
Abstract
Hypophysitis is a rare disorder of childhood, often associated with diabetes insipidus and hypopituitarism. It is hallmarked by inflammation of the adenohypophysis, neurohypophysis, or both. The exact cause has not been clearly delineated. The five hypophysitis types are based on histologic findings: lymphocytic, granulomatous, xanthomatous, IgG4-related, and necrotizing. Lymphocytic is the most common in children. Diabetes insipidus, and less often growth failure, are typically seen prior to hypophysitis diagnosis. Imaging studies by and large reveal pituitary stalk thickening. Pituitary biopsy might be recommended to exclude germinoma, Langerhans cell histiocytosis (LCH), lymphoma and craniopharyngioma, as well as secondary causes of hypophysitis that can be intracranial or systemic. Children need to be closely followed as oncologic disorders can still develop over time. Diabetes insipidus often persists and pituitary deficiencies frequently develop. Hypophysitis in children is a complex but poorly understood condition and warrants further study to understand its true incidence and implications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Pituitary
- Hypophysitis
- Pituitary stalk
- Diabetes insipidus
- Pituitary stalk thickening
- Anterior pituitary hormone deficiency
- Langerhans cell histiocytosis
- Germinoma
Vignette
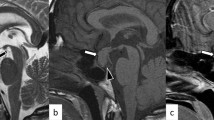
SJ is a 9-year-old male presenting with a 6-week history of polyuria and polydipsia, and 4 months of intermittent headaches alleviated with vomiting. He denies any appetite, visual, skin, or stooling changes. There has been no weight gain in the past 6 months, but his height has consistently tracked the 55th percentile over the past 7 years. Laboratory investigations are notable for a urine specific gravity of 1.000 and he is referred to Endocrinology. Overnight fasting evaluation shows morning urine and blood work consistent with diabetes insipidus (elevated sodium and serum osmolality with inappropriately low urine osmolality). Anterior pituitary hormone investigations are normal. Magnetic resonance imaging of the brain and pituitary demonstrates homogeneous enhancement of a thickened pituitary stalk, measuring 5 mm in antero-posterior dimension, abutting the optic chiasm without compression, and a T1 hyperintensity of posterior pituitary representing the neurohypophysis bright spot. Nightly oral desmopressin therapy is started, 0.05 mg, with resolution of polyuria and polydipsia, and concentrated urine osmolality and normalized serum osmolality.
His neuro-oncologic evaluation shows a normal skeletal survey, normal serum and cerebrospinal fluid (CSF) alpha-1-feto protein (AFP), beta-human chorionic gonadotropin (β-hCG), and angiotensin converting enzyme levels, and a normal CSF immunoglobulin G level. He undergoes pituitary stalk biopsy, which demonstrates an inflammatory infiltrate consisting of mainly T and B lymphocytes, and histiocytes. No Langerhans or germ cells are identified. A comprehensive solid tumor panel (tumor/normal pair) analysis of the biopsy shows no clinically significant variants, and BRAF analysis is negative. He recovers in the intensive care unit without incident and is discharged on oral desmopressin therapy.
About 2 weeks after biopsy, he develops fatigue and bradycardia, with a decreased morning cortisol of 5.5 mcg/dL and a low free thyroxine on direct analysis and equilibrium dialysis (0.6 and 0.7 ng/dL, respectively). He is started on hydrocortisone therapy followed several days later by thyroxine replacement, with clinical and laboratory improvements. Follow-up MRIs over the next 12 months show no new lesions or changes in pituitary stalk thickening, and no changes in serum tumor markers.
However, he experiences growth failure with decreasing insulin-like growth factor-1 levels. Subsequent stimulation testing with arginine and glucagon agents, off hydrocortisone therapy for several days, demonstrates a peak cortisol of 8.7 mcg/dL and peak growth hormone of 1.89 ng/mL. Hydrocortisone is restarted, and growth hormone is started soon thereafter.
Chapter Write-Up
The first case of hypophysitis was described in 1962 and became increasingly recognized throughout the 1980s as a cause of hypopituitarism, mostly in women with an intrasellar mass or hypopituitarism symptoms during late pregnancy or the postpartum period [1, 2]. Histologic study of this first case showed lymphocytic infiltration of the anterior pituitary without giant cells or granulomas. These findings were felt to be different than the more commonly reported phenomenon of Sheehan’s syndrome seen in post-partum women: healed pituitary necrosis and acellular fibrosis, typically found in a part of the pituitary susceptible to ischemia. Some of the reports described the degree of hypopituitarism as out of proportion to the pituitary mass size seen on imaging or intra-operatively, suggesting cellular pituitary destruction as the cause of pituitary destruction, rather than compression [2]. Although these early adult studies could not elucidate a precise hypophysitis etiology, autoimmunity was felt to play a role as many of the subjects were female with concurrent autoimmune disorders [1,2,3]. This hypothesis was further supported by animal model studies demonstrating that lymphocytic hypophysitis could be induced in rats injected with both homologous and heterologous pituitary tissue [4]. While most of the first hypophysitis cases were described in post-partum or pre-menopausal females, subjects outside of this demographic were reported more frequently over the next 30 years in the medical literature, including children.
Description/Incidence
Hypophysitis is an uncommon disorder in children with less than 100 cases reported to date [5, 6]. Both acute and chronic inflammation of the pituitary gland has been described in these cases, which can lead to hypopituitarism. Dysfunction or deficiency of the adenohypophysis, neurohypophysis, and hypothalamus, or a combination of the three, can be seen in adults and children [7]. A comparison of these three subtypes in adults has proven challenging as the classifications in the literature have been heterogeneously derived from an endocrinological, pathological, and radiological standpoint [8]. The cause can be primary (isolated to the pituitary gland) which is often idiopathic, or secondary to infiltration, local tumor extension, distant metastases, medications, infection, or systemic inflammatory disorders [9, 10]. The diagnosis of hypophysitis is not always certain during the first several months or years of a subject’s presenting complaints, and other etiologies, such as germinoma and histiocytosis, have to be monitored for vigilantly. An annual incidence of 2.4 in 10 million has been reported for adult surgical and conservative hypophysitis cases, and 1 in 9 million for surgical cases [11, 12]. Given the rarity of hypophysitis in children, and a dearth of studies reporting such cases, ascertaining the true incidence of pediatric hypophysitis is difficult.
Classifications
There have been five histologic types of hypophysitis described to date: (1) lymphocytic, (2) granulomatous, (3) xanthomatous, (4) IgG4-related, and (5) necrotizing. Of these five sub-types, lymphocytic is the most common in children, with the latter four rarely reported in children. Adult lymphocytic hypophysitis typically occurs in adult females, associated with pregnancy or autoimmune disorders such as Hashimoto’s thyroiditis, Graves’ disease, type I diabetes, and systemic lupus erythematosus. In children, no gender predisposition has been established [6]. Whether or not these autoimmune conditions put a child at greater risk for hypophysitis has not yet been determined.
Difference from Adults
In children, symptoms of diabetes insipidus and signs of growth hormone deficiency (GHD) are typically the most common presenting complaints or concerns, both prior to hypophysitis diagnosis and any surgical procedures or interventions [6, 13]. While many adults with hypophysitis often report to visual disturbances and headaches, this is seldom seen in the pediatric population [6]. Many of the adults described in earlier studies suffered from partial or panhypopituitarism at some point prior to surgical interventions, including adrenocorticotrophic hormone (ACTH), gonadotropin (luteinizing and follicle-stimulating hormones), and thyrotropin (TSH) deficiencies [2, 6]. Symptoms of neurohypophysis dysfunction (polydipsia and polyuria) and permanent diabetes insipidus were less seldom seen, with the first case not reported until 1991 [14]. Growth hormone deficiency occurs less often in adults. This has led some authors to speculate that anterior pituitary hormone deficiencies (APHDs) develop in reverse order in hypophysitis when compared to the timeline of deficiencies seen in pituitary adenomas (ACTH and TSH deficiencies first in hypophysitis, followed by GH and gonadotropin deficiencies) [11]. Total hypophysectomies performed on adults with hypophysitis resulted in permanent hypopituitarism for most; however, several who underwent partial hypophysectomies continued to have hypopituitarism if they had significant pre-operative pituitary deficiencies [2, 15]. This would suggest that ongoing pituitary inflammation develops over time in subjects with hypophysitis, both pre-operatively and post-operatively, regardless of the surgical pituitary procedure they undergo. Children are therefore likely at a greater hypopituitarism risk post-surgical interventions, particularly those with one or more APHDs at the time of initial presentation.
Laboratory and Imaging Studies
A stepwise approach should be taken in pediatric hypophysitis investigations, with a goal to promptly and definitively establish the diagnosis, combining the utility of clinical assessment, and laboratory and imaging studies, prior to biopsy. Anterior pituitary hormone levels should be analyzed first, along with serum and urine osmolality. The timing of pituitary MRI with and without contrast is often done concomitantly or after initial blood and urine tests. Tumor markers (AFP, β-hCG) and angiotensin-converting enzyme (ACE) level can be drawn next, along with complete blood count, metabolic panel, erythrocyte sedimentation rate, and C-reactive protein. Skeletal survey, or cranial X-ray, should be obtained to rule out extra-cranial lesions, seen typically in LCH. Serologic studies for syphilis, tuberculosis-specific testing, and bacterial and fungal cultures can also be considered [13]. Upon collaboration with Neuro-Oncology specialists, bone marrow aspirate can be considered as well.
MR imaging at the time of preliminary workup is often beneficial and sometimes diagnostic. One large-scale prospective study of children and young adults in Italy showed a yield of 28% of etiologic diagnosis in 85 patients with central diabetes insipidus [16]. Pituitary stalk thickening and enlargement has been shown to be the hallmark of hypophysitis imaging [8, 14, 17, 18]. A marked contrast enhancement of the hypophysitis is also typically seen, as well as an area of ring-like enhancement, in line with central necrosis [19, 20]. Normal-sized or slightly enlarged pituitary fossa is also often observed [18, 21, 22].
Histologic Findings
Many of the first adult subjects described were noted to have pituitary glands depicted as unusually firm. Lymphocytic infiltration was often noted on adenohypophysis microscopy, along with a small number of plasma cells, and varying degrees of edema and fibrosis [2]. In children, lymphoid follicles with coinciding plasma cells are the characteristic histologic finding in lymphocytic hypophysitis, throughout part of, or the entire, pituitary gland. Eosinophils, neutrophils, and macrophages are occasionally observed. Fibrosis typically replaces normal pituitary tissue [23]. Current literature on adult lymphocytic hypophysitis cases has shown two entities, both demonstrating a preponderance for T lymphocytes over B lymphocytes [24, 25]. In the first pattern, Th17+ lymphocytes are found in greater numbers than T reg cells, with pituitary infiltration characterized by higher numbers of macrophages, monocytes, granulocytes, and natural killer cells. CD20+ B cell production of lymphocytes is seen in the second entity, surrounded by CD3+ T cells and numerous T reg cells, taking on a lymphoid tissue pattern similar to the types seen in the immune tolerant phase of chronic infections [25].
Natural History
The natural history of this condition in children is often unpredictable: sometimes with spontaneous recovery and complete resolution of clinical, laboratory, and radiological abnormalities; and other cases resulting in permanent hypopituitarism with or without neurologic impairment. It had previously been suggested that the pituitary typically enlarges with inflammation and edema early in the course. This stage can be subclinical for some individuals, while others might suffer from mass-effect symptoms or partial APHDs. Fibrosis then occurs as previous pituitary tissue atrophies, with resolution of pituitary growth or mass and subsequent hypopituitarism. However, some regain pituitary function, suggesting that certain individuals with prior symptoms of pituitary deficiencies suffered compression of surrounding pituitary tissue from their respective inflammatory lesion [2].
Differential
The differential for those with suspected hypophysitis includes: primary hypophysitis, germinoma, histiocytosis (Langerhans, and non-Langerhans), lymphoma, craniopharyngiomas, pituitary adenomas, other tumors, metastases, medications, sarcoidosis, abscesses, syphilis, Tuberculosis, fungal, viral, and bacterial infections (e.g., group B streptococcus, Haemophilus influenza). New onset diabetes insipidus with or without pituitary stalk thickening is generally the presenting symptom within the pediatric age group. As the germinomas and histiocytosis can progress over time, an early diagnostic certainty of hypophysitis is not always straightforward and is often a diagnosis of exclusion over time. Children should be followed closely for several years as a germinoma can be diagnosed up to 3 years after initial hypophysitis diagnosis, and sometimes even after longer periods, particularly those in the prepubertal age range [6, 26]. Pituitary stalk biopsies are typically reserved for those with progressive or worsening stalk thickening, evolving APHDs, or increased intracranial pressure or optic chiasm compression (see Chap. 4) [26,27,28,29,30].
Preliminary investigations to help exclude germinoma should include serum and CSF AFP and β-hCG analyses. Serum and CSF ACE levels should also be considered to rule out sarcoidosis. Pituitary MRI findings seen on germinoma and LCH can be unique among children, with the former group showing a more proximal thickening, and the latter demonstrating a central thickening [31, 32]. Of those children presenting with diabetes insipidus, the exact time to oncologic diagnosis varies: germinoma diagnosis within the first 1–18 months, and an LCH diagnosis typically within the first through twelfth months, but sometimes not until several years later [27, 33]. Pituitary stalk biopsy is often the gold standard in differentiating the conditions, although germinomas can show lymphocytic infiltrate with local inflammation, without a visible pituitary mass, making the diagnosis more challenging [34].
Treatment
Pituitary hormone replacement is the mainstay of treatment for children in both the acute and chronic phases of hypopituitarism secondary to hypophysitis. As mentioned previously, surgical exploration is reserved with those manifesting signs of optic nerve compression or increased intracranial pressure. Corticosteroids have been used for treatment at some adult centers with mixed results. A recurrence of symptoms while on corticosteroid therapy, or during a taper, has also been reported [14, 19, 35,36,37]. Since the inflammatory process is typically self-limited in hypophysitis, corticosteroids are unlikely going to provide any curative benefit. The treatment benefits of corticosteroid therapy in children with hypophysitis outweighs the risks has not been proven to date.
Immunosuppressants (azathioprine, methotrexate, and cyclosporine) have been used in several adult cases of corticosteroid-resistant hypophysitis, but their long-term benefits and effectiveness have yet to be fully studied [7]. Certain immunotherapies are now being trialed in adults, including infliximab and rituximab treatments, as well as stereotactic radiosurgery and fractionated radiotherapy in select patients with corticosteroid-resistant cases [7, 38, 39]. These novel treatments are still reserved for select adults with challenging hypophysitis cases and are not yet standard of care in children.
Utility of Autoantibodies
The usefulness of antipituitary and antihypothalamus antibodies (APA, AHA) is still disputed in light of conflicting results obtained with varying laboratory assays, and low sensitivity and specificity [10, 40,41,42]. The timing of APA and AHA measurement may also play a role in their utility as they are unlikely to be produced after pituitary gland destruction secondary to an autoimmune attack [23]. Positive APAs can be found in healthy subjects as well, and other pituitary disorders, such as pituitary adenomas, Sheehan’s syndrome, and empty sella syndrome [10, 23, 40]. Lastly, the pathogenic role of these antibodies has not been studied extensively in children with idiopathic hypopituitarism [43, 44].
Imaging Findings Over Time
Pituitary stalk thickening is the classic finding in pediatric hypophysitis. The precise timing of onset, persistence, and resolution of this finding on serial MRI scans has not been well defined. One retrospective study in Belgium looked at nine children, ages 3–14, over a 15-year time span, who initially presented with signs of diabetes insipidus and pituitary stalk thickening on imaging, with subsequent MRI studies. The stalk thickening resolved in 6, regressed in 2, and persisted in 1 over a mean follow-up of 5.4 (1.5–15) years, with a change in stalk thickness defined by at least 20% variation on larger axis on two consecutive examinations [45]. An earlier retrospective pediatric study from 2000 looked at 79 subjects with central diabetes insipidus. Fifty percent of these subjects had idiopathic DI, and the authors suggested these children did not have hypophysitis given their young age, presence of APHDs, and progressive stalk thickening over time. The remainder had intracranial tumor (23%), LCH (15%), familial disease (6%), skull fracture (3%), and autoimmune polyendocrinopathy (1%). Twenty-nine of these 79 subjects demonstrated pituitary stalk thickening at some point on MRI. Six of these showed normalization of stalk size (median time span 1.3, range 1.0–5.7 years), one with a decrease in thickness size (over 2.1 years of follow up), seven with further thickening (median 1.6, range 0.8–10.3 years), and four with new onset thickening (median 0.8, range 0.2–3.0 years) [46].
MRI studies can also sometimes provide more information regarding ongoing tumor development, and prospective anterior pituitary hormone deficiency (APHD) risks. It has been proposed that increased anterior pituitary size on surveillance MRI screening more strongly supports the diagnosis of neoplastic and infiltrative disorders (e.g., germinoma, histiocytosis), while reduction in anterior pituitary size supports a hypophysitis diagnosis [26]. For those subjects in the 2000 retrospective review with idiopathic DI, there was a statistically significant greater risk of APHD development with a smaller than expected anterior pituitary on MRI [46].
The precise timing of pituitary MRI surveillance has not been clearly defined for children with suspected hypophysitis based on diabetes insipidus diagnosis and imaging studies. Some groups advocate for every 3 months early on, while others recommend every 6 months during the first 2 years of diagnosis, and then yearly for the next 3 years [26]. More studies are needed to gain the necessary clinical information on how to best follow children with pituitary stalk thickening and suspected hypophysitis. Most institutions with pituitary centers follow these children 2–4 times annually during the initial years of presentation.
Anterior Pituitary Hormone Deficiency (APHD) Over Time
With regard to the development of anterior pituitary hormone deficiencies (APHDs) over time, studies have shown conflicting results. The retrospective Belgian study looking at 9 children with DI and pituitary stalk thickening at presentation demonstrated one subject with an APHD (central hypothyroidism) at DI onset, with only one patient developing APHDs over the mean follow-up of 5.4 years, with subsequent resolution of stalk thickening (and it is not explicitly stated if the patient with hypopituitarism was the one with central hypothyroidism at onset). DI remained in all subjects [45].
Of the 79 pediatric subjects with central diabetes insipidus analyzed in the 2000 retrospective study, 48 displayed anterior pituitary hormone deficiencies (APHDs) with median onset of 0.6 (range 0.1–18.0) years after DI diagnosis, with GH deficiency being the most common, followed by TSH, gonadotropin, and ACTH deficiencies. Of the idiopathic DI patients in this study, 49% developed APHDs, with a median onset of first deficiency at 0.6 (range 0.1–18) years after DI diagnosis, and the median onset of final deficiency at 7.7 (range 0.1–18) years. Growth hormone deficiency was the most common finding. Seventy-five percent of the patients with LCH developed APHDs with the median onset of 3.5 (range 0.1–6.0) years after DI diagnosis. The mean estimated probability of developing one anterior pituitary hormone deficit within 6 years of DI diagnosis was 81% in LCH patients and 49% in those with idiopathic DI, with no difference of developing additional deficiencies between the two groups. The study further explores the association of stalk thickening with the development of APHD between the LCH and idiopathic DI groups. Seventeen of the eighteen idiopathic DI patients with thickened stalk developed APHDs, while only 2 of the 19 idiopathic cases without stalk thickening developed APHDs. The risk of APHDs in LCH patients was independent of the pituitary stalk size [46]. Demonstration of the precise timing and definitive risk of single or multiple APHDs in children with hypophysitis has yet to be studied in depth.
Conclusion
Lymphocytic hypophysitis is an uncommon disorder in children but carries a significant clinical risk of hypopituitarism. There are differences in both clinical presentation and laboratory findings between adults and children, and the approach to the differential also requires special consideration in children. Recognizing the signs of this disorder and establishing a thorough differential with appropriate preliminary investigations is essential prior to the gold standard of pituitary biopsy. Frequent follow-up and MRI surveillance is needed in children, with prompt attention to MRI changes. Endocrinologic follow-up is necessary as anterior pituitary hormone deficits are often already occurring at presentation in some children or develop over time. Further studies of hypophysitis are needed to devise a more thorough clinical guideline for the pediatric population.
References
Goudie RB, Pinkerton PH. Anterior hypophysitis and Hashimoto’s disease in a young woman. J Pathol Bacteriol. 1962;83:584–5.
Cosman F, et al. Lymphocytic hypophysitis. Report of 3 new cases and review of the literature. Medicine (Baltimore). 1989;68(4):240–56.
Carpenter CC, et al. Schmidt’s syndrome (thyroid and adrenal insufficiency). A review of the literature and a report of fifteen new cases including ten instances of coexistent diabetes mellitus. Medicine (Baltimore). 1964;43:153–80.
Levine S. Allergic adenohypophysitis: new experimental disease of the pituitary gland. Science. 1967;158(3805):1190–1.
Gellner V, et al. Lymphocytic hypophysitis in the pediatric population. Childs Nerv Syst. 2008;24(7):785–92.
Kalra AA, Riel-Romero RM, Gonzalez-Toledo E. Lymphocytic hypophysitis in children: a novel presentation and literature review. J Child Neurol. 2011;26(1):87–94.
Bellastella G, et al. Revisitation of autoimmune hypophysitis: knowledge and uncertainties on pathophysiological and clinical aspects. Pituitary. 2016;19(6):625–42.
Honegger J, et al. Diagnosis of primary Hypophysitis in Germany. J Clin Endocrinol Metab. 2015;100(10):3841–9.
Faje A. Hypophysitis: evaluation and management. Clin Diabetes Endocrinol. 2016;2:15.
Caturegli P, et al. Autoimmune hypophysitis. Endocr Rev. 2005;26(5):599–614.
Buxton N, Robertson I. Lymphocytic and granulocytic hypophysitis: a single centre experience. Br J Neurosurg. 2001;15(3):242–5. discussion 245-6
Khare S, et al. Primary (autoimmune) hypophysitis: a single centre experience. Pituitary. 2015;18(1):16–22.
Romano A, Rigante D, Cipolla C. Autoimmune phenomena involving the pituitary gland in children: new developing data about diagnosis and treatment. Autoimmun Rev. 2019;18(10):102363.
Nussbaum CE, Okawara SH, Jacobs LS. Lymphocytic hypophysitis with involvement of the cavernous sinus and hypothalamus. Neurosurgery. 1991;28(3):440–4.
Honegger J, et al. Lymphocytic and granulomatous hypophysitis: experience with nine cases. Neurosurgery. 1997;40(4):713–22. discussion 722-3
Di Iorgi N, et al. Central diabetes insipidus in children and young adults: etiological diagnosis and long-term outcome of idiopathic cases. J Clin Endocrinol Metab. 2014;99(4):1264–72.
Lee JH, et al. Lymphocytic hypophysitis: occurrence in two men. Neurosurgery. 1994;34(1):159–62. discussion 162-3
Vanneste JA, Kamphorst W. Lymphocytic hypophysitis. Surg Neurol. 1987;28(2):145–9.
Beressi N, et al. Pseudotumoral lymphocytic hypophysitis successfully treated by corticosteroid alone: first case report. Neurosurgery. 1994;35(3):505–8. discussion 508
Supler ML, Mickle JP. Lymphocytic hypophysitis: report of a case in a man with cavernous sinus involvement. Surg Neurol. 1992;37(6):472–6.
Hashimoto M, et al. Lymphocytic adenohypophysitis: an immunohistochemical study. Surg Neurol. 1991;36(2):137–44.
Higuchi M, et al. Pituitary granuloma and chronic inflammation of hypophysis: clinical and immunohistochemical studies. Acta Neurochir. 1993;121(3–4):152–8.
Caturegli P, et al. Pituitary autoimmunity: 30 years later. Autoimmun Rev. 2008;7(8):631–7.
Guaraldi F, et al. Pituitary Autoimmunity. Front Horm Res. 2017;48:48–68.
Mirocha S, et al. T regulatory cells distinguish two types of primary hypophysitis. Clin Exp Immunol. 2009;155(3):403–11.
Di Iorgi N, Morana G, Maghnie M. Pituitary stalk thickening on MRI: when is the best time to re-scan and how long should we continue re-scanning for? Clin Endocrinol. 2015;83(4):449–55.
Mootha SL, et al. Idiopathic hypothalamic diabetes insipidus, pituitary stalk thickening, and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab. 1997;82(5):1362–7.
Leger J, et al. Thickened pituitary stalk on magnetic resonance imaging in children with central diabetes insipidus. J Clin Endocrinol Metab. 1999;84(6):1954–60.
Al-Agha AE, et al. Acquired central diabetes insipidus in children: a 12-year Brisbane experience. J Paediatr Child Health. 2001;37(2):172–5.
Beni-Adani L, et al. Surgical implications of the thickened pituitary stalk accompanied by central diabetes insipidus. J Neurosurg. 2005;103(2 Suppl):142–7.
Edouard T, et al. Isolated lymphocytic infiltration of pituitary stalk preceding the diagnosis of germinoma in 2 prepubertal children treated with growth hormone. Horm Res. 2009;72(1):57–62.
Schmitt S, et al. Pituitary stalk thickening with diabetes insipidus preceding typical manifestations of Langerhans cell histiocytosis in children. Eur J Pediatr. 1993;152(5):399–401.
Prosch H, et al. Central diabetes insipidus: is it Langerhans cell histiocytosis of the pituitary stalk? A diagnostic pitfall. Pediatr Blood Cancer. 2006;46(3):363–6.
Gutenberg A, et al. Pituitary and systemic autoimmunity in a case of intrasellar germinoma. Pituitary. 2011;14(4):388–94.
Ahmed SR, et al. Necrotizing infundibulo-hypophysitis: a unique syndrome of diabetes insipidus and hypopituitarism. J Clin Endocrinol Metab. 1993;76(6):1499–504.
Pestell RG, Best JD, Alford FP. Lymphocytic hypophysitis. The clinical spectrum of the disorder and evidence for an autoimmune pathogenesis. Clin Endocrinol. 1990;33(4):457–66.
Reusch JE, et al. Preoperative diagnosis of lymphocytic hypophysitis (adenohypophysitis) unresponsive to short course dexamethasone: case report. Neurosurgery. 1992;30(2):268–72.
Ray DK, et al. Gamma knife surgery for lymphocytic hypophysitis. J Neurosurg. 2010;112(1):118–21.
Selch MT, et al. Stereotactic radiotherapy for the treatment of lymphocytic hypophysitis. Report of two cases. J Neurosurg. 2003;99(3):591–6.
Crock PA. Cytosolic autoantigens in lymphocytic hypophysitis. J Clin Endocrinol Metab. 1998;83(2):609–18.
De Bellis A, Bizzarro A, Bellastella A. Pituitary antibodies and lymphocytic hypophysitis. Best Pract Res Clin Endocrinol Metab. 2005;19(1):67–84.
Ricciuti A, et al. Detection of pituitary antibodies by immunofluorescence: approach and results in patients with pituitary diseases. J Clin Endocrinol Metab. 2014;99(5):1758–66.
Cocco C, et al. The hypothalamic-pituitary axis and autoantibody related disorders. Int J Mol Sci. 2017;18(11):2322.
De Bellis A, et al. Detection of antipituitary and antihypothalamus antibodies to investigate the role of pituitary or hypothalamic autoimmunity in patients with selective idiopathic hypopituitarism. Clin Endocrinol. 2011;75(3):361–6.
Schaefers J, et al. Clinical presentation and outcome of children with central diabetes insipidus associated with a self-limited or transient pituitary stalk thickening, diagnosed as infundibuloneurohypophysitis. Clin Endocrinol. 2017;87(2):171–6.
Maghnie M, et al. Central diabetes insipidus in children and young adults. N Engl J Med. 2000;343(14):998–1007.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Gibson, C. (2021). Hypophysitis. In: Alter, C.A. (eds) Diabetes Insipidus in Children. Springer, Cham. https://doi.org/10.1007/978-3-030-83248-3_5
Download citation
DOI: https://doi.org/10.1007/978-3-030-83248-3_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-83247-6
Online ISBN: 978-3-030-83248-3
eBook Packages: MedicineMedicine (R0)