
Abstract
Diabetes Mellitus (DM) is a disorder characterized by hyperglycemia, which arises as a consequence of a deficiency in the secretion and/or insulin action, probably due to disturbances of various molecular pathways. The two main forms of DM are type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). There is a body of evidence showing that oxidative stress can be a crucial factor implicated in the pathophysiology of DM, involving a series of molecular changes. Several studies have reported that patients with DM have elevated levels of oxidative stress markers, DNA damage, and oxidative lesions, in addition to an impaired antioxidant response and DNA repair ability. This chapter reviews the complex relationship between such mechanisms, which are strictly linked to oxidative stress in DM; in addition, it also describes results obtained on high-throughput large-scale studies, indicating that patients with T1DM and T2DM show several transcriptional changes (mRNAs and microRNAs) regarding the responses to oxidative stress, DNA repair, antioxidant system, endoplasmic reticulum stress, and mitochondrial dysfunction. All of these processes reflect the typical picture of physiological changes that occur during the development and progression of the disease, demonstrating the need for adequate glycemic control, in an attempt to delay the progression of DM and thus prevent the consequent complications of diabetes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Diabetes Mellitus (DM)
- Type 1 diabetes mellitus (T1DM)
- Type 2 diabetes mellitus (T2DM)
- Oxidative stress
- DNA damage
- Oxidative lesions
- Antioxidant response
- DNA repair
- Transcriptional changes
- Antioxidant system
- Endoplasmic reticulum stress
- Mitochondrial dysfunction
- Long-term complications
- Glycated hemoglobin levels
- Plasma glucose levels
- Reactive oxygen species (ROS)
- Reduction-oxidation (redox) reactions,
- Superoxide dismutase (SOD)
- Ascorbic acid (vitamin C)
- α-tocopherol (vitamin E)
- Reduced glutathione (GSH)
- Carotenoids
- Lycopene
- Minerals
- Omega-3 and 6
- Flavonoids
- Vitamin E
- Antioxidant enzymes
- Redox homeostasis
- Lipid peroxidation
- Protein oxidation
- Mitochondrial electron transport chain
- Gene expression profiles
- Peripheral blood mononuclear cells
- HLA
- Insulin (INS)
- Transcriptional expression profiles
1 Introduction
Diabetes Mellitus (DM) is a known important worldwide public health problem with the number of cases increasing every year. According to the International Diabetes Federation, there were approximately 463 million people between the ages of 20 and 79 years with diabetes worldwide in 2019. This number is projected to reach 578 million by 2030, and 700 million by 2045 (International Diabetes Federation 2019). The two major forms of DM are type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). T1DM is an autoimmune disorder that occurs predominantly in young individuals, resulting from a destruction of the insulin-producing β-cells by immune cells, which culminates in insulin deficit. T2DM is more common in middle-aged people or above (Newsholme et al. 2016).
Therefore, DM is a chronic metabolic disease that arises from a deficiency of the organism in insulin secretion and/or insulin resistance in the peripheral tissues, which, in turn, leads to chronic high blood glucose levels or hyperglycemia. Ultimately, chronic hyperglycemia has been implicated in long-term complications involving a variety of organs, including kidneys, eyes, heart, nerves, and blood vessels (International Diabetes Federation 2019). Individuals are diagnosed with diabetes when displaying the following clinical characteristics: glycated hemoglobin levels (HbA1C) ≥6.5% (48 mmol/mol), fasting plasma glucose levels (FPG) ≥126 mg/dL (7.0 mmol/L), 2-h plasma glucose levels after 75 g glucose load ≥200 mg/dL (11.1 mmol/L) (in the absence of unequivocal hyperglycemia, these three parameters should be confirmed by retaking the test), or for individuals with classic hyperglycemic symptoms/hyperglycemic crisis, casual plasma glucose levels ≥200 mg/dL (11.1 mmol/L) (International Diabetes Federation 2019).
There is evidence of an association between oxidative stress and both types of DM. Interestingly, while oxidative stress can be a consequence of these disorders due to hyperglycemia, it can also be a contributing factor to the pathogenesis of both T1DM and T2DM, considering that reactive molecules play a crucial role in pancreatic β-cell damage, in addition to the damage to other tissues and biological macromolecules.
1.1 Oxidative Stress
The advancement of age imposes a progressive increase in the production of reactive oxygen species (ROS), and also a reduced antioxidant defense capacity of the body (Cui et al. 2012). ROS encompass a group of reactive molecules produced from molecular oxygen by reduction–oxidation (redox) reactions. These molecules can be divided into non-radicals and free radical species. The superoxide anion radical (O2•–) and the hydroxyl radical (OH•) are commonly referred to as free radicals because they have at least one free electron. Hydrogen peroxide (H2O2), singlet molecular oxygen (1O2), and hypochlorous acid (HOCl) are some major known non-radical species (Collin 2019; Sies and Jones 2020). ROS are normal byproducts of cellular metabolism generated by various endogenous and exogenous sources. The main endogenous sources are the mitochondrial electron transport chain and nicotinamide adenine dinucleotide phosphate (NADPH) oxidases. However, ROS can also be produced in different cell compartments (such as the endoplasmic reticulum [ER], lysosomes, peroxisomes, and membranes) by the action of various stimuli (nutrients, growth factors, hormones, cytokines, and others) (He et al. 2017; Sies and Jones 2020). In addition to intracellular endogenous sources, ROS can be generated by exogenous physical agents (ultraviolet light, X-rays, and γ-rays), air pollutants, tobacco smoke, heavy metals, and certain drugs (Moldogazieva et al. 2019).
Under normal conditions and appropriate levels, ROS play important roles in several physiological processes, participating in cell signaling pathways, glucose uptake, memory function, and cell–cell interactions (Collin 2019; Sies and Jones 2020). On the opposite, although moderate amounts of ROS have positive effects (such as killing of invading pathogens, wound healing, and repairing processes), the misregulation of ROS production can cause oxidative damage to macromolecules, as well as mitochondrial dysfunction and cell death (Fig. 15.1) (Peoples et al. 2019). When present at very high concentrations, ROS react with lipids, proteins, and DNA, and can be severely detrimental to cells, thus causing a condition of redox imbalance. This condition leads to the concept of oxidative stress that arises from the imbalance between prooxidant species and the antioxidant defense (Moldogazieva et al. 2019; Sies and Jones 2020). Moreover, given the fact that superoxide anion and hydrogen peroxide levels are significantly higher in the mitochondrial matrix than in cytosolic and nuclear spaces, mitochondria become primary targets for ROS-induced damage (Luo et al. 2020). The excessive ROS can react with several mitochondrial proteins, compromising its function and dynamics and also disrupt the mitochondrial permeability transition pore, which facilitates the escape of electrons from the electron transport chain generating ROS and directly contributing to the release of ROS to the cytosol (He et al. 2017; Sies and Jones 2020). Mitochondrial dysfunction affects several biological processes, including nuclear genomic stability and cellular bioenergetics (Luo et al. 2020). Besides, it have been associated with several diseases, such as diabetes (Bhansali et al. 2017) and neurodegenerative diseases (Delbarba et al. 2016).
The constant exposure to oxidants triggers many enzymatic and nonenzymatic mechanisms, as well as adaptive responses to counteract ROS, ultimately leading to the reestablishment of the cellular oxidant/antioxidant homeostasis (Kalyanaraman 2013). The enzymatic mechanisms include the action of superoxide dismutase (SOD ), catalase (CAT), glutathione peroxidase (GPx), and thioredoxin system. SOD enzymes (cytosolic copper/zinc SOD, mitochondrial manganese SOD, and extracellular SOD) are involved in the normal dismutation of O2•− in O2 and H2O2; H2O2 can be detoxified in H2O + O2 mainly by CAT, when H2O2 levels are low, and by GPx when these levels are high. GPx also metabolizes other lipid peroxides (LOOH) (Fig. 15.1) (Kalyanaraman 2013; He et al. 2017). Regarding the mechanism of adaptive response to ROS, there is a signaling pathway under cell exposure to the oxidative challenge, with the activation of genes encoding antioxidant enzymes responsible for the maintenance of redox homeostasis in eukaryotes. A crucial gene is the nuclear factor erythroid 2-related factor 2 gene (NRF2) , which is a master regulator of the antioxidant response (He et al. 2020).
On the other hand, there are also nonenzymatic detoxification mechanisms, which include the small molecular weight antioxidants: ascorbic acid (vitamin C), α-tocopherol (vitamin E), reduced glutathione (GSH), carotenoids, lycopene, some minerals (zinc, manganese, and selenium), omega-3 and 6, flavonoids, among others. While vitamin C reacts rapidly with several ROS types, such as superoxide, hydrogen peroxide, and hydroxyl radical, vitamin E can halt lipid peroxidation (He et al. 2017).
Nevertheless, stress conditions that impair those adaptive mechanisms, reduce the concentrations of antioxidants, or affect the action of antioxidant enzymes can lead to oxidative stress by disrupting the redox homeostasis due to excessive ROS production, leading to ROS-mediated damage of important organelles and biomolecules (He et al. 2017). Therefore, when there is a redox imbalance, in which the levels of prooxidants exceed those of antioxidants, a consequent induction of damage to macromolecules can occur, such as lipid peroxidation, protein oxidation, and DNA damage, leading to disease (Kalyanaraman 2013; He et al. 2017), as illustrated in Fig. 15.1.

ROS and oxidative damage . ROS can be generated by endogenous and exogenous sources. There are antioxidant mechanisms that maintain ROS homeostasis; however, overproduction of ROS can occur causing a prooxidant redox imbalance, which leads to oxidative damage and mitochondrial dysfunction. Furthermore, mitochondrial dysfunction increases even more ROS production. Excessive ROS levels can induce lipid peroxidation, protein oxidation, and DNA damage, thus leading to disease. ROS: reactive oxygen species; O●−: superoxide radical; OH●: hydroxyl radical; OH−: hydroxyl ions; H2O2: hydrogen peroxide; O−: superoxide radical; ONOO−: peroxynitrite; NO●: nitric oxide; SOD: superoxide dismutase; GPx: glutathione peroxidase enzyme; CAT: catalase
In the DNA, ROS can react with both purines and pyrimidines, generating a number of modified DNA base products. Guanine exhibits a low redox potential and, it is preferentially oxidized, with 8-oxo-7,8-dihydroguanine (8-oxoguanine; 8-oxoG) being the most extensively oxidized DNA lesion that has been measured in several studies (Storr et al. 2013; Dizdaroglu et al. 2017). Nucleotides are also prone to oxidation by ROS, and the oxidized bases, deoxynucleotide triphosphates, (especially oxidized dGTP and dATP), could be erroneously incorporated into the DNA during the replication (Sun et al. 2015). Following DNA replication, these lesions can cause a change from GC to TA (transversion), thus causing mutations (Włodarczyk and Nowicka 2019). Therefore, DNA damage can exert an impact on DNA replication fidelity, leading to mutations, and imposing a risk to cell metabolism and survival (Włodarczyk and Nowicka 2019). ROS not only induce oxidized bases, abasic (AP) sites, and single-strand breaks (SSBs) (Hegde et al. 2012), but can also cause DNA intrastrand and interstrand crosslinks, DNA–protein crosslinks, double-strand breaks (DSBs), as well as damage to the DNA sugar moiety (Storr et al. 2013; Dizdaroglu et al. 2017).
DNA repair mechanisms play a crucial role in the maintenance of genome integrity against a large variety of endogenous and exogenous ROS, and also chemical and physical agents, guaranteeing the fidelity of DNA replication, thus preventing mutations and their consequences (Hanawalt and Wilson 2016). Base excision repair (BER) is a well-known major mechanism involved in the repair of ROS-induced oxidative lesions and SSBs in the DNA. BER requires several proteins; the key enzymes are DNA glycosylases, which remove different damaged bases by cleavage of the N-glycosylic bonds between the bases and the deoxyribose moieties of the nucleotide residues. Briefly, several enzymes participate in a sequential pathway, performing the initial lesion recognition, removal of oxidized bases by DNA glycosylases, such as 8-oxoguanine DNA glycosylase (OGG1), followed by the DNA incision by the apurinic/apyrimidinic endonuclease 1 (APE1), and subsequent recruitment of DNA polymerase β (Pol β), which perform gap filling; subsequently, ligase 1 or a complex of X-ray repair complementing protein 1 (XRCC1) and ligase IIIα seals the resulting gap to complete the repair process (Svilar et al. 2011; Krokan and Bjørås 2013; Cadet and Davies 2017).
BER is the major pathway for the repair of oxidative base damage, but other repair processes exist in eukaryotes, such as nucleotide excision repair (NER), mismatch DNA repair (MMR), translesion synthesis (TS), homologous recombination (HR), and nonhomologous end-joining (NHEJ); these repair processes are important mechanisms implicated in the repair of several types of DNA lesions (base damages, SSBs, DSBs, crosslinks, adducts, intercalation, among others) (Krokan and Bjørås 2013; Cadet and Davies 2017), being integrated with several cellular processes, such as cell cycle regulation, apoptosis, transcription, and replication. They may also be activated in response to oxidative damage, as an alternative repair pathway (Slupphaug 2003; Surova and Zhivotovsky 2013). Interestingly, Souza-Pinto et al. (2009) demonstrated that RAD52 (recombination protein) cooperates with OGG1 to repair oxidative DNA damage, suggesting a coordinated action between these proteins.
1.2 Diabetes Mellitus and Oxidative Stress
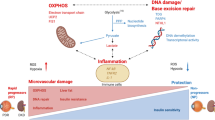
Hyperglycemia may lead to increased oxidative stress by the direct production of ROS, or by changes in the redox homeostasis through the disruption of a variety of mechanisms. The production of ROS in DM can be induced by endothelial and vascular smooth muscle cells, NADPH oxidase, xanthine oxidase, cyclooxygenase, and uncoupled NOS, while nonenzymatic sources include the generation of superoxide by the mitochondrial respiratory chain, advanced glycation end products (AGEs), activation of protein kinase C, glucose autoxidation process, and activated polyol pathway (Ahmad et al. 2017). All of these pathways can trigger redox imbalance (elevated oxidative stress), leading to damaged macromolecules that ultimately, are implicated in the development and progression of DM (Fig. 15.2) (Sifuentes-Franco et al. 2017).

Hyperglycemia-induced oxidative stress . Oxidative stress is a critical contributing factor to diabetes. A chronic state of hyperglycemia can promote an increase in glycolysis, which, in turn, acts on several signaling pathways, such as polyol, advanced glycosylation end products (AGEs), hexosamine, and protein kinase C (PKC) pathways. All of these pathways can produce oxidative stress. Consequently, the stress signaling and the activation of mitochondrial metabolism lead to increased nitric oxide synthases (NOS), cytokines, and NADPH oxidase and generation of superoxide by the mitochondrial respiratory chain. These pathways, when deregulated, promote the production of ROS, redox imbalance (a condition of oxidative stress), increasing lipid peroxidation, damage to proteins and DNA, consequently leading to diabetes. O●−: superoxide radical; ONOO−: peroxynitrite; NO●: nitric oxide
T1DM and T2DM are metabolic disorders, apparently with distinct mechanisms, but in both diseases, there is a significant loss of insulin-producing β-cells due to cell death. High acute or chronic glucose levels in diabetic patients promote an increase in the glycolytic flux and, consequently, an increase in the mitochondrial metabolism, which exacerbate ROS production (Gerber and Rutter 2017; Volpe et al. 2018). Thus, hyperglycemia induces excessive production of superoxide anion in the mitochondrial electron transport chain, formation of AGEs, as well secretion of proinflammatory cytokines, leading to numerous metabolic abnormalities and activation of pathways involved in the development of DM and diabetes complications (Djeli et al. 2019). Particularly, ROS are associated with the activation of inflammatory pathways that lead to apoptosis in β-cells (Hurrle and Hsu 2017), reduction of glucose transporters, inhibition of insulin receptor, and decreased insulin gene expression, which are associated with insulin resistance (Boucher et al. 2014; DeFronzo et al. 2015). This picture is compatible with alterations in gene expression profiles, as observed in peripheral blood mononuclear cells (PBMCs) of diabetic patients who display chronic hyperglycemia (Xavier et al. 2015).
2 Type 1 Diabetes Mellitus
T1DM is a consequence of the autoimmune destruction of the insulin-producing pancreatic β-cells, which eventually ceases insulin production and hence the glucose uptake by the tissues of the body, and culminates in hyperglycemia (Katsarou et al. 2017). Approximately 5–10% of all diabetic patients have T1DM, which can occur at any age, although it usually arises during childhood and adolescence (International Diabetes Federation 2019). The patients with T1DM commonly present classical symptoms as ketoacidosis, excessive thirst, blurred vision, bedwetting, frequent urination, fatigue, constant hunger, and weight loss (International Diabetes Federation 2019). They require daily insulin injections to control glucose levels and avoid life-threatening hypoglycemia; metformin, glucagon-like peptide-1 receptor (GLP4-1R) agonists, sodium-glucose cotransporter-2 (SGLT2) inhibitors, dipeptidyl peptidase-4 (DPP4) inhibitors may also be used in some cases (International Diabetes Federation 2019).
The exact cause of T1DM has not been elucidated, but it might be a result of a complex combination of several susceptibility genes (with functions on metabolism and immune system), as well as environmental factors (DiMeglio et al. 2018). Over 90% of T1DM cases have the characteristic presence of autoantibodies for glutamate decarboxylase (GAD65), islet antigen-2 (IA-2), tetraspanin-7, and zinc transporter 8 (ZNT8) that have been usually used as biomarkers of the disease (Katsarou et al. 2017; DiMeglio et al. 2018).
Individuals with the HLA class 2 haplotypes, HLA DRB1*0301-DQA1*0501-DQ*B10201 (DR3) and HLA DRB1*0401-DQA1*0301-DQB1*0301 (DR4-DQ8), located on chromosome 6, have the highest genetic risk for T1DM, which are related to the development of β-cell-targeted autoimmunity (Katsarou et al. 2017); in addition, non-HLA genes including those encoding insulin (INS), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), non-receptor protein tyrosine phosphatase type 22 (PTPN22), and interleukin 2 receptor alpha (IL2RA) have been identified by genome-wide association studies (GWAS) as having strong associations with the disorder (Nyaga et al. 2018). Regarding environmental factors, viral infections or toxins, as well as climate and diet have been suggested to contribute to T1DM onset (International Diabetes Federation 2019).
2.1 Oxidative Stress and DNA Damage in T1DM Patients
There is evidence that oxidative stress plays a crucial role in the increased inflammation and release of cytokines, which promotes the destruction of β-cells and the development of T1DM (Nassima et al. 2014). In this context, it has been investigated the levels of antioxidants, markers of oxidative stress, and DNA damage in patients suffering from T1DM relative to healthy subjects.
Studies with T1DM children and adults have consistently shown an increased oxidative stress condition, evidenced by increased total antioxidant status (TAS) (Gheni et al. 2020), high levels of lipid peroxidation (malondialdehyde (MDA), lipoperoxides (LPO), and 8-iso-prostaglandin F2α (8-iso-PGF2α)), protein oxidation, DNA oxidation (8-OhDG), and carbonylated proteins (Goodarzi et al. 2010; Codoñer-Franch et al. 2010; Nassima et al. 2014; Altincik et al. 2016). Moreover, it has been reported that glucose fluctuations may potentiate oxidative stress in non-obese T1DM children (Meng et al. 2015). On the other hand, the activity of antioxidant enzymes (GPx, glutathione reductase (GR) and SOD), in addition to lipophilic antioxidants (α-tocopherol and β-carotene), was found significantly decreased in T1DM patients compared to healthy subjects (Codoñer-Franch et al. 2010; Nassima et al. 2014). In fact, at the early onset of the disease, T1DM children already show glutathione depletion compared to healthy subjects (Pastore et al. 2012).
Furthermore, it has been reported an association between increased oxidative stress and impaired antioxidant status with lower levels of Magnesium and Zinc (Zn) and increased levels of Copper (Cu), in particular, in poorly controlled (with HbA1c ≥ 9%) children (Salmonowicz et al. 2014) and adults (Lin et al. 2014) with T1DM. Abnormalities in Zn and Cu levels seem to be associated with increased oxidative stress and diabetes complications (Bjørklund et al. 2019). Interestingly, Salmonowicz et al. (2014) and Lin et al. (2014) found an increase in CAT and SOD activity in T1DM patients, respectively, which might be compensating for the excessive ROS generation in these individuals, but the lower total antioxidant status might indicate a deficiency of the antioxidant system in those patients. Moreover, the disruption of thiol/disulfide homeostasis, known to play an important role in antioxidant responses, is also associated with oxidative damage, and it is considered another approach to evaluate oxidative stress (Ates et al. 2016). Durmus et al. (2019) and Ates et al. (2016) have shown that T1DM patients have a shift of thiol/disulfide homeostasis toward disulfide direction, an indication of oxidative stress, associated with chronic inflammation, which is followed by high levels of c-reactive protein, besides hyperglycemia.
Additionally, it has been demonstrated that men and women with T1DM have significantly more DNA damage and oxidized DNA damage (measured by Fpg-sensitive sites) in comparison with their corresponding controls (Dinçer et al. 2003). Even those T1DM patients with acceptable glycemic control reported significantly elevated rates of DNA damage (Hannon-Fletcher et al. 2000). Chronic hyperglycemia-induced cellular damage and oxidative stress are strongly associated with micro- and macrovascular complications of diabetes. Recent studies have shown a correlation between increased oxidative stress (high MDA and nitric oxid (NO) levels) and hematologic alterations in T1DM patients (Abdel-Moneim et al. 2020). Abnormalities in erythrocytes have been suggested to play a pivotal role in the development of microvascular complications (Abdel-Moneim et al. 2020). High levels of lipid peroxidation and NO have been linked to greater severity of retinopathy (Ruia et al. 2016). The glycemic control and oxidative stress management is an important issue to be addressed. It has been reported that glycemic variability in T1DM patients induces alterations in erythrocyte membrane stability (Rodrigues et al. 2018); recently, a correlation has been found between low levels of antioxidants enzymes (SOD and glutathione), high levels of oxidative stress (MDA), and impairment of bone formation, in children with T1DM (El Amrousy et al. 2021). Collectively, these studies suggest an impairment of the antioxidant defense system and an increase in oxidative stress and DNA damage in T1DM patients, which are associated with T1DM progression and later complications.
2.2 Transcriptional Expression Profiles of Oxidative Stress and DNA Repair Genes in T1DM Patients
A wide interest has been directed to the investigation of molecular pathways underlying the development and progression of T1DM and T2DM and their interactions. Within this context, studies at a genomic scale have been providing a large volume of data that can help in understanding and clarifying the etiopathogenesis of diabetes, with the possibility of contributing to the development of new therapeutic strategies.
Since the initial work reported by Schena et al. (1995), the microarray technique became a common and important tool in medical and biological research. Over more than two decades, several studies developed at a large scale regarding transcriptional profiling have been performed to compare expression profiles displayed by patients (for several diseases) relative to healthy subjects. Recently, Gastol et al. (2020) have performed a microarray study in T1DM patients to look for alterations in molecular pathways, by analyzing differentially expressed genes (DEGs) in blood cells. Interestingly, they found that T1DM patients showed an upregulation of genes related to DNA repair (APEX1, ERCC3, ERCC5, PARP1, PARP4, MLH1, XPC), antioxidant enzymes (PRDX1, SOD1, SOD2), ER-stress response (ATF6, PRDX6, GCLC, TXNRD1), proteasome and autophagosome formation (ATG3, ULK1, BECN1, DNAJB1, SQSTM1), apoptosis (caspases, TNF family factors, and their receptors), inflammation (NFKB, Il-10, Il-1b), and activation of inflammatory pathways. On the other hand, the patients presented a downregulation of genes involved in glucose transport (SLC2A11), glutathione synthesis (GCLM), expression of mitochondrial proteins of complexes I and III, and proteolytic enzymes (cathepsins, FRAP1, ATG10, GABARAPL2). Possibly, the inhibition of mitochondrial proteins and proteins involved in glutathione synthesis might be responsible for the induction of oxidative stress, which can be related to the activation of DNA repair and antioxidant pathways.
There is evidence that ER stress is involved in the destruction of pancreatic β-cells, triggering the development of both T1DM and T2DM. The ER is a major organelle responsible for regulating protein synthesis, folding, maturation, and transport and has a key role in insulin synthesis. The ER maintains a controlled balance between the synthesis and proper protein folding. However, several conditions can break this homeostatic balance, such as excess of nutrients, insulin resistance, increased levels of ROS, and inflammation related to obesity. The disturbance of this homeostasis leads to an accumulation of misfolded proteins in the organelle, either by an increased rate of protein synthesis or by alterations in the ER milieu, compromising the efficiency of protein folding. Regardless of the case, the unfolded protein response (UPR) is triggered to restore protein homeostasis. In patients with diabetes, hyperglycemia triggers the ER to produce an excessive amount of insulin, which overloads the ER, leading to the accumulation of misfolded and unfolded proteins. The ER overload induces a stress condition that activates the UPR, and under pathological conditions and excessive ER stress, the promotion of cell death may occur (Cao et al. 2020). Mainly three proteins are responsible for the activation of UPR: inositol-requiring protein-1α (IRE1α), protein kinase RNA (PKR)-like ER kinase (PERK), and activating transcription factor 6 (ATF6) (Cao et al. 2020). Accordingly, Gastol et al. (2020) observed an increased expression of ATF6, which has been linked to the activation of autophagy (Walter et al. 2018). In addition, it has been suggested that ER stress induces the release of proinflammatory cytokines, which was confirmed by the increased plasma levels of IL-6 and activation of inflammatory genes. The authors also discussed that despite the upregulation of proteasome and autophagosome formation in T1DM, the removal of damaged proteins can be compromised by a concomitant downregulation of lysosomal proteolytic enzymes, which has been associated with ER stress (Cao et al. 2020).
Irvine et al. (2012) investigated whether there were differences in gene expression of purified peripheral blood CD14+ monocytes between recently diagnosed T1DM children and adult healthy controls by applying the whole-genome microarrays, followed by validation of some genes by quantitative polymerase chain reaction (qPCR). The authors showed that the monocyte expression profiles exhibited by the patients were clustered into two subgroups, with one of them (group B) clustering separate from the other patient subgroup and the healthy controls. At diagnosis, both subgroups of patients were clinically identical, however, group B presented increased levels of HbA1c 3 and 6 months after diagnosis and needed significantly higher insulin doses during the first year of the disease. Expression profiles in monocytes from patients belonging to group B showed an upregulation of genes related to the UPR, which results from ER stress (IRE1, GRP78, DDIT3, XBP1), HIF1A, which is a major mediator of oxidative stress, and several of its targets (DDIT4, PFKFB3, and ADM); while genes that play a role in mitochondrial oxidative phosphorylation (PDHB, MDH1, IDH1, SDHC, ACLY) and cellular antioxidant pathways (CAT, G6PD, OXR1, PRDX1, PRDX3) were found downregulated, indicating perturbation of protective systems (Irvine et al. 2012). Moreover, mitochondrion was the most significantly enriched cellular component term for the downregulated genes in group B. The two biological processes, oxidative and ER stresses were found closely associated. Oxidative stress can promote ER stress, and in response to that, ER activates the UPR transcriptional program. UPR failure may lead to prolonged ER stress, which, in turn, triggers apoptosis and inflammation (Cao et al. 2020). Accordingly, genes controlling apoptosis were enriched in monocytes from group B patients. Hence, collectively, these findings imply that the group B monocytes are intrinsically susceptible to stress or exist in a stressful environment, as well as indicate the persistence of ER stress (Irvine et al. 2012).
Intriguingly, Stechova and co-workers (2012) compared gene expression profiles of freshly isolated PMBCs from T1DM patients, their first-degree relatives with higher genetic risk of developing the disease, and nondiabetic individuals by the microarray technology. They observed a clear difference between the expression profiles of relatives of patients (in particular the autoantibody-negative ones) and healthy controls. Moreover, the highest number of differentially activated cell signaling processes (99 pathways), including DNA damage and oxidative stress pathways was reported in the comparison between the relatives, regardless of autoantibody status, and the control group. Thus, these findings showed that nondiabetic relatives of T1DM patients also present alterations in gene expression. Caramori et al. (2015) performed a transcriptional profiling study in skin fibroblasts taken from 100 T1DM patients and found that longstanding T1DM patients (without diabetic nephropathy) displayed upregulation of DNA repair pathways, DNA replication, cell cycle, and RNA degradation compared to T1DM patients with nephropathy, and also compared to healthy controls. The authors suggest that the increased expression of repair pathways may be involved in preventing or delaying the onset of nephropathy.
Another study investigated gene expression profiles of endothelial progenitor cells (EPC), which were in vitro differentiated from PBMCs, from T1DM patients pre- and post-supplementation with folic acid (FA, a B-vitamin with antioxidant properties) and nondiabetic individuals (van Oostrom et al. 2009). The authors found 1591 DEGs between pre-FA treatment T1DM patients and the control group. These genes were associated with several processes including response to stress and response to hypoxia. Among the upregulated genes (related to these two terms) detected in EPC from T1DM patients were dual oxidase 2 (DUOX2), a NADPH oxidase that can produce superoxide, nitric oxide synthase 2A (NOS2A) that is capable of generating NO , thioredoxin reductase 2 (XNRD2), a major enzyme involved in the control of the intracellular redox balance, lactoperoxidase (LPO) and NADPH oxidase organizer 1 (NOXO1), which is associated with the generation of ROS. Importantly, after FA treatment the gene expression profiles (513 of the 1591 DEGs) in diabetic EPC normalized to levels similar to those exhibited by healthy individuals. As expected, FA altered the expression of oxidative stress-associated genes in EPC, with four (DUOX2, NOS2A, NOXO1, and LPO) being included among the 513 normalized genes. In addition, another differentially expressed gene (down-regulated) in T1DM patients that was normalized by FA treatment was the transcription factor V-maf musculoaponeurotic fibrosarcoma oncogene homolog F (MAFF). This transcription factor can bind to NRF2 , which, in turn, plays a crucial role in the antioxidant defense (Golpour et al. 2020).
Recent advances have brought novel high-throughput multi-omics approaches to provide a deep comprehensive understanding between disease state and the molecular profiles of healthy individuals. Instead of a single omics analysis, Balzano-Nogueira et al. (2021) applied an integrative approach to evaluate gene expression profiles, metabolomics, and dietary biomarkers to establish a multi-omics signature in children up to 12 months before T1DM development. Interestingly, before T1DM development, the children displayed upregulation of several genes associated with glucose utilization, energy metabolism, DNA repair, ROS scavenging, ER-protein processing, and apoptosis compared to the children who did not develop T1DM. Additionally, arachidonate-lipoxygenase genes (ALOX12, ALOX15, ALOX15B, and PTGS1), which are known to be activated by ROS and to increase the release of proinflammatory and pro-angiogenic molecules, were found upregulated. On the other hand, immune system pathways (regulation of natural killer immunity, CXCR4 signaling, TGFβ signaling, FOXO signaling) became downregulated just before the development of T1DM. At 0–3 months before T1DM diagnosis, pathways associated with antigen presentation (NF-kB signaling and insulin signaling) became strongly activated. Altogether, the authors pointed out molecular profiles following the progression of T1DM, supporting the hypothesis that the increased oxidative stress and inflammation do occur even several months before the onset of T1DM, being related to the increased activity of proinflammatory cytokines, activation of pathways that favors autoimmunity and cellular damage, concomitantly with abnormalities in lipid metabolism and nutrient uptake.
Regarding the expression of noncoding protein genes, microRNAs (miRNAs) have been indicated both as potential biomarkers for the earlier diagnosis of diabetes and as therapeutic targets for the treatment of this disorder (Assmann et al. 2017). MiRNAs are endogenous noncoding RNA molecules of approximately 22 nucleotides that are involved in the posttranscriptional regulation of protein-coding gene expression by base-pairing to specific sites generally in 3ʼ untranslated regions (UTRs) of the messenger RNA (mRNA) targets; in this way, miRNAs lead to the degradation and/or translational downregulation of their targets (Agbu and Carthew 2021). Takahashi et al. (2014) compared the miRNA expression profiles displayed by PBMCs from T1DM patients with those from healthy nondiabetic controls by performing microarray experiments. The authors identified a set of 44 differentially expressed miRNAs (35 upregulated and nine downregulated) that clearly distinguish T1DM patients from healthy subjects. After target prediction, results pointed to 10,827 and 6,636 potential targets of the up- and downregulated miRNAs, respectively; of note, a total of 85 and 75 genes implicated in DNA repair and response to oxidative stress, respectively, are potential targets of the 44 differentially modulated miRNAs in T1DM. Furthermore, Assmann et al. (2017) performed a systematic review of several miRNA studies performed in different tissues (serum, plasma, PBMCs, or pancreas) from T1DM patients compared to the controls. They found several circulating miRNAs (miR-21-5p, miR-24-3p, miR-148a-3p, miR-181a-5p, miR-210-5p, and miR-375) that were upregulated and some miRNAs (miR-146a-5p, miR-150-5p, miR-342-3p, miR-1275, and miR-100-5p) that were found downregulated in T1DM patients compared to the nondiabetic controls. Regarding the upregulated miR-21-5p, in a previous report in the literature, its function was implicated in anti-inflammatory process by inhibition of the NF-kB signaling pathway (Sheedy et al. 2010); regarding other relevant functions attributed to miRNAs, miR-24 and miR-148 activate insulin expression (Agbu and Carthew 2021), while miR-148a-3p is a regulator of β-cell self-tolerance and autoimmunity (Gonzalez-Martin et al. 2016); and miR-210-5p targets include genes related to mitochondrial metabolism, DNA repair, angiogenesis, and cell survival (Devlin et al. 2011).
Taken together, studies on the whole-transcript expression showed important alterations related to the expression of DNA repair and antioxidant genes, ER stress response, UPR, apoptosis, mitochondrial genes, and inflammation in T1DM patients, as well as in their relatives and also in children before T1DM development. Moreover, those genes are putative targets of a set of miRNAs that clearly distinguished T1DM patients from healthy individuals. These data support the hypothesis that patients with T1DM respond to the increased oxidative stress and DNA lesions by means of changes in their gene expression profiles, which probably, may affect several biological processes, and may explain the physiological alterations in the course of the disease.
3 Type 2 Diabetes Mellitus
T2DM is the most common type of DM, accounting for approximately 90% of all diagnosed cases of diabetes (International Diabetes Federation 2019). The disease is mainly characterized by hyperglycemia resulting from resistance to insulin action and/or by a deficiency in the secretion of this hormone, presenting a great correlation with an unhealthy lifestyle, aging, obesity, and lack of physical activity (DeFronzo et al. 2015). The symptoms of T2DM are similar to those of T1DM but less acute or intense. The majority of T2DM cases are symptomless and remained undiagnosed for a long period, until the hyperglycemia starts to trigger a series of complications, including diabetic retinopathy, nephropathy, neuropathy, cardiovascular diseases (International Diabetes Federation 2019), and more recently, it has been reported risk to the development of dementia (Mittal and Katare 2016; Chatterjee and Mudher 2018).
The biochemical mechanisms and physiological processes that characterize T2DM are not well understood. Nevertheless, over 500 genomic regions and some susceptibility genes have been identified by GWAS, including PPARG, KCNJ, CAPN10, FTO, CDKN2A/B, CDKAL1, TCF7L2, and IGFBP2; in addition, several identified genes are associated with diabetes complications, such as GJA8 and SLC18A2 (for retinopathy), UMOD and TENM3 (for nephropathy), NRP2 (for neuropathy), and SORT1 (for coronary heart disease); furthermore, SCN3A and SV2A genes, which are potential targets for therapeutic purpose, were also identified (Vujkovic et al. 2020). Additionally, some studies have been exploring epigenetic alterations in diabetes, since they may also contribute to the genetic susceptibility to T2DM (Basile et al. 2014; Kwak and Park 2016).
The main metabolic alterations in T2DM are insulin resistance, β-cell dysfunction, and chronic inflammation (DeFronzo et al. 2015). Obesity is a great contributor to those alterations, causing a chronic inflammatory response in the adipose tissue, characterized by abnormal production of cytokines, which include mostly molecules playing roles in stress response processes and activation of inflammatory pathways, such as the JNK and NF-kB pathways (Tsalamandris et al. 2019). Among the released cytokines are the Tumor Necrosis Factor-alpha (TNF-α) and Interleukin 6 (IL-6), which are released at large amounts by adipocytes and act inhibiting the tyrosine phosphorylation of insulin receptor substrate (IRS-1) impairing the insulin signaling pathway and leading to insulin resistance (Chen et al. 2017). Besides, excessive body fat leads to increased circulation of free fatty acids (FFA), known to impair pancreatic β-cell function and decrease insulin secretion, in addition to increasing the release of TNF-α and IL-6. Under this condition, there is a preferential use of lipids as an energy source, especially by muscles, which prevents glucose utilization and glycogen synthesis, leading to hyperglycemia. Furthermore, there is an increase in insulin secretion to compensate for the insulin receptor resistance, and this condition gradually leads to the development of the disease (Huang et al. 2018). Interestingly, a whole-blood transcriptome study in a large cohort (comprising 1977 nondiabetic obese subjects) reported a correlation between increased body mass index (BMI) and downregulation of several genes involved in insulin signaling (IRS2, PIK3CD, PIK3R4, PDPK1, AKT1, PTEN, PTPN1), DNA repair (ATM) and defense against ROS, including target genes and regulators of NRF2 (SOD2, NFE2L2, TXNRD1, MGST2, GSTM2, NQO2), suggesting that these alterations may contribute to T2DM development in obese subjects (Homuth et al. 2015).
As already mentioned, in diabetes, the chronic increase in glucose levels leads to overproduction of ROS and AGEs, and consequently, oxidative stress. ROS and oxidative stress activate pathways linked to increased release of proinflammatory cytokines, growth factors, adhesion molecules, and procoagulant factors, all culminating in β-cell dysfunction, insulin resistance, endothelial dysfunction, and T2DM progression with micro- and macrovascular complications (Akash et al. 2013).
Currently, the first line of therapy for T2DM patients is the recommendation of changes in lifestyle concomitantly with diet and weight management, in addition to regular physical activity and the use of glucose-lowering medicaments, such as metformin, which is the preferred drug as the initial pharmacological treatment. Depending on the progression of the disease, SGLT2 inhibitors, GLP-1 RA, DPP-4 inhibitors, sulphonylureas, thiazolidinediones, and insulin have also been recommended for glycemic control in T2DM patients (ADA 2020). Treatment for T2DM aims to reduce hyperglycemia by two main mechanisms: increased secretion of insulin by the pancreas or decreased production of glucose by the liver. However, T2DM is a progressive condition, that makes its treatment complicated, mainly due to the lack of control in insulin secretion and progressive cell death that leads to β-cell dysfunction, which should adjust the amount of insulin secreted in accordance with the needs of the organism. Therefore, patients often have episodes of hypoglycemia and hyperglycemia, both linked to serious complications of diabetes.
3.1 Oxidative Stress, Mitochondrial Dysfunction, and DNA Damage in T2DM Patients
In T2DM, hyperglycemia contributes significantly to the production of ROS, especially due to an overproduction of superoxide and hydrogen peroxide by the mitochondrial electron-transport chain (Dodson et al. 2013). Hyperglycemia can also induce the formation of AGEs, from protein glycation, which in turn contribute even more to ROS generation, thus aggravating the oxidative stress condition and leading to oxidative damage (Reddy et al. 2013; Pugazhenthi et al. 2017). In this context, several studies evaluating T2DM patients have shown increased levels of oxidative stress markers, measured by high levels of lipid peroxidation (thiobarbituric acid reactive substances (TBARS) and MDA), oxidized proteins, and AGEs (Abou-Seif and Youssef 2004; Strom et al. 2017) when compared to healthy individuals. The increased oxidative stress in T2DM patients is accompanied by decreased levels of antioxidant enzymes (GSH , SOD, and CAT) and total antioxidant status (Abou-Seif and Youssef 2004; Jiménez-Osorio et al. 2014; Strom et al. 2017). NRF2 , a key protein involved in the transcription of genes belonging to the antioxidant response system, was also found in decreased levels in T2DM patients compared to healthy individuals (Jiménez-Osorio et al. 2014; Sireesh et al. 2018). Actually, newly diagnosed T2DM patients display decreased NRF2 mRNA expression levels and reduced levels of its downstream target genes (SOD , HO-1, GPx, and CAT), increased mRNA expression levels of oxidative stress markers (p22Phox, TRPC6, and SOCS3), and increased levels of inflammatory cytokines (IL-4, IL-10, IL-13, IFN-γ, TNF-α, and GM-CSF). The reduced levels of NRF2 , and consequently, low efficiency of the antioxidant response observed in T2DM patients can further aggravate the oxidative stress condition, contributing to the development of diabetes complications (Jiménez-Osorio et al. 2014). For instance, poor renal function in T2DM patients is associated with increased levels of lipid peroxidation (8-iso-PGF2α and MDA) (Sauriasari et al. 2015).
For this purpose, Golpour et al. (2020) performed a double-blind randomized placebo-controlled clinical trial and observed that a 10-week supplementation with fish oil n-3 PUFAs containing eicosapentaenoic (EPA) and docosahexaenoic (DHA) acids increased NRF2 gene expression, as well as the total antioxidant status, and decreased lipid peroxidation (MDA) in T2DM patients compared to the placebo group. Another randomized clinical trial has shown that an eight-week supplementation with resveratrol increased the expression of both NRF2 and SOD genes, increasing the total antioxidant capacity and decreasing the levels of carbonylated proteins, besides significantly reducing weight, BMI, and blood pressure levels in T2DM patients, compared to those who did not receive any supplementation (Seyyedebrahimi et al. 2018). Thus, NRF2 upregulation additionally with the reduction of oxidative stress markers may have beneficial effects for T2DM patients.
It has been reported that T2DM patients show increased levels of oxidized bases in DNA (urinary 8-OHdG) (Tatsch et al. 2015) and in the nucleotide pool (serum 8-oxodG) (Sun et al. 2015), compared to healthy individuals, and this can also be a consequence of oxidative stress. Furthermore, high levels of 8-OHdG in T2DM patients have been accompanied by high levels of proinflammatory cytokines and higher insulin resistance, suggesting a relationship between inflammation, insulin resistance, and oxidative-induced damage in T2DM (Tatsch et al. 2015).
Concerning the importance of glycemic control, higher levels of protein oxidation and lipid peroxidation, and decreased antioxidant status are reported in hyperglycemic T2DM patients in comparison with non-hyperglycemic T2DM group of patients (Çakatay 2005; Lodovici et al. 2008; Bigagli et al. 2012). The high level of protein oxidation in T2DM patients without any comorbidity indicates that oxidative stress is related to hyperglycemia and may not be exclusively a consequence of complications of the disease. Besides, it is well known that oxidative stress induces DNA damage. Studies with T2DM patients have reported that hyperglycemic patients exhibit high levels of DNA damage and oxidative DNA damage compared to those of non-hyperglycemic T2DM patients (Lodovici et al. 2008; Xavier et al. 2015). In this line, Xavier et al. (2014) have shown that a one-week intervention to control glucose levels is efficient to significantly reduce DNA damage levels in T2DM patients compared to healthy individuals. Since high DNA damage levels are associated with the development of diabetes complications (Giacco and Brownlee 2010; Kumar et al. 2020), it is plausible to suggest that proper glycemic control may delay the progression of the disease and later complications.
Moreover, besides the evidence that T2DM patients show higher DNA damage than healthy subjects, when cells from T2DM patients were in vitro exposed to mutagens, it was found a lower efficiency of DNA repair mechanisms (Blasiak et al. 2004; Merecz et al. 2015). Curiously, Merecz et al. (2015) showed that T2DM patients with polymorphisms in APE1 gene (a key gene in BER) showed different DNA repair capacities, and higher DNA damage levels compared to those without the polymorphism.
Since cellular respiration in mitochondria makes this organelle the site of increased production of ROS inside the cell (Peoples et al. 2019), some studies have evaluated different mitochondrial parameters in patients with diabetes mellitus. Bhansali et al. (2017) have shown that T2DM patients present high levels of mitochondrial ROS and several mitochondrial alterations, such as membrane depolarization, reduced mass, and morphological alterations, all of them being indicative of mitochondrial dysfunction. They also showed a downregulation of both mRNA and proteins (PINK1, MFN2, NIX, PARKIN, and LC3-II) associated with mitophagy, suggesting that an impaired mitophagy favors the accumulation of dysfunctional mitochondria and increases ROS production. Additionally, RNA sequencing in blood cells of T2DM patients has shown a downregulation of several mitochondrial genes, such MT-ATP6, MT-ND1, MT-ND2, MT-ND4, MT-ND4L, MT-ND5, MT-ND6 (Ustinova et al. 2020; Lv et al. 2020) related to mitochondrial oxidative phosphorylation and mitochondrial energy transduction compared to healthy subjects. Furthermore, the quantification of mtDNA copy number (mtDNA-CN) has been explored as a marker to assess mitochondrial function, suggesting that a greater number of mtDNA-CN is related to a better mitochondrial function. Accordingly, it has been found that T2DM patients show lower mtDNA-CN when compared to healthy subjects (Cho et al. 2017; Constantin-Teodosiu et al. 2020; Latini et al. 2020; Fazzini et al. 2021; Memon et al. 2021) and Latini et al. (2020) have suggested that diabetes complications are also associated with lower mtDNA-CN.
Therefore, these studies suggest that hyperglycemia is an important factor involved in oxidative stress, oxidative damage, and mitochondrial dysfunction, indicating the requirement of proper control of blood glucose levels and ROS production, in an attempt to reduce their detrimental effects on different macromolecules, such as nucleic acids, lipids and proteins, and avoiding later diabetes complications.
3.2 Transcriptional Gene Expression Profiles in T2DM and Alterations in Oxidative Stress and DNA Repair Genes
Despite the number of studies regarding T2DM, the molecular mechanisms involved in the development and progression of the disease still requires elucidation. In the last years, many studies have used large-scale transcriptomic analysis (microarray, RNA sequencing, single-cell RNA sequencing) to analyze mRNA, microRNA, long noncoding RNAs, and circulatory RNAs expression profiles exhibited by T2DM patients to reveal the main genes and pathways associated with the pathophysiological changes described in T2DM. Manoel-Caetano et al. (2012) conducted a study comparing the transcriptional expression patterns exhibited by PBMCs from T2DM patients compared with healthy subjects. The authors obtained a list of 92 differentially expressed genes (52 upregulated and 40 downregulated) in diabetic patients compared to the control group; among them, genes related to oxidative stress responses and hypoxia (OXR1, SMG1, and UCP3) were highly upregulated, possibly in an attempt to deal with increased oxidative stress. Regarding the downregulated genes, many were involved in inflammation, immune response, and DNA repair (including SUMO1, ATRX, and MORF4L2). The downregulation of several DNA repair genes is in agreement with the decreased efficiency of DNA repair reported for T2DM patients (Blasiak et al. 2004; Merecz et al. 2015). A study performed by Xavier et al. (2015) compared the mRNA transcriptional expression profiles of PBMCs from hyperglycemic, non-hyperglycemic T2DM patients and healthy individuals. Among the results, they found 478 genes (261 upregulated and 217 downregulated) differentially expressed related to several processes including upregulation of the inflammatory response process and the regulation of DNA repair, downregulation of response to superoxide, and response to the ER stress for the comparison between the hyperglycemic and nonhyperglycemic T2DM patients. Xavier et al. (2015) also found several differentially expressed miRNAs, such as hsa-miR-186, hsa-miR-222, and hsa-miR-29b, when comparing T2DM patients versus healthy controls, and these miRNAs were related to the development of β-islets, cell cycle regulation, and insulin resistance, respectively. The authors further searched for possible interactions between the miRNAs and the differentially expressed mRNAs providing new information to the pathogenesis of T2DM and the importance of adequate glycemic control.
The skeletal muscle also represents an important target tissue in the study of T2DM pathogenesis. A large RNA-seq based transcriptome study of human skeletal muscle of T2DM patients reported a significant downregulation of key genes involved in insulin signaling (such as MTOR, PIK3CA, MAPK9, SLC2A4, PPARA, IRS2), which is suggestive of impaired insulin action; oxidative phosphorylation, indicative of mitochondrial dysfunction and related to increased ROS generation; ER protein processing, which may be associated with ER stress; and upregulation of genes related to apoptosis, TP53 signaling, TNF-receptor family members and NF-kB signaling, indicative of increased cell death and inflammation in T2DM (Wu et al. 2017).
Another approach to the comprehension of T2DM etiopathogenesis has been reported in β-cells of pancreatic islets from human donors by Marselli et al. (2020), using RNA sequencing; the authors showed that T2DM islets have several molecular changes regarding upregulation of ROS activity, intracellular calcium regulation, apoptotic pathways, and metabolism of FFAs, whereas processes related to the mitochondrial respiratory chain and translational control were downregulated compared to the islets of healthy donors. Accordingly, Lundberg et al. (2018) showed that T2DM islets displayed downregulation of genes related to mitochondrial function, while genes associated with oxidative stress and UPR were upregulated. Pancreatic β-cell death has been associated with ER stress and UPR response, due to the high insulin demand, which causes an increased dependence on ER functioning to ensure proper synthesis and insulin folding (Cao et al. 2020). Komura et al. (2010) detected elevated expression of ER stress markers, comparing the transcriptional expression profiles of PBMCs from T2DM patients versus healthy individuals. Furthermore, Iwasaki et al. (2014) provided evidence that ATF4 (a transcription factor activated after metabolic stresses, including ER stress) was activated by FFAs in macrophages. Back et al. (2009) showed that the absence of eIF2α phosphorylation (responsible for activating ATF4) in mice β-cells caused dysregulated proinsulin translation, increased oxidative damage, and defective ER trafficking of proteins and apoptosis. In this context, Lytrivi et al. (2020) provided a comprehensive perspective of transcriptional changes in β-cells induced by FFAs. They found changes in lipid metabolism, ER stress, cell cycle, oxidative stress, and cAMP/PKA signaling implicated in the lipotoxicity of β-cells. Similarly, Bikopoulos et al. (2008) showed that aside from pancreatic islets chronically exposed to FFAs having a significantly reduced glucose-stimulated insulin secretion and increased ROS generation, they also presented altered expression of 40 genes mainly related to the FFAs metabolism, inflammation, and also to antioxidant defense (which were upregulated), highlighting the importance of FFAs as risk factors for the development of T2DM.
Another advanced technology, the single-cell RNA sequencing (scRNA-seq), has been used to analyze transcriptional profiles of T2DM patients at the β-cell level. Bosi et al. (2020) have made an integrative analysis of large scRNA-seq studies of human islets to identify molecular alterations and provide new relevant information for T2DM pathogenesis. They identified 226 differentially expressed genes (210 upregulated and 16 downregulated) that included upregulation of pathways linked to lysosome activity and ER stress/UPR and downregulation of pathways associated with regulation of ROS, DNA repair, and also regulation of DNA damage checkpoint, among others. Curiously, 25 of the differentially expressed genes (mainly linked to β-cell damage, increased oxidative stress, ER stress, impaired insulin action, and autophagy) had not been previously associated with T2DM, which help to understand β-cell dysfunction in T2DM.
Regarding diabetes complications, Massaro et al. (2019) found several miRNAs associated with specific diabetes complications. The miR-144-3p, for example, was found differentially expressed in both T1DM and T2DM, and its targets are linked to impaired insulin signaling pathway (IRS1, TGF-β1, and PTEN). These findings strongly correlate with insulin resistance and diabetes development (White 2014). Moreover, the gene atlas reported 650 nonredundant genes related to specific complications of diabetes (Rani et al. 2017), and seven genes (AGER, TNFRSF11B, CRK, PON1, CRP, and NOS3) were reported to be associated with cardiovascular diseases, nephropathy, retinopathy, and neuropathy, which are complications of the disease. Furthermore, the authors also reported miRNAs associated with diabetes complications; the hsa-miR-107, for instance, common to all complications, is associated with ER stress-induced lipid accumulation. Other miRNAS, such as mir-802, mir-181, mir-34a, and mir-24a, have been suggested as novel potential therapeutic targets, associated with impaired glucose metabolism, insulin resistance, and β-cell death and dysfunction, respectively (Rani et al. 2017).
Taken together, information in the literature on transcriptional expression profiles highlights not only a serious picture of changes in different molecular signaling pathways in T2DM (such as inflammation, oxidative stress response, DNA repair, apoptosis, antioxidant response, mitochondrial function, immune response, and ER stress, among others) but also establishes a link and integration between biological processes, which are clearly related to the physiological changes presented by patients. In addition, the use of microarrays and other advanced techniques for the study of large-scale transcriptional profiles brings an immense amount of data, revealing altered pathways still unknown in T2DM, thus expanding knowledge about the disease and also providing valuable data for new therapeutic and diagnostic possibilities.
4 Conclusions
Diabetes mellitus is a worldwide public health problem characterized by disturbances in the control of glucose levels, generating hyperglycemia and serious consequences for the body. Several multidisciplinary studies have shown that hyperglycemia has a major impact on the onset of oxidative stress and mitochondrial dysfunction, which is strongly correlated with damage to β-cells and the progression of the disease toward various complications. There is evidence that in both types, T1DM and T2DM, there is an increase of oxidative stress (as it can be detected by various molecular and biochemical markers), oxidative damage to macromolecules, as well as decreased antioxidant response and impaired DNA repair capacity. Studies on a genomic scale have shown that patients with T1DM and T2DM present important transcriptional changes related to a series of biological processes, especially regarding responses to oxidative stress, DNA repair, inflammation, immune response, ER stress, and mitochondrial alterations, among other processes, which reflects the characteristic picture of physiological changes that occur during development and progression of the disease. Therefore, all these changes described for diabetes show the need for adequate control of glycemia, in an attempt to reduce the deleterious effects over the years of chronic disease, as well as to delay its progression and thus avoiding subsequent complications of diabetes.
References
Abdel-Moneim A, Zanaty MI, El-Sayed A et al (2020) Relation between oxidative stress and hematologic abnormalities in children with type 1 diabetes. Can J Diabetes 44:222–228
Abou-Seif MA, Youssef AA (2004) Evaluation of some biochemical changes in diabetic patients. Clin Chim Acta 346:161–170
ADA (2020) Standards of medical care in diabetes—2021 abridged for primary care providers. Clin Diabetes cd21as01
Agbu P, Carthew RW (2021) MicroRNA-mediated regulation of glucose and lipid metabolism. Nat Rev Mol Cell Biol:1–14
Ahmad W, Ijaz B, Shabbiri K et al (2017) Oxidative toxicity in diabetes and Alzheimer’s disease: mechanisms behind ROS/ RNS generation. J Biomed Sci 24:1–10
Akash MSH, Rehman K, Chen S (2013) Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J Cell Biochem 114:525–531
Altincik A, Tuǧlu B, Demir K et al (2016) Relationship between oxidative stress and blood glucose fluctuations evaluated with daily glucose monitoring in children with type 1 diabetes mellitus. J Pediatr Endocrinol Metab 29:435–439
Assmann TS, Recamonde-Mendoza M, De Souza BM, Crispim D (2017) MicroRNA expression profiles and type 1 diabetes mellitus: systematic review and bioinformatic analysis. Endocr Connect 6:773–790
Ates I, Kaplan M, Yuksel M et al (2016) Determination of thiol/disulphide homeostasis in type 1 diabetes mellitus and the factors associated with thiol oxidation. Endocrine 51:47–51
Back SH, Scheuner D, Han J et al (2009) Translation attenuation through eIF2α phosphorylation prevents oxidative stress and maintains the differentiated state in β cells. Cell Metab 10:13–26
Balzano-Nogueira L, Ramirez R, Zamkovaya T et al (2021) Integrative analyses of TEDDY Omics data reveal lipid metabolism abnormalities, increased intracellular ROS and heightened inflammation prior to autoimmunity for type 1 diabetes. Genome Biol 22:39
Basile KJ, Johnson ME, Xia Q, Grant SFA (2014) Genetic susceptibility to type 2 diabetes and obesity: Follow-up of findings from genome-wide association studies. Int J Endocrinol 2014:769671
Bhansali S, Bhansali A, Walia R et al (2017) Alterations in mitochondrial oxidative stress and mitophagy in subjects with prediabetes and type 2 diabetes mellitus. Front Endocrinol (Lausanne) 8:347
Bigagli E, Raimondi L, Mannucci E et al (2012) Lipid and protein oxidation products, antioxidant status and vascular complications in poorly controlled type 2 diabetes. Br J Diabetes Vasc Dis 12:33–39
Bikopoulos G, da Silva PA, Lee SC et al (2008) Ex vivo transcriptional profiling of human pancreatic islets following chronic exposure to monounsaturated fatty acids. J Endocrinol 196:455–464
Bjørklund G, Dadar M, Pivina L et al (2019) The role of zinc and copper in insulin resistance and diabetes mellitus. Curr Med Chem 27:6643–6657
Blasiak J, Arabski M, Krupa R et al (2004) DNA damage and repair in type 2 diabetes mellitus. Mutat Res Mol Mech Mutagen 554:297–304
Bosi E, Marselli L, De Luca C et al (2020) Integration of single-cell datasets reveals novel transcriptomic signatures of-cells in human type 2 diabetes. NAR Genomics Bioinforma 2:lqaa097
Boucher J, Kleinridders A, Kahn CR (2014) Insulin receptor signaling in normal. Cold Spring Harb Perspect Biol 6:a009191
Cadet J, Davies KJA (2017) Oxidative DNA damage & repair: an introduction. Free Radic Biol Med 107:2–12
Çakatay U (2005) Protein oxidation parameters in type 2 diabetic patients with good and poor glycaemic control. Diabetes Metab 31:551–557
Cao ZH, Wu Z, Hu C et al (2020) Endoplasmic reticulum stress and destruction of pancreatic β cells in type 1 diabetes. Chin Med J 133:68–73
Caramori ML, Kim Y, Goldfine AB et al (2015) Differential gene expression in diabetic nephropathy in individuals with type 1 diabetes. J Clin Endocrinol Metab 100:E876–E882
Chatterjee S, Mudher A (2018) Alzheimer’s disease and type 2 diabetes: a critical assessment of the shared pathological traits. Front Neurosci 12:383
Chen Z, Yu R, Xiong Y et al (2017) A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis 16:1–9
Cho SB, Koh I, Nam HY et al (2017) Mitochondrial DNA copy number augments performance of A1C and oral glucose tolerance testing in the prediction of type 2 diabetes. Sci Rep 7:43203
Codoñer-Franch P, Pons-Morales S, Boix-García L, Valls-Bellés V (2010) Oxidant/antioxidant status in obese children compared to pediatric patients with type 1 diabetes mellitus. Pediatr Diabetes 11:251–257
Collin F (2019) Chemical basis of reactive oxygen species reactivity and involvement in neurodegenerative diseases. Int J Mol Sci 20:2407
Constantin-Teodosiu D, Constantin D, Pelsers MM et al (2020) Mitochondrial DNA copy number associates with insulin sensitivity and aerobic capacity, and differs between sedentary, overweight middle-aged males with and without type 2 diabetes. Int J Obes 44:929–936
Cui H, Kong Y, Zhang H (2012) Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct 2012:1–13
de Souza-Pinto NC, Maynard S, Hashiguchi K et al (2009) The recombination protein RAD52 cooperates with the excision repair protein OGG1 for the repair of oxidative lesions in mammalian cells. Mol Cell Biol 29:4441–4454
DeFronzo RA, Ferrannini E, Groop L et al (2015) Type 2 diabetes mellitus. Nat Rev Dis Prim 1:15019
Delbarba A, Abate G, Prandelli C et al (2016) Mitochondrial alterations in peripheral mononuclear blood cells from Alzheimer’s disease and mild cognitive impairment patients. Oxidative Med Cell Longev 2016:5923938
Devlin C, Greco S, Martelli F, Ivan M (2011) MiR-210: more than a silent player in hypoxia. IUBMB Life 63:94–100
DiMeglio LA, Evans-Molina C, Oram RA (2018) Type 1 diabetes. Lancet 391:2449–2462
Dinçer Y, Akçay T, Ilkova H et al (2003) DNA damage and antioxidant defense in peripheral leukocytes of patients with Type I diabetes mellitus. Mutat Res - Fundam Mol Mech Mutagen 527:49–55
Dizdaroglu M, Coskun E, Jaruga P (2017) Repair of oxidatively induced DNA damage by DNA glycosylases: mechanisms of action, substrate specificities and excision kinetics. Mutat Res - Rev Mutat Res 771:99–127
Djeli N, Radakovi M, Dimirijevi V et al (2019) Oxidative stress and DNA damage in peripheral blood mononuclear cells from normal, obese, prediabetic and diabetic persons exposed to adrenaline in vitro. Mutat Res Toxicol Environ Mutagen 843:81–89
Dodson M, Darley-Usmar V, Zhang J (2013) Cellular metabolic and autophagic pathways: Traffic control by redox signaling. Free Radic Biol Med 63:207–221
El Amrousy D, El-Afify D, Shabana A (2021) Relationship between bone turnover markers and oxidative stress in children with type 1 diabetes mellitus. Pediatr Res 89:878–881
Fazzini F, Lamina C, Raftopoulou A et al (2021) Association of mitochondrial DNA copy number with metabolic syndrome and type 2 diabetes in 14176 individuals. J Intern Med 290:190–202
Gastol J, Polus A, Biela M et al (2020) Specific gene expression in type 1 diabetic patients with and without cardiac autonomic neuropathy. Sci Rep 10:1–8
Gerber PA, Rutter GA (2017) The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxidants Redox Signal 26:501–518
Gheni DA, Al-Maamori JA, Ghali KH (2020) The impact of oxidative stress and some endogenous antioxidants on type 1 diabetes mellitus. Eur J Mol Clin Med 7:4295–4310
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107:1058–1070
Golpour P, Nourbakhsh M, Mazaherioun M et al (2020) Improvement of NRF2 gene expression and antioxidant status in patients with type 2 diabetes mellitus after supplementation with omega-3 polyunsaturated fatty acids: A double-blind randomised placebo-controlled clinical trial. Diabetes Res Clin Pract 162:108120
Gonzalez-Martin A, Adams BD, Lai M et al (2016) The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol 17:433–440
Goodarzi MT, Navidi AA, Rezaei M, Babahmadi-Rezaei H (2010) Oxidative damage to DNA and lipids: correlation with protein glycation in patients with type 1 diabetes. J Clin Lab Anal 24:72–76
Hanawalt PC, Wilson SH (2016) Cutting-edge Perspectives in Genomic Maintenance III: Preface. DNA Repair (Amst) 44:1–3
Hannon-Fletcher MPA, O’Kane MJ, Moles KW et al (2000) Levels of peripheral blood cell DNA damage in insulin dependent diabetes mellitus human subjects. Mutat Res - DNA Repair 460:53–60
He F, Ru X, Wen T (2020) NRF2, a transcription factor for stress response and beyond. Int J Mol Sci 21:1–23
He L, He T, Farrar S et al (2017) Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem 44:532–553
Hegde ML, Izumi T, Mitra S (2012) Oxidized base damage and single-strand break repair in mammalian genomes: Role of disordered regions and posttranslational modifications in early enzymes. Prog Mol Biol Transl Sci 110:123–153
Homuth G, Wahl S, Müller C et al (2015) Extensive alterations of the whole-blood transcriptome are associated with body mass index: Results of an mRNA profiling study involving two large population-based cohorts. BMC Med Genet 8:65
Huang X, Liu G, Guo J, Su ZQ (2018) The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci 14:1483–1496
Hurrle S, Hsu WH (2017) The etiology of oxidative stress in insulin resistance. Biom J 40:257–262
International Diabetes Federation (2019) IDF Diabetes Atlas, 9th edn. International Diabetes Federation, Brussels
Irvine KM, Gallego P, An X et al (2012) Peripheral blood monocyte gene expression profile clinically stratifies patients with recent-onset type 1 diabetes. Diabetes 61:1281–1290
Iwasaki Y, Suganami T, Hachiya R et al (2014) Activating transcription factor 4 links metabolic stress to interleukin-6 expression in macrophages. Diabetes 63:152–161
Jiménez-Osorio AS, Picazo A, González-Reyes S et al (2014) Nrf2 and redox status in prediabetic and diabetic patients. Int J Mol Sci 15:20290–20305
Kalyanaraman B (2013) Teaching the basics of redox biology to medical and graduate students: Oxidants, antioxidants and disease mechanisms. Redox Biol 1:244–257
Katsarou A, Gudbjörnsdottir S, Rawshani A et al (2017) Type 1 diabetes mellitus. Nat Rev Dis Prim 3:1–17
Komura T, Sakai Y, Honda M et al (2010) CD14+ monocytes are vulnerable and functionally impaired under endoplasmic reticulum stress in patients with type 2 diabetes. Diabetes 59:634–643
Krokan HE, Bjørås M (2013) Base excision repair. Cold Spring Harb Perspect Biol 5:1–22
Kumar V, Agrawal R, Pandey A et al (2020) Compromised DNA repair is responsible for diabetes-associated fibrosis. EMBO J 39:e103477
Kwak SH, Park KS (2016) Recent progress in genetic and epigenetic research on type 2 diabetes. Exp Mol Med 48:e220
Latini A, Borgiani P, De Benedittis G et al (2020) Mitochondrial DNA copy number in peripheral blood is reduced in type 2 diabetes patients with polyneuropathy and associated with a MIR499A gene polymorphism. DNA Cell Biol 39:1467–1472
Lin CC, Huang HH, Hu CW et al (2014) Trace elements, oxidative stress and glycemic control in young people with type 1 diabetes mellitus. J Trace Elem Med Biol 28:18–22
Lodovici M, Giovannelli L, Pitozzi V et al (2008) Oxidative DNA damage and plasma antioxidant capacity in type 2 diabetic patients with good and poor glycaemic control. Mutat Res - Fundam Mol Mech Mutagen 638:98–102
Lundberg M, Stenwall A, Tegehall A et al (2018) Expression profiles of stress-related genes in islets from donors with progressively impaired glucose metabolism. Islets 10:69–79
Luo J, Mills K, le Cessie S et al (2020) Ageing, age-related diseases and oxidative stress: what to do next? Ageing Res Rev 57:100982
Lv B, Bao X, Li P et al (2020) Transcriptome sequencing analysis of peripheral blood of type 2 diabetes mellitus patients with thirst and fatigue. Front Endocrinol (Lausanne) 11:558344
Lytrivi M, Ghaddar K, Lopes M et al (2020) Combined transcriptome and proteome profiling of the pancreatic β-cell response to palmitate unveils key pathways of β-cell lipotoxicity. BMC Genomics 21:590
Manoel-Caetano FS, Xavier DJ, Evangelista AF et al (2012) Gene expression profiles displayed by peripheral blood mononuclear cells from patients with type 2 diabetes mellitus focusing on biological processes implicated on the pathogenesis of the disease. Gene 511:151–160
Marselli L, Piron A, Suleiman M et al (2020) Persistent or transient human β cell dysfunction induced by metabolic stress: specific signatures and shared gene expression with type 2 diabetes. Cell Rep 33:108466
Massaro JD, Polli CD, Silva MCE et al (2019) Post-transcriptional markers associated with clinical complications in Type 1 and Type 2 diabetes mellitus. Mol Cell Endocrinol 490:1–14
Memon AA, Sundquist J, Hedelius A et al (2021) Association of mitochondrial DNA copy number with prevalent and incident type 2 diabetes in women: A population-based follow-up study. Sci Rep 11:4608
Meng X, Gong C, Cao B et al (2015) Glucose fluctuations in association with oxidative stress among children with T1DM: comparison of different phases. J Clin Endocrinol Metab 100:1828–1836
Merecz A, Markiewicz L, Sliwinska A et al (2015) Analysis of oxidative DNA damage and its repair in Polish patients with diabetes mellitus type 2: Role in pathogenesis of diabetic neuropathy. Adv Med Sci 60:220–230
Mittal K, Katare DP (2016) Shared links between type 2 diabetes mellitus and Alzheimer’s disease: A review. Diabetes Metab Syndr Clin Res Rev 10:S144–S149
Moldogazieva NT, Mokhosoev IM, Mel’Nikova TI et al (2019) Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxidative Med Cell Longev 2019:3085756
Nassima M, Djamila AA, Baya G, Hafida M (2014) Oxidative stress biomarkers during Type 1 diabetes in Algerian children. Clin Biochem 47:776–777
Newsholme P, Cruzat VF, Keane KN et al (2016) Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J 473:4527–4550
Nyaga DM, Vickers MH, Jefferies C et al (2018) The genetic architecture of type 1 diabetes mellitus. Mol Cell Endocrinol 477:70–80
Pastore A, Ciampalini P, Tozzi G et al (2012) All glutathione forms are depleted in blood of obese and type 1 diabetic children. Pediatr Diabetes 13:272–277
Peoples JN, Saraf A, Ghazal N et al (2019) Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med 51:1–13
Pugazhenthi S, Qin L, Reddy PH (2017) Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta Mol basis Dis 1863:1037–1045
Rani J, Mittal I, Pramanik A et al (2017) T2DiACoD: a gene atlas of type 2 diabetes mellitus associated complex disorders. Sci Rep 7:6892
Reddy VP, Perry G, Cooke MS et al (2013) Mechanisms of DNA damage and repair in Alzheimer disease. In: Madame curie bioscience database. Landes Bioscience, Austin
Rodrigues R, de Medeiros LA, Cunha LM et al (2018) Correlations of the glycemic variability with oxidative stress and erythrocytes membrane stability in patients with type 1 diabetes under intensive treatment. Diabetes Res Clin Pract 144:153–160
Ruia S, Saxena S, Prasad S et al (2016) Correlation of biomarkers thiobarbituric acid reactive substance, nitric oxide and central subfield and cube average thickness in diabetic retinopathy: a cross-sectional study. Int J Retin Vitr 2:8
Salmonowicz B, Krzystek-Korpacka M, Noczyńska A (2014) Trace elements, magnesium, and the efficacy of antioxidant systems in children with type 1 diabetes mellitus and in their siblings. Adv Clin Exp Med 23:259–268
Sauriasari R, Andrajati R, Azizahwati et al (2015) Marker of lipid peroxidation related to diabetic nephropathy in Indonesian type 2 diabetes mellitus patients. Diabetes Res Clin Pract 108:193–200
Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470
Seyyedebrahimi S, Khodabandehloo · Hadi, Ensieh ·, et al (2018) The effects of resveratrol on markers of oxidative stress in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled clinical trial. Acta Diabetol 55:341–353
Sheedy FJ, Palsson-Mcdermott E, Hennessy EJ et al (2010) Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol 11:141–147
Sies H, Jones DP (2020) Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 21:363–383
Sifuentes-Franco S, Pacheco-Moisés FP, Rodríguez-Carrizalez AD, Miranda-Díaz AG (2017) The role of oxidative stress, mitochondrial function, and autophagy in diabetic polyneuropathy. J Diabetes Res 2017:1673081
Sireesh D, Dhamodharan U, Ezhilarasi K et al (2018) Association of NF-E2 Related Factor 2 (Nrf2) and inflammatory cytokines in recent onset Type 2 Diabetes Mellitus. Sci Rep 8:1–10
Slupphaug G (2003) The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res Mol Mech Mutagen 531:231–251
Stechova K, Kolar M, Blatny R et al (2012) Healthy first-degree relatives of patients with type 1 diabetes exhibit significant differences in basal gene expression pattern of immunocompetent cells compared to controls: Expression pattern as predeterminant of autoimmune diabetes. Scand J Immunol 75:210–219
Storr SJ, Woolston CM, Zhang Y, Martin SG (2013) Redox environment, free radical, and oxidative DNA damage. Antioxidants Redox Signal 18:2399–2408
Strom A, Kaul K, Brüggemann J et al (2017) Lower serum extracellular superoxide dismutase levels are associated with polyneuropathy in recent-onset diabetes. Exp Mol Med 49:e394
Sun J, Lou X, Wang H et al (2015) Serum 8-hydroxy-2′-deoxyguanosine (8-oxo-dG) levels are elevated in diabetes patients. Int J Diabetes Dev Ctries 35:368–373
Surova O, Zhivotovsky B (2013) Various modes of cell death induced by DNA damage. Oncogene 32:3789–3797
Svilar D, Goellner EM, Almeida KH, Sobol RW (2011) Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxidants Redox Signal 14:2491–2507
Takahashi P, Xavier DJ, Evangelista AF et al (2014) MicroRNA expression profiling and functional annotation analysis of their targets in patients with type 1 diabetes mellitus. Gene 539:213–223
Tatsch E, Carvalho JAMD, Hausen BS et al (2015) Oxidative DNA damage is associated with inflammatory response, insulin resistance and microvascular complications in type 2 diabetes. Mutat Res - Fundam Mol Mech Mutagen 782:17–22
Tsalamandris S, Antonopoulos AS, Oikonomou E et al (2019) The role of inflammation in diabetes: Current concepts and future perspectives. Eur Cardiol Rev 14:50–59
Ustinova M, Ansone L, Silamikelis I et al (2020) Whole-blood transcriptome profiling reveals signatures of metformin and its therapeutic response. PLoS One 15:e0237400
van Oostrom O, de Kleijn DPV, Fledderus JO et al (2009) Folic acid supplementation normalizes the endothelial progenitor cell transcriptome of patients with type 1 diabetes: a case-control pilot study. Cardiovasc Diabetol 8:47
Volpe CMO, Villar-Delfino PH, Dos Anjos PMF, Nogueira-Machado JA (2018) Cellular death, reactive oxygen species (ROS) and diabetic complications review-article. Cell Death Dis 9:119
Vujkovic M, Keaton JM, Lynch JA et al (2020) Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet 52:680–691
Walter F, O’Brien A, Concannon CG et al (2018) ER stress signaling has an activating transcription factor 6 (ATF6)-dependent “off-switch”. J Biol Chem 293:18270–18284
White MF (2014) IRS2 integrates insulin/IGF1 signalling with metabolism, neurodegeneration and longevity. Diabetes Obes Metab 16:4–15
Włodarczyk M, Nowicka G (2019) Obesity, DNA damage, and development of obesity-related diseases. Int J Mol Sci 20:1146
Wu C, Xu G, Tsai SYA et al (2017) Transcriptional profiles of type 2 diabetes in human skeletal muscle reveal insulin resistance, metabolic defects, apoptosis, and molecular signatures of immune activation in response to infections. Biochem Biophys Res Commun 482:282–288
Xavier DJ, Takahashi P, Evangelista AF et al (2015) Assessment of DNA damage and mRNA/miRNA transcriptional expression profiles in hyperglycemic versus non-hyperglycemic patients with type 2 diabetes mellitus. Mutat Res Mol Mech Mutagen 776:98–110
Xavier DJ, Takahashi P, Manoel-Caetano FS et al (2014) One-week intervention period led to improvements in glycemic control and reduction in DNA damage levels in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 105:356–363
Yaşar Durmuş S, Şahin NM, Ergin M et al (2019) How does thiol/disulfide homeostasis change in children with type 1 diabetes mellitus? Diabetes Res Clin Pract 149:64–68
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Lima, J.E.B.F., Moreira, N.C.S., Takahashi, P., Xavier, D.J., Sakamoto-Hojo, E.T. (2022). Oxidative Stress, DNA Damage, and Transcriptional Expression of DNA Repair and Stress Response Genes in Diabetes Mellitus. In: Passos, G.A. (eds) Transcriptomics in Health and Disease. Springer, Cham. https://doi.org/10.1007/978-3-030-87821-4_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-87821-4_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-87820-7
Online ISBN: 978-3-030-87821-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)