
Abstract
Recycling electronic scrap is a significant source of rare earth metals. Whereas traditional recycling routes for some electronic scrap emphasize the recovery of silver and gold, value can be attained by recovering of rare earth elements from unique feed streams. This paper describes a hydrometallurgical process for the recovery of rare earth elements from hard disk drives using HCl as a re-usable extraction medium. The mixture was selectively leached using HCl to remove the magnet alloy coating from shredded hard disk drives. The dissolved rare earth elements were precipitated using sodium sulfate, recovered as the sodium double salt, and subsequentially converted to hydroxides. The recovery of rare earth elements is consistent with amounts predicted using a thermodynamic model based on the MSE (Mixed-Solvent Electrolyte) framework of precipitated double salts. The effect of HCl concentration was measured upon the magnet dissolution rate. In addition, the leaching rates for steel were evaluated and found to be three orders of magnitude lower than the magnet alloy. An automated system was used to control leachate pH. The magnet and steel dissolution rate were examined for various HCl concentrations. The recovery of rare earth hydroxides was over 80%.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
Introduction
Electronic waste (e-waste) or waste electrical and electronic equipment (WEEE) is a growing feed stream of materials which were not recycled until the EU WEEE legislative directive was approved in 2002 [1, 2]. The challenges to e-waste recycling include the fact that manual separation and shredding are labor and equipment intensive, and there are few economically relevant recycling strategies for many types of e-waste [3]. As of 2016, 20% of e-waste was recycled worldwide in documented, proper manner, but rest is either dumped or shipped to other companies where it is recycled in a crude manner. In addition, rare earth elements (REEs) are not recovered to any significant extent [4]. In fact, less than 5% of rare metals were recycled from WEEE as of 2019 [5]. REEs are contained in displays, speakers, cell phones, motors, hard disk drives (HDDs), voice coil actuators, and other materials. Alternatively, according to Adamas Intelligence, the demand for rare earth oxides used in electric cars is predicted to increase by fivefold by 2030 [6]. The United States Department of Energy (DOE) considered neodymium (Nd), dysprosium (Dy), europium (Eu), terbium (Tb), and yttrium (Y) as REE critical materials [7]. The overall crustal abundance of the rare earth is similar to copper (Cu), but due to the technical challenges associated with separation, the enrichment factor required for a rare earth mine must be significantly higher than Cu [8]. For example, the cutoff grade for Cu might be 0.6%, but the cutoff grade for rare earth mine at Mt. Weld Mine is 4% [9,10,11]. When the most valuable elements are dilute, additional ore must be processed to meet the demand for those dilute metals needed for clean energy. In particular, the magnet REEs (Nd, Dy, Pr) have increasing importance as they are required for clean energy technologies, such as electric motors and wind turbines which justify the criticality. Fortunately, REEs are found in HDD magnets, particularly Nd in Nd2Fe14B (Nd-Fe-B) magnets [12]. Reviews of REE reserves, suppliers, uses, and potential recycling sources are available [12, 13]. The recovery of REEs from e-waste could meet future criticality and bolster economic growth [14]. If more recycled metals are recovered for value, less mining is required, and waste is minimized.
Because few recycling methods for REEs have reached the commercial scale and most secondary sources of REEs are not being recycled, there is an increased interest in recycling research. The literature has outlined potential REE recycling feedstocks, existing recycling technologies and future needs [15,16,17]. A review of REE recycling specific to Nd-Fe-B magnets was published, including both pyrometallurgical and hydrometallurgical processes [15]. However, a more recent review outlines more avant-garde methods for treating e-waste for REEs from HDD magnets [18]. Given this recent review, the summary below will focus on the studies most relevant to this paper.
Hydrometallurgical methods for the extraction of REEs are supported by their easy dissolution by mineral acids. The dissolution of scrap as the Nd-Fe-B magnets using sulfuric acid dissolution and subsequent pH adjustment to capture was described in a patent by Lyman in 1992 [19]. Nd-Fe-B magnet alloys react quickly with protons (H+) with copious evolution of H2 gas [20]. Due to reactivity, sintered magnets are typically coated with nickel (Ni) or layers of Ni and Cu. Selection of the acid for dissolution (H2SO4, HCl, etc.) depends on the downstream recovery processes [21]. For example, downstream solvent extraction methods are supported by hydrochloric and nitric acid leaching and selective precipitation methods prefer sulfuric acid leaching [15].
To recover REEs from solution, selective precipitation reactions have frequently been reported to isolate dissolved REEs. All REEs form sparingly soluble trihydroxides in basic medium [22], which can be used to precipitate REEs. However, a pH shift to approximately pH 6 is higher than precipitation of co-dissolved metals such as iron (Fe), nickel (Ni), and zinc (Zn). The REEs are highly soluble in chloride and nitrate while having low solubility in sulfate which is pH dependent [23]. From moderately concentrated H2SO4, a slight increase in pH to a value slightly over 1 will precipitate the REEs as double salt solids ((RE)2(SO4)3·Na2SO4·xH2O using NaOH). As this pH is below that for precipitation of Fe [22], this method was used to capture REEs from nickel-metal hydride (Ni-MH) batteries [24, 25]. Pietrelli et al. used 2 M H2SO4 and obtained over 90% REE release, followed by pH adjustment to 1.5 using NaOH to recover over 70% of REE content. The REE recovery from magnet scrap (swarf) using the H2SO4-NaOH route have also been reported [26]. Phosphoric acid dissolution followed by pH increase has been used to precipitate REEs from Ni-MH battery leachate solutions [26].
Recently, this group has focused on a comprehensive recovery process that can produce different value streams from e-waste [20, 27]. An economic evaluation of the comprehensive process has concluded that a cost-efficient recovery of REEs from electronic scrap can only be achieved if other metals are recovered for value [14]. Specifically, for the recovery of REEs, the ferrous fraction (magnetically separated) of mobile phones was treated using H2SO4 followed by pH increase to separate REEs [27]. While use of H2SO4 is efficient for dissolution and recovery of REEs, the presence of sulfate and sodium (Na) in the extraction media could complicate material separation if the leaching solution was intended to be re-used.
This paper describes a modification to the chemistry used in that work, where an HCl-based system is employed to dissolve REEs from mixed magnet-steel mixtures followed Na2SO4 addition to precipitate as the sodium double salt. The resultant double salt is then converted to rare earth hydroxide (RE(OH)3) using NaOH. While previous work included data on the recovery of REEs, this work is dedicated to the REE recovery process using HCl-based dissolution which includes a more thorough assessment of recovery in comparison with the model predictions of recovery, additional purification of the powder product, and an assessment of the possibility of leachate re-use.
Materials and Methods
Materials
Leaching solutions were prepared in deionized water (18 MΩ-cm) using ACS grade or better reagents. To examine the dissolution process, uncoated “Nd” (Nd-Fe-B) disk magnets (1.3 cm diameter, 0.3 cm height) were used (McMaster Carr). Shredded HDD material was obtained from Oak Ridge National Laboratory (ORNL). This material was hand sorted using a coated Nd-Fe-B magnet to remove steel and magnet fragments from aluminum (Al), plastic, and circuit board material. This ferrous material was used for recovery experiments. Magnetic particles and chunks were magnetically attached to steel or tightly bound together onto steel as described previously [15]. Due to the small quantity of non-homogenous scrap attained from Oak Ridge National Laboratory, the team was unable to obtain a statistically relevant analysis of the feed material.
Magnet and Steel Dissolution Studies
Steel coupons (1.6 × 1.6 cm) were cut from C-1100 shim steel. Air or N2 gas were purged at 0.02 m3/h into an open beaker at ambient temperature (20–22 °C). Experiments were performed for 6–24 h in 100 mL of 1 M HCl. Uncoated magnets were exposed to a range of diluted HCl solutions at ambient temperature (20–22 °C). A solution volume of 250 mL was chosen to avoid significant concentration change during the test. Stirring was not employed, although bubbles of H2 produced from the reaction agitated the solution. Magnets were carefully balanced at an angle against the side of the beaker to minimize contact area. Weight of the magnets was recorded three times before and after exposure. Using the weight change, known density (7.4 g/cm3), surface area (3.88 cm2), and exposure time (1 h), the uniform dissolution rate was calculated. A uniform rate was used due to the lack of evidence for localized attack.
Leaching System
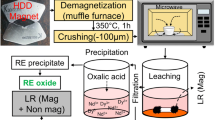
A diagram of the system used to process material is shown in Fig. 1. The separated HDD material was placed in the reaction column, and a pump was used to continuously flow the acid solution through the vessel containing the material. A pH probe was inserted in-line to inform a pH controller to release acid as needed. Dissolution was performed at normal room temperature (18–22 °C). Solution was pumped through the reaction column at approximately 250 mL/min using a diaphragm pump. While the system was automated, operation occurred under supervision during normal work hours as a precaution. The pH controller operated a low flow peristaltic pump to deliver 5 M HCl to the leach reservoir. The pH was controlled to maintain a value below 0. The addition of acid diluted the leach solution over time. Solution was removed periodically and precipitated individually.

Diagram of the system to perform REE dissolution. (Color figure online)
Recovery of Dissolved REEs
After dissolution, REEs were precipitated from the leachate as sulfate double salts (NaRE(SO4)2·xH2O) by adding solid Na2SO4. Samples of the leachate, before and after REE precipitation, were taken for chemical analysis. The precipitated double salt was converted to RE(OH)3 (REOH) by reaction in 2 M NaOH for 2 h at 70 °C [26]. The final REOH product was filtered, rinsed, and dried. In some cases, powder was further purified by a second treatment in 10 M NaOH at 70 °C to completely react remaining sulfate double salt and to leach metal impurities.
Analysis
A Bruker (S2 PICOFOX) bench-top total reflection X-ray fluorescence spectrometer (TXRF) was used to analyze solutions and powders. The REOH products were dissolved in HCl before analysis. An aliquot of solution was pipetted onto the instrument sample holders and dried. A selenium (Se) internal standard was used for analysis. The concentrations of Pr, Nd, Dy, Gd in the final powder products were analyzed by an inductive coupled plasma mass spectrometer (ICP-MS) Thermo Scientific iCAP Q. The analysis of Na, S, Fe, and Zn was performed using an iCAP Series 6000 inductively coupled plasma optical emission spectrophotometer (ICP-OES) from Thermo Scientific. Calibration of ICP-MS and ICP-OES was performed using commercially prepared standards (VGH and Spex).
Powders were analyzed using X-ray diffraction (XRD) in a Bruker D8 Advance diffractometer operated at 40 kV and 40 mA, with a cobalt (Co) target (K = 1.78897A) being used for the characterization of the REE deposits formed. The XRD spectra were obtained for the REE sulfate double salts and hydroxides by scanning from 5 to 70° 2θ with a step size of 0.02° 2θ.
Thermodynamic Modelling
Aqueous systems containing REEs were modelled using the previously developed mixed-solvent electrolyte (MSE) model [27, 28]. The model parameters were determined for REE sulfate-containing systems using the procedures that were described in a previous work [30]. These procedures ensure that the model matches the available experimental data for solid–liquid equilibria, vapor–liquid equilibria, and caloric properties. Subsequently, the model was used to calculate the solubility of REE—sodium sulfate double salts and to predict on a thermodynamic basis the amounts of the solids that are expected to precipitate in the experiments.
Results and Discussion
Dissolution Rate Determination
As described in previous work, the magnet alloy was reactive to acid solutions with visible H2 evolution [20]. Due to challenges of separating magnets from electronic devices, this process examined dissolution from steel-magnet mixtures. As described in Eq. 1, the alloys react directly with H+ in strong acids to form H2, as shown in this proposed reaction.
The H2 gas produced by the dissolution reaction would need to be dealt with in an industrial process through simple flaring, energy recovery or through dilution with air. Produced H2 acts as an indicator of reaction progress as does the time between pH doses (H+ consumption).
To selectively dissolve the magnet alloy, previous work employed a N2 gas purge to reduce the concentration of dissolved O2 [25]. This was in attempt to control steel corrosion, as described in the well-known iron corrosion reaction (Eq. 2), by reducing the concentration of dissolved oxygen.
To assess the corrosion of mild steel, coupons of mild steel were exposed to 1 M HCl with either N2 gas or air bubbling into the solution. The corrosion rate measured over 24 h was 6.82 × 10–4 mm/h in air and decreased to 1.31 × 10–4 mm/h in N2. Purging with an inert gas to remove O2 reduces the rate as Eq. 2 suggests.
Uncoated disk magnets were used to measure the dissolution rate of REE magnets as a function of HCl concentration. The rate of dissolution increased almost linearly with HCl concentration (slope = 0.109 mm/h/M), with the rate at 1 M HCl being 0.170 mm/h, three orders of magnitude greater than that of steel. The dissolution rates are rapid demonstrating the reactive nature of the magnet alloy.
However, as will be described below, even in the presence of oxygen, the dissolution rate is three orders of magnitude lower than the rate for the Nd-Fe-B magnet alloy. Thus, it was decided to not purge O2 from the dissolution reactor, as the O2 removal may not be critical to control dissolution selectivity. It is anticipated that steel corrosion will be even lower than measured for two reasons: (1) Dissolved hydrogen from magnet dissolution should shift the corrosion potential of the steel to prevent active corrosion and (2) the internally produced H2 should act to purge O2.
Recovery of REEs from HDDs
The REE recovery was performed using 200 g of magnetically separated HDD material. Although the dissolution rate was demonstrated to increase with HCl concentration, a pH of 0 (1 M HCl) was selected as the control value. The system was operated over several days for a total of 52 h. A total of 545 mL of 5 M HCl was added during operation. Solutions were pulled periodically (five total solutions) and each processed individually to produce REOH powders. Figure 2 shows the elemental weight, measured with TXRF, for all five solutions (4 pulls and the remains) before and after sodium sulfate precipitation. The data was calculated using concentration and measured volume followed by adding each solution. The inset in Fig. 2 shows the % of mass decrease for the REEs in the solutions after precipitation (recovery). The Nd showed the greatest drop in weight along with Y which was present in very small quantities. The REEs such as Pr and Dy showed intermediate recovery while La showed very poor recovery. The lower recovery was presumably due to the lower starting concentrations close to the solubility limit for the double salt. Note that Fe largely remains in solution while Zn appears to carry over to the precipitate. There was some carry-over of Cl as well. The S increases due to addition of excess Na2SO4. Measurement of Na was not reliable using TXRF and was excluded from Fig. 2.

Total weight of elements before and after precipitation for all five solutions. Inset figure shows the percent change for REEs after precipitation. (Color figure online)
Table 1 provides recovery data as well as compositions of the recovered REOH powders. The total weight of REOH product was 5.176 g. The % recovery was calculated using the solution analysis before precipitation and compared to the powder analysis and weight of powder recovered. Recoveries of REOH exceeded 80% except for Solution 2. The composition of the powder was determined using various analysis methods described in the table.
In determining the composition %, REEs were assumed to be RE(OH)3, as supported by the XRD data to be discussed below. Using a weighted average (based on fraction of total powder collected), the REOH powder was over 77 wt% Nd(OH)3 and 81 wt% RE(OH)3. Note that the total wt%, which accounts for all analyzed components, averaged 85%. It would be assumed the missing weight would be due to O and H in waters of hydration, hydrous oxides, or sulfates. The adjusted % RE(OH)3 values, obtained by ratio of the total REOH % to the total wt%, show an average of 96%. The presence of significant Na and S is likely carry-over from the Na2SO4 in solution 5. The primary transition metal contaminate is Zn which averages 1.3%. Well-known from cementation post-processing, Zn readily dissolves in HCl. As shown in Fig. 2, most Zn reports to the precipitate. The Fe was present at much lower levels, averaging 0.03%. This also agrees with Fig. 3 where most of the Fe remained in solution. Other significant impurities were Ca and Cl. The origin of Ca impurities is not known.

The XRD spectrum of the solid obtained from solution #5. (Color figure online)
Another way to estimate recovery is through acid consumption. If it is assumed that all the acid consumed (0.545 mol H+) was due to magnet dissolution using Eq. 1, 0.0321 mol of REOH are expected to be in solution. Using the assumption that the powder product was entirely Nd(OH)3 that equates to 0.0265 mol Nd(OH)3. Thus, approximately 0.0056 mol were not recovered (~17%). Considering that the powder product contains some water, this agrees well with the % recovery reported in Table 1.
A sample of the REOH powder recovered from solution 5 was analyzed by XRD as shown in Fig. 3. The powder was identified as a match for Nd(OH)3 with no other phases observed.
Comparison of Recovery to Model
The MSE model predicts the thermodynamic equilibrium state for the reaction of Na, REE, sulfate ion, and water in the following equation:
Spedding and Jaffe [29] first described the relationship between REE sulfate solubility and ionic radius. Recently, Das et al. [30] developed a comprehensive parameterization of the MSE model for both REE sulfates and Na-REE double sulfate salts by analyzing various experimental thermodynamic data across the REE series. Based on this parameterization, the MSE model was used as the thermodynamic basis for determining the recovery of REEs as sodium double salts from the leaching solution. The analysis of Das et al. [30] revealed a characteristic, non-monotonic trend in the solubilities of the sulfates as a function of the ionic radius. The non-monotonic or “two-series” behavior is in fact a pervasive feature of REE salt solutions. It has been attributed to the combined effect of the change in the REE radius (which decreases smoothly across the REE series) and the variation in the number of water molecules in the first coordination sphere of REE ions. In particular, it was demonstrated that the hydration number for REEs varies in the REE series [32,33,34]. La through Nd have nine water molecules of hydration and Tb to Lu have eight. Pm to Gd have an intermediate number of water molecules which causes a change in expected chemical property relationships. Consequently, for many salt solutions, the transition in the hydration behavior gives rise to non-monotonic behavior of thermodynamic properties [30]. As a result, thermodynamic properties of REEs often have maxima or minima at Pr or Nd. In the case of Na-REE double sulfates, this effect manifests itself in a pronounced minimum in the solubility for Pr. Starting with Nd, the solubility increases as a function of crystalline radius and becomes substantially higher for heavy REEs. Based on the analysis of Das et al., the solubilities of different Na2RE2(SO4)4 salts were calculated at 25 °C for different Na2SO4 concentrations and are presented in Fig. 4 as a function of crystal cationic radii of REEs [31]. In particular, the solubility of Dy is over an order of magnitude higher. The model also shows a decrease in the solubility with the amount of Na2SO4 added which is congruent with what Le Chatelier's Principle would predict. This is primarily due to the common ion effect and is in agreement with the experimental data reviewed by Das et al. [30].

Prediction of Na2REE2(SO4)4 solubility in 0.1 m (blue circles), 0.5 m (pink triangles), and 1 m (green squares) Na2SO4 at 25 °C as a function of crystal cationic radii [21] of REEs (listed in reverse atomic radius). (Color figure online)
Recovery of REEs can be predicted based on the MSE model using the concentration prior to precipitation and the weight of Na2SO4 added. Then, the predicted recovery can be compared with the XRF data. A comparison of the model and experimental data is shown in Fig. 5. The actual recovery values for Nd were somewhat lower than predicted by the MSE model, which suggests that nearly all Nd should be removed in the precipitation step. The inset shows the detail for Pr recovery, where the predicted recovery was somewhat higher than the actual recovery for sample S1 and moderately lower for samples S2–S4. In the case of Pr, the initial concentration was much lower than for Nd and, therefore, near-complete recovery was not predicted because a non-negligible fraction of Pr had to remain in solution according to double salt solubility. Finally, the recovery of Dy was not predicted by the MSE model yet significant recovery was observed. This is due to the very small initial concentrations of Dy, which were lower than the solubility of the double salts. While this observation could not be reconciled based on solubility calculations, the predictions for Nd and Pr were reasonable. The reason for the apparent discrepancy for Dy may lie in the co-precipitation of Dy and Nd due to the overwhelmingly larger amount of Nd in the samples. It is well known that rare earth salts have a strong propensity to form solid solutions containing two or more rare earth elements. This has been observed for multiple classes of compounds, including REE oxides [35,36,37,38,39], chlorides [40, 41], nitrates [40], and cuprates [42]. Although the formation of solid solutions has not been reported for REE—Na double sulfate salts—it can be reasonably presumed that solid solutions are also possible for this class of compounds. If this is the case, then a relatively small amount of Dy may be incorporated into a Nd-dominated double salt, thus leading to the recovery of Dy together with Nd. Also, note that XRF is a semiquantitative measurement method for the rapid measurement of many elements. Future work will include more robust quantitative measurements for the REEs.

Comparison of recovery predicted by MSE model versus experimental data by XRF. The green bars represent the initial concentration of REEs in the leaching solution, while the blue and red bars give the experimental and predicted recovery, respectively. The symbols S1–S5 denotes samples 1–5. (Color figure online)
Recovery of REEs from HDDs Using Recycled Solution
To assess the potential for recycling the leachate solution, a portion of the remaining solution (after precipitation) was recycled with the aim of minimizing water usage and waste volume. A second benefit of this approach is that REEs not precipitated previously would carry over and thus could reduce overall losses. Acid is the primary reactant in dissolving the magnets (Reaction 1), and this is not significantly improved by recycling. Table 2 shows results of recovered REEs. Results were similar to the previous experiment using fresh HCl; however, recovery values were about 10% lower. This could be due to a lower magnet composition in this feed material (compositional variation) and thus a smaller percentage dropped out in the precipitation step. A smaller quantity of 5 M HCl was added (451 mL) compared with the previous experiment, and the final REOH product contained much less Zn than the previous extraction. Although not shown, the powder obtained from solution 3 indicated Na2SO4 phase in the XRD pattern. This indicates that too much Na2SO4 was added and that additional processing might be needed to purify the product. It is interesting that about 1/3 less Zn was observed compared with powders described in Sect. 3.2.
Purification of Recovered Powders
With the aim to improve the purity of the REOH product by removing the unreacted sulfate and other metal impurities, REOH powder 4 (Table 2) was subjected to a second alkaline digestion in 10 M KOH. The purification results shown in Table 3 indicate a 12% increase in the REOH composition after the second alkaline treatment. Elemental analysis performed with XRF did not detect sulfur in the re-processed sample, which indicates that sulfates are no longer present in the refined REOH. Regarding metal impurities, reductions of 50% and 65% were achieved for Fe and Zn, respectively, while Co was completely removed from the REOH product.
Both, Co and Zn can re-dissolve at high pH forming dicobaltite (HCoO2−) and zincate (ZnO2−) [22]. The purified REOH product obtained reached a REE purity of 99.1% metal basis.
Conclusions
Electronic scrap offers a significant source of REEs and the efficient recovery of REEs could be important to clean energy technologies. This paper was built upon a foundation of previous work regarding the recovery of REEs from mixed steel-Nd-Fe-B alloy material. This mixture is a feed from shredded HDDs as output from data destruction services. Experiments examined HCl concentration upon the rate of dissolution of uncoated Nd-Fe-B magnets. It was determined that the HCl concentration had a linear effect upon leaching rate of magnets. The rate for steel was found to be much lower and was not of significant consequence in the process. A pH-controlled flowing dissolution system was used to dissolve Nd-Fe-B magnet fragments from shredded HDDs. After dissolution, the REEs were precipitated using sodium sulfate and subsequently converted to REOH powder. Analysis demonstrated over 80% of dissolved REEs were recovered. The product showed Na, S, Fe, and Zn were major impurities. The purity level was decreased significantly by a second NaOH treatment. Therefore, a REOH intermediate product with a purity of 99.1% (metal basis) was obtained from processed HDD. Recycling of the acidic dissolution solution resulted in slightly lower recovery of about 70%. It is uncertain if the decreased recovery was due to the recycling of acidic solution or simply a lower Nd-Fe-B composition in the second batch.
References
Mihai FC, Gnoni MG, Meidiana C, Ezeah C, Elia V (2019) Waste electrical and electronic equipment (WEEE): Flows, quantities, and management-a global scenario, no. 2010. Elsevier Inc.
Balde CP, Forti V, Gray V, Kuehr R, Stegmann P (2017) The global e-waste monitor 2017
Tansel B (2017) From electronic consumer products to e-wastes: global outlook, waste quantities, recycling challenges. Environ Int 98:35–45. https://doi.org/10.1016/j.envint.2016.10.002
Yang Y et al (2017) REE Recovery from end-of-life NdFeB permanent magnet scrap: a critical review. J Sustain Metall 3(1):122–149. https://doi.org/10.1007/s40831-016-0090-4
Linnenkoper K (2019) Is it now or never for rare earth recycling? Recycling International
Adamas Intelligence (2020) Rare earth magnet market outlook to 2030. https://www.adamasintel.com/rare-earth-magnet-market-outlook-to-2030/
Mckittrick M, Bauer D, David D, Mckittrick M (2011) U.S. department of energy critical materials strategy. https://doi.org/10.1017/CBO9781107415324.004
Haxel GB, Hedrick JB, Orris GJ, Sound S, Of M, Mineral OUR (2002) Rare earth elements—critical resources for high technology. United States Geol Surv Fact Sheet 087:4
Dold B (2008) Sustainability in metal mining: from exploration, over processing to mine waste management. Rev Environ Sci Biotechnol, SPEC. ISS7(4):275–285. https://doi.org/10.1007/s11157-008-9142-y
Hellman PL, Duncan RK (2014) Evaluation of rare earth element deposits. Trans Institutions Min Metall Sect B Appl Earth Sci 123(2):107–117. https://doi.org/10.1179/1743275814Y.0000000054
Buchert D, Manhart M, Bleher A, Pingel D (2012) Recycling critical raw materials from waste electronic equipment Commissioned by the North Rhine- Westphalia State agency for nature, environment and consumer protection authors 49(0):30–40
Report F, Greens T, Group EFA, Parliament E (2011) Study on rare earths and their recycling 49:30–40
Nguyen RT, Diaz LA, Imholte DD, Lister TE (2017) Economic assessment for recycling critical metals from hard disk drives using a comprehensive recovery process. Jom 69(9):1546–1552. https://doi.org/10.1007/s11837-017-2399-2
Binnemans K et al (2013) Recycling of rare earths: a critical review. J Clean Prod 51:1–22. https://doi.org/10.1016/j.jclepro.2012.12.037
Anand T, Mishra B, Apelian D, Blanpain B (2011) The case for recycling of rare earth metals-a CR3 communication. Jom 63(6):8–9. https://doi.org/10.1007/s11837-011-0098-y
Zhang Y, Gu F, Su Z, Liu S, Anderson C, Jiang T (2020) Hydrometallurgical recovery of rare earth elements from ndfeb permanent magnet scrap: a review. Metals (Basel) 10(6):1–34. https://doi.org/10.3390/met10060841
Lyman J, Palmer G (1992) Scrap treatment method for rare earth transition metal alloys. US Patent 5,129,945
Lister TE, Wang P, Anderko A (2014) Recovery of critical and value metals from mobile electronics enabled by electrochemical processing. Hydrometallurgy 149(2014):228–237. https://doi.org/10.1016/j.hydromet.2014.08.011
Binnemans K et al (2013) Recycling of rare earth: a critical review. J Clean Prod 51:1–22. 10.1016. https://doi.org/10.1016/j.jclepro.2012.12.037
Pourbaix M (1974) Atlas of electrochemical equilibria in aqueous solutions, 2nd edn. NACE International, Houston, TX
Stevenson PC et al (1961) The radiochemistry of the rare earths, scandium, yttrium, and actinium. https://library.lanl.gov/cgi-bin/getfile?rc000021.pdf
Pietrelli L, Bellomo B, Fontana D, Montereali MR (2002) Rare earths recovery from NiMH spent batteries. Hydrometallurgy 66(1–3):135–139. https://doi.org/10.1016/S0304-386X(02)00107-X
Bertuol DA, Bernardes AM, Tenório JAS (2009) Spent NiMH batteries-The role of selective precipitation in the recovery of valuable metals. J Power Sources 193(2):914–923. https://doi.org/10.1016/j.jpowsour.2009.05.014
Lyman JW, Palmer G (1995) Hydrometallurgical treatment of nickel-metal hydride battery electrodes
Diaz LA, Lister TE, Parkman JA, Clark GG (2016) Comprehensive process for the recovery of value and critical materials from electronic waste. J Clean Prod 125:236–244. https://doi.org/10.1016/j.jclepro.2016.03.061
Abreu RD, Morais CA (2010) Purification of rare earth elements from monazite sulphuric acid leach liquor and the production of high-purity ceric oxide. Miner Eng 23(6):536–540. https://doi.org/10.1016/j.mineng.2010.03.010
Wang P, Anderko A, Young RD (2002) A speciation-based model for mixed-solvent electrolyte systems. Fluid Phase Equilib 203(1–2):141–176. https://doi.org/10.1016/S0378-3812(02)00178-4
Das G, Lencka MM, Eslamimanesh A, Anderko A, Riman RE (2017) Rare-earth elements in aqueous chloride systems: thermodynamic modeling of binary and multicomponent systems in wide concentration ranges. Fluid Phase Equilib 452:16–57. https://doi.org/10.1016/j.fluid.2017.08.014
Spedding FH, Jaffe S (1954) Conductances, solubilities and ionization constants of some rare earth sulfates in aqueous solutions at 25 °C. J Am Chem Soc 76(3):882–884
Das G et al (2019) Rare earth sulfates in aqueous systems: thermodynamic modeling of binary and multicomponent systems over wide concentration and temperature ranges. J Chem Thermodyn 131:49–79. https://doi.org/10.1016/j.jct.2018.10.020
Shannon RD, Prewitt CT (1969) Effective ionic radii in oxides and fluorides . Acta Crystallogr Sect. B Struct Crystallogr Cryst Chem 25(5):925–946. https://doi.org/10.1107/s0567740869003220
Habenschuss A, Spedding FH (1979) The coordination (hydration) of rare earth ions in aqueous chloride solutions from x-ray diffraction. II. LaCI3, PrCI3, and NdCI3. J Chem Phys 70(8):3758–3763. https://doi.org/10.1063/1.437928
Habenschuss A, Spedding FH (1979) The coordination (hydration) of rare earth ions in aqueous chloride solutions from x-ray diffraction. I. TbCI3, DyCI3, ErCI3, TmCI3, and LuCI3. J Chem Phys 70(6):2797–2806. https://doi.org/10.1063/1.437866
Habenschuss A, Spedding FH (1980) The coordination (hydration) of rare earth ions in aqueous chloride solutions from x-ray diffraction. III. SmCl3, EuCl3, and series behavior. J Chem Phys 73(1):442–450. https://doi.org/10.1063/1.439895
McCullough JD (1950) An X-ray study of the rare-earth oxide systems: CeIV—NdIII, CrIV—PrIII, CeIV—PrIV and PrIV—NdIII. J Am Chem Soc 72(3):1386–1390
Schneider SJ, Roth RS (1960) Phase equilibria in systems involving the rare-earth oxides. Part II. Solid state reactions in trivalent rare-earth oxide systems. J Res Natl Bureau of Standards. Sect A, Phys Chem 64(4):317–332
Reddy BM, Katta L, Thrimurthulu G (2010) Novel nanocrystalline Ce1−x Lax O2−δ (x= 0.2) Solid solutions: structural characteristics and catalytic performance. Chem Mater 22(2):467–475
Reddy BM, Vinodkumar T, Durgasri DN, Rangaswamy A (2017) Synthesis and characterization of nanostructured Ce0.8M0.2O2− δ (M= Sm, Eu, and Gd) solid solutions for catalytic CO oxidation. Proc Natl Acad Sci, India, Sect A 87(1):155–161
Han X, Wang Y, Hao H, Guo R, Hu Y, Jiang W (2016) Ce1–xLaxOy solid solution prepared from mixed rare earth chloride for soot oxidation. J Rare Earths 34(6):590–596
Nikolaev AV, Sorokina AA, Vilenskaya AY, Tsubanov VG (1967) Solubility in ternary mixtures containing lanthanide salts and water. Izv Sibir Otd Akad Nauk SSSR Ser Khim Nauk 14:5–21
Sokolova NP, Bagryantseva LL, Komissarova LN (1983) Solid solutions in the systems NdCl3-LnCl3-H2O at low temperatures. Izv Sibir Otd Akad Nauk SSSR Ser Khim Nauk 4:89–92
Trusova EM, Popov VP, Tikhonov PA, Glushkova VB (2002) Solid solutions of rare-earth cuprates and their electrical properties. Glass Phys Chem 28(4):264–267
Acknowledgements
This work is supported by the Critical Materials Institute, an Energy Innovation Hub funded by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Manufacturing Office. This manuscript has been authored by Battelle Energy Alliance, LLC under Contract No. DE-AC07-05ID14517. We thank Timothy McIntire (Oak Ridge National Laboratory) for supplying shredded HDD material. We also thank Byron White and Arnold Erickson for providing analytical services that supported this work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Minerals, Metals & Minerals Society
About this paper

Cite this paper
Lister, T.E. et al. (2021). Recovery of Rare Earth Elements from Recycled Hard Disk Drive Mixed Steel and Magnet Scrap. In: Azimi, G., et al. Rare Metal Technology 2021. The Minerals, Metals & Materials Series. Springer, Cham. https://doi.org/10.1007/978-3-030-65489-4_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-65489-4_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-65488-7
Online ISBN: 978-3-030-65489-4
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)