2016). In addition, three months of rapamycin treatment directly mitigates dynapenia as measured by improved grip strength and rotarod performance in already aged mice (Bitto et al. 2016). Further, 5 months of calorie restriction in 21-month old rats improves ATP production and grip strength compared to ad libitum-fed rats (Zangarelli et al. 2006). Calorie restriction also mitigates oxidative stress, preserving the neuromuscular junction and contributing to the maintenance of skeletal muscle function (Zangarelli et al. 2006; Valdez et al. 2010). Therefore, despite inhibition of growth, both calorie restriction and mTOR inhibition via rapamycin treatment attenuate dynapenia.
While it may seem untenable to translate interventions such as caloric restriction or rapamycin treatment in older adults, these interventions provide insight into potential mechanisms that may improve or maintain muscle function. There is a current clinical trial using metformin, an AMPK activator and mTOR inhibitor, to augment strength training (Long et al. 2017). The trial highlights a strategy of improving metabolic health and bioenergetics to improve muscle function and potential to gain muscle mass. It may seem counterintuitive to restrict growth as a means of improving skeletal muscle function; however, it is important to remember that muscle size is not the sole determinant of function. Muscle quality (i.e., force divided by cross-sectional area) is determined by such factors as neuronal integrity, oxidatively modified proteins, and AGE accumulation (Clark and Manini 2008; Brocca et al. 2017). Further, restricting growth by calorie restriction or rapamycin treatment does not constrain mitochondrial ATP production, but rather puts in motion a series of stress-related mechanisms that preserve energy production to maintain cellular integrity (Drew et al. 2003; Lanza et al. 2012; Miller et al. 2014).
Conclusion
Skeletal muscle mitochondrial function declines because of aging, constraining the energetic budget available for cellular processes. As a result, cellular energy allocation to proteostatic mechanisms, and consequently somatic maintenance, declines resulting in dynapenia. Therefore, the maintenance of mitochondrial proteostasis has a central role in preventing dynapenia in two ways; preventing damage to cellular components, and improving the efficient production of ATP for elastic cellular processes. Future studies should focus on the importance of protein turnover, independent of hypertrophy, for mitigating dynapenia. Further, there should be an effort to translate interventions that target skeletal muscle mitochondria to specifically target dynapenia in humans. Finally, aerobic exercise training should be viewed as an important adjunct or even primary form of exercise to help maintain skeletal muscle function with aging.
Abbreviations
- AGE:
-
Advanced glycation end-product
- AMPK:
-
5′ adenosine monophosphate-activated protein kinase
- ATP:
-
Adenosine triphosphate
- DAG:
-
Diacylglyceride
- mTOR:
-
Mechanistic target of rapamycin
- NADH:
-
Reduced nicotinamide adenine dinucleotide
- ROS:
-
Reactive oxygen species
References
Amara CE, Shankland EG, Jubrias SA et al (2007) Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci 104:1057–1062. doi:10.1073/pnas.0610131104
Anderson EJ, Lustig ME, Boyle KE et al (2009) Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 119:573–581. doi:10.1172/JCI37048
Ayyadevara S, Balasubramaniam M, Suri P et al (2016) Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging 8:3486–3497. doi:10.18632/aging.101141
Balch WE, Morimoto RI, Dillin A, Kelly JW (2008) Adapting proteostasis for disease intervention. Science 319:916–919. doi:10.1126/science.1141448
Barreiro E, Hussain SNA a (2010) Protein carbonylation in skeletal muscles: impact on function. Antioxid Redox Signal 12:417–429. doi:10.1089/ars.2009.2808
Batsis JA, Mackenzie TA, Lopez-Jimenez F, Bartels SJ (2015) Sarcopenia, sarcopenic obesity, and functional impairments in older adults: National Health and Nutrition Examination Surveys 1999–2004. Nutr Res 35:1031–1039. doi:10.1016/j.nutres.2015.09.003
Baumgartner RN, Koehler KM, Gallagher D et al (1998) Epidemiology of sarcopenia among the elderly in New Mexico. Am J Epidemiol 147:755–763
Befroy DE, Petersen KF, Dufour S et al (2008) Increased substrate oxidation and mitochondrial uncoupling in skeletal muscle of endurance-trained individuals. Proc Natl Acad Sci U S A 105:16701–16706. doi:10.1073/pnas.0808889105
Berthon P, Freyssenet D, Chatard J-C et al (1995) Mitochondrial ATP production rate in 55 to 73-year-old men: effect of endurance training. Acta Physiol Scand 154:269–274. doi:10.1111/j.1748-1716.1995.tb09908.x
Bhatti JS, Bhatti GK, Reddy PH (2016) Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. doi:10.1016/j.bbadis.2016.11.010
Bitto A, Ito TK, Pineda VV et al (2016) Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 5:1–17. doi:10.7554/eLife.16351
Brand MD (1990) The contribution of the leak of protons across the mitochondrial inner membrane to standard metabolic rate. J Theor Biol 145:267–286. doi:10.1016/S0022-5193(05)80131-6
Brand MD (1997) Regulation analysis of energy metabolism. J Exp Biol 200:193–202
Brocca L, McPhee JS, Longa E et al (2017) Structure and function of human muscle fibres and muscle proteome in physically active older men. J Physiol 0:1–22. doi:10.1113/JP274148
Buttgereit F, Brand MD (1995) A hierarchy of ATP-consuming processes in mammalian cells. Biochem J 312(Pt 1):163–167. doi:10.1210/er.2008-0019
Civitarese AE, Carling S, Heilbronn LK et al (2007) Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med 4:e76. doi:10.1371/journal.pmed.0040076
Clark BC, Manini TM (2008) Sarcopenia != dynapenia. J Gerontol Ser A Biol Sci Med Sci 63:829–834. doi:10.1093/gerona/63.8.829
Clark BC, Manini TM (2012) What is dynapenia? Nutrition 28:495–503. doi:10.1016/j.nut.2011.12.002
Coen PM, Dubé JJ, Amati F et al (2010) Insulin resistance is associated with higher intramyocellular triglycerides in type I but not type II myocytes concomitant with higher ceramide content. Diabetes 59:80–88. doi:10.2337/db09-0988
Conley KE, Jubrias SA, Esselman PC (2000) Oxidative capacity and ageing in human muscle. J Physiol 526:203–210. doi:10.1111/j.1469-7793.2000.t01-1-00203.x
Corcoran MP, Lamon-Fava S, Fielding RA (2007) Skeletal muscle lipid deposition and insulin resistance: effect of dietary fatty acids and exercise. Am J Clin Nutr 85:662–677
Cruz-Jentoft AJ, Baeyens JP, Bauer JM et al (2010) Sarcopenia: European consensus on definition and diagnosis. Age Ageing 39:412–423. doi:10.1093/ageing/afq034
Cruz-Jentoft AJ, Landi F, Schneider SM et al (2014) Prevalence of and interventions for sarcopenia in ageing adults: a systematic review. Report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing 43:748–759. doi:10.1093/ageing/afu115
Dai D-F, Chiao YA, Marcinek DJ et al (2014) Mitochondrial oxidative stress in aging and healthspan. Longev Heal 3:6. doi:10.1186/2046-2395-3-6
Dalal M, Ferrucci L, Sun K et al (2009) Elevated serum advanced glycation end products and poor grip strength in older community-dwelling women. J Gerontol Ser A Biol Sci Med Sci 64A:132–137. doi:10.1093/gerona/gln018
Dennis M, Bier (1999) The energy costs of protein metabolism: lean and mean on Uncle Sam’s team. In: Research Institute of Medicine (US) Committee on Military Nutrition (ed) The role of protein and amino acids in sustaining and enhancing performance. National Academies, Washington, D.C.
Distefano G, Standley RA, Dubé JJ et al (2016) Chronological age does not influence ex-vivo mitochondrial respiration and quality control in skeletal muscle. J Gerontol Ser A Biol Sci Med Sci 0:glw102. doi:10.1093/gerona/glw102
Doherty TJ (2003) Invited review: aging and sarcopenia. J Appl Physiol 95:1717–1727. doi:10.1152/japplphysiol.00347.2003
Drake JC, Bruns DR, Peelor FF et al (2014) Long-lived crowded-litter mice have an age-dependent increase in protein synthesis to DNA synthesis ratio and mTORC1 substrate phosphorylation. Am J Physiol Endocrinol Metab 307:E813–E821. doi:10.1152/ajpendo.00256.2014
Drake JC, Wilson RJ, Yan Z (2016) Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J 30:13–22. doi:10.1096/fj.15-276337
Drenth H, Zuidema S, Bunt S et al (2016) The contribution of advanced glycation end product (AGE) accumulation to the decline in motor function. A systematic review. Eur Rev Aging Phys Act. doi:10.1186/s11556-016-0163-1
Drew B, Dirks PA, Selman C et al (2003) Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol 284:R474–R480. doi:10.1152/ajpregu.00455.2002
Dumitru CA, Zhang Y, Li X, Gulbins E (2007) Ceramide: a novel player in reactive oxygen species-induced signaling? Antioxid Redox Signal 9:1535–1540. doi:10.1089/ars.2007.1692
European Commission Directorate-General for Economic and Financial Affairs (2015) The 2015 Ageing Report Economic and budgetary projections for the 28 EU Member States (2013–2060)
Fielding RA, Vellas B, Evans WJ et al (2011) Sarcopenia: an undiagnosed condition in older adults. Current Consensus definition: prevalence, etiology, and consequences. International working group on sarcopenia. J Am Med Dir Assoc 12:249–256. doi:10.1016/j.jamda.2011.01.003
Figueiredo PA, Powers SK, Ferreira RM et al (2009) Aging impairs skeletal muscle mitochondrial bioenergetic function. J Gerontol Ser A Biol Sci Med Sci 64:21–33. doi:10.1093/gerona/gln048
Fischer KE, Gelfond JAL, Soto VY et al (2015) Health effects of long-term rapamycin treatment: the impact on mouse health of enteric rapamycin treatment from four months of age throughout life. PLoS One 10:1–18. doi:10.1371/journal.pone.0126644
Gavrilov LA, Gavrilova NS (2002) Evolutionary theories of aging and longevity. Sci World J 2:339–356. doi:10.1100/tsw.2002.96
Gomez-Cabrera M-C, Domenech E, Viña J (2008) Moderate exercise is an antioxidant: upregulation of antioxidant genes by training. Free Radic Biol Med 44:126–131. doi:10.1016/j.freeradbiomed.2007.02.001
Gonzalez-Freire M, De Cabo R, Bernier M et al (2015) Reconsidering the role of mitochondria in aging. J Gerontol Ser A Biol Sci Med Sci 70:1334–1342. doi:10.1093/gerona/glv070
Goodpaster BH, He J, Watkins S, Kelley DE (2001) Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86:5755–5761. doi:10.1210/jcem.86.12.8075
Greggio C, Jha P, Kulkarni SS et al (2017) Enhanced respiratory chain supercomplex formation in response to exercise in human skeletal muscle. Cell Metab 25:301–311. doi:10.1016/j.cmet.2016.11.004
Gregory TR (2001) Coincidence, coevolution, or causation? DNA content, cellsize, and the C-value enigma. Biol Rev 76:65–101. doi:10.1111/j.1469-185X.2000.tb00059.x
Hamilton KL, Miller BF (2017) Mitochondrial proteostasis as a shared characteristic of slowed aging: the importance of considering cell proliferation. J Physiol. doi:10.1113/JP274335
Hancock CR, Han DH, Higashida K et al (2011) Does calorie restriction induce mitochondrial biogenesis? A reevaluation. FASEB J 25:785–791. doi:10.1096/fj.10-170415
Harber MP, Konopka AR, Douglass MD et al (2009) Aerobic exercise training improves whole muscle and single myofiber size and function in older women. AJP Regul Integr Comp Physiol 297:R1452–R1459. doi:10.1152/ajpregu.00354.2009
Harber MP, Konopka AR, Undem MK et al (2012) Aerobic exercise training induces skeletal muscle hypertrophy and age-dependent adaptations in myofiber function in young and older men. J Appl Physiol 113:1495–1504. doi:10.1152/japplphysiol.00786.2012
Haus JM, Carrithers JA, Trappe SW, Trappe TA (2007) Collagen, cross-linking, and advanced glycation end products in aging human skeletal muscle. J Appl Physiol 47306:2068–2076. doi:10.1152/japplphysiol.00670.2007
Heeman B, Van den Haute C, Aelvoet S-AS-A et al (2011) Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci 124:1115–1125. doi:10.1242/jcs.078303
Holloszy JO (1967) Biochemical adaptations in muscle. J Biol Chem 242:2278–2282
Hou C (2013) The energy trade-off between growth and longevity. Mech Ageing Dev 134:373–380. doi:10.1016/j.mad.2013.07.001
Hou C, Zuo W, Moses ME et al (2008) Energy uptake and allocation during ontogeny. Science 322:736–739. doi:10.1126/science.1162302
Jacobs RA, Boushel R, Wright-Paradis C et al (2013) Mitochondrial function in human skeletal muscle following high-altitude exposure. Exp Physiol 98:245–255. doi:10.1113/expphysiol.2012.066092
Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R (2004) The healthcare costs of sarcopenia in the United S… [J Am Geriatr Soc. 2004]—PubMed result. J Am Geriatr Soc 52:80–85. doi:10.1111/j.1532-5415.2004.52014.x
Jensen MB, Jasper H (2014) Mitochondrial proteostasis in the control of aging and longevity. Cell Metab 20:214–225
Kapahi P (2010) Protein synthesis and the antagonistic pleiotropy hypothesis of aging. Adv Exp Med Biol 694:30–37
Kent JA, Fitzgerald LF (2016) In vivo mitochondrial function in aging skeletal muscle: capacity, flux, and patterns of use. J Appl Physiol 121:996–1003. doi:10.1152/japplphysiol.00583.2016
Konopka AR, Harber MP (2014) Skeletal muscle hypertrophy after aerobic exercise training. Exerc Sport Sci Rev 42:53–61. doi:10.1249/JES.0000000000000007
Konopka AR, Trappe TA, Jemiolo B et al (2011) Myosin heavy chain plasticity in aging skeletal muscle with aerobic exercise training. J Gerontol Ser A Biol Sci Med Sci 66A:835–841. doi:10.1093/gerona/glr088
Kruse SE, Karunadharma PP, Basisty N et al (2016) Age modifies respiratory complex I and protein homeostasis in a muscle type-specific manner. Aging Cell 15:89–99. doi:10.1111/acel.12412
Lanza IR, Zabielski P, Klaus KA et al (2012) Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metab 16:777–788. doi:10.1016/j.cmet.2012.11.003
Larsen S, Nielsen J, Hansen CN et al (2012) Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590:3349–3360. doi:10.1113/jphysiol.2012.230185
Levine RL, Stadtman ER (2001) Oxidative modification of proteins during aging. Exp Gerontol 36:1495–1502 pii]
Long DE, Peck BD, Martz JL et al (2017) Metformin to augment strength training effective response in seniors (MASTERS): study protocol for a randomized controlled trial. Trials 18:192. doi:10.1186/s13063-017-1932-5
Lynch M, Marinov GK (2015) The bioenergetic costs of a gene. Proc Natl Acad Sci 112:201514974. doi:10.1073/pnas.1514974112
Madeira VMC (2012) Overview of mitochondrial bioenergetics. Methods Mol Biol 810:1–6
Marcinek DJ, Schenkman K a, Ciesielski W a et al (2005) Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol 569:467–473. doi:10.1113/jphysiol.2005.097782
Martin BT, Zimmer EI, Grimm V, Jager T (2012) Dynamic energy budget theory meets individual-based modelling: a generic and accessible implementation. Methods Ecol Evol 3:445–449. doi:10.1111/j.2041-210X.2011.00168.x
Menshikova EV, Ritov VB, Fairfull L et al (2006) Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci 61:534–540
Miller BF, Robinson MM, Bruss MD et al (2012a) A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 11:150–161. doi:10.1111/j.1474-9726.2011.00769.x
Miller BF, Robinson MM, Reuland DJ et al (2012b) Calorie restriction does not increase short-term or long-term protein synthesis. J Gerontol A Biol Sci Med Sci 68:1–9. doi:10.1093/gerona/gls219
Miller BF, Drake JC, Naylor B et al (2014) The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res Rev 18:106–111. doi:10.1016/j.arr.2014.09.005
Moghaddas S, Hoppel CL, Lesnefsky EJ (2003) Aging defect at the QO site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Arch Biochem Biophys 414:59–66
Morrow RM, Picard M, Derbeneva O et al (2017) Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc Natl Acad Sci. doi:10.1073/pnas.1700997114
Mortimore GE, Pösö a R (1987) Intracellular protein catabolism and its control during nutrient deprivation and supply. Annu Rev Nutr 7:539–564. doi:10.1146/annurev.nutr.7.1.539
Murton AJ (2015) Muscle protein turnover in the elderly and its potential contribution to the development of sarcopenia. Proc Nutr Soc 74:1–10. doi:10.1017/S0029665115000130
Nair KS (2005) Aging muscle. Am J Clin Nutr 81:953–963
Nisbet RM, Jusup M, Klanjscek T, Pecquerie L (2012) Integrating dynamic energy budget (DEB) theory with traditional bioenergetic models. J Exp Biol 215:892–902. doi:10.1242/jeb.059675
Nisoli E, Tonello C, Cardile A et al (2005) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310:314–317. doi:10.1126/science.1117728
Paddon-Jones D, Rasmussen BB (2009) Dietary protein recommendations and the prevention of sarcopenia. Curr Opin Clin Nutr Metab Care 12:86–90. doi:10.1097/MCO.0b013e32831cef8b
Picard M, Ritchie D, Wright KJ et al (2010) Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell 9:1032–1046. doi:10.1111/j.1474-9726.2010.00628.x
Pikosky MA, Gaine PC, Martin WF et al (2006) Aerobic exercise training increases skeletal muscle protein turnover in healthy adults at rest. J Nutr 136:379–383
Poppek D, Grune T (2005) Protein repair and degradation. In: Grune T (ed) Reactions, processes: oxidants and antioxidant defense systems. Springer Berlin Heidelberg, Berlin, pp 177–201
Porter C, Wall BT (2012) Skeletal muscle mitochondrial function: is it quality or quantity that makes the difference in insulin resistance? J Physiol 590:5935–5936. doi:10.1113/jphysiol.2012.241083
Rivas DA, Morris EP, Haran PH et al (2012) Increased ceramide content and NFκB signaling may contribute to the attenuation of anabolic signaling after resistance exercise in aged males
Rivas DA, Mcdonald DJ, Rice NP et al (2016) Diminished anabolic signaling response to insulin induced by intramuscular lipid accumulation is associated with inflammation in aging but not obesity. Am J Physiol Regul Integr Comp Physiol ajpregu. doi:10.1152/ajpregu.00198.2015
Robinson MM, Dasari S, Konopka AR et al (2017) Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans clinical and translational report enhanced protein translation underlies improved metabolic and physical adapta. Cell Metab 25:581–592. doi:10.1016/j.cmet.2017.02.009
Rolfe DF, Brown GC (1997) Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev 77(3):731–758
Romanello V, Sandri M (2015) Mitochondrial quality control and muscle mass maintenance. Front Physiol 6:422. doi:10.3389/fphys.2015.00422
Ryu D, Mouchiroud L, Andreux PA et al (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22:879–888. doi:10.1038/nm.4132
Salminen A, Ojala J, Kaarniranta K, Kauppinen A (2012) Mitochondrial dysfunction and oxidative stress activate inflammasomes: impact on the aging process and age-related diseases. Cell Mol Life Sci 69:2999–3013. doi:10.1007/s00018-012-0962-0
Scalzo RL, Peltonen GL, Binns SE et al (2014) Greater muscle protein synthesis and mitochondrial biogenesis in males compared with females during sprint interval training. FASEB J 28:2705–2714. doi:10.1096/fj.13-246595
Semba RD, Nicklett EJ, Ferrucci L (2010) Does Accumulation of advanced glycation end products contribute to the aging phenotype? J Gerontol Ser A Biol Sci Med Sci 65A:963–975. doi:10.1093/gerona/glq074
Shanley DP, Kirkwood TB (2000) Calorie restriction and aging: a life-history analysis. Evol Int J Org Evol 54:740–750
Siegel MP, Kruse SE, Percival JM et al (2013) Mitochondrial-targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 12:763–771. doi:10.1111/acel.12102
Snow LM, Fugere NA, Thompson LV (2007) Advanced glycation end-product accumulation and associated protein modification in type II skeletal muscle. With Aging 62:1204–1210
Stadtman ER, Oliver CN, Levine RL et al (1988) Implication of protein oxidation in protein turnover, aging, and oxygen toxicity. Basic Life Sci 49:331–339
Stern M (2017) Evidence that a mitochondrial death spiral underlies antagonistic pleiotropy. Aging Cell 16:1–9. doi:10.1111/acel.12579
Suarez RK (1998) Oxygen and the upper limits to animal design and performance. J Exp Biol 1072:1065–1072
Suarez RK, Suarez KR (2012) Energy and metabolism. In: Comprehensive physiology. Wiley, Hoboken
Toyama BH, Hetzer MW (2013) Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol 14:55–61. doi:10.1038/nrm3496
Valdez G, Tapia JC, Kang H et al (2010) Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci USA 107:14863–14868. doi:10.1073/pnas.1002220107
van Leeuwen IMM, Vera J, Wolkenhauer O (2010) Dynamic energy budget approaches for modelling organismal ageing. Philos Trans R Soc B Biol Sci 365:3443–3454. doi:10.1098/rstb.2010.0071
Wanagat J, Cao Z, Pathare P, Aiken JM (2001) Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J 15:322–332. doi:10.1096/fj.00-0320com
Waterlow JC (1984) Protein turnover with special reference to man. Q J Exp Physiol 69:409–438. doi:10.1113/expphysiol.1984.sp002829
Wibom R, Hultman E, Johansson M (1992) Adaptation of mitochondrial ATP production in human skeletal muscle to endurance training and detraining. J Appl Physiol 73:2004–2010
Wiley CD, Velarde MC, Lecot P et al (2016) Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab 23:303–314. doi:10.1016/j.cmet.2015.11.011
Xue Q-L, Leng S (2016) Rapamycin increases grip strength and attenuates age-related decline in maximal running distance in old low capacity runner rats. Aging 8:1–8. doi:10.18632/aging.100929
Zampieri S, Pietrangelo L, Loefler S et al (2015) Lifelong physical exercise delays age-associated skeletal muscle decline. J Gerontol Ser A Biol Sci Med Sci 70:163–173. doi:10.1093/gerona/glu006
Zangarelli A, Chanseaume E, Morio B et al (2006) Synergistic effects of caloric restriction with maintained protein intake on skeletal muscle performance in 21-month-old rats: a mitochondria-mediated pathway. FASEB J 20:2439–2450. doi:10.1096/fj.05-4544com
Acknowledgements
The authors would like to thank members of the Translational Research on Aging and Chronic Disease lab for their collective contributions that have led to the ideas and knowledge presented in this review.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Michael Lindinger.
Karyn L. Hamilton and Benjamin F. Miller: Co-Senior Authors.
Rights and permissions
About this article

Cite this article
Musci, R.V., Hamilton, K.L. & Miller, B.F. Targeting mitochondrial function and proteostasis to mitigate dynapenia. Eur J Appl Physiol 118, 1–9 (2018). https://doi.org/10.1007/s00421-017-3730-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-017-3730-x