
Abstract
Lung carcinoids (LCs) are rare tumors, whose incidence has increased during the last decades; however, some controversies still exist on their diagnosis and management, also given the scarcity of prospective data. Surgery remains the gold standard for locoregional disease, while for advanced or progressive disease, no standard treatment or therapeutic algorithm is currently available, and multidisciplinary management is always required. Medical therapy represents the gold standard in the avanced setting, being somatostatin analogues (SSAs), everolimus, chemotherapies, peptide receptor radiotherapy (PRRT) treatments the therapeutic armamentarium. SSAs represent a viable option for both symptom and disease control, as well as PRRT with radiolabeled SSAs is an option in patients with well-differentiated low-intermediate grade neuroendocrine tumors (NETs) expressing high levels of somatostatin receptors (SSTRs). Systemic chemotherapy is reserved for those cases of locally advanced or metastatic disease, even if the standard regimen has not yet been defined. The role of targeted therapies in LCs remains still limited with the exception of the only approved drug (everolimus) in clinical practice. Regarding immunotherapy, in low-grade LCs, pembrolizumab and spartalizumab had shown preliminary promising results in terms of both safety and clinical outcome, but data from phase II study are disappointing. However, as the majority of studies exploring the efficacy of available treatments are retrieved from studies on gastroenteropancreatic NETs and/or are retrospective in nature, further studies, specifically dedicated to LCs, are needed to draw more robust conclusions, particularly to clarify the best sequence and timing of systemic drugs in the management of this subgroup of rare neoplasms.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Lung carcinoids
- Bronchial neuroendocrine tumors
- Typical carcinoid
- Atypical carcinoid
- Epidemiology
- Diagnosis
- Treatment
- Therapy
1 Introduction and Epidemiology
Neuroendocrine tumors (NETs) are malignant neoplasms arising from neuroendocrine system, mainly located in the gastrointestinal tract and lung [1]. An estimated 25–30% of all NETs have their origin in the bronchial tract and lungs. Lung NETs account for less than 1–2% of all pulmonary neoplasms [1,2,3,4]. Their incidence rate, which is 0.2–2/100000 population/year in Europe and United States, has dramatically risen over the past 30 years [1, 5, 6]. NETs of the lung comprise a heterogeneous population of tumors ranging from well-differentiated bronchial NETs to highly malignant and poorly differentiated small-cell lung cancer (SCLC) and large-cell neuroendocrine carcinoma (LCNEC) [7].
Well-differentiated NETs of the lung are also named lung carcinoids (LCs), including typical (TCs) and atypical carcinoids (ACs). TCs are more common than ACs, accounting for 90% of all LCs [5]. Both the subtypes arise mainly in female and in Caucasian, Hispanic, and Asian people [5, 8,9,10]. The median age at diagnosis is 45 years for TCs and 55 for ACs [5, 8,9,10,11].
Surgery is the gold standard in earlier stages. The 5-year overall survival (OS) for limited disease ranges from 87–90% for TCs to 44–78% for ACs, respectively [12,13,14,15,16]. Conversely, the patients with advanced stages show a poor prognosis with a median OS of 17 months and a 5-year survival rate of about 27% [17].
Given their rarity, only few available data from prospective studies are available, and no global consensus exists in regards to therapeutic management of LCs. Target agents such as everolimus, somatostatin analogues (SSAs), and chemotherapy treatments represent the options of choice in the advanced setting [5]. Peptide receptor radiotherapy (PRRT) and immunotherapy are emerging options. A multidisciplinary approach is strongly suggested in all clinical scenarios. Herein, we will provide a comprehensive literature review on diagnosis and management of advanced LCs.
2 Etiology and Classification
The majority of LCs are sporadic neoplasms [18, 19]. Differently from high-grade lung NETs, no relationship between LCs and smoking habit has been proved so far [5]. In approximately 5% of the patients, the development of these neoplasms occurs in the context of multiple endocrine neoplasia type 1 (MEN1) syndrome [20,21,22,23], and the association to a rare pulmonary carcinoid tumor genetic syndrome is reported in sporadic cases as well [19]. Although rare, some patients may present with multiple lung nodules or tumorlets and widespread peripheral airway neuroendocrine cell hyperplasia. In this case, a diagnosis of diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH) can be made [24]. The mechanisms underlying progression and/or onset of LCs from DIPNECH are still unclear [7, 25].
3 Histopathological Features and WHO Classification
The availability of adequate tissue sample is required to distinguish lung NET subtypes.
The 2015 WHO classification has defined four lung NETs subtypes: TC, AC, LCNEC, and SCLC. SCLC and LCNEC are poorly differentiated, aggressive neoplasms, while LCs are well differentiated, indolent tumors (Table 12.1) [7].
The definition of LCs include lesions of more than 5 mm in diameter, while lung lesions of 5 mm or less are still classified as carcinoid tumorlets (DIPNECH) [7].
Two aspects are crucial to define lung NETs subtypes: the presence of necrosis and the number of mitoses. In particular, TCs have no evidence of necrosis and less than two mitoses per 2 mm2 in the tumor area, while ACs are characterized by focal necrosis and 2–10 mitoses per 2 mm2 [7]. However, the distinction between well- and poorly differentiated lung NETs based on the number of mitoses (less or more than 10/mm2, respectively) is still debated [5, 7, 26]. Other biomarkers like synaptophysin, chromogranin A (CgA), and CD56 have to be combined to the WHO classification to confirm lung NET diagnosis [7].
The prognostic role of Ki-67 cell proliferation index by immunohistochemistry (IHC) has not been well defined in lung NETs, and its role is currently under debate. According to the WHO of 2015, a Ki-67 value lower than 20% characterizes LCs (≤5% in the TC and ≤20% in the AC), while a value >40% is typical of the high-grade pulmonary NETs [7].
Although the Ki-67 index is not currently accredited with lung NET subtyping due to some overlap of cut-off thresholds among biologically adjacent tumors (TC versus AC, AC versus LCNEC, LCNEC versus SCLC), its differential distribution between low- to intermediate-grade and high-grade tumors has made it an exceptional discriminator especially on biopsy/cytology samples, being its determination recommended on surgical specimens as well [7, 27, 28].
The reproducibility and clinical usefulness of Ki-67 in lung NETs is still under debate, and there is no current diagnostic role for Ki-67, whereas the mitotic count has remained the only proliferation criterion in tumor classifications over time [7].
4 Diagnostic Workup
4.1 Clinical Presentation
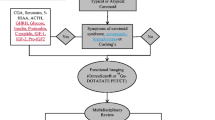
TCs are usually located in the central parenchyma of the lung, while ACs often develop peripherally. TCs and ACs are usually diagnosed in earlier stages, but liver and bone metastases are common in advanced stages [5, 24]. Patients with LCs are often asymptomatic or present unspecific symptoms like hemoptysis, dyspnea, cough, or chest pain [24, 29]. Functional LCs are rare.
Although typical carcinoid syndrome occurs in only 10% of the cases [13, 30], NETs of the lung may secrete various hormones and vasoactive peptides.
In particular, LCs have been more often associated with adrenocorticotropic hormone (ACTH) production and can cause Cushing’s syndrome [31, 32].
4.2 Imaging
Computed tomography (CT) is the gold standard to diagnose and stage LCs. A single round lesion is the most frequent radiological aspect [17, 24], but multiple calcified lesions may also be present [33]. Magnetic resonance imaging (MRI) may help in the detection of bone or liver metastases [5]. Approximately 80–90% of LCs express somatostatin receptors (SSTRs), thus functional imaging based on radiolabeled SSA plays a key role in both staging and defining treatment strategy [13, 29, 34, 35]. The octreoscan or somatostatin receptor scintigraphy (111In-pentetreotide/octreotide scan) demonstrates 93% sensitivity and 87% specificity, respectively, in LC diagnosis [34, 36]. However, more recently, the 68Ga-DOTATATE PET/CT has been introduced in lung NET management.
Considering the binding to SSTRs, 68Ga-DOTATATE showed a tenfold higher affinity than octreotide [37, 38]. A sensitivity of 81% and a specificity of 90% have been reported by Haug and collaborators, and in another study, the 68Ga-DOTATATE PET/CT detected a significantly larger number of NET lesions expressing type 2 SSTR than octreoscan (p < 0.001) [39,40,41].
Also taken into account the reduced scanning time and the better imaging resolution, PET/CT 68Ga-DOTATATE should be preferred in LC management [42, 43].
The role of fluoro-deoxy-glucose (FDG)-PET/CT in LCs is still debated. TCs are usually characterized by low or no uptake on FDG scan, whereas a higher uptake is possible in ACs with a high proliferative index [44]. In a small retrospective series of 20 LCs, Bongiovanni et al. showed a shorter progression-free survival (PFS) in LCs with positive 18-FDG PET/CT scan than in those with negative 18-FDG-PET/CT scan (p = 0.015) [45]. These data could support the possible prognostic role of 18-FDG-PET/CT and its potential ability in suggesting different therapeutic strategies for LCs according to the different FDG uptake.
5 Surgery
Surgical resection, with preservation of as much normal lung tissue as possible, is still the gold standard in case of limited and resectable disease [5, 24].
Regarding TCs, surgery is associated with excellent outcomes, with 5- and 10-year survival rates of ∼90% and ∼80%, respectively, and very low recurrence rates (3–5%) [46, 47]. In detail, recent analyses of the European Association of Thoracic Surgeons (ESTS) Neuroendocrine Tumours Working Group revealed that surgically resected TCs are associated with a 5-year survival rate of 94% [48]. On the other hand, 5- and 10-year survival rates following surgery in AC patients are lower (about 70% and 50%, respectively), given the higher rate of relapse (∼25%) [46, 47, 49].
According to the Surveillance, Epidemiology, and End Results (SEER) database, lobectomy is the most common procedure (51.2%), compared to sublobar resection (wedge resection or segmentectomy, 24.1% of cases). Other less-used procedures are pneumonectomy, bronchoplasty, extended resection, and bronchoscopic ablation [50].
As a rule, the surgical technique of choice is lobectomy. On the other hand, given the indolent nature of TCs, sublobar resections may be taken into account in this setting as similar outcomes compared to lobectomy have been reported [51, 52]. Furthermore, segmentectomy and wedge resections should be considered in patients with compromised pulmonary function and/or severe comorbidities [53]. Further randomized studies are needed to better assess the differences in terms of perioperative outcome, long-term survival, and disease recurrence between the two approaches.
A minimally invasive approach, such as video-assisted thoracoscopic surgery (VATS), is recommended in experienced centers due to fewer complications and potentially increased survival rates [53, 54]. Some authors reported promising results also in the setting of minimally invasive bronchoplastic procedures [55].
The natural history of these tumors is strictly related to the lymph-node status. It has been reported that lymph-node metastases may be present in up to 25% of the cases in TCs and >50% in ACs, respectively [56]. Node-negative and N1 patients have similar outcomes. Conversely, N2 tumors have been reported especially in ACs and are associated with a dismal prognosis [16, 57]. Furthermore, in the majority of the patients, lymph-node involvement does not modify surgical indication, given that the neoadjuvant treatment does not improve the resectability rate or survival in LCs [58]. Thus, although the need for lymph-node dissection is still poorly defined in TCs, a complete radical lymphadenectomy should be performed in all cases.
From the surgical point of view, unknown lung lesions (that exhibit NET radiological features) undergoing upfront surgery should undergo wedge excisional biopsy. If intraoperative frozen section is consistent with NET and the margins are negative, systematic lymph-node dissection should be performed [59]. If the patient is node-negative, a completion lobectomy is not required. In node-positive patients with adequate pulmonary reserve, lobectomy should be performed regardless of histology [5, 59]. If atypical features are found during pathologic evaluation, an interval completion lobectomy may be considered in fit patients [5, 49]. When there is suspicion of N2 nodal involvement or after mediastinoscopy/endobronchial ultrasound (EBUS) showing N2 disease, multimodality treatment might be required [60].
Approximately 20% of all LCs present as pure endobronchial polyp-like lesions without gross radiologically detectable involvement of the bronchial wall and lung parenchyma [60]. Rarely, N1 lymph nodes may involve the bronchial takeoff. In both cases, bronchoplastic procedures (bronchial sleeve resection or wedge) with or without parenchymal resection are indicated [61] since they protect from the detrimental effects of pneumonectomy on respiratory functions as well as on quality of life. In the literature, the incidence of sleeve resections in the different series varies from 1.4% to 41% of cases [62]. Given the indolent behavior of these tumors, a complete resection with disease-free margins (∼5 mm) is mandatory [62]. In rare cases, a pulmonary artery reconstruction can be associated as well [63]. Bronchial sleeve resections have been also described for more peripheral lesions involving the segmental bronchi [64], although this procedure is still debated. In order to assess the best surgical strategy, fiberoptic bronchoscopy should always be performed by the operating surgeon to have a precise idea of the anatomic details. The technical procedures are identical to those of bronchial sleeve resections performed for lung cancer. As a rule, the anastomosis can be encircled by intercostals pedicle flap, thymic, or mediastinal tissue in order to favor protection/revascularization of the bronchial anastomosis, to separate it from the arterial side when a combined bronchovascular reconstruction is performed (avoiding broncho-arterial fistulas) and to contain a small dehiscence [62]. Concerning the perioperative outcome, one study only [65] compared bronchoplastic procedures between LCs and primary lung cancer, reporting less frequent anastomotic and nonsurgical complications in the LC group (probably because of the younger age of these patients).
Surgery is still considered a curative treatment for LCs, also in the metastatic setting [65]. Pulmonary resection is often recommended in patients with limited hepatic metastases, with ∼20% achieving a cure [66]. If complete resection is not possible, palliative debulking surgery may be taken into account in particular cases to relieve symptoms or prevent complications (i.e., pneumonitis). However, resection of the primary tumor is not indicated in case of unresectable metastases when the primary site is relatively stable.
6 Endobronchial Resection
Due to their indolent clinical course, bronchoscopic excision can be taken into account in those cases presenting with centrally located intraluminal LCs. In the literature, a variety of endobronchial procedures have been reported as an effective alternative to surgical treatment such as YAG laser, diode laser, cryo or electrosurgery, argon-plasma coagulation, and mechanical debulking [67,68,69,70,71]. To date, these techniques are still considered suboptimal approaches and reserved for selected cases only. As a rule, extra-luminal tumor growth, larger tumor diameter, and suspected locoregional/distant metastases are generally considered contraindication for bronchoscopic excision [68]. Furthermore, bronchoscopic treatment is not always effective, especially when LCs extend to the segmental bronchi and when the tumor is in either the upper left or right lobes.
As reported by a recent study assessing prognostic factors for endobronchial ablation, only small intraluminal tumors smaller than 2 cm are suitable for this kind of procedure, whereas all other tumors should be treated with conventional surgery [68]. Another indication is the desobstruction and recanalization of the involved bronchus to obtain resolution of post-obstructive pneumonia [72] as well as to limit the extension of the subsequent surgical resection [73]. A recent systematic review reported excellent outcome results for TCs (5-year survival ranging from 89 to 94% in the literature) and a low rate of locoregional and distant recurrence in bronchoscopic-treated patients, ranging from 0–5% to 0–4% respectively [74]. Although these results seem promising, these studies are biased by the patients’ selection (small tumors, young patients, usually indolent histology). Further prospective-randomized studies are, therefore, needed to better address this issue.
7 Somatostatin Analogues
For low-proliferating lung neuroendocrine tumors, treatment with SSAs is an option for functional tumors with clinical symptoms. SSAs constitute the gold standard for symptomatic control with >50% improvement in both flushing and diarrhea in gastroenteropancreatic (GEP) and LCs [75, 76]. Patients who have metastatic NETs and carcinoid syndrome should be treated with SSAs such as octreotide or lanreotide [77]. The long-acting release (LAR) formulation of octreotide is commonly used for the chronic management of symptoms in patients with carcinoid syndrome. Standard doses of octreotide LAR are 30 mg intramuscularly every 28 days. Dose and frequency may be further increased for symptom control as needed. Short-acting octreotide (usually 150–250 mcg subcutaneously three times daily) can be added to octreotide LAR for rapid relief of symptoms or for breakthrough symptoms [78, 79].
Lanreotide Autogel has a similar mechanism of action as octreotide, but is administered as a deep subcutaneous injection at the dose of 120 mg every 28 days [80, 81].
The more recent multicentric phase III ELECT trial randomized 115 patients with carcinoid syndrome (including LCs) who were either naïve or responsive to octreotide to receive 120 mg of lanreotide or placebo and evaluated the number of days patients required use of rescue octreotide. Patients in the lanreotide arm required less-frequent rescue octreotide than those in the placebo arm (33.7% vs. 48.5%; P = 0.017), supporting the use of lanreotide for symptom control [82].
Moreover, interesting results have been reported by a specific study focused on advanced, functioning LCs, describing complete symptom control and normalization of urinary 5-hydroxyindoleacetic acid (5-HIAA) in 7 out of 126 ACs patients who received short-acting octreotide injections [83].
In nonfunctioning tumors, the use of SSAs is still controversial, but after the results reported by the PROMID and the CLARINET study indicating antitumor efficacy of octreotide LAR and Lanreotide Autogel drugs in GEP-NETs, it is now also widely accepted for nonfunctioning tumors of other origins [84, 85].
However, studies specifically focused on LCs are scanty. In a recent retrospective study, Bongiovanni et al. [45] investigated the efficacy of SSAs as first-line treatment of 30 metastatic nonfunctioning LCs and reported that, out the 30 patients, one patient (3.3%) achieved a partial response (PR) and 26 (86.6%) showed stable disease (SD), thus highlighting the antitumor activity of SSAs with a satisfiable safety profile.
A randomized double-blind, phase 3 study (SPINET trial, NCT02683941, clinicaltrial.gov) evaluating the efficacy and safety of lanreotide-autogel versus placebo is ongoing in patients with well-differentiated, metastatic, and/or unresectable TCs and ACs.
Due to the very limited level of evidence about the use of SSAs in nonfunctioning, advanced LCs, no clear consensus exists on the timing of octreotide or lanreotide initiation in asymptomatic patients with metastatic well-differentiated LCs. However, if patients with advanced low-grade LCs present with clinically significant tumor burden, initiation of octreotide and lanreotide may be considered [86].
8 Peptide Receptor Radiotherapy
PRRT with radiolabeled SSAs is an option in patients with NETs expressing high levels of SSTRs, namely well-differentiated forms [34, 87, 88].
Well-differentiated LCs frequently express subtype 2 of the SSTR family, and this can be identified by 68Ga-DOTATATE PET/CT scans, which constitute predictors of response [89].
PRRT with either 90Y- or 177Lu-peptides is generally well tolerated, and reported results are promising, even if mainly focused on gastrointestinal neoplasms.
So far, no prospective studies were performed with PRRT specifically in lung NETs, but only few retrospective study including LC patients are published [88,89,90,91,92,93,94].
A recent study examined the long-term efficacy, survival, and toxicity of 177Lu-dotatate in a group of 610 Dutch patients with metastatic GEP and lung NETs [95]. PFS and overall survival (OS) for all patients were 29 months and 63 months, respectively.
Other smaller studies also found improved OS (58.8 months) [90] and median PFS (20.1 months with TCs and 15.7 months with ACs) with PRRT treatment in patients with advanced LCs [93].
The phase III study NETTER-1 evaluated the efficacy and safety of 177Lu-dotatate in 229 patients with advanced, progressive, SSTR–positive midgut NETs who were randomly assigned to receive either 177-Lu-dotatate plus best supportive care (BSC) including octreotide LAR or octreotide LAR alone. Results of this study showed that treatment with 177Lu-dotatate was associated with a significant improvement in PFS (not reached vs. 8.4 months; P < 0.0001). Objective tumor responses were observed in 18% of patients who received 177Lu-dotatate versus 3% in the control group (P < 0.001). According to this trial, 177Lu-dotatate resulted in markedly longer PFS than high-dose octreotide LAR and was associated with limited acute toxic effects. Unfortunately, no LCs were included [96].
Given the results of this landmark trial, PRRT with 177Lu-dotatate was approved by the Food and Drug Administration (FDA) in January 2018 for the treatment of patients with unresectable, low- or intermediate-grade, locally advanced, or metastatic GEP-NETs.
The National Comprehensive Cancer Network (NCCN) 2019 guidelines [86] recommend to consider PRRT with 177Lu-dotatate as a treatment option for some patients with advanced and/or metastatic gastrointestinal tract and lung NETs that are SSTR-positive on imaging and show disease progression while taking SSAs, if the tumor is either low-grade (typical) with clinically significant tumor burden or intermediate-grade (atypical).
In summary, only few, mainly retrospective, studies specifically dedicated on PRRT in LCs are available, but, based on data reported in literature, particularly in the setting of gastrointestinal NETs, PRRT might be considered as an effective option in progressive LCs with strong expression of SSTRs and high tumor burden.
9 Chemotherapy
No randomized trials on chemotherapy are available, and there is no currently an established standard regimen, being the role of chemotherapy in LCs under debate. Because of their low proliferative capacity, LCs are generally considered as chemoresistant neoplasms [97].
In general, available chemotherapy regimens for TCs and ACs include the use of streptozotocin plus 5-fluorouracil/doxorubicin, or capecitabine/oxaliplatin, temozolomide, dacarbazine, doxorubicin, etoposide, and cyclophosphamide [5, 26, 29, 98,99,100,101].
The objective response rates (ORR) with single-agent chemotherapy is generally not >20%, reserving this approach to pretreated patients or to patients with poor performance status [5, 26, 29, 101].
Poly-chemotherapy regimens (i.e., platinum-based chemotherapy, temozolomide combined with capecitabine or bevacizumab, capecitabine plus oxaliplatin) have demonstrated greater activity and are the best option in patients with good performance status and significant tumor burden. Patients are treated with poly-chemotherapy regimens SD in 30–50%, PR in 5–10%, and symptomatic response in 40–60% of the cases. However, these results derived from studies conducted on patients with NETs of any sites including only few patients with LCs [5, 26, 29, 98,99,100,101,102,103].
Among all the chemotherapy agents evaluated, temozolomide and oxaliplatin have shown the best clinical benefit in patients with LCs so far.
Temozolomide is a well-known alkylating agent, used in different types of cancer. The oral administration, the ability to cross the blood-brain barrier, and the possibility of being associated with other cytostatics represent the main strengths of this drug [104, 105]. Ekeblad and colleagues performed a retrospective analysis of 36 patients with histologically confirmed metastatic or inoperable malignant NETs treated with oral temozolomide (100–200 mg/m2 for 5 days every 28 days). The study group included ten patients with TCs and three with ACs. After a median follow-up of 7 months (range 2–17 months), 31% of patients with LCs had SD and 31% showed a PR [106]. The most relevant toxicity was grade 3 and 4 thrombocytopenia in 14% of the cases.
Another retrospective study evaluated the activity of temozolomide in 31 patients affected by metastatic LCs, reporting PR in 14% of the cases and SD in 52% of the cases. The most common toxicity was, again, grade 3 and 4 thrombocytopenia [107].
Given these premises, a phase 2 study with the aim to evaluate the efficacy and safety of Lanreotide Autogel plus temozolomide in patients with advanced or unresectable LCs or thymus carcinoids is currently ongoing (ATLANT-NCT 02698410, clinicaltrial.gov).
Some data correlated the role of methylguanine DNA methyltransferase (MGMT) as predictor of response to temozolomide [108, 109]. MGMT is an enzyme promoting repair of DNA damage caused by alkylating agents. High levels of intracellular enzyme reduce the alkylating agents activity, whereas MGMT gene methylation reduces the levels of the intracellular enzyme.
Kulke et al. reported data about 95 patients with advanced NETs treated with temozolomide and showed that MGMT gene methylation is more common in pancreatic NETs compared with LCs.
This resulted in higher overall response rate with temozolomide in pancreatic NETs compared with LCs (34% versus 2% PR, respectively) [110]. Moreover, the recent study by Campana et al. evaluated the correlation between the outcome of 95 advanced NETs treated with temozolomide and the MGMT promoter methylation status. The authors showed an ORR of 51.8 and 17.7% in patients with or without MGMT promoter methylation, respectively, suggesting that the presence of MGMT promoter methylation represented a strong predictive factor for temozolomide response in NETs [109].
Finally, oxaliplatin has been reported as an active and potentially effective agent in retrospective analyses of patients with metastatic well-differentiated lung NETs alone or combined with other primary sites, treated with XELOX, GEMOX, CAPOX, or FOLFOX regimens [100, 110, 111].
Conversely, the use of cisplatin/carboplatin plus etoposide schedule, recommended by the international guidelines for the treatment of poorly differentiated lung NECs (SCLC and LCNEC), is not currently suggested as a treatment of choice in advanced LCs [34, 98, 101].
10 Targeted Therapy
10.1 mTOR (Mammalian Target of Rapamycin) Inhibitors
Although lung NETs are typically poorly represented in clinical trials of NET treatments, two phase III trials (RADIANT-2 and RADIANT-4), evaluating the efficacy of everolimus in advanced NETs, have recently reported specific results for LCs [112, 113].
In RADIANT-2, which evaluated the impact of combination therapy with the oral mammalian target of rapamycin (mTOR) inhibitor everolimus and the SSA octreotide LAR in patients with advanced NET and carcinoid symptoms, among them 6.9% of patients in the experimental group and 2.3% of patients in the control group were diagnosed with lung NETs. Overall, patients were randomly assigned to receive octreotide LAR 30 mg intramuscularly every 28 days combined with everolimus 10 mg per day (N = 216) or octreotide LAR plus a placebo (N = 213). Treatment with everolimus combined with octreotide was associated with longer PFS: 16.4 months in patients treated with everolimus and octreotide versus 11.3 months in control patients (P = 0.026); patients with lung NETs showed trend to improve PFS with everolimus plus octreotide (P = 0.228) [113, 114].
Based on these promising findings, a subsequent RADIANT-4 trial evaluated progressive, nonfunctioning, well-differentiated NETs, including LCs, where patients were treated with everolimus plus BSC versus placebo plus BSC [113]. Out of a total of 302 patients who were randomized to receive either everolimus or placebo (n = 97), the primary endpoint was PFS. In total, 175 patients had gastrointestinal NET and 90 had lung disease. Everolimus-treated patients showed a prolonged median PFS, as compared with those receiving placebo (11.0 vs. 3.9 months, HR 0.48; p < 0.00001). This benefit in PFS was consistent in all subgroup analyses: in particular, there was a 50% improvement in PFS for patients with lung tumors and a 44% benefit for those with gastrointestinal NETs [113, 115].
Finally, a trial specifically looking at lung NETs, the LUNA phase II trial, where patients were randomly assigned to everolimus, the SSA pasireotide (a novel multi-receptor ligand SSA with higher affinity for SSTR1, SSTR3, and SSTR5 than octreotide, but a lower affinity for SSTR2) [116], or the combination of both, was associated with antitumor activity and an acceptable safety profile. A total of 112 patients with LCs were included. The LUNA study achieved the preplanned statistical objective of a 9-month PFS rate >20% in all the three arms, supporting the efficacy of everolimus in lung NETs [117].
Since the results from these three phase II-III prospective trials have been published, everolimus, a selective mTOR inhibitor, has been approved for the treatment of unresectable or metastatic, well-differentiated, nonfunctional NETs of lung origin in patients with progressive disease.
10.2 Antiangiogenic Agents
The potential role of antiangiogenic agents in LCs is still far from being clearly understood. Sunitinib is an orally administered kinase inhibitor small molecule with activity against a number of tyrosine kinase inhibitors including vascular endothelial growth factor receptor (VEGFR)-1, −2, −3, platelet-derived growth factor receptor (PDGFR)-a and -b [118]. A phase II study evaluated the activity of sunitinib in 109 NET patients including 41 with carcinoids of whom 14 were foregut including LCs and observed that sunitinib had antitumor activity in pancreatic forms, while its activity against carcinoid tumors could not be definitively determined [119].
The PAZONET trial [120] of pazopanib as a sequencing treatment in progressive metastatic NETs, including patients with LCs, observed a clinical benefit in 85% of patients treated with pazopanib.
Moreover, bevacizumab, an anti-VEGF monoclonal antibody, is also being evaluated for LCs. In the phase II study by Yao et al. [121] including 44 patients with advanced NETs of different origins (four were LCs), patients were randomized to either bevacizumab or pegylated interferon (IFN). The PFS rates after 18 weeks were 95% with bevacizumab versus 68% with pegylated IFN.
11 Immunotherapy
Immune checkpoint inhibitors have changed the clinical practice of different types of malignant neoplasms [122, 123]. The main evidence of the efficacy of immunotherapy in NETs is currently exclusively limited to poorly differentiated NETs of the skin (Merkel Cell Carcinomas), while results from clinical trials are disappointing for well-differentiated tumors [124]. In low-grade LCs, two immunotherapy agents have been evaluated in clinical studies so far, namely pembrolizumab and spartalizumab.
Pembrolizumab is a highly selective, humanized anti-programmed cell death protein 1 (PD-1) antibody. Antitumoral activity of pembrolizumab in carcinoids (lung and gut) and well-differentiated pancreatic NETs was initially evaluated in the phase 1b KEYNOTE-028 study at dose of 10 mg/kg, every 2 weeks. Out of 25 treated patients with LCs, three (12%) had objective response rate. Durations of response were 6.9, 9.2, and 11.1 months for the three LC responders, respectively. Stable disease rate was 60% (15 patients) [125]. The KEYNOTE-158 phase 2 basket study investigated the antitumor activity and safety of pembrolizumab (200 mg intravenously every 3 weeks) in different types of cancer, progressed after standard-of-care systemic therapy. One-hundred-seven patients with different types of NETs were enrolled and 14 of them were LCs. Median follow-up was 24.2 months (range: 0.6–33.4). ORR was 3.7%, with 0 complete responses and 4 PR (three pancreatic and one rectal). All the responses were in patients with PD-L1-negative tumors. Median PFS and median OS were 4.1 months and 24.2 months, respectively. Treatment-related grade 3–5 adverse events occurred in 21.5% of the patients. The authors concluded that pembrolizumab showed limited efficacy in pretreated advanced well-differentiated NETs [126].
Spartalizumab (PDR001), a high-affinity, humanized, anti-PD-1 antibody, was evaluated in a phase II, multicenter study in well- and poorly differentiated NETs. Primary endpoint was the ORR, and secondary endpoints included duration of response, biomarker analyses, and safety. In this study, PDR001 was administered at a flat dose of 400 mg, every 4 weeks. Of the 116 patients enrolled, 30 were thoracic NETs, 33 pancreatic NETs, 32 gastrointestinal NETs, and 21 GEP-NECs. After a median follow-up of 7.6 months in NET and 6 months in GEP-NEC, ORR was 7.4% in well-differentiated NETs and 4.8% in poorly differentiated NECs.
Patients with thoracic NETs had a higher ORR (20%) compared to GEP-NETs (1.5%) and GEP-NECs (5%). Most common grade 3/4 adverse events (>2.5%) were hypertension, dyspnea, anemia, abdominal and back pain. Interestingly, PD-L1 expression was generally low, with a higher proportion of PD-L1 expression in immune cells >1% in GEP-NECs (43%) compared to thoracic NETs (19%) and gastrointestinal NETs (33%). These preliminary results might suggest clinical activity of PDR001 in thoracic NETs [127].
12 Summary and Conclusions
LCs are rare tumors with an incidence of 2% and 0.2% for TCs and ACs, respectively. However, due to the improvement in screening and imaging techniques and increased disease awareness, LC incidence has increased during the last three decades [1, 5, 6]. Given the disease rarity and the scarcity of prospective studies, some controversies still exist on diagnosis and management of LCs. A multidisciplinary approach is always recommended.
Surgery with preservation of as much normal lung tissue as possible remains the gold standard in case of limited and resectable disease. Bronchoscopic excision might be also taken into account in those cases presenting with centrally located intraluminal LCs, even though further prospective studies are warranted to validate this more conservative approach.
For advanced or progressive disease, no standard treatment or therapeutic algorithm is currently available. In advanced metastatic LCs, medical therapy represents the milestone and SSA, everolimus, chemotherapy, and PRRT treatments constitute the therapeutic armamentarium in this setting, being generally reserved to well-differentiated tumors with low proliferative index and SSTR positivity.
The majority of the studies exploring the efficacy of SSA therapy for both symptom and tumor growth control are retrieved from studies on GEP-NETs; however, the LUNA trial is the only prospective study dedicated exclusively to LCs [117], which reported that long-acting pasireotide, everolimus, or combination therapy with both agents was associated with antitumor activity and an acceptable safety profile. Furthermore, the SPINET trial is ongoing, evaluating the efficacy and safety of lanreotide versus placebo for the treatment of well-differentiated, metastatic and/or unresectable nonfunctioning LCs.
PRRT with radiolabeled SSAs is an option in patients with well-differentiated low-intermediate grade NETs expressing high levels of SSTRs; however, only few, mainly retrospective, studies specifically dedicated to PRRT in LCs are available. Systemic chemotherapy is reserved for those cases of locally advanced or metastatic disease; however, the standard chemotherapy regimen to be recommended in clinical practice is still unclear, and the choice of chemotherapy should be made by taking into account the characteristics of both patient and tumor.
The role of targeted therapies in LCs remains still limited with the exception of the only approved drug (everolimus) in clinical practice. Everolimus represents a therapeutic option in patients with progressive disease with advanced LCs.
The RADIANT 4 study will likely have a major impact on clinical practice, especially for lung NETs [115]. Indeed, it was the first randomized study to specifically show that everolimus is significantly effective in patients with LCs. We believe that this study represents a major breakthrough, as, to date, there has not been any properly established treatment algorithm for LCs. In particular, we think that everolimus may be particularly suitable for patients with more aggressive and rapidly progressing disease such as those with ACs, in whom upfront treatment can be suggested. Moreover, data on second-line therapy with everolimus are even more grounded, also compared with those available for chemotherapy and PRRT, which are mostly derived from retrospective series or nonrandomized studies in a mixed population of patients with TCs and subjects with ACs.
Conversely, there is a lack of studies focused on the potential role of antiangiogenic agents in LCs. Finally, immune checkpoint inhibitors have shown promising results in different tumors, and the main evidence of the efficacy of immunotherapy in the neuroendocrine setting is only in Merkel Cell Carcinomas but not in well-differentiated forms, even though [124] in low-grade LCs, pembrolizumab and spartalizumab had shown preliminary promising results in terms of both efficacy and safety.
Further studies, specifically dedicated to LCs, are needed to draw more robust conclusions, particularly to better clarify the most adequate sequence and timing of systemic drugs in the management of this subgroup of rare neoplasms.
References
Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, Abdalla EK, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26(18):3063–72.
Rekhtman N. Neuroendocrine tumors of the lung an update. Arch Pathol Lab Med. 2010;134(11):1628–38.
Korse CM, Taal BG, van Velthuysen ML, Visse O. Incidence and survival of neuroendocrine tumours in the Netherlands according to histological grade: experience of two decades of cancer registry. Eur J Cancer. 2013;49(8):1975–83.
Naalsund A, Rostad H, Strom EH, Lund MB, Strand TE. Carcinoid lung tumors--incidence, treatment and outcomes: a population-based study. Eur J Cardiothorac Surg. 2011;39(4):565–9.
Caplin ME, Baudin E, Ferolla P, Filosso P, Garcia-Yuste M, Lim E, et al. Pulmonary neuroendocrine (carcinoid) tumors: European Neuroendocrine Tumor Society expert consensus and recommendations for best practice for typical and atypical pulmonary carcinoid. Ann Oncol. 2015;26:1604–20.
Petursdottir A, Sigurdardottir J, Fridriksson BM, Johnsen A, Isaksson HJ, Hardardottir H, et al. Pulmonary carcinoid tumours: incidence, histology, and surgical outcome. A population-based study. Gen Thorac Cardiovasc Surg. 2020;68(5):523–9.
Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 World Health Organization Classification of Lung Tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10:1243–60.
Faggiano A, Ferolla P, Grimaldi F, Campana D, Manzoni M, Davì MV, et al. Natural history of gastro-entero-pancreatic and thoracic neuroendocrine tumors. Data from a large prospective and retrospective Italian Epidemiological study: the NET MANAGEMENT study. J Endocrinol Investig. 2012;35:817–23.
Hassan MM, Phan A, Li D, Dagohoy CG, Leary C, Yao JC. Risk factors associated with neuroendocrine tumors: a U.S.-based case-control study. Int J Cancer. 2008;123:867–73.
Filosso PL, Rena O, Donati G, Casadio C, Ruffini E, Papalia E, et al. Bronchial carcinoid tumors: surgical management and long-term outcome. J Thorac Cardiovasc Surg. 2002;123:303–9.
Zuetenhorst JM, Taal BG. Metastatic carcinoid tumors: a clinical review. Oncologist. 2005;10:123–31.
Ferguson MK, Landreneau RJ, Hazelrigg SR, Altorki NK, Naunheim KS, Zwischenberger JB, et al. Long-term outcome after resection for bronchial carcinoid tumors. Eur J Cardiothorac Surg. 2000;18:156–61.
Pusceddu S, Lo Russo G, Macerelli M, Proto C, Vitali M, Signorelli D, et al. Diagnosis and management of typical and atypical lung carcinoids. Crit Rev Oncol Hematol. 2016;100:167–76.
Filosso PL, Oliaro A, Ruffini E, Bora G, Lyberis P, Asioli S, et al. Outcome and prognostic factors in bronchial carcinoids: a single-center experience. J Thorac Oncol. 2013;8(10):1282–8.
Skuladottir H, Hirsch FR, Hansen HH, Olsen JH. Pulmonary neuroendocrine tumors: incidence and prognosis of histological subtypes. A population-based study in Denmark. Lung Cancer. 2002;37(2):127–35.
Cardillo G, Sera F, Di Martino M, Graziano P, Giunti R, Carbone L, et al. Bronchial carcinoid tumors: nodal status and long-term survival after resection. Ann Thorac Surg. 2004;77(5):1781–5.
Raz DJ, Nelson RA, Grannis FW, Kim JY. Natural history of typical pulmonary carcinoid tumors: a comparison of nonsurgical and surgical treatment. Chest. 2015;147(4):1111–7.
De Giorgi U, Fanini F, Amadori D, Cancellieri A, Fiorentini G, Poletti V, et al. Tumorlets in familial history of bronchopulmonary carcinoid. J Thorac Oncol. 2011;6(9):1613–4.
Oliveira AM, Tazelaar HD, Wentzlaff KA, Kosugi NS, Hai N, Benson A, et al. Familial pulmonary carcinoid tumors. Cancer. 2001;91:2104–9.
Leotlela PD, Jauch A, Holtgreve-Grez H, Thakker RV. Genetics of neuroendocrine and carcinoid tumors. Endocr Relat Cancer. 2003;10:437–50.
Matsuda KM, Nobrega R, Quezado M, Schrump DS, Filie AC. Melanocytic bronchopulmonary carcinoid tumor in a patient with multiple endocrine neoplasia syndrome type 1: a case report with emphasis on intraoperative cytological findings. Diagn Cytopathol. 2010;38:669–74.
Sachithanandan N, Harle RA, Burgess JR. Bronchopulmonary carcinoid in multiple endocrine neoplasia type 1. Cancer. 2005;103:509–15.
Ferolla P, Daddi N, Urbani M, Semeraro A, Ribacchi R, Giovenali P, et al. Tumorlets, multicentric carcinoids, lymphnodal metastases, and long-term behavior in bronchial carcinoids. J Thorac Oncol. 2009;4:383–7.
Travis WD. Advances in neuroendocrine lung tumors. Ann Oncol. 2010;21(Suppl 7):vii65–71.
Wirtschafter E, Walts AE, Liu ST, Marchevsky AM. Diffuse idiopathic pulmonary neuroendocrine cell hyperplasia of the lung (DIPNECH): current best evidence. Lung. 2015;193(5):659–67.
Filosso PL, Ferolla P, Guerrera F, Ruffini E, Travis WD, Rossi G, et al. Multidisciplinary management of advanced lung neuroendocrine tumors. JThorac Dis. 2015;7(Suppl 2):S163–71.
Volante M, Gatti G, Papotti M. Classification of lung neuroendocrine tumors: lights and shadows. Endocrine. 2015;50(2):315–9.
Pelosi G, Papotti M, Rindi G, Scarpa A. Unraveling tumor grading and genomic landscape in lung neuroendocrine tumors. Endocr Pathol. 2014;25:151–64.
Öberg K, Hellman P, Ferolla P, Papotti M. Neuroendocrine bronchial and thymic tumors: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(suppl 7):vii120–3.
Halperin DM, Shen C, Dasari A, Xu Y, Chu Y, Zhou S, et al. Frequency of carcinoid syndrome at neuroendocrine tumour diagnosis: a population-based study. Lancet Oncol. 2017;18:525–34.
Alexandraki KI, Grossman AB. The ectopic ACTH syndrome. Rev Endocr Metab Disord. 2010;11(2):117–26.
Neary NM, Lopez-Chavez A, Abel BS, Boyce AM, Schaub N, Kwong K, et al. Neuroendocrine ACTH-producing tumor of the thymus--experience with 12 patients over 25 years. J Clin Endocrinol Metab. 2012;97(7):2223–30.
Meisinger QC, Klein JS, Butnor KJ, Gentchos G, Leavitt BJ. CT features of peripheral pulmonary carcinoid tumors. AJR Am J Roentgenol. 2011;197(5):1073–80.
Righi L, Volante M, Tavaglione V, Billè A, Daniele L, Angusti T, et al. Somatostatin receptor tissue distribution in lung neuroendocrine tumours: a clinicopathologic and immunohistochemical study of 218 clinically aggressive cases. Ann Oncol. 2010;21(3):548–55.
Fisseler-Eckhoff A, Demes M. Neuroendocrine tumors of the lung. Cancers (Basel). 2012;4:777–98.
Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9(1):61–72.
Mojtahedi A, Thamake S, Tworowska I, Ranganathan D, Delpassand ES. The value of (68)Ga-DOTATATE PET/CT in diagnosis and management of neuroendocrine tumors compared to currentFDA approved imaging modalities: a review of literature. Am J Nucl Med Mol Imaging. 2014;4(5):426–34. eCollection 2014
Reubi JC, Schär JC, Waser B, Wenger S, Heppeler A, Schmitt JS, et al. Affinity profiles for human somatostatin receptor subtypes SST1-SST5 of somatostatin radiotracers selected for scintigraphic and radiotherapeutic use. Eur J Nucl Med Mol Imaging. 2000;27:273–82.
Haug AR, Cindea-Drimus R, Auernhammer CJ, Reincke M, Wangler B, Uebleis C, et al. The role of 68Ga-DOTATATE PET/CT in suspected neuroendocrinen tumors. J Nucl Med. 2012;53:1686–92.
Haug AR, Cindea-Drimus R, Auernhammer CJ, Reincke M, Beuschlein F, Wängler B, et al. Neuroendocrine tumor recurrence: diagnosis with 68Ga-DOTATATE PET/CT. Radiology. 2014;270:517–25.
Srirajaskanthan R, Kayani I, Quigley AM, Soh J, Caplin ME, Bomanji J. The role of 68Ga-DOTATATE PET in patients with neuroendocrine tumors and negative or equivocal findings on 111In-DTPA-octreotide scintigraphy. J Nucl Med. 2010;51(6):875–82.
Krausz Y, Freedman N, Rubinstein R, Lavie E, Orevi M, Tshori S, et al. 68Ga-DOTA-NOC PET/CT imaging of neuroendocrine tumors: comparison with 111In-DTPA-octreotide (OctreoScan®). Mol Imaging Biol. 2011;13:583–93.
Hörsch D, Schmid KW, Anlauf M, Darwiche K, Denecke T, Baum RP, et al. Neuroendocrine tumors of the bronchopulmonary system (typical and atypical carcinoid Tumors): current strategies in diagnosis and treatment. Conclusions of an expert meeting February 2011 in Weimar, Germany. Oncol Res Treat. 2014;37:266–76.
Abgral R, Leboulleux S, Déandreis D, Aupérin A, Lumbroso J, Dromain C, et al. Performance of (18) fluorodeoxyglucose-positron emission tomography and somatostatin receptor scintigraphy for high Ki67 (≥10%) well-differentiated endocrine carcinoma staging. J Clin Endocrinol Metab. 2011;96(3):665–71.
Bongiovanni A, Recine F, Riva N, Foca F, Liverani C, Mercatali L, et al. Outcome analysis of first-line somatostatin analog treatment in metastatic pulmonary neuroendocrine tumors and prognostic significance of 18FDG-PET/CT. Clin Lung Cancer. 2017;18(4):415–20.
Aydin E, Yazici U, Gulgosteren M, Agackiran Y, Kaya S, Gulhan E, et al. Longterm outcomes and prognostic factors of patients with surgically treated pulmonary carcinoid: our institutional experience with 104 patients. Eur J Cardiothorac Surg. 2011;39:549–54.
Cañizares MA, Matilla JM, Cueto A, Algar J, Muguruza I, Moreno-Mata N, et al. Atypical carcinoid tumours of the lung: prognostic factors and patterns of recurrence. Thorax. 2014;69:648–53.
Filosso PL, Guerrera F, Evangelista A, Welter S, Thomas P, Casado PM, et al. Prognostic model of survival for typical bronchial carcinoid tumours: analysis of 1109 patients on behalf of the European Association of Thoracic Surgeons (ESTS) Neuroendocrine Tumours Working Group. Eur J Cardiothorac Surg. 2015;48:441–7. discussion 447
Okoye CC, Jablons DM, Jahan TM, Kukreja J, Cardozo S, Yom SS. Divergent management strategies for typical versus atypical carcinoid tumors of the thoracic cavity. Am J Clin Oncol. 2014;37:350–5.
Fox M, Van Berkel V, Bousamra M, Sloan S, Martin RCG. Surgical management of pulmonary carcinoid tumors: sublobar resection versus lobectomy. Am J Surg. 2013;205(2):200–8.
Yendamuri S, Gold D, Jayaprakash V, Dexter E, Nwogu C, Demmy T. Is sublobar resection sufficient for carcinoid tumors? Ann Thorac Surg. 2011;92(5):1774–8.
Afoke J, Tan C, Hunt I, Zakkar M. Is sublobar resection equivalent to lobectomy for surgical management of peripheral carcinoid? Interact Cardiovasc Thorac Surg. 2013;16(6):858–63.
Detterbeck FC, Lewis SZ, Diekemper R, Addrizzo-Harris D, Alberts WM. Executive summary: diagnosis and management of lung cancer, 3rd ed. American College of Chest Physicians evidence-based clinical practice guidelines. Chest. 2013;143(5 Suppl):7S–37S.
Mei J, Guo C, Xia L, Liao H, Pu Q, Ma L, et al. Long-term survival outcomes of video-assisted thoracic surgery lobectomy for stage I-II non-small cell lung cancer are more favorable than thoracotomy: a propensity score-matched analysis from a high-volume center in China. Transl Lung Cancer Res. 2019;8(2):155–66.
Yu D, Han Y, Zhou S, Song X, Li Y, Xiao N, et al. Video-assisted thoracic bronchial sleeve lobectomy with bronchoplasty for treatment of lung cancer confined to a single lung lobe: a case series of Chinese patients. J Cardiothorac Surg. 2014;9:67.
Lim E, Yap YK, De Stavola BL, Nicholson AG, Goldstraw P. The impact of stage and cell type on the prognosis of pulmonary neuroendocrine tumors. J Thorac Cardiovasc Surg. 2005;130:969–72.
Travis WD, Rush W, Flieder DB, Falk R, Fleming MV, Gal AA, et al. Survival analysis of 200 pulmonary neuroendocrine tumors with clarification of criteria for atypical carcinoid and its separation from typical carcinoid. Am J Surg Pathol. 1998;22:934–44.
Wolin EM. Advances in the diagnosis and management of well-differentiated and intermediate-differentiated neuroendocrine tumors of the lung. Chest. 2017;151(5):1141–6.
Wurzt A, Benhamed L, Conti M, Bouchindhomme B, Porte H. Results of systematic nodal dissection in typical and atypical carcinoid tumors of the lung. J Thorac Oncol. 2009;4(3):388–94.
Detterbeck FC. Management of carcinoid tumors. Ann Thorac Surg. 2010;89:998–1005.
El Jamal M, Nicholson AG, Goldstraw P. The feasibility of conservative resection for carcinoid tumors: is pneumonectomy ever necessary for uncomplicated cases? Eur J Cardiothorac Surg. 2000;18:301–6.
Anile M, Diso D, Rendina EA, Venuta F. Bronchoplastic procedures for carcinoid tumors. Thorac Surg Clin. 2014;24(3):299–303.
Venuta F, Ciccone AM, Anile M, Ibrahim M, De Giacomo T, Coloni GF, et al. Reconstruction of the pulmonary artery for lung cancer: long term results. J Thorac Cardiovasc Surg. 2009;138:1185–91.
Okada M, Nishio W, Sakamoto T, Uchino K, Yuki T, Nakagawa A, et al. Sleeve segmentectomy for non small cell lung carcinoma. J Thorac Cardiovasc Surg. 2004;128:420–4.
Lemaitre J, Mansour Z, Kochetkova EA, Koriche C, Ducrocq X, Wihlm JM, et al. Bronchoplastic lobectomy: do early results depend on the underlying pathology? A comparison between typical carcinoids and primary lung cancer. Eur J Cardiothorac Surg. 2006;30:168–71.
Phan AT, Oberg K, Choi J, Harrison LH, Hassan MM, Strosberg JR, et al. NANETS consensus guideline for the diagnosis and management of neuroendocrine tumors well-differentiated neuroendocrine tumors of the thorax (includes lung and thymus). Pancreas. 2010;39:784–98.
Cavaliere S, Foccoli P, Toninelli C. Curative bronchoscopic laser therapy for surgically resectable tracheobronchial tumors: personal experience. J Bronchol. 2002;9(2):90–5.
Reuling EMBP, Dickhoff C, Plaisier PW, Coupé VMH, Mazairac AHA, Lely RJ, et al. Endobronchial treatment for bronchial carcinoid: patient selection and predictors of outcome. Respiration. 2018;95(4):220–7.
Dalar L, Ozdemir C, Abul Y, Sokucu SN, Karasulu L, Urer HN, et al. Endobronchial treatment of carcinoid tumors of the lung. Thorac Cardiovasc Surg. 2016;64(2):166–71.
Brokx HA, Paul MA, Postmus PE, Sutedja TG. Long-term follow-up after firstline bronchoscopic therapy in patients with bronchial carcinoids. Thorax. 2015;70(5):468–72.
Orino K, Kawai H, Ogawa J. Bronchoscopic treatment with argon plasma coagulation for recurrent typical carcinoids: report of a case. Anticancer Res. 2004;24(6):4073–7.
Venuta F, Rendina EA, De Giacomo T, Mercadante E, Francioni F, Pugliese F, et al. Nd:YAG laser resection of lung cancer invading the airway as a bridge to surgery and palliative treatment. Ann Thorac Surg. 2002;74:995–8.
Neuberger M, Hapfelmeier A, Schmidt M, Gesierich W, Reichenberger F, Morresi-Hauf A, et al. Carcinoid tumours of the lung and the ‘PEPPS’ approach: evaluation of preoperative bronchoscopic tumour debulking as preparation for subsequent parenchyma-sparing surgery. BMJ Open Respir Res. 2015;2(1):e000090.
Reuling EMBP, Dickhoff C, Plaisier PW, Bonjer HJ, Daniels JMA. Endobronchial and surgical treatment of pulmonary carcinoid tumors: a systematic literature review. Lung Cancer. 2019;134:85–95.
Oberg K, Ferone D, Kaltsas G, Knigge UP, Taal B, Plöckinger U, et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: biotherapy. Neuroendocrinology. 2009;90:209–13.
Modlin IM, Pavel M, Kidd M, Gustafsson BI. Review article: somatostatin analogues in the treatment of gastroenteropancreatic neuroendocrine (carcinoid) tumours. Aliment Pharmacol Ther. 2010;31:169–88.
Oberg K, Kvols L, Caplin M, Delle Fave G, de Herder W, Rindi G, et al. Consensus report on the use of somatostatin analogs for the management of neuroendocrine tumors of the gastroenteropancreatic system. Ann Oncol. 2004;15:966–73.
Janson ET, Oberg K. Long-term management of the carcinoid syndrome. Treatment with octreotide alone and in combination with alpha- interferon. Acta Oncol. 1993;32:225–9.
Kvols LK, Moertel CG, O’Connell MJ, Schutt AJ, Rubin J, Hahn RG. Treatment of the malignant carcinoid syndrome. Evaluation of a long-acting somatostatin analogue. N Engl J Med. 1986;315:663–6.
Ruszniewski P, Ish-Shalom S, Wymenga M, O’Toole D, Arnold R, Tomassetti P, et al. Rapid and sustained relief from the symptoms of carcinoid syndrome: results from an open 6-month study of the 28-day prolonged-release formulation of lanreotide. Neuroendocrinology. 2004;80:244–51.
Ruszniewski P, Valle JW, Lombard-Bohas C, Cuthbertson DJ, Perros P, Holubec L, et al. Patient-reported outcomes with lanreotide autogel/depot for carcinoid syndrome: an international observational study. Dig Liver Dis. 2016;48:552–8.
Vinik AI, Wolin EM, Liyanage N, Gomez-Panzani E, Fisher GA, ELECT Study Group. Evaluation of lanreotide depot/autogel efficacy and safety as a carcinoid syndrome treatment (elect): a randomized, double-blind. Placebo-Controll Trial Endocr Pract. 2016;22:1068–80.
Filosso PL, Ruffini E, Oliaro A, Papalia E, Donati G, Rena O. Long-term survival of atypical bronchial carcinoids with liver metastases, treated with octreotide. Eur J Cardiothorac Surg. 2002;21:913–7.
Rinke A, Müller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27:4656–63.
Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláčková E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. 2014;371(3):224–33.
Shah MH, Goldner WS, Benson AB, Bergsland E, Berlin JD, Blaszkowsky LS et al. Neuroendocrine and adrenal tumors. NCCN Clinical Practice Guidelines in Oncology. 2019.
Hörscha D, Schmid KW, Anlauf M, Darwiche K, Denecke T, Baum RP, et al. Neuroendocrine tumors of the bronchopulmonary system (typical and atypical carcinoid tumors): current strategies in diagnosis and treatment conclusions of an expert meeting February 2011 in Weimar, Germany. Oncol Res Treat. 2014;37:266–76.
Bodei L, Cremonesi M, Kidd M, Grana CM, Severi S, Modlin IM, et al. Peptide receptor radionuclide therapy for advanced neuroendocrine tumors. Thorac Surg Clin. 2014;24:333–49.
Waldherr C, Pless M, Maecke HR, Haldemann A, Mueller-Brand J. The clinical value of [90Y-DOTA]-D-Phe1-Tyr3-octreotide (90Y-DOTATOC) in the treatment of neuroendocrine tumours: a clinical phase II study. Ann Oncol. 2001;12:941–5.
Mariniello A, Bodei L, Tinelli C, Baio SM, Gilardi L, Colandrea M, et al. Long-term results of PRRT in advanced bronchopulmonary carcinoid. Eur J Nucl Med Mol Imaging. 2016;43(3):441–52.
Imhof A, Brunner P, Marincek N, Briel M, Schindler C, Rasch H, et al. Response, survival, and long-term toxicity after therapy with the radiolabeled somatostatin analogue [90YDOTA]-TOC in metastasized neuroendocrine cancers. J Clin Oncol. 2011;29(17):2416–23.
Bodei L, Cremonesi M, Grana C, Rocca P, Bartolomei M, Chinol M, et al. Receptor radionuclide therapy with 90Y-[DOTA]0-Tyr3-octreotide (90Y-DOTATOC) in neuroendocrine tumours. Eur J Nucl Med Mol Imaging. 2004;31:1038–46.
Ianniello A, Sansovini M, Severi S, Nicolini S, Grana CM, Massri K, et al. Peptide receptor radionuclide therapy with 177Lu-DOTATATE in advanced bronchial carcinoids: prognostic role of thyroid transcription factor 1 and 18F-FDG PET. Eur J Nucl Med Mol Imaging. 2016;43(6):1040–6.
van Essen M, Krenning EP, Bakker WH, de Herder WW, van Aken MO, Kwekkeboom DJ. Peptide receptor radionuclide therapy with 177Lu-octreotate in patients with foregut carcinoid tumours of bronchial, gastric and thymic origin. Eur J Nucl Med Mol Imaging. 2007;34:1219–27.
Brabander T, van der Zwan WA, Teunissen JJM, Kam BLR, Feelders RA, de Herder WW, et al. Long-term efficacy, survival, and safety of (177)Lu-DOTA(0),Tyr(3)octreotate in patients with gastroenteropancreatic and bronchial neuroendocrine tumors. Clin Cancer Res. 2017;23(16):4617–24.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of 177Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376:125–35.
Lim E, Goldstraw P, Nicholson AG, Travis WD, Jett JR, Ferolla P, et al. Proceedings of the IASLC international workshop on advances in pulmonary neuroendocrine tumors 2007. J Thorac Oncol. 2008;3(10):1194–201.
Granberg D, Eriksson B, Wilander E, Grimfjärd P, Fjällskog ML, Oberg K, et al. Experience in treatment of metastatic pulmonary carcinoid tumors. Ann Oncol. 2001;12(10):1383–91.
Turner NC, Strauss SJ, Sarker D, Gillmore R, Kirkwood A, Hackshaw A, et al. Chemotherapy with 5-fluorouracil, cisplatin and streptozotocin for neuroendocrine tumours. Br J Cancer. 2010;102(7):1106–12.
Bajetta E, Catena L, Procopio G, De Dosso S, Bichisao E, Ferrari L, et al. Are capecitabine and oxaliplatin (XELOX) suitable treatments for progressing low-grade and high-grade neuroendocrine tumours? Cancer Chemother Pharmacol. 2007;59:637–42.
Kvols LK, Brendtro KL, North American Neuroendocrine Tumor Society (NANETS). The North American Neuroendocrine Tumor Society (NANETS) guidelines: mission, goals, and process. Pancreas. 2010;39(6):705–6.
Wirth LJ, Carter MR, Jänne PA, Johnson BE. Outcome of patients with pulmonary carcinoid tumors receiving chemotherapy or chemoradiotherapy. Lung Cancer. 2004;44:213–20.
Chan JA, Stuart K, Earle CC, Clark JW, Bhargava P, Miksad R, et al. Prospective study of bevacizumab plus temozolomide in patients with advanced neuroendocrine tumors. J Clin Oncol. 2012;30(24):2963–8.
Pavel M, Grossman A, Arnold R, Perren A, Kaltsas G, Steinmüller T, et al. ENETS consensus guidelines for the management of brain, cardiac and ovarian metastases from neuroendocrine tumors. Neuroendocrinology. 2010;91:326–32.
Koumarianou A, Kaltsas G, Kulke MH, Oberg K, Strosberg JR, Spada F, et al. Temozolomide in advanced neuroendocrine neoplasms: pharmacological and clinical aspects. Neuroendocrinology. 2015;101(4):274–88.
Ekeblad S, Sundin A, Janson ET, Welin S, Granberg D, Kindmark H, et al. Temozolomide as monotherapy is effective in treatment of advanced malignant neuroendocrine tumors. Clin Cancer Res. 2007;13:2986–91.
Crona J, Fanola I, Lindholm DP, Antonodimitrakis P, Öberg K, Eriksson B, et al. Effect of temozolomide in patients with metastatic bronchial carcinoids. Neuroendocrinology. 2013;98:151–5.
Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009;15:338–45.
Campana D, Walter T, Pusceddu S, Gelsomino F, Graillot E, Prinzi N, et al. Correlation between MGMT promoter methylation and response to temozolomide-based therapy in neuroendocrine neoplasms: an observational retrospective multicenter study. Endocrine. 2018;60(3):490–8.
Walter T, Planchard D, Bouledrak K, Scoazec JY, Souquet PJ, Dussol AS, et al. Evaluation of the combination of oxaliplatin and 5-fluorouracil or gemcitabine in patients with sporadic metastatic pulmonary carcinoid tumors. Lung Cancer. 2016;96:68–73.
Spada F, Antonuzzo L, Marconcini R, Radice D, Antonuzzo A, Ricci S, et al. Oxaliplatin-based chemotherapy in advanced neuroendocrine tumors: clinical outcomes and preliminary correlation with biological factors. Neuroendocrinology. 2016;103(6):806–14.
Pavel ME, Hainsworth JD, Baudin E, Peeters M, Hörsch D, Winkler RE, et al. RADIANT-2 Study Group. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378(9808):2005–12.
Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastro-intestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2016;387(10022):968–77.
Fazio N, Granberg D, Grossman A, Saletan S, Klimovsky J, Panneerselvam A, et al. Everolimus plus octreotide long-acting repeatable in patients with advanced lung neuroendocrine tumors: analysis of the phase 3, randomized, placebo-controlled RADIANT-2 study. Chest. 2013;143(4):955–62.
Fazio N, Buzzoni R, Delle Fave G, Tesselaar ME, Wolin E, Van Cutsem E, et al. Everolimus in advanced, progressive, well-differentiated, non-functional neuroendocrine tumors: RADIANT-4 lung subgroup analysis. Cancer Sci. 2018;109(1):174–81.
Cives M, Strosberg J. The expanding role of somatostatin analogs in gastroenteropancreatic and lung neuroendocrine tumors. Drugs. 2015;75:847–58.
Ferolla P, Brizzi MP, Meyer T, Mansoor W, Mazieres J, Do Cao C, et al. Efficacy and safety of long-acting pasireotide or everolimus alone or in combination in patients with advanced carcinoids of the lung and thymus (LUNA): an open-label, multicentre, randomised, phase 2 trial. Lancet Oncol. 2017;18(12):1652–64.
Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501–13.
Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26(20):3403–10.
Grande E, Capdevila J, Castellano D, Teulé A, Durán I, Fuster J, et al. Pazopanib in pretreated advanced neuroendocrine tumors: a phase II, open-label trial of the Spanish Task Force Group for Neuroendocrine Tumors (GETNE). Ann Oncol. 2015;26(9):1987–93.
Yao JC, Phan A, Hoff PM, Chen HX, Charnsangavej C, Yeung SC, et al. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26(8):1316–23.
Califano R, Kerr K, Morgan RD, Lo Russo G, Garassino M, Morgillo F, et al. Immune checkpoint blockade: a new era for non-small cell lung cancer. Curr Oncol Rep. 2016;18(9):59.
Onitilo AA, Wittig JA. Principles of immunotherapy in melanoma. Surg Clin North Am. 2020;100(1):161–73.
Femia D, Prinzi N, Anichini A, Mortarini R, Nichetti F, Corti F, et al. Treatment of advanced Merkel cell carcinoma: current therapeutic options and novel immunotherapy approaches. Target Oncol. 2018;13(5):567–82.
Mehnert JM, Rugo HS, O’Neil BH, Santoro A, Schellens JHM, Cohen RB, et al. Pembrolizumab for patients with PD-L1-positive advanced carcinoid or pancreatic neuroendocrine tumors: results from the KEYNOTE-028 study. Ann Oncol. 2017;28(Suppl_5):v142–57. https://doi.org/10.1093/annonc/mdx36.
Strosberg JR, Mizuno N, Doi T, Grande E, Delord JP, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced neuroendocrine tumors: results from the phase 2 KEYNOTE-158 study. Clin Cancer Res. 2020;26(9):2124–30.
Yao JC, Strosberg J, Fazio N, Pavel ME, Ruszniewski P, Bergsland E, et al. Activity & safety of spartalizumab (PDR001) in patients (pts) with advanced neuroendocrine tumors (NET) of gastro-intestinal (GI), or thoracic (T) origin & gastroenteropancreatic neuroendocrine carcinoma (GEP NEC) who have progressed on prior treatment (TX). Ann Oncol. 2018;29(suppl_8):viii467–78. https://doi.org/10.1093/annonc/mdy293.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Prinzi, N., Rossi, R.E., Leuzzi, G., Pusceddu, S. (2021). NETs of the Lung. In: Beretta, G., Berruti, A., Bombardieri, E., Fazio, N., Goletti, O. (eds) Neuroendocrine Neoplasia Management. Springer, Cham. https://doi.org/10.1007/978-3-030-72830-4_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-72830-4_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-72829-8
Online ISBN: 978-3-030-72830-4
eBook Packages: MedicineMedicine (R0)