
Abstract
The gut microbiota is established in the newborn period and plays a pivotal role in the development of the mucosal tissue, immune maturation, and host metabolism. Distortions in the assembly and maturation of the microbiota during this critical time-window can therefore have profound effects on future health and the susceptibility to non-communicable diseases.
In this chapter, we provide an overview of the ecological processes involved in the establishment of the indigenous microbial communities during infancy and childhood. Moreover, we summarize the current knowledge on the disruptive effects of lifestyle changes on gut microbiota assembly and maturation. Finally, we highlight important areas for further research in order to identify approaches to revert the deprivation of our microbiota.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Ecological theory
- Microbiota assembly
- Birth mode
- Breastfeeding
- Complementary food
- Social interactions
- Natural environment
- Antibiotics
1 Introduction
The indigenous microbiota of the human gut has long been recognized to contribute to health and disease by influencing gut and immune maturation, host nutrition, and protection against pathogen invasion [1].
In line with this, perturbations in the development and composition of the gut microbiota have been associated with the development of obesity [2,3,4], allergies [5,6,7], inflammatory bowel diseases [8,9,10], and many other non-communicable diseases [11, 12].
Infancy sets the stage for intestinal microbial assembly, diversification, and maturation. Moreover, this period is a critical time-window during which the microbiota provides a stimulus for the ontogeny of the enteric mucosal tissue and immune system with persistent local as well as systemic effects [13, 14]. Insights into the processes that drive the first inoculum to differentiate into a highly individualized [6, 15,16,17,18] and stable microbial ecosystem, as established after the first years of life, will therefore have a direct impact on our ability to manage and maintain human health [19, 20]. To develop successful strategies to restore or maintain a healthy microbiota, we thus need to refine our understanding of the processes driving the inter-individual variation in microbial composition and assembly. It is increasingly being recognized that the principles of ecological theory can help us to understand and predict community variations in the human microbiota [19].
In this chapter we will discuss the development of the gut microbiota during infancy and childhood from an ecological theoretical perspective, and describe how lifestyle changes may distort the natural development of microbial assembly and maturation.
2 Ecological Principles of Microbial Community Assembly
The ecologist Mark Vellend has synthesized various concepts of community assembly by categorizing the underlying processes into four groups: dispersal, selection, drift, and diversification [21, 22].
Dispersal
of bacteria from the meta-community is an important process to seed the initially sterile infant gut but is a process that also plays an important role thereafter. The meta-community consists of numerous local communities, some of which are host-related (e.g., maternal skin, gut, and vaginal microbiota), while others are not (e.g., soil, food-born, building environment microbial communities) [23].
In this respect, priority effects describe how the order and timing of dispersal from the meta-community might alter how diversification, drift, and selection affect the assembly of the infant gut microbiota. In other words, the impact that particular species can have on the community assembly in the infant gut would depend on the timing and order in which they arrive (history) [23, 24].
Environmental selection
or niche-based interaction is the deterministic process that, based upon the conditions in a given habitat, drives differences in growth and death rates among microbial taxa based upon their fitness and niche-differences [23]. Diet, host metabolites, and the immune system are primary sources that drive environmental selection. From the perspective of environmental selection, the human body can, for example, be seen as a “habitat filter”—a collection of conditions and resources that nourish the growth of some microbes, but not others. This view implies that the interaction is unidirectional; the host shapes the microbiota. However, in many circumstances of environmental filtering, the host-microbe interactions are bi-directional [19]. The interactions between the host immune system and the microbiota are a classical example, but also the interactions between host metabolites such as bile acids and the microbiota [25] exemplify such bi-directional selection processes. Also, the abrupt shifts in the composition and structure of the microbiota that are commonly observed during an infectious episode or a course of antibiotics are examples of environmental selection processes.
Ecological drift
is a completely stochastic (chance-driven) event in which changes in species population size occur regardless of species identity [23, 26]. The effect of drift is expected to be stronger on low abundant species as slight negative changes in their abundance could already push them stochastically to local extinction, unless they have (or gain, e.g., via diversification) a competitive advantage or become replenished by dispersal from outside the local community [19, 27].
It is however almost impossible to distinguish the effect of drift from the effect of other ecological processes except when studied in an experimentally controlled setting [26].
Diversification
is the process of the generation of new genetic variants, often as a result of a persistent selective pressure. Highly abundant and dense microbial populations, rapid growth rates, and strong selective pressures, conditions that are all met in the adult human gastrointestinal tract, can fuel microbial adaptation via mutation or recombination (e.g., via horizontal gene transfer) [19]. However, as the selective regimes during infancy frequently shift as a result of, among other factors, a developing host and alterations in feeding regimes, the degree to which diversification is involved in the infant gut during assembly remains largely unknown [23].
Neutral Community Assembly
A theoretical framework of community assembly which assumes that dispersal, diversification, and ecological drift are completely stochastic processes is the neutral community model (NCM). According to this model, neither environmental selection nor inherent species differences in their ability to disperse or diversify play a role in the community assembly. Although such models do not account for deterministic factors and make many simplifying assumptions, they have among others successfully been applied to predict the structures of aquatic and respiratory microbial communities [28, 29]. Such models moreover are important for gaining insight into the importance of neutral dispersal in shaping the structure of microbial communities, to identify conditions that lead to divergence from neutral dynamics or to identify microbial taxa that do not assemble in a neutral manner [30]. NCM has also been applied to the assembly of gut microbial communities. In a study on the zebrafish intestinal microbial communities from larvae to adulthood, the importance of non-neutral processes increased as the host matured [31]. A recent study among the Tsimane, an indigenous Bolivian population, also underscored the importance of neutral forces in shaping microbiota assembly in early life and to a lesser extent in adulthood [30]. These observations suggest that a significant amount of diversity in microbial community structures between individuals could be explained by neutral processes of drift and dispersal [31].
Glossary of Terms and Definitions Used in This Review
Terminology | Description | Reference |
|---|---|---|
Community | A group of potentially interacting species that live together in a specified place and time | |
Dispersal | Movement of microorganisms across space | |
Ecological drift | Stochastic changes in the relative abundance of different microbial taxa within a community throughout time as a result of birth, death, and reproduction | |
Environmental selection | Changes in the microbial community structure as a result of deterministic fitness differences between microbial taxa | |
In situ diversification | Generation of new genetic variants by mutation or recombination (e.g., via horizontal gene transfer) | |
Meta-community | A set of local communities that are linked by dispersal of multiple potentially interacting species | [33] |
Neutral assembly theory/neutral community model | Theory/model assuming that dispersal, diversification, and ecological drift are completely stochastic processes and that neither environmental selection nor species traits play a role in community assembly | |
Perturbation | An external event/stressor that causes a distinct selective pressure on the ecosystem, also called disturbance | |
Priority effects/historical contingency | The order in which species arrive at local sites (e.g., in the infant gut) dictates the effect of species on one another | |
Resilience | The property of a microbial ecosystem that defines how fast, and to what extent it will recover its initial functional or taxonomical composition following perturbation | |
Resistance | The power of an ecosystem to remain unchanged upon a perturbation |
3 Establishment of the Microbiome
As compared to the microbiome of adults, which has been suggested to be relatively stable and resilient [37,38,39], the microbiome in infants is highly dynamic. The microbial richness and diversity gradually increase from early infancy to childhood, while the variation in microbial composition between children decreases [6, 39].
The first colonizers of the infant gut microbiota are typically facultative anaerobes, particularly Enterobacteriaceae, but also facultative anaerobic genera within the Firmicutes phylum including staphylococci, streptococci, lactobacilli, and enterococci [6, 40,41,42,43]. Although members of the Enterobacteriaceae were shown to be specific for the infant gut, most of the initial colonizers are homogeneously distributed across different body sites [44]. During the following months, obligate anaerobes, including Bifidobacterium, Bacteroides, Veillonella, and Clostridium, start to dominate [6, 43]. While Enterobacteriaceae wane, they still have an important share during the first half year of life after which their abundance further decreases [6, 40, 41, 43]. Under the impact of weaning and transition to an adult-like diet, Veillonella and Bifidobacterium start to decrease, Bacteroides further increase, and members of the Lachnospiraceae (e.g., Blautia and Roseburia) and Ruminococcaceae (e.g., Faecalibacterium, Ruminococcus) rise to become dominant members of the gut microbiota [6, 41,42,43]. Many of these bacteria are butyrate-producers, and a concomitant increase in butyrate levels has indeed been observed [45, 46].
Finally, some prokaryotes, including the archaeal genus Methanobrevibacter and the bacterial genera Desulfovibrio, Bilophila, and members of the Christensenellaceae family, only colonize after infancy and keep rising in prevalence and abundance beyond the age of 5 years [39]. This indicates that the adequate niche has first to be created before these genera can settle. These niche formations are partly driven by the prior arrival of other microbial taxa for necessary cross-feeding interactions, but also on a fully reduced environment and the availability of complex dietary carbohydrates.
Several studies have aimed to identify distinct phases of microbiome progression [6, 39, 43, 47]. Although the exact period differed between studies, it is evident that most rapid changes in microbiome development occur within the first 6–12 months of life. Microbiome maturation thereafter continues in a less profound manner, but the exact age at which a stable adult-like microbial community structure is reached is still a matter of debate. While it has previously been suggested that stabilization of the microbiome occurs around the age of 2–3 years [47, 48], differences could still be observed in the microbiome of Swedish 5-year-old [39] and Dutch 6–9-year-old children [49] as compared to Swedish and Dutch adults, respectively. A recent meta-analysis on metagenomic data from over 1900 fecal samples from nine studies confirmed that the microbiome could predict a child’s chronological age well beyond the first 3 years of life [42]. In part, this controversy can be attributed to the sparseness of data on the microbiome of children beyond the age of 3 years [37].
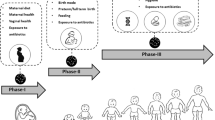
4 Determinants of Infant Gut Microbiome Assembly and the Impact of Lifestyle Changes (Fig. 1)
4.1 Seeding of Newborn Ecosystem
The microbial species that colonize first play a crucial role in the development of the ecosystem in the gut, potentially influencing the ultimate composition and functionality of the microbiota for life. However, at what stage the first microbes start to seed and colonize the gut is still a matter of debate. In 2014, Agaard and colleagues reported the existence of a microbiome in the placenta of healthy women [50]. Since then, many other human studies detected microbial signals in amniotic fluid, fetal intestine, cord blood, placenta, or meconium samples, thereby suggesting in utero colonization and challenging the concept that seeding of the gut ecosystem initiates at birth [51,52,53,54]. In utero dispersal of microbes would have tremendous implications for fetal and neonatal health and development, but objections were raised against most of these studies (see review [52]). First, the majority of studies relied on the detection of bacterial DNA which does not confirm the existence of a living microbial community [55]. Second, no consistent microbial profile was detected across the different studies (i.e., the dominant bacterial taxa varied widely between studies Micrococcus, Lactobacillus, Staphylococcus). Lastly, it appeared that results were biased by contamination, i.e., the detected bacteria in low-biomass samples were also identified in negative controls [56, 57]. As a result, many other studies that appropriately controlled for contamination could not provide evidence of microbial taxa in utero [58,59,60,61,62]. On the other hand, recent studies demonstrated the existence of viable bacteria-like morphology in the fetal intestine and placenta by culture and microscopy [53, 63] albeit debated as well [64, 65]. In addition, oral administration of trackable bacteria (e.g., genetically labeled Enterococcus faecium) to pregnant female mice could be recovered from amniotic fluid and meconium of the offspring [66, 67]. Today, the debate still continues, and it is still questionable whether a prenatal intrauterine microbiome exists.

Infographic depicting the main ecological processes and deterministic factors shaping the microbiota throughout infancy and childhood. Main sources of microbial dispersal to the infant gut comprise the infant’s mother and other family members, pets and peers, food sources, and the natural environment (depicted in blue). The order at which microbial taxa arrive in the infant gut may dictate the colonization success of subsequent incoming bacteria (depicted in orange). Additionally, the environment of the infant gastrointestinal tract may select for or exclude specific microbial taxa (depicted in green). This environmental selection is driven by among others the substrate availability, oxygen concentration, pH, and host genetics
Birth is likely the first step in seeding the newborn gut and definitely the most dramatic one, given the amount and diversity of microbes to which a newborn gets exposed. The passage through the birth canal has long been recognized as the major transmission of first microbes, i.e., Lactobacillus and Prevotella [68, 69]. However, studies comparing microbial strain profiles of infant fecal to both maternal vaginal and rectal samples revealed that mother-to-child transmission mainly occurs for rectal rather than vaginal strains [42, 70]. In particular, as consistently shown in numerous studies, maternal Bacteroides strains are most frequently transferred to the intestine of neonates born via a natural delivery [6, 70,71,72].
The mode of delivery evidently impacts the dispersal of microorganisms from the mother and results in distinct microbial profiles between infants born vaginally and via Cesarean section (C-section). The microbiota of C-section delivered infants is characterized by delayed colonization of mainly Bacteroides and Bifidobacterium compared to vaginally delivered infants [40, 43, 47, 70, 71, 73,74,75]. In a recent study, we longitudinally monitored the establishment of the human infant gut microbiota and further supported that birth mode most strongly affects members of the genus Bacteroides [6]. The decreased levels of Bacteroides in infants born by C-section (Cesarean section) remained significant after careful adjustment for other confounders and persisted for at least 31 weeks [6]. In addition, other studies demonstrated that this founder effect could even last until 4-years postpartum [47, 76]. Although C-section is accompanied by prophylactic antibiotic administration to the mother, the effect of delivery mode on the infant gut microbiota has recently been demonstrated to be independent of intrapartum antibiotic prophylaxis (IAP) [77]. In this study, IAP was administered only after clamping of the umbilical cord in mothers undergoing C-section, enabling the researchers to examine the impact of the birth mode on the neonatal microbiota in the absence of antibiotic exposure to the baby. The latter study confirmed the lower levels of Bacteroides spp. and Bifidobacterium spp. in C-section delivered infants. Altogether, this suggests that dispersal of maternal microbes during vaginal delivery is crucial to acquire certain microbial species in early life.
Cesarean section delivery is perhaps one of the factors most commonly linked to distortions in the microbiota establishment in early life [40, 71]. C-section numbers are rising worldwide [78]. Nowadays one out of five babies is delivered by C-section with numbers having almost doubled since 2000, while in Brazil rates have risen up to 45%. Notably, the Netherlands has one of the lowest C-section rates (17%) among developed countries, especially when compared to neighboring countries such as Germany (30%) [78]. The latter might be because pregnant women at low risk of birth complications get unique midwife-led care in the Netherlands which is associated with lower intervention rates [79, 80]. Although in some cases it might be a life-saving procedure, the high numbers of C-sections without medical reason (i.e., elective C-section) are worrisome.
Many efforts have recently been made to reverse the dispersal limitation in C-section delivered neonates by exposing them to the maternal vaginal microbiota immediately upon delivery, a procedure termed “vaginal seeding” or “bacterial baptism” [81]. The recent evidence that maternal fecal rather than vaginal bacteria are depleted in the intestinal microbiota of C-section delivered neonates [42, 70], however, challenges the efficacy of vaginal seeding to abolish the dispersal limitation during C-section. In a recent proof-of-principle study by Korpela et al., fecal microbiota transplantation (FMT) was therefore used to restore the microbiota in infants born by C-section [82]. An enhancement of Bacteroidaceae and Bifidobacteriaceae in FMT-treated as compared to untreated infants was observed without any short-term adverse effects. Although FMT might be a more successful strategy to restore maternal transfer in C-section delivered infants, concerns remain on the risks and uncertainties of this treatment (i.e., dispersal of pathogens or overstimulation). It is therefore crucial to obtain further insight into the maternal microbial species that C-section delivered infants fail to achieve and the corresponding health implications in order to move towards more controlled attempts to restore the initial microbial colonization of newborns, e.g., in the form of synthetic microbial transplants.
4.2 Infant Diet—Environmental Selection and Dispersal of Milk Microbes
The World Health Organization (WHO) and the United Nations International Children’s Emergency Fund (UNICEF) advocate exclusive breastfeeding (EBF) for the first 6 months of life [83]. Despite these global recommendations, breastfeeding prevalence, particularly EBF, is low in Europe and the Americas with dramatic disparities between countries [84].
Low rates and early cessation of breastfeeding have important adverse public health impacts. Besides nutrients, breast milk contains a wide variety of bioactive factors (e.g., lysozyme, lactoferrin, and immunoglobulins) that support the development and maturation of the infant gut, the innate and adaptive immunity, and systemic metabolism [85].
Moreover, breastfeeding plays a crucial role in the establishment of the infant gut microbiome both directly, by the dispersal of living bacteria in breast milk, and indirectly, by environmental selection in the form of prebiotic nutrient substrates and bioactive components.
Breastfeeding directly impacts the infant microbiome by dispersal of viable microorganisms in human milk. An exclusively breastfed infant will daily consume the significant amount of approximately 105–107 commensal bacteria while suckling [86]. Facultative anaerobic skin and throat bacteria, such as members of the genera Staphylococcus, Streptococcus, and Cutibacterium, are the predominant bacteria detected in breast milk by traditional culturing approaches [86, 87]. At lower concentrations lactic acid bacteria, including Lactobacillus and Enterococcus, members of the gram-negative Enterobacteriaceae, as well as obligate anaerobes, such as Bifidobacterium and Veillonella have been frequently isolated from breast milk [86,87,88]. The application of culture-independent molecular approaches has revealed a human milk microbial diversity beyond expectancy, including major gut-associated obligate anaerobes such as Bacteroides and several members of Clostridia, including the butyrate-producing Faecalibacterium and Roseburia, which are important for colonic health [87]. The origin of the microbes in breast milk is not fully understood, but likely involved the maternal skin, the infant’s oral cavity during suckling, and potentially even the mother’s gut via the entero-mammary pathway [89]. The exact composition of the human milk microbiome is highly variable and potentially influenced by geography, birth mode, lactation stage, as well as diet, health status, medication use, and genotype of the mother [89, 90]. Numerous studies have shown the importance of breast milk microbiota as an important source of successfully seeding the infant gut. Identical strains of Bifidobacterium, Lactobacillus, and Staphylococcus have been isolated from breast milk and infant feces in mother-infant dyads [91,92,93]. Transmission of bifidobacterial strains from breastmilk to the infant gut has additionally been demonstrated by a combination of gene marker-based amplicon sequencing, metagenomic shotgun sequencing, and strain isolation followed by genomic analysis [91, 94].
The dispersal of milk microbes to the infant gut obviously contributes to the profound differences in the microbial communities in breastfed as compared to formula-fed infants. The indigenous microbiota of exclusively breastfed infants is low in diversity as a result of the predominance of bifidobacteria, which can account for up to 70–80% of all bacteria in the stools of breastfed infants [6, 95]. In addition, lactobacillus species that are commonly used as probiotics have also been found to be enriched among breastfed infants [73]. Although the absolute bifidobacterium counts in infants receiving formula feeding tend to be as high as in breastfed infants [96,97,98,99,100], the microbiota of formula-fed infants is characterized by a much higher diversity and increased numbers of Bacteroides, Clostridium, and Enterobacteriaceae, including opportunistic pathogens such as Clostridioides difficile and E. coli [6, 99, 101,102,103]. Moreover, the Bifidobacterium composition at the species level differs according to feeding type with B. catenulatum, and B. adolescentis, species that are commonly found in adults, being relatively more abundant in formula-fed infants [73, 104]. In breastfed infants, bifidobacterial species that thrive on human milk oligosaccharides (HMOs), including B. longum, B. bifidum, and B. breve, prevail [37, 47, 73]. Over 200 different types of HMOs have been known to be present in breastmilk, and the composition is affected by genetic factors, such as secretor genotype, suggesting that the maternal genome can affect the infant microbiota [37]. Indeed, Lacto-N-fucopentaose I and 2′-fucosyllactose, which are dominant HMOs in secretor women, but absent in non-secretors, are associated with the infant microbiota composition [105]. Next to the important selective pressure of HMOs, other bioactive compounds, including lysozyme, lactoferrin, and immunoglobulins, may also impact the infant microbiota [106,107,108].
4.3 Introduction of Complementary Foods
Weaning is a critical next step in microbiota maturation as the indigenous microbiota becomes exposed to a variety of food components, including plant- and animal-derived glycans. Upon weaning the microbial diversity increases as a result of the complementation and gradual replacement of HMO-utilizing bacteria, such as bifidobacteria, by a more complex ecosystem consisting of specialists such as members of Bacteroides, Lachnospiraceae, and Ruminococcaceae which are more capable of degrading complex plant-derived carbohydrates and starch [37, 73, 109]. The major pectin-degrading enzyme, pectinesterase, has, for example, been shown to be enriched in infants by the age of 1 year, most likely resulting from the increased intake of pectin-rich foods as compared to younger infants [73]. In line with the increased capacity to ferment complex dietary carbohydrates, levels of fecal short-chain fatty acids increase [37, 45]. Additionally, the microbiome shows an increased capacity to produce amino acids and vitamins and metabolize xenobiotics following the introduction of solid foods [37, 73, 110].
The particular effects of complementary foods, however, strongly depend on the geography in line with the major differences in dietary habits around the world. The intestinal microbiota of non-Western populations, known for their high consumption of dietary fibers, has consistently been shown to be more diverse, enriched in Prevotella and depleted in Bacteroides when compared to populations in Western countries consuming a diet high in simple sugars, starch, and animal fat and protein [111]. Along with other profound differences, this trade-off between Prevotella and Bacteroides was also reported in a study comparing the microbiota of children living in rural Africa and Europe [112]. However, among children that were still being breastfed, these differences between both populations were not yet apparent, highlighting that both cessation of breastfeeding in combination with the subsequent dietary pattern drive the geographical differences in the intestinal microbiota composition.
Next to the selective pressure of dietary substrate availability, food is also a major source of allochthonous bacteria, ranging from 104 to 109 bacteria per gram of food with fermented food having the highest bacterial counts [113]. Interestingly, a diet meal plan as recommended by the US Department of Agriculture (emphasizing fruits and vegetables, lean meat, dairy, and whole grains) was found to contain a thousandfold higher numbers of viable bacteria than an average American diet [113]. Reduced consumption of fresh fruits and vegetables and increased consumption of ultra-processed foods (e.g., preserved meats, refined grains, hydrogenated oils) as observed in typical western diets thus substantially reduces the ingestion of food-borne microbes.
Knowledge on the fate of food-borne microbes is largely limited to probiotic bacteria from the genera Lactobacillus and Bifidobacterium and shows only a transient integration of such bacteria in the intestinal microbiome [113]. However, this may be different for microbial specialists that can colonize intestinal niches that emerge in later infancy or childhood. The archaeon Methanobrevibacter smithii, which only colonizes after infancy [39], has, for example, been shown to be more prevalent among school-aged children consuming organic dairy products [114]. Molecular analysis confirmed the presence of M. smithii in milk products suggesting that this may be a source of archaea colonization in children.
4.4 Dispersal from Siblings, Peers, and Pets
Social interactions with siblings and peers (e.g., during daycare attendance) may result in dispersal of microbes. Singletons have indeed been shown to have a distinct colonization pattern when compared to infants that grow up together with older siblings. The Canadian Healthy Infant Longitudinal Development (CHILD) cohort showed a lower abundance of Clostridioides difficile and its family Peptostreptococcaceae in 4-month-old infants with older siblings as compared to singletons [115]. A longitudinal German study reported that exposure to older siblings was associated with an increased diversity as well as increased levels in several genera within the phylum of Actinobacteria (Bifidobacterium and Corynebacterium at 5 weeks and Eggerthella at 21 weeks) and a higher microbial diversity at 31 weeks of age [6]. Several other studies also observed a higher abundance of bifidobacteria among children with older siblings [99, 116]. A higher richness and diversity in children with siblings have been observed in some [117] but not all studies [47, 115]. Within the large TEDDY study, infants with older siblings had a different microbial community structure and accelerated microbiome maturation as compared to infants growing up in the absence of older siblings. Both species level microbial community structure as well as microbiota maturity, however, only started to significantly differ between children with and without siblings after the first months of life [47]. This delayed effect of older siblings might reflect more close interactions and dispersal when infants grow older or the opening of a niche that allows colonization by specific strains from household members. Alternative explanations for this sibling-effect, such as an altered vaginal microbiota or breastmilk composition in multiparous as compared to primiparous women, appear less likely as the strongest effect would in such cases be expected in earliest infancy.
Companion animals might also have an impact on the infant microbiome development as an altered microbial environment, indicated by different microbial composition of household dust or surfaces in the homes, has consistently been observed among households with indoor pets [118,119,120,121,122]. The influence of pets on the environmental microbiome might subsequently impact both the immune and microbiome development of infants [120]. In particular the intestinal microbial gene content of dogs shows striking similarities with the human gut microbiome [123]. Dogs were the first animals to be domesticated in modern human history and frequently shared food resources with humans, which has likely contributed to the co-evolution of the human and canine gut microbiome. However, the limited studies on the impact of pet exposure on the infant gut microbiome have so far not differentiated between the effect of different pet species. Results from the TEDDY study revealed that infants living with furry pets had an altered microbial community structure and accelerated maturation of the microbiome when compared to infants growing up in the absence of furry pets [47]. In another study, the animal-specific Bifidobacterium pseudolongum was detected in a significantly larger proportion of 1-month-old pet-exposed as compared to non-exposed infants [124]. Moreover, increased levels of Ruminococcus and Oscillospira were observed among infants with furry pets in their households in the CHILD-study [125]. However, not all studies did observe an impact of furry pets on the establishment and maturation of the infant gut microbiota [115], likely due to the more subtle effects as compared to the effects of siblings [47].
Large studies on the identification of determinants of infant gut microbiota development have so far not observed an association between daycare attendance and gut microbial diversity and community structure [47, 115]. A recent study for the first time compared the microbiota of infants before and 4 weeks after entering center-based childcare to that of infants being fully cared for by the parents at home. In line with the previous studies, this study also found the infant gut microbiota not to be affected in a uniform way by center-based childcare [126]. These studies do however not rule out an impact of daycare attendance on an individual level, e.g., by dispersal of microbial taxa between peers. In fact, studies showing the spread of fecal multi-drug resistant E. coli strains in daycare centers prove the existence of such dispersal events and underscore the importance of further investigation. Dispersal between socially interacting individuals is further supported by results from the Wisconsin Longitudinal Study in which spouses had a more similar microbiota and shared more bacterial taxa than siblings (adult siblings not living together) and unrelated pairs. Moreover, married individuals, especially those reporting close relationships, harbor microbial communities of greater diversity and richness relative to those living alone [127].
4.5 Dispersal of Microbes from the Natural Environment
The intestinal microbial ecology also differs between infants from various geographical regions. Even within different Western-European countries, differences in the infant microbiota have been observed. Infants from Northern European countries were characterized by an overrepresentation of Bifidobacterium, while an increased microbial diversity was found in infants born in Southern European countries [103]. More pronounced differences are observed when comparing the microbiota of infants living in western countries to that of children born in low- and middle-income countries, with the latter group being characterized by a higher diversity and enhanced levels of Prevotella and decreased abundance of Bacteroides in early life [18, 128,129,130]. Diet, with a typical Western diet being low in plant-derived carbohydrates and high in animal protein, sugar, starch, and fat as compared to more agrarian societies, is likely the most important driver for geographical variations in microbiota composition via environmental selection of microbes that can benefit from the substrate availability [111]. However, besides selective pressures such as diet and host genetics, the unique natural environment might at least also partly explain geographic variations in the indigenous microbial community structures [131]. According to the biodiversity hypothesis [132], reduced contact with diverse microbiota and macrobiota in our natural environments adversely affects the assembly and composition of the human indigenous microbiota and in turn to inadequate immune stimulation and ultimately increased susceptibility to non-communicable diseases. Several recent studies do suggest that these natural and human microbial ecosystems are indeed interrelated. Most evidence so far stems from studies that show associations between the skin microbiota and living near natural environments [133,134,135], but the first studies linking living in proximity to natural environments to the gut microbiota also start to emerge. Preliminary results from the Wisconsin Infant Study Cohort (WISC) showed that the microbiota of infants raised in farming versus non-farming environments differed modestly, yet significantly from each other [136]. Within the context of the Canadian CHILD-study, the association between living near to natural environments in the urban context and the gut microbiota in 4-month-old infants was examined. Although proximity to a natural environment was associated with an altered microbiota composition, this could only be observed for formula-fed infants who were exposed to pets in their homes [137]. This suggests that both environmental selection (e.g., breastfeeding prevents the colonization of environmental microbes) and indirect dispersal of environmental microbes via pets as vectors may be involved. In the PASTURE birth cohort, growing up on a family-run farm, and particular farm exposures such as visits to animal sheds and consumption of eggs or milk directly from the farm, influenced the maturation of the gut microbiota during the time window from 2 to 12 months. This accelerated microbiota maturation could moreover explain a substantial proportion of the well-known protective farm effect on asthma. Interestingly, growing up close to green areas (forest and agriculture land) has also been shown to reduce the risk of atopic sensitization, supporting the hypothesis of a strong environmental effect on the commensal microbiota [138]. The most direct and causal evidence in support of the biodiversity hypothesis, however, comes from intervention studies in which exposure to natural environments is stimulated. In a Finish trial, the environmental biodiversity of urban daycare centers was enriched by covering their yards with forest floor and sod [139]. Not only did the skin microbial (Proteobacteria) diversity increase among participating 3–5-year-old children, also the gut microbiota composition changed with a decreased relative abundance of Clostridiales and an increased diversity after as compared to before the intervention. Another example is the “Play & Grow” program from the University of Hong Kong, a family-oriented early environmental education program aimed to reconnect preschoolers to nature and induce changes in health behaviors and outcomes by having outdoor activities that promote exposure to nature [140]. In a proof-of-principle study of this “Play & Grow” program exposure to nature activities resulted in a decreased Bacteroidetes richness and increased Proteobacteria richness in the gut microbiota of children in the intervention group [141].
Together these results demonstrate the interrelatedness of the microbial ecosystems of our natural environments and our indigenous microbiota. Reduced contact of people with the natural environment as well as biodiversity loss of our wider environment will thus inevitably impact the biodiversity and composition of our intestinal microbiota.
4.6 Perturbations by Antibiotic Exposure
Broad-spectrum antibiotics, particularly beta-lactam antibiotics (e.g., amoxicillin, amoxicillin/clavulanate, cephalosporins) and macrolides (e.g., azithromycin), are by far the most commonly administered drugs during infancy and early childhood. Prescription rates, however, vary widely across the globe, reflecting differences in medication policies, preferences of health-care providers and mothers, infection rates, access to care, as well as over-the-counter sales of antibiotics [142, 143]. A significant portion of broad-spectrum antibiotics are still being prescribed for upper respiratory infections, which are mostly self-limiting and of viral origin with little evidence for clinical benefit [144, 145]. The negative impact of such misuse and overuse of antibiotics is substantial as it not only drives the development and dissemination of antimicrobial resistance but also results in profound perturbations on the indigenous microbiota [40, 43, 75, 99, 146]. A reduction in microbial diversity and decrease in obligate anaerobes, including Bifidobacterium and Bacteroides, and increase in Proteobacteria are commonly observed in the fecal microbiota of infants exposed to antibiotics, although perturbations vary according to type of antibiotics administered [147]. While in adults the microbiota shows a high level of resilience upon antibiotic exposure, the infant microbiota is far less resistant and resilient. In previously antibiotic-naive infants, a single course of amoxicillin was found to profoundly disrupt the microbiota composition. Rather than returning to the original composition after the antibiotic course, an accelerated maturation towards a depletion of bifidobacteria and enrichment of clostridia was observed [147]. This demonstrates that even an antibiotic pulse has a lasting effect on the maturation process of the infant microbial ecology.
Maternally administered antibiotics could potentially also impact the infant microbiota assembly either by altering the maternal vaginal and intestinal microbiota and thus dispersal (limitation) of maternal microbes [148], or by placental transfer of antibiotics [149]. The first route is likely most important for antibiotics administered in the third trimester, while the second route might come into play for intrapartum antibiotic prophylaxis (IAP) to prevent maternal wound and neonatal Group B Streptococcus (GBS) infections. IAP is not routinely practiced across the globe, but recent adjustments of international guidelines will lead to a further increase of prophylactic antibiotic administration during delivery and consequently increased antibiotic exposure to the infant [150].
A profound impact of IAP on the infant gut microbiota has been consistently observed across studies, with a decreased diversity and relative abundances of Actinobacteria and Bacteroidetes and concomitant increased levels of Proteobacteria belonging to the most robust findings [150,151,152,153,154,155]. Far fewer studies have been examining the impact of antibiotic exposure earlier during pregnancy and results are so far contradictory, likely due to the heterogeneity in study designs and potential confounding factors [150].
5 Conclusions
Human lifestyle changes have profoundly affected our indigenous microbiome and depleted microbial diversity at an unprecedented pace [156, 157]. Given the pivotal role of the infant microbiota as a stimulus for the ontogeny of the enteric mucosal tissue and immune system [13, 14], lifestyle changes that distort the assembly of the microbiota in early life can particularly impact future health and disease susceptibility. From an ecological perspective, lifestyle changes could both affect priority effects and other types of dispersal (limitation) as well as environmental selection. Delayed or limited exposure to maternal vaginal or fecal microbes during C-section delivery is a key example of dispersal limitation. Microbial dispersal is further limited as a result of reduced family sizes, reduced contact with the natural environment, and loss of biodiversity in our natural ecosystems. Reduced breastfeeding and increased consumption of ultra-processed foods reduce the dispersal of human milk and food-borne microorganisms. Moreover, changed dietary habits (e.g., more animal fat, simple sugars, and reduced dietary fibers) cause a distinct selective pressure on the indigenous microbial ecosystem of Western populations. These perturbating external events can be classified as pulses which are short-term perturbations (e.g., a course of antibiotics), or as presses which are long-term or continuous perturbations (e.g. dietary habits) [35]. As such, besides our western dietary habits, even relatively discrete, short-term perturbations (pulses), such as antibiotics, can profoundly and persistently alter the assembly and maturation of the dynamic and low resilient microbiota in early life [147].
Despite the considerable advances in our understanding of the microbiota maturation during infancy over the past decades, a large part of inter-individual variation in microbiota composition remains unexplained. This suggests that either important determinants have so far been overlooked or that other ecological processes such as historical contingency or stochastics effects play a major role [158]. Further research should take these ecological phenomena into account, but also focus on unravelling other so far unknown deterministic factors that might contribute to the variation and acquisition of the microbiota, and thereby the vital biological processes in life.
Many studies are ongoing to examine the impact of targeted treatments, including specific probiotics, prebiotics, and post-biotics (e.g., products of bacterial metabolism) to strengthen the natural development of the intestinal microbiota or restore its disruption. In addition, maternal FMT has recently been shown to be able to overcome the limited dispersal of maternal microbes and to postnatally restore the microbiota of C-section delivered infants. Although promising, data are still scarce and FMT requires careful screening of donor stools. It should therefore not yet be offered as standard care and only be used within the context of well-controlled experimental settings. Ultimately, synthetic multi-microbial substitutes of FMT will likely be an inevitable further development to make this a viable treatment strategy [159].
Next to the abovementioned dietary or clinical interventions, re-connecting to nature might be another approach to stimulate a healthy microbiome maturation. The rapid urbanization of our living environment and reduced exposure to natural environments might have impeded the beneficial health effects including the dispersal of bacteria and the maturation and homeostasis of immunological responses. In contrast, outdoor activity in a natural biodiverse environment may improve the microbial colonization, and in turn decrease the risk of non-communicable diseases and improve children’s general well-being. Human intervention studies tackling this concept of “microbiota re-wilding” are scarce, but the few initiatives that have been undertaken so far are promising [139, 141]. To further increase our understanding on the impact of our (natural) environment, observational studies embedding the infant gut microbiota development into a broader framework of environmental exposure are highly warranted [131]. Moreover, additional human interventional strategies should be explored to examine if strengthening connections to nature as part of everyday life indeed positively influences microbiota development during infancy.
Finally, education of the general public, healthcare professionals, and policy makers on the importance of our gut microbiota and the damaging health consequences of distortion of microbiota assembly in early life should be one of the top priorities. Refraining from unnecessary antibiotic use, informing on the negative consequences of elective C-section delivery, stimulating breastfeeding rates, and a diet high in fiber and low in animal fat and protein are all key to prevent the impoverishment of the indigenous microbiota during this critical period of life.
References
Dethlefsen L, et al. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21(9):517–23.
Le Chatelier E, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500(7464):541–6.
Meijnikman AS, et al. Distinct differences in gut microbial composition and functional potential from lean to morbidly obese subjects. J Intern Med. 2020;288(6):699–710.
Ridaura VK, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214.
Bridgman SL, et al. Gut microbiota and allergic disease in children. Ann Allergy Asthma Immunol. 2016;116(2):99–105.
Galazzo G, et al. Development of the microbiota and associations with birth mode, diet, and atopic disorders in a longitudinal analysis of stool samples, collected from infancy through early childhood. Gastroenterology. 2020;158(6):1584–96.
Stokholm J, et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018;9(1):141.
Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15(3):382–92.
Ray K. IBD. Understanding gut microbiota in new-onset Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2014;11(5):268.
Galazzo G, et al. Faecal microbiota dynamics and their relation to disease course in Crohn’s disease. J Crohns Colitis. 2019;13(10):1273–82.
Noce A, et al. Impact of gut microbiota composition on onset and progression of chronic non-communicable diseases. Nutrients. 2019;11(5):1073.
Thaiss CA, et al. The microbiome and innate immunity. Nature. 2016;535(7610):65–74.
Al Nabhani Z, Eberl G. Imprinting of the immune system by the microbiota early in life. Mucosal Immunol. 2020;13(2):183–9.
Renz H, et al. The neonatal window of opportunity-early priming for life. J Allergy Clin Immunol. 2018;141(4):1212–4.
Human Microbiome Project, C. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–14.
Palmer C, et al. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5(7):e177.
Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–8.
Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–7.
Costello EK, et al. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336(6086):1255–62.
Martinez I, Muller CE, Walter J. Long-term temporal analysis of the human fecal microbiota revealed a stable core of dominant bacterial species. PLoS One. 2013;8(7):e69621.
Vellend M. Conceptual synthesis in community ecology. Q Rev Biol. 2010;85(2):183–206.
Vellend M. The theory of ecological communities (MPB-57). Princeton, NJ: Princeton University Press; 2016.
Sprockett D, Fukami T, Relman DA. Role of priority effects in the early-life assembly of the gut microbiota. Nat Rev Gastroenterol Hepatol. 2018;15(4):197–205.
Fukami T. Historical contingency in community assembly: integrating niches, species pools, and priority effects. Annu Rev Ecol Evol Syst. 2015;46:1–23.
van Best N, et al. Bile acids drive the newborn’s gut microbiota maturation. Nat Commun. 2020;11(1):3692.
Fodelianakis S, et al. Direct quantification of ecological drift at the population level in synthetic bacterial communities. ISME J. 2021;15(1):55–66.
Nemergut DR, et al. Patterns and processes of microbial community assembly. Microbiol Mol Biol Rev. 2013;77(3):342–56.
Ostman O, et al. Regional invariance among microbial communities. Ecol Lett. 2010;13(1):118–27.
Venkataraman A, et al. Application of a neutral community model to assess structuring of the human lung microbiome. MBio. 2015;6(1):e02284.
Sprockett DD, et al. Microbiota assembly, structure, and dynamics among Tsimane horticulturalists of the Bolivian Amazon. Nat Commun. 2020;11(1):3772.
Burns AR, et al. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016;10(3):655–64.
Vannette RL, Fukami T. Historical contingency in species interactions: towards niche-based predictions. Ecol Lett. 2014;17(1):115–24.
Leibold MA, et al. The metacommunity concept: a framework for multi-scale community ecology. Ecol Lett. 2014;7(7):601–13.
Zhou J, Ning D. Stochastic community assembly: does it matter in microbial ecology? Microbiol Mol Biol Rev. 2017;81(4):e00002.
Fassarella M, et al. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut. 2021;70(3):595–605.
Sommer F, et al. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. 2017;15(10):630–8.
Derrien M, Alvarez AS, de Vos WM. The gut microbiota in the first decade of life. Trends Microbiol. 2019;27(12):997–1010.
Faith JJ, et al. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439.
Roswall J, et al. Developmental trajectory of the healthy human gut microbiota during the first 5 years of life. Cell Host Microbe. 2021;29(5):765–776 e3.
Bokulich NA, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med. 2016;8(343):343ra82.
Nagpal R, et al. Ontogenesis of the gut microbiota composition in healthy, full-term, vaginally born and breast-fed infants over the first 3 years of life: a quantitative bird’s-eye view. Front Microbiol. 2017;8:1388.
Podlesny D, Fricke WF. Strain inheritance and neonatal gut microbiota development: a meta-analysis. Int J Med Microbiol. 2021;311(3):151483.
Yassour M, et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci Transl Med. 2016;8(343):343ra81.
Chu DM, et al. Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat Med. 2017;23(3):314–26.
Differding MK, et al. Timing of complementary feeding is associated with gut microbiota diversity and composition and short chain fatty acid concentrations over the first year of life. BMC Microbiol. 2020;20(1):56.
Nilsen M, et al. Butyrate levels in the transition from an infant- to an adult-like gut microbiota correlate with bacterial networks associated with Eubacterium rectale and Ruminococcus gnavus. Genes (Basel). 2020;11(11):1245.
Stewart CJ, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2018;562(7728):583–8.
Arrieta MC, et al. The intestinal microbiome in early life: health and disease. Front Immunol. 2014;5:427.
Zhong H, et al. Impact of early events and lifestyle on the gut microbiota and metabolic phenotypes in young school-age children. Microbiome. 2019;7(1):2.
Aagaard K, et al. The placenta harbors a unique microbiome. Sci Transl Med. 2014;6(237):237ra65.
He Q, et al. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes. 2020;12(1):1794266.
Hornef M, Penders J. Does a prenatal bacterial microbiota exist? Mucosal Immunol. 2017;10(3):598–601.
Rackaityte E, et al. Viable bacterial colonization is highly limited in the human intestine in utero. Nat Med. 2020;26(4):599–607.
Stinson LF, et al. The not-so-sterile womb: evidence that the human fetus is exposed to bacteria prior to birth. Front Microbiol. 2019;10:1124.
Kliman HJ. Comment on “the placenta harbors a unique microbiome”. Sci Transl Med. 2014;6(254):254le4.
Bushman FD. De-discovery of the placenta microbiome. Am J Obstet Gynecol. 2019;220(3):213–4.
de Goffau MC, et al. Recognizing the reagent microbiome. Nat Microbiol. 2018;3(8):851–3.
de Goffau MC, et al. Human placenta has no microbiome but can contain potential pathogens. Nature. 2019;572(7769):329–34.
Kennedy KM, et al. Fetal meconium does not have a detectable microbiota before birth. Nat Microbiol. 2021;6:865.
Kuperman AA, et al. Deep microbial analysis of multiple placentas shows no evidence for a placental microbiome. BJOG. 2020;127(2):159–69.
Lauder AP, et al. Comparison of placenta samples with contamination controls does not provide evidence for a distinct placenta microbiota. Microbiome. 2016;4(1):29.
Leiby JS, et al. Lack of detection of a human placenta microbiome in samples from preterm and term deliveries. Microbiome. 2018;6(1):196.
Seferovic MD, et al. Visualization of microbes by 16S in situ hybridization in term and preterm placentas without intraamniotic infection. Am J Obstet Gynecol. 2019;221(2):146 e1–146 e23.
de Goffau MC, et al. Batch effects account for the main findings of an in utero human intestinal bacterial colonization study. Microbiome. 2021;9(1):6.
Rackaityte E, et al. Corroborating evidence refutes batch effect as explanation for fetal bacteria. Microbiome. 2021;9(1):10.
Jimenez E, et al. Isolation of commensal bacteria from umbilical cord blood of healthy neonates born by cesarean section. Curr Microbiol. 2005;51(4):270–4.
Jimenez E, et al. Is meconium from healthy newborns actually sterile? Res Microbiol. 2008;159(3):187–93.
Dominguez-Bello MG, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–5.
Romero R, et al. The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome. 2014;2(1):4.
Mitchell CM, et al. Delivery mode affects stability of early infant gut microbiota. Cell Rep Med. 2020;1(9):100156.
Shao Y, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature. 2019;574(7776):117–21.
Wampach L, et al. Birth mode is associated with earliest strain-conferred gut microbiome functions and immunostimulatory potential. Nat Commun. 2018;9(1):5091.
Backhed F, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703.
Reyman M, et al. Author correction: impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat Commun. 2019;10(1):5352.
van Best N, et al. On the origin of species: factors shaping the establishment of infant’s gut microbiota. Birth Defects Res C Embryo Today. 2015;105(4):240–51.
Fouhy F, et al. Perinatal factors affect the gut microbiota up to four years after birth. Nat Commun. 2019;10(1):1517.
Reyman M, et al. Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat Commun. 2019;10(1):4997.
Boerma T, et al. Global epidemiology of use of and disparities in caesarean sections. Lancet. 2018;392(10155):1341–8.
Offerhaus PM, et al. Change in primary midwife-led care in the Netherlands in 2000-2008: a descriptive study of caesarean sections and other interventions among 807,437 low-risk births. Midwifery. 2015;31(6):648–54.
Zhao Y, et al. Modest rise in caesarean section from 2000-2010: the Dutch experience. PLoS One. 2016;11(5):e0155565.
Mueller NT, et al. ‘Vaginal seeding’ after a caesarean section provides benefits to newborn children: FOR: Does exposing caesarean-delivered newborns to the vaginal microbiome affect their chronic disease risk? The critical need for trials of ‘vaginal seeding’ during caesarean section. BJOG. 2020;127(2):301.
Korpela K, et al. Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: a proof-of-concept study. Cell. 2020;183(2):324–334 e5.
World Health Organization, U. Global nutrition targets 2025: breastfeeding policy brief. Geneva: WHO; 2014.
Bagci Bosi AT, et al. Breastfeeding practices and policies in WHO European Region Member States. Public Health Nutr. 2016;19(4):753–64.
O’Sullivan A, Farver M, Smilowitz JT. The influence of early infant-feeding practices on the intestinal microbiome and body composition in infants. Nutr Metab Insights. 2015;8(Suppl 1):1–9.
Heikkila MP, Saris PE. Inhibition of Staphylococcus aureus by the commensal bacteria of human milk. J Appl Microbiol. 2003;95(3):471–8.
Jost T, et al. Assessment of bacterial diversity in breast milk using culture-dependent and culture-independent approaches. Br J Nutr. 2013;110(7):1253–62.
Solis G, et al. Establishment and development of lactic acid bacteria and bifidobacteria microbiota in breast-milk and the infant gut. Anaerobe. 2010;16(3):307–10.
Gomez-Gallego C, et al. The human milk microbiome and factors influencing its composition and activity. Semin Fetal Neonatal Med. 2016;21(6):400–5.
Demmelmair H, et al. Maternal and perinatal factors associated with the human milk microbiome. Curr Dev Nutr. 2020;4(4):nzaa027.
Duranti S, et al. Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome. 2017;5(1):66.
Laursen MF, Bahl MI, Licht TR. Settlers of our inner surface - factors shaping the gut microbiota from birth to toddlerhood. FEMS Microbiol Rev. 2021;45:fuab001.
Martin V, et al. Sharing of bacterial strains between breast milk and infant feces. J Hum Lact. 2012;28(1):36–44.
Milani C, et al. Exploring vertical transmission of bifidobacteria from mother to child. Appl Environ Microbiol. 2015;81(20):7078–87.
Goldsmith F, et al. Lactation and intestinal microbiota: how early diet shapes the infant gut. J Mammary Gland Biol Neoplasia. 2015;20(3–4):149–58.
Adlerberth I, Wold AE. Establishment of the gut microbiota in Western infants. Acta Paediatr. 2009;98(2):229–38.
Fallani M, et al. Determinants of the human infant intestinal microbiota after the introduction of first complementary foods in infant samples from five European centres. Microbiology (Reading). 2011;157(Pt 5):1385–92.
Harmsen HJ, et al. Analysis of intestinal flora development in breast-fed and formula-fed infants by using molecular identification and detection methods. J Pediatr Gastroenterol Nutr. 2000;30(1):61–7.
Penders J, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118(2):511–21.
Penders J, et al. Quantification of Bifidobacterium spp., Escherichia coli and Clostridium difficile in faecal samples of breast-fed and formula-fed infants by real-time PCR. FEMS Microbiol Lett. 2005;243(1):141–7.
Azad MB, et al. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ. 2013;185(5):385–94.
Bezirtzoglou E, Tsiotsias A, Welling GW. Microbiota profile in feces of breast- and formula-fed newborns by using fluorescence in situ hybridization (FISH). Anaerobe. 2011;17(6):478–82.
Fallani M, et al. Intestinal microbiota of 6-week-old infants across Europe: geographic influence beyond delivery mode, breast-feeding, and antibiotics. J Pediatr Gastroenterol Nutr. 2010;51(1):77–84.
Haarman M, Knol J. Quantitative real-time PCR assays to identify and quantify fecal Bifidobacterium species in infants receiving a prebiotic infant formula. Appl Environ Microbiol. 2005;71(5):2318–24.
Borewicz K, et al. Correlating infant faecal microbiota composition and human milk oligosaccharide consumption by microbiota of one-month old breastfed infants. Mol Nutr Food Res. 2019;63:e1801214.
Maga EA, et al. Consumption of lysozyme-rich milk can alter microbial fecal populations. Appl Environ Microbiol. 2012;78(17):6153–60.
Minami J, et al. Lysozyme in breast milk is a selection factor for bifidobacterial colonisation in the infant intestine. Benefic Microbes. 2016;7(1):53–60.
Rogier EW, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A. 2014;111(8):3074–9.
Laursen MF, et al. Infant gut microbiota development is driven by transition to family foods independent of maternal obesity. mSphere. 2016;1(1):e00069.
Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4578–85.
Salonen A, de Vos WM. Impact of diet on human intestinal microbiota and health. Annu Rev Food Sci Technol. 2014;5:239–62.
De Filippo C, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107(33):14691–6.
Lang JM, Eisen JA, Zivkovic AM. The microbes we eat: abundance and taxonomy of microbes consumed in a day’s worth of meals for three diet types. PeerJ. 2014;2:e659.
van de Pol JA, et al. Gut colonization by methanogenic archaea is associated with organic dairy consumption in children. Front Microbiol. 2017;8:355.
Azad MB, et al. Infant gut microbiota and the hygiene hypothesis of allergic disease: impact of household pets and siblings on microbiota composition and diversity. Allergy, Asthma Clin Immunol. 2013;9(1):15.
Hasegawa K, et al. Household siblings and nasal and fecal microbiota in infants. Pediatr Int. 2017;59(4):473–81.
Laursen MF, et al. Having older siblings is associated with gut microbiota development during early childhood. BMC Microbiol. 2015;15:154.
Dunn RR, et al. Home life: factors structuring the bacterial diversity found within and between homes. PLoS One. 2013;8(5):e64133.
Kettleson EM, et al. Key determinants of the fungal and bacterial microbiomes in homes. Environ Res. 2015;138:130–5.
Kim H, et al. Birth mode, breastfeeding, pet exposure, and antibiotic use: associations with the gut microbiome and sensitization in children. Curr Allergy Asthma Rep. 2019;19(4):22.
Maier RM, et al. Environmental determinants of and impact on childhood asthma by the bacterial community in household dust. Appl Environ Microbiol. 2010;76(8):2663–7.
Sitarik AR, et al. Dog introduction alters the home dust microbiota. Indoor Air. 2018;28(4):539–47.
Coelho LP, et al. Similarity of the dog and human gut microbiomes in gene content and response to diet. Microbiome. 2018;6(1):72.
Nermes M, et al. Furry pets modulate gut microbiota composition in infants at risk for allergic disease. J Allergy Clin Immunol. 2015;136(6):1688–1690 e1.
Tun HM, et al. Exposure to household furry pets influences the gut microbiota of infant at 3-4 months following various birth scenarios. Microbiome. 2017;5(1):40.
Hermes GDA, et al. Does entry to center-based childcare affect gut microbial colonization in young infants? Sci Rep. 2020;10(1):10235.
Dill-McFarland KA, et al. Close social relationships correlate with human gut microbiota composition. Sci Rep. 2019;9(1):703.
Clemente JC, et al. The microbiome of uncontacted Amerindians. Sci Adv. 2015;1(3):e1500183.
De Filippo C, et al. Diet, environments, and gut microbiota. A preliminary investigation in children living in rural and urban Burkina Faso and Italy. Front Microbiol. 2017;8:1979.
Grzeskowiak L, et al. Distinct gut microbiota in southeastern African and northern European infants. J Pediatr Gastroenterol Nutr. 2012;54(6):812–6.
Tasnim N, et al. Linking the gut microbial ecosystem with the environment: does gut health depend on where we live? Front Microbiol. 2017;8:1935.
von Hertzen L, et al. Helsinki alert of biodiversity and health. Ann Med. 2015;47(3):218–25.
Hanski I, et al. Environmental biodiversity, human microbiota, and allergy are interrelated. Proc Natl Acad Sci U S A. 2012;109(21):8334–9.
Ruokolainen L, et al. Significant disparities in allergy prevalence and microbiota between the young people in Finnish and Russian Karelia. Clin Exp Allergy. 2017;47(5):665–74.
Ruokolainen L, et al. Green areas around homes reduce atopic sensitization in children. Allergy. 2015;70(2):195–202.
Thorsen J, et al. Evaluating the effects of farm exposure on infant gut microbiome. J Allergy Clin Immunol. 2019;143(2):AB299.
Nielsen CC, et al. Natural environments in the urban context and gut microbiota in infants. Environ Int. 2020;142:105881.
Depner M, et al. Maturation of the gut microbiome during the first year of life contributes to the protective farm effect on childhood asthma. Nat Med. 2020;26(11):1766–75.
Roslund MI, et al. Biodiversity intervention enhances immune regulation and health-associated commensal microbiota among daycare children. Sci Adv. 2020;6(42):eaba2578.
Sobko T, Tse M, Kaplan M. A randomized controlled trial for families with preschool children - promoting healthy eating and active playtime by connecting to nature. BMC Public Health. 2016;16:505.
Sobko T, et al. Impact of outdoor nature-related activities on gut microbiota, fecal serotonin, and perceived stress in preschool children: the Play & Grow randomized controlled trial. Sci Rep. 2020;10(1):21993.
Rogawski ET, et al. Use of antibiotics in children younger than two years in eight countries: a prospective cohort study. Bull World Health Organ. 2017;95(1):49–61.
Stam J, et al. Antibiotic use in infants in the first year of life in five European countries. Acta Paediatr. 2012;101(9):929–34.
Hersh AL, et al. Antibiotic prescribing in ambulatory pediatrics in the United States. Pediatrics. 2011;128(6):1053–61.
Smith SM, Smucny J, Fahey T. Antibiotics for acute bronchitis. JAMA. 2014;312(24):2678–9.
Fouhy F, et al. High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob Agents Chemother. 2012;56(11):5811–20.
Korpela K, et al. Antibiotics in early life associate with specific gut microbiota signatures in a prospective longitudinal infant cohort. Pediatr Res. 2020;88(3):438–43.
Mueller NT, et al. The infant microbiome development: mom matters. Trends Mol Med. 2015;21(2):109–17.
Pacifici GM. Placental transfer of antibiotics administered to the mother: a review. Int J Clin Pharmacol Ther. 2006;44(2):57–63.
Dierikx TH, et al. The influence of prenatal and intrapartum antibiotics on intestinal microbiota colonisation in infants: a systematic review. J Infect. 2020;81(2):190–204.
Aloisio I, et al. Evaluation of the effects of intrapartum antibiotic prophylaxis on newborn intestinal microbiota using a sequencing approach targeted to multi hypervariable 16S rDNA regions. Appl Microbiol Biotechnol. 2016;100(12):5537–46.
Azad MB, et al. Impact of maternal intrapartum antibiotics, method of birth and breastfeeding on gut microbiota during the first year of life: a prospective cohort study. BJOG. 2016;123(6):983–93.
Coker MO, et al. Specific class of intrapartum antibiotics relates to maturation of the infant gut microbiota: a prospective cohort study. BJOG. 2020;127(2):217–27.
Nogacka A, et al. Impact of intrapartum antimicrobial prophylaxis upon the intestinal microbiota and the prevalence of antibiotic resistance genes in vaginally delivered full-term neonates. Microbiome. 2017;5(1):93.
Stearns JC, et al. Intrapartum antibiotics for GBS prophylaxis alter colonization patterns in the early infant gut microbiome of low risk infants. Sci Rep. 2017;7(1):16527.
Moeller AH, et al. Rapid changes in the gut microbiome during human evolution. Proc Natl Acad Sci U S A. 2014;111(46):16431–5.
Wibowo MC, et al. Reconstruction of ancient microbial genomes from the human gut. Nature. 2021;594:234.
Litvak Y, Baumler AJ. The founder hypothesis: a basis for microbiota resistance, diversity in taxa carriage, and colonization resistance against pathogens. PLoS Pathog. 2019;15(2):e1007563.
Baron TH, Kozarek RA. Fecal microbiota transplant: we know its history, but can we predict its future? Mayo Clin Proc. 2013;88(8):782–5.
Acknowledgments
We would like to thank Mayk Lucchesi for designing the artwork in this chapter.
Compliance with Ethical Standards
Funding: This chapter was financially supported by a grant from the Joint Programming Initiative A healthy diet for a healthy life (HDHL) Joint Action Intestinal Microbiomics (project number 50–52905–98–599).
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Penders, J., van Best, N. (2022). The Development of the Gut Microbiota in Childhood and Its Distortion by Lifestyle Changes. In: Rook, G.A.W., Lowry, C.A. (eds) Evolution, Biodiversity and a Reassessment of the Hygiene Hypothesis. Progress in Inflammation Research, vol 89. Springer, Cham. https://doi.org/10.1007/978-3-030-91051-8_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-91051-8_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-91050-1
Online ISBN: 978-3-030-91051-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)