
Abstract
The prostate is a tubulo-alveolar gland and part of both the reproductive and urinary system. The prostate is, next to the seminal vesicales and the bulbourethral glands, part of the male accessory sex glands and produces seminal fluids which, during ejaculation, mix with sperm cells to neutralize the acid vaginal milieu and nourish spermatozoa. Lower urinary tract symptoms associated with benign prostatic hyperplasia (LUTS/BPH) and prostate cancer (PCa) are the most common prostatic diseases. Knowledge of the anatomy, physiology and receptors of the prostate is crucial for effective treatment of LUTS/BPH and PCa.
α1-Adrenergic receptor antagonists (α1-blockers) competitively bind to α1-adrenoceptors at the neuro-muscular junction of sympathetic nervous system, reduce the smooth muscle cell tone of the prostate and bladder outlet, decrease urethral resistance, and improve LUTS/BPH quickly and effectively. α1-blockers also significantly increase the chance of spontaneous voiding in patients with acute urinary retention. Phosphodiesterase type 5 inhibitors (PDE5i) act on the non-adrenergic, non-cholinergic neurotransmitter pathway by blocking the conversion of active cyclic guanosine monophosphate (cGMP) to inactive guanosine monophosphate, thereby prolonging the muscle relaxing effects of protein kinase G. PDE5i quickly and effectively ameliorate LUTS/BPH and also concomitant erectile dysfunction. 5α-reductase inhibitors (5ARIs) interfere with the intracellular metabolism of androgens and block the conversion from testosterone to dihydrotestosterone by blocking 5α-reductase type 2. Treatment with 5ARIs slowly reduces prostate size, improves LUTS/BPH and decreases disease progression, thereby reducing the chance of acute urinary retention and need for prostate surgery. Anti-androgens block androgenic effects and estrogens as well as agonists or antagonists of gonadotropin releasing hormone reduce androgen production to effectively treat androgen-sensitive PCa.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Prostate
- Urinary physiology
- Prostate-specific antigen
- Sympathetic nervous system
- Adrenergic alpha blockers
- Nitric oxide
- Phosphodiesterase 5 inhibitors
- 5 Alpha reductase inhibitors
- Androgen receptor
Introduction
The prostate is located in the lower pelvis and positioned between the bladder neck and pelvic floor. The prostate is a tubulo-alveolar gland, part of both the reproductive and urinary system, and universally present in mammals. The urethra of humans runs in cranio-caudal direction through the prostate (i.e. prostatic urethra) and the ejaculatory ducts run on both sides from dorso-cranial to ventro-caudal direction towards the prostatic urethra. The prostate grows and matures during puberty under the influence of testosterone from a small gland of 2–5 cm3 in childhood to a gland volume of approximately 20–25 cm3 in early adolescence [1]. Approximately 15–30 ejaculatory ducts disembogue at the verumontanum which is located on the dorsal side halfway through the prostatic urethra. Based on the position of the verumontanum, the prostatic urethra can be divided into a proximal and distal part which has in the young healthy adult a length of approx. 1 cm each. Starting at the verumontanum and ventrally directed, the proximal prostatic urethra has an angle of approximately 35° [2, 3].
A total of 30–50 glands are embedded in the prostate. The glandular tubes contain epithelial cells which coat the lumen and are responsible for the secretion during ejaculation. Prostatic glands are surrounded by interstitial tissue which accommodates smooth muscle cells, fibrocytes, elastic and collagen fibers, blood and lymph vessels, as well as nerves (Fig. 8.1). According to the structure and alignment of the glands as well as the microscopic shape of epithelial cells, the prostate can be divided into four distinct zones (Fig. 8.2) [4, 5] which can be visualized by ultrasound [7] or MRI [8, 9]:
-
1.
Anterior zone: this area does not have glands and only contains muscular and fibrotic cells. The aglandular prostate forms the ventral surface of the prostate and is inseparable fused with the glandular prostate. The function of this area remains unknown but strong muscular components suggest contracting abilities.
-
2.
Central zone: constitutes approximately 25% of the glandular prostate in young, healthy adults, has a conical shape with the tip of the cone directed towards the verumontanum and the base towards the seminal vesicles. Glandular tubes run parallel to the intra-prostatic ejaculatory duct and disembogue in the center of the verumontanum. The glands are oriented in a coronal plane and contain wide chambers. The epithelium of the glands has multiple rows.
-
3.
Peripheral zone: constitutes approximately 70% of the glandular prostate in young, healthy adults and surrounds the central zone from the base to the apex. The ducts radiate laterally from the urethra and disembogue lateral and distal of the verumontanum. The glands have a uniform configuration, only a few branches and a round lumen. The epithelial cells are long and positioned in a single row and have a uniform shape as well as a light cytoplasm.
-
4.
Transition zone: constitutes approximately 5% of the glandular prostate in young, healthy adults and is positioned on the dorsal side of the proximal prostatic urethra. The transition zone only has a few ducts which run parallel to the urethral axis. The glands are straight and the epithelial cells have a similar shape compared to those of the peripheral zone; however, the interstitial tissue is irregular.

TUR specimen of a patient with benign prostatic hyperplasia demonstrating the different tissue components of the transition zone (hematoxylin eosin staining, enlargement 150-fold). Prostatic glands with single layer epithelial cells appear blue and the lumen white (without secretion, washed out during fixation). The glands are separated by interstitial tissue with a large amount of smooth muscle cells (red). A glandular duct with single layer epithelial cells runs in vertical direction through the tissue. Elastic and collagen fibers as well as lymphatic vessels and nerves are not stained and remain invisible on this image (courtesy of Dr. S. Afram, Pathological Institute Gronau, Germany)

Schematic illustration of the gross anatomy of the prostate, imaged 45 degrees from the left (zonal anatomy according to McNeal [4, 5]). The anterior fibromuscular stroma (AFS) is positioned ventrally. The central zone is located dorsally of the urethra and is surrounded by the peripheral zone. The transition zone is positioned at the proximal urethra (proximal of the verumontanum, i.e. the location where the ejaculatory ducts (ED) disembogue into the urethra). The seminal vesicles (SV) are located dorso-laterally of the prostate on both sides (modified according to Roehrborn et al. [6])
The different morphology of the prostatic glands and different appearances of epithelial cells in different zones indicate different functions within the glandular prostate [10, 11]; however, it remains largely unknown which zones are responsible for particular functions or secretions in the context of ejaculation and reproduction.
Different diseases develop in different zones. Prostate cancer and prostatitis begin in the peripheral zone [12], whereas benign prostatic hyperplasia (BPH) originates in the transition zone [13]. However, the cause of BPH remains largely unknown. The prevalence of BPH increases with aging [1] and may result in enlargement of the transition zone and secondarily in enlargement of the entire prostate (i.e. benign prostatic enlargement, BPE), elongation of the proximal prostatic urethra (>1 cm), increase of the ventrally directed angle of the proximal prostatic urethra (>35°), increased urethral resistance of urine flow (i.e. benign prostatic obstruction, BPO) and lower urinary tract symptoms (LUTS) [2, 3, 14, 15].
Physiology of the Prostate
The prostate is—next to the seminal vesicales and the bulbourethral glands (Cowper)—part of the male accessory sex glands and produces seminal fluids. Smooth muscle cells of the prostate contract during orgasm in order to expel ejaculate through the urethra [16]. Epithelial cells of the prostatic glands produce a secretion that empties during ejaculation through the glandular ducts into the prostatic urethra where they mix with semen from the testes and epididymes as well as with secretions from the seminal vesicales [16].
Prostatic secretion is an important component of the ejaculate and constitutes approximately 25–30% of the ejaculate volume (~0.5 mL). The prostatic secretions are a milky white mixture of simple sugars (e.g. fructose and glucose), proteins, minerals and alkaline chemicals which protect and nourish sperm. The sugars secreted by the prostate function as nutrition for the spermatozoa while they pass into the female body to fertilize eggs [16]. Protein content is less than 1% of the total prostatic secretion and includes proteolytic enzymes, prostate-specific antigen (PSA), prostatic acid phosphatase and β-micro-seminoprotein [17]. PSA, a glycoprotein and member of the kallikrein-related peptidase family produced and secreted in prostatic epithelial cells, and other enzymes break down seminal proteins to free spermatozoa from the viscous semen, thereby making semen thinner and allowing sperm cells to swim freely. Additionally, PSA can also dissolve cervical mucus in order to give sperm cells free passage to the uterus to fertilize eggs. The secretions also contain zinc with a concentration 500–1000 times the concentration in blood [16]. The alkaline chemicals in prostatic secretions (~pH 7.8) neutralize acidic vaginal secretions to promote the survival and prolong the lifespan of spermatozoa in the female body, thereby increasing their chance of successfully fertilizing an egg [16].
The prostate needs hormones (androgens) to function properly. The predominant male sex hormone is testosterone which is mainly produced by the Leydig cells of the testes, to a lesser amount also in the adrenal glands. Testosterone is metabolized into the active hormone dihydrotestosterone (DHT) inside prostatic epithelial cells by the enzyme 5α-reductase (see Section “Androgens”).
Pharmacology of the Prostate
The prostate contains several tissue types (especially smooth muscle cells and epithelium) which are rich of receptors and enzymes. Targeting these receptors/enzymes helps treating LUTS/BPH or PCa. LUTS/BPH is the fourth most common disease in men aged ≥50 years and prostate cancer is the most common malignancy in males [18]. Therefore, the following targets and the selective manipulation of these became most important for all urologists and also general practitioners.
Adrenergic Receptors (Adrenoceptors)
The Sympathetic Nerve System
The general principal in the human body is that the tone of the smooth musculature is under control of the autonomic (vegetative) nervous system [19, 20]. Depolarization of sympathetic nerves leads to noradrenaline release at the terminal postganglionic neuron into the synaptic gap where adrenoceptors are located at the pre- and postsynaptic membrane (Fig. 8.3). Two main types of adrenoceptors exist, the α- and β-adrenoceptors, which can further be divided into α1- and α2- as well as β1-, β2- and β3-adrenoceptors [22]. Stimulation of the adrenoceptors at the postsynaptic membrane results in g-protein induced activation of phospholipase C and production of inositol-triphosphate and diacetylglycerol (second messengers) which release Ca2+ from the sarcoplasmatic reticulum, thereby initiating smooth muscle cell contraction [23]. Because different organs in the human body typically express different subtypes and concentrations of adrenoceptors, stimulation of these postsynaptic adrenoceptors results in various responses in different organs (e.g. prostate, internal urethral sphincter, blood vessels, heart, gastro-intestinal tract). Noradrenaline in the synaptic gap inhibits further noradrenaline release at the presynaptic membrane via α2-adrenoceptors (negative feedback), thereby terminating the sympathetic response at the effector organ. Learmonth could already show in the year 1931 that stimulation of the pre-sacral (hypogastricus) nerve leads to a contraction of the prostatic musculature [24], and organ bath studies demonstrated a contraction of prostate strips after addition of noradrenaline [25].

Schematic illustration of the sympathetic nervous system (left) and enlargement of the neuromuscular connection (right). After depolarization of the postganglionic neuron, noradrenaline (NA) is released into the synaptic gap. NA molecules bind to α- and β-adrenoceptors on the postsynaptic membrane where they initiate effects (e.g. smooth muscle cell contraction). NA release is terminated via a negative feedback mechanism mediated by α2-andrenoceptors on the presynaptic membrane (modified according to Oelke et al. [21])
α1-Adrenergic Receptors
The human prostate predominantly contains α1-adrenoceptors, to a lesser amount also α2-adrenoceptors [26,27,28,29]. α1-Adrenoceptors are located on smooth muscle cells (proportion of α1:α2 = 4:1), whereas α2-adrenoceptors are mainly located in the epithelium and blood vessels [26, 28, 30]. Functional studies clarified that smooth muscle cell contraction in the prostate is mediated by α1-adrenoceptors [31,32,33,34,35]. Hyperplastic prostatic tissue (BPH) has more smooth muscle cells (absolute and in percentage) and carries more α-adrenoceptors than normal prostate tissue [34, 36, 37]. However, the content of α1-adrenoceptors in the prostate of BPH-patients with LUTS is identical to BPH-patients without LUTS; therefore α1-adrenoceptors do not seem to be the origin of LUTS [32].
Pharmacologically interesting for the treatment of LUTS/BPH are especially the α1-adrenoceptors of which three subtypes have been identified, the α1A-, α1B- and α1D-adrenoceptors [38,39,40]. Historically, the α1C-adrenoceptor was also described but turned out to be identical with the α1A-subtype; therefore, the exact specification of this α1-adrenoceptor subtype was abandoned [41, 42]. Approximately 70% of α1-adrenoceptors in the prostate are α1A-subtypes [43] which are predominantly located on smooth muscle cells [41]. The α1A-adrenoceptors increase numerically and the proportion of α1A:α1B:α1D adrenoceptor subtypes changes from 63:6:31 in normal prostatic tissue to 85:1:14 in BPH tissue [44]. Inhibition of the α1A-adrenoceptor subtype results in smooth muscle cell relaxation. The α1B-adrenoceptor is mainly located in blood vessels and inhibition of this adrenoceptor subtype is associated with vasodilation [45, 46]. The α1D-adrenoceptor is predominantly expressed in the bladder, peripheral ganglia and spinal cord; inhibition of this adrenoceptor subtype results in direct or indirect (peripheral ganglia, spinal cord) relaxation of the detrusor [47,48,49].
α1-Adrenergic Receptor Antagonists (α1-Blockers)
It was shown in patients with BPH that smooth muscle cells account for ~39% of the cellular volume of the prostate and ~51% of the stroma volume [50]. The aim of the treatment of LUTS/BPH is to relax smooth muscle cells of the prostate and bladder neck by inhibition of α1A-adrenoceptors which eventually decrease urethral resistance. Inhibition of the adrenergic innervation of the prostate can reduce the intra-prostatic urethral pressure by approx. 47% [51,52,53] and can improve the detrusor pressure at maximum urine flow (Pdet.Qmax) by approximately 11.4 cm H2O, maximum urine flow (Qmax) by 2.3 mL/s as well as bladder outlet obstruction index (BOOI = Pdet.Qmax − 2Qmax) by 14.2 cm H2O [54].
Initially, the unselective α1-/α2-adrenoceptor antagonists phenoxybenzamine and phentolamine were used to treat LUTS/BPH (Fig. 8.4); although significant and relevant LUTS reduction was achieved with both α-blockers in clinical trials, adverse events and especially blood pressure decrease were too pronounced for routine clinical use [55, 56]. Prazosin was the first selective α1-adrenoceptor antagonist used and licensed for the treatment of LUTS/BPH [57]. Later, the more α1A-adrenoceptor subtype specific α1-blockers doxazosin and terazosin were introduced to treat LUTS/BPH but these drugs still showed relevant blood pressure decrease, the reason why these two α1-blockers were also used to treat arterial hypertension alone or in combination with LUTS/BPH. The strategy to treat both diseases with one α1-blocker was abandoned in the year 2000 because the ALHAT (Antihypertensive and Lipid-Lowering treatment to prevent Heart ATack) trial demonstrated that doxazosin cannot prevent cardio-vascular events (angina pectoris, myocardial infarction or heart insufficiency); therefore, α1-blockers were considered inappropriate to treat arterial hypertension alone [58,59,60,61]. Later, the “uroselective” α1-blockers alfuzosin and tamsulosin were developed which showed more pronounced α1A-adrenoceptor inhibition and especially a lower incidence of arterial hypotension. The development of α1-blockers has currently ended with the development of specific antagonists of the α1A-adrenoceptor subtype (silodosin) and the α1D-adrenoceptor subtype (naftopidil; only licensed in Asia).

Commercially available α-adrenoceptors antagonists and their subtype selectivity. Only the α1-adrenoceptor antagonists in blue color are currently licensed for the treatment of “symptoms or signs of BPH” (modified according to Oelke et al. [21])
The pharmacokinetic profiles of currently available α1-blockers to treat LUTS/BPH are listed in Table 8.1. Once-daily formulations (extended release, modified release, gastrointestinal therapeutic system [GITS] or oral controlled absorption system [OCAS]) aim to slowly release the α1-blocker into the intestinal tract which results in a more stable and consistent serum concentration, avoiding serum peaks and unwarranted side effects such as arterial hypotension [63,64,65]. Additionally, once-daily formulations are likely to increase the intake compliance and adherence to the drug [66].
Clinical Efficacy of α1-Blockers
Although LUTS and urine flow improvements take a few weeks to fully develop, statistically significant and clinically relevant differences compared to placebo were already documented within a few hours or days after first drug intake [67]. α1-Blockers show a similar efficacy, expressed as percent improvement in the International Prostate Symptom Score (IPPS) questionnaire, in patients with mild, moderate or severe LUTS [68]. RCTs demonstrated that α1-blockers reduce LUTS (both storage and voiding symptoms) by ~30–40% and increase Qmax by ~20–25% after the placebo run-in period [62]. In observational studies (without a placebo run-in period), IPSS improved by ~50% and Qmax by ~40%. Indirect comparisons and limited head-to-head comparisons between α1-blockers indicate that all α1-blockers have a similar efficacy when used in appropriate doses [69, 70]. However, a recent network meta-analysis suggested that some α1-blockers reduce LUTS to a greater extent than others [71]; mean IPSS reduction was −7.1 points for doxazosin and −6.8 points for terazosin, whereas mean IPSS reduction for silodosin, alfuzosin, tamsulosin and naftopidil was −5.8, −5.5, −5.5 and − 5.4 points, respectively. This preliminary data needs confirmation by independent analyses, especially considering the different doses and formulations of the individual α1-blockers.
α1-Blockers do not reduce prostate size and, additionally, prostate size does not affect α1-blocker efficacy in studies with follow-up period of ≤1 year [62, 72]. Qmax at baseline does not have relevant influence on LUTS reduction [73]. Long-term efficacy of α1-blockers was documented for a period of longer than 4 years [74]. Nevertheless, α1-blockers are not able to prevent acute urinary retention (AUR) in the long run, especially in patients with a prostate volume >40 cm3 (see Section “Androgens”) [74, 75].
Men with BPH can develop AUR due to increased bladder outlet resistance (BPO) and/or insufficient detrusor function (detrusor underactivity). The incidence rate is approximately 2.2 cases/1000 patient-years in asymptomatic and 18.3–35.9 cases/1000 patient-years in symptomatic men [76]. The use of α1-blockers in these patients allows approx. 60% of men to void spontaneously again compared to approx. 38% using placebo (successful trial without catheter, TWOC), which measures up to a 55% increase in the success rate [77].
For further reduction of LUTS, α1-blockers can be combined with drugs of other drugs classes, e.g. with phosphodiesterase type 5 inhibitors (PDE5i; see Section “Phosphodiesterases”) [78,79,80], 5α-reductase inhibitors (5ARIs; see Section “Androgens”) [75, 81], muscarinic receptor antagonists (antimuscarinics) [82, 83] or β3-receptor agonists [84].
Adverse Event Profile of α1-Blockers
Although alfuzosin, doxazosin and terazosin have a similar molecular structure (quinazoline-based derivatives) and do not selectively inhibit specific α1-adrenoceptor subtypes, the adverse event profile of alfuzosin is more similar to tamsulosin (non-arylamine sulfonamide derivative) than to doxazosin or terazosin. The mechanisms underlying such differential tolerability are not fully understood but may involve better penetration and/or tissue distribution by alfuzosin and tamsulosin [62]. Other factors, such as subtype selectivity and the pharmacokinetic profiles of certain formulations, may also contribute to the adverse event profile of individual drugs.
The most frequently reported adverse events of α1-blockers are asthenia, dizziness and (orthostatic) hypotension which can be explained by the inhibition of α1B-adrenoceptor subtypes in vessels, leading to vasodilation and blood pressure decrease [85]. At least some of the observed asthenia and dizziness can be attributed to blood pressure decrease. In particular, patients with cardiovascular co-morbidity and/or vasoactive co-medication appear to be prone to vasodilatation during α1-blocker treatment [86]. This includes anti-hypertensive drugs, such as diuretics, Ca2+-channel blockers, β-blockers, angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists but also phosphodiesterase type 5 inhibitors (PDE5i) prescribed for erectile dysfunction or male LUTS. Vasodilatation is more pronounced with immediate-release doxazosin and terazosin but less common for alfuzosin or tamsulosin (odds ratio for vascular-related adverse events 3.3, 3.7, 1.7 and 1.4, respectively; the latter two not reaching statistical significance [87]). In contrast, blood pressure decrease or prevalence of orthostatic hypotension with the α1A-selective blocker silodosin is comparable with placebo [85, 88] and, therefore, this drug can safely be used with anti-hypertensive co-medication [89].
The intraoperative floppy iris syndrome (IFIS) was only discovered in 2005 in the context of cataract surgery [90]. IFIS consists of a typical triad of the following intraoperative characteristics: (1) a flaccid iris stroma leading to fluttering and billowing of the iris, (2) a tendency for the floppy iris stroma to prolapse through the surgical incision and (3) a progressive pupil constriction despite standard perioperative pharmacologic measures for prevention. Most patients, however, manifest an incomplete form of this triad displaying only one or two signs [85]. The basis of IFIS is thought to be antagonism of the α1-adrenoceptor subtype located in the iris dilator muscle. The incidence varies between 30 and 88% for tamsulosin, 15 and 70% for alfuzosin, and 2 and 45% for doxazosin users. In a meta-analysis, alfuzosin, doxazosin, tamsulosin and terazosin had an increased risk for IFIS [91]. The odds-ratio for IFIS was 393 for tamsulosin, 9.7 for alfuzosin, 6.4 for doxazosin and 5.5 for terazosin. Silodosin has yet not been associated with IFIS [85]. α1-Blocker treatment should not be initiated prior to scheduled cataract surgery, and the ophthalmologist should be informed about previous or current α1-blocker use [85]. Important to mention is that the occurrence of IFIS may complicate cataract surgery and make it technically more challenging but no reports about increased health risks of these patients have been published.
A systematic review concluded that α1-blockers do not adversely affect libido, sometimes have a small beneficial effect on erectile function (e.g. doxazosin) but can cause abnormal ejaculation [92]. Originally, abnormal ejaculation was thought to be retrograde but more recent data demonstrate that it is due to (relative) anejaculation (i.e. reduction or absence of seminal fluids during ejaculation). Abnormal ejaculation seems to be caused by α1A-adrenoceptor subtype selective inhibition of smooth muscle cells in the prostate, seminal vesicles, ejaculatory ducts and spermatic cords and is more frequently observed with silodosin and tamsulosin than with other α1-blockers. LUTS reduction was shown to be higher in men with abnormal ejaculation compared to men without [93]. The apparently greater risk for abnormal ejaculation with tamsulosin is intriguing as even the more α1A-selective drug silodosin carries an even greater risk [94, 95]. However, all α1-blockers are dosed to block α1A-adrenoceptors effectively; therefore, the mechanism underlying abnormal ejaculation still remains to be elucidated.
A dose-dependent increased risk of dementia for tamsulosin was only recently described (2018) in a cohort analysis of more than 250,000 US-American patients after a follow-up of 20 months (hazard ratio 1.11–1.20; 38.8 cases/1000 patient years) [96]. In contrast, an increased risk for dementia was not seen for other α1-blockers (alfuzosin, doxazosin or terazosin) or 5ARIs (finasteride or dutasteride). Another recently published cohort study (2019) with approx. 60,000 Korean patients with a follow-up of 52 months could not confirm the results seen in the North American study [97]. However, the daily standard dose for tamsulosin in Asian patients is only 0.2 mg, and differences in pharmacokinetics, metabolism and genetic/ethnicity may have contributed to these controversial results. Therefore, the association between α1-blocker use and dementia still needs to be clarified in future studies.
Phosphodiesterases
The NO/NOS: Phosphodiesterase System
Nitric oxide (NO) is a universal and important non-adrenergic, non-cholinergic neurotransmitter which is involved in signal transmission in the human lower urinary tract [98]. NO is synthesized from the amino acid L-arginine by NO synthases (NOS) [99]. NO diffuses into cells and stimulates the synthesis of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP), which are synthesized from the corresponding nucleoside triphosphates by adenylyl- or guanylyl-cyclases (Fig. 8.5). The intracellular increase of cAMP or cGMP triggers a signal transduction cascade involving activation of cyclic nucleotide-dependent protein kinases (PKA or PKG), subsequent phosphorylation of the actin-myosin system, as well as Ca2+ channels and ATP-driven Ca2+ pumps located in the outer cell membrane or the membrane of the sarcoplasmatic reticulum [98]. This cascade leads to a reduction in cytosolic Ca2+ and, finally, to smooth muscle relaxation [102]. NO may also be involved in the micturition cycle by inhibiting reflex pathways in the spinal cord and neurotransmission in the urethra, prostate and urinary bladder [103]. The effects of cAMP and cGMP are terminated by PDE isoenzymes, a heterogeneous group of enzymes which catalyze the hydrolysis of cAMP or cGMP into their inactive forms.

Cellular responses (e.g. smooth muscle cell relaxation) after activation of protein kinase A (PKA) by cyclic adenosine monophosphate (cAMP-pathway, left) or protein kinase G (PKG) by cyclic guanosine monophosphate G (cGMP-pathway, right). The effect of cAMP is terminated by phosphodiesterase type 4 (PDE 4) and the effect of cGMP is terminated by phosphodiesterase type 5 (PDE 5). PDE 4 inhibitors (PDE 4i; e.g. rolipram, Ro 20-1724 or RP 73401) block the degradation and thus inactivation of cAMP. PDE 5 inhibitors (PDE 5i; e.g. avanafil, sildenafil, tadalafil, udenafil or vardenafil) block the degradation and thus inactivation of cGMP, which results in accumulation of cyclic monophosphates and prolonged cellular responses (modified according to Ghofrani et al. [100] and Lincoln et al. [101]). AMP adenosine monophosphate, cAMP cyclic adenosine monophosphate, ATP adenosine triphosphate, GMP guanosine monophosphate, cGMP cyclic guanosine monophosphate, GTP guanosine triphosphate, NO nitric oxide, NOS nitrite oxide synthases, PDE phosphodiesterase, PDEi phosphodiesterase inhibitor, PKA protein kinase A, PKG protein kinase G
PDEs are classified according to their preferences for cAMP or/and cGMP, kinetic parameters of cyclic nucleotide hydrolysis, sensitivity to the inhibition by various compounds, allosteric regulation by other molecules, and chromatographic behavior on anion exchange columns [99]. In total, 11 families of PDE isoenzymes have been identified in humans (PDEs 1–11). Some of these isoenzyme families have more than one gene and some genes are alternatively spliced so that more than 50 isoenzymes or variants have been identified. Some PDE genes are also variably expressed in different tissues [104]; therefore, the distribution and functional significance of PDE isoenzymes vary in different organs and, consequently, isoenzyme-selective inhibitors have the potential to exert specific effects on target tissues. In the lower urinary tract, the cAMP- and/or cGMP-specific PDE isoenzymes PDE 1, PDE 2, PDE 4, PDE 5 and PDEs 7–10 have been identified of which PDE4 (cAMP-specific PDE) and PDE5 (cGMP-specific PDE) are the predominant ones in the prostate, bladder and urethra [105]. Immunohistochemistry showed that PDEs 4 and 5 are mainly located in stromal and glandular tissues of the transition zone. A clinical study with 30 men with moderate to severe LUTS awaiting transurethral resection of the prostate (TURP) were randomly divided to receive 20 mg tadalafil or 200 mg udenafil 1 h prior to TURP or TURP without the preceding administration of a PDE5i. The concentrations of the PDE5i and cGMP were measured in the plasma and resected prostate specimen and, afterwards, the prostate tissue/plasma ratio was calculated. The authors showed that (1) the PDE5i was predominantly distributed in the prostate (transition zone) and (2) tadalafil and udenafil significantly increased cGMP levels in both the plasma and prostate tissue.
Organ bath studies demonstrated that, after α1-adrenergic stimulation of smooth muscle cells with noradrenaline, the tension of prostate tissue strips was dose-dependently reversed by PDE4 inhibitors (e.g. rolipram, Ro 20-1724 and RP 73401) and PDE5 inhibitors (e.g. sildenafil, tadalafil and vardenafil) and accompanied by an increase of cAMP or cGMP levels in the tissue [105,106,107]. These results indicate that cAMP and cGMP as well as PDEs 4 and 5 are involved in relaxation of prostatic smooth muscle cells.
The NO/NOS- and cAMP/cGMP-systems can also relax smooth muscle cells outside the lower urinary tract, for example in arteries, leading to vasodilatation and increased blood circulation. This is an important hint because it has been postulated that hyperplasia of myocytes, fibrocytes and epithelial cells—as seen in BPH tissue—may be induced by hypoxia resulting from an age-related impairment of blood flow in the small pelvis. Consequently, a key role of urogenital ageing and subsequent alterations in the blood supply of the prostate has been suggested for the development of BPH [108]. Transrectal contrast-enhanced color Doppler ultrasound studies demonstrated that perfusion of the transition zone of the prostate was significantly lower and mean flow resistance index significantly higher in men with BPH than in healthy controls [109]. Thus, it seems likely that regular administration of PDE5i may, to a certain degree, overcomes ischemia due to vascular damage and increase local blood flow by relaxation of smooth muscle cells in pelvic arteries [110].
Phosphodiesterase Type 5 Inhibitors (PDE5i)
PDE5i have a similar structure to cGMP and inhibit the breakdown of NO-derived cGMP molecules by competitively binding the catalytic site of PDE5, thereby causing accumulation of cGMP in the cell (cytoplasm) for continuous activation of the NO/cGMP system and, hence, prolonged (prostatic) muscle cell relaxation (Fig. 8.5).
Four selective oral PDE5i (avanafil, sildenafil, tadalafil and vardenafil) have been licensed in Europe for the treatment of erectile dysfunction (udenafil has only been licensed in Asia) and two PDE5i for the treatment of pulmonary arterial hypertension (sildenafil and tadalafil) [62, 111]. However, only tadalafil has yet been licensed for the treatment of LUTS/BPH, although clinical trials with sildenafil, udenafil and vardenafil in men with LUTS were also conducted and showed similar favorable effects (Table 8.2). The available PDE5i differ primarily in their pharmacokinetic profiles; therefore, clinical differences between these PDE5Is are mainly related to the time to onset and duration of action [62]. All PDE5i are rapidly resorbed from the gastrointestinal tract, have a high protein binding in plasma, are metabolized primarily in the liver and eliminated predominantly into the feces. However, the half-lives of PDE5i differ substantially, reaching from approx. 4 h with sildenafil to more than 17 h with tadalafil. Therefore, tadalafil is suitable for once-daily use (5 mg), whereas other PDE5i must be taken two or three times daily to result in similar intracellular cGMP and PKG concentrations.
Clinical Efficacy of PDE5i
PDE5i were developed to treat pulmonary arterial hypertension and erectile dysfunction [113]. However, a post hoc analysis of patients with erectile dysfunction treated with sildenafil demonstrated that PDE5i was also able to significantly improve concomitant LUTS/BPH and LUTS-related QoL [114, 115]. The improvement of LUTS was independent on the improvement of erectile function. Consequently, RCTs on the efficacy of sildenafil, tadalafil, udenafil and vardenafil were conducted and investigated the key parameters LUTS (IPSS), uroflowmetry (Qmax), and post-void residual urine [79, 116, 117]. Significant LUTS reduction was documented for all PDE5i, and both storage and voiding LUTS decreased during treatment [62, 79, 118]. Symptom bother directly impacts QoL and improvement of storage LUTS is mainly responsible for improved bother and QoL during tadalafil treatment [119]. A Cochrane meta-analysis showed that monotherapy with PDE5i in patients with LUTS/BPH was associated with a significant improvement of IPSS (mean difference compared with placebo −1.9 points) and BPH-Impact Index (mean difference compared with placebo −0.52 points) [120]. Tadalafil was equally effective in reducing LUTS compared to the α1-blocker tamsulosin [121] but treatment satisfaction with tadalafil was significantly greater, most probably due to improved erectile function [122].
Significant effects on LUTS were detected for tadalafil as early as 1 week of treatment [121, 123]. In total, 70% of tadalafil-treated patients had a clinically relevant LUTS reduction of ≥3 IPSS points; 60% of responders passed this threshold already after 1 week and 80% after 4 weeks of treatment [123]. Significant and continuous LUTS reduction was documented for 52 weeks in an open 12-month trial [124]. On the standardized and validated 15-item International Index of Erectile Function (IIEF) questionnaire, tadalafil treatment resulted in improvements of erectile function of 6.0 points (IIEF-questions 1–5 + 15; range 0–30), ejaculation of 0.8 points (IIEF-9; range 0–5), orgasmic function of 1.5 points (IIEF-questions 9 + 10; range 0–10), satisfaction with sexual intercourse of 2.3 points (IIEF 6 + 7 + 8; range 0–15) and overall sexual satisfaction of 2.0 points (IIEF-questions 13 + 14; range 0–10), whereas the IIEF domains remained stable or even deteriorated with tamsulosin [125]. Qmax was not consistently improved in the individual studies but a post hoc analysis of all tadalafil trials demonstrated a significant mean increase of 1.1 mL/s with tadalafil 5 mg once daily compared to 0.4 mL/s with placebo [126].
PDE5i have no effects on prostate volume and prostate volume does not affect treatment response or efficacy of PDE5i. Post-void residual urine remains unchanged during PDE5i treatment. Efficacy with tadalafil differed between patients aged <75 vs. ≥75 years, with significant efficacy only in the <75-year age group. The older age group suffered of more concomitant diseases and used more drugs which seem to reduce tadalafil efficacy [127]. Patients with a history of more than one drug for arterial hypertension have a significantly lower LUTS response compared to men without; otherwise, cardio-vascular risk factors/comorbidities do not affect the magnitude of LUTS/BPH improvement during tadalafil treatment [128]. Placebo-adjusted least squares (LS) mean improvements in total IPSS were −1.2 in men using >1 antihypertensive drugs vs. −3.3 in men using only ≤1 antihypertensive drug.
Only a few trials with durations of 6–12 weeks compared the efficacy of PDE5i or α1-blocker monotherapies against PDE5i + α1-blocker combination therapy. The drug combination frequently improved IPSS, Qmax, and post-void residual urine to a greater extent than the single drug class alone. A meta-analysis on PDE5i trials demonstrated significantly improved LUTS/BPH (IPSS) when using α1-blocker + PDE5i combination vs. α1-blocker or PDE5i alone but the differences between mono- and combination therapies were small [118]. Therefore, it is still debatable whether combination therapy is justified in routine patients with LUTS/BPH, especially when considering the price for both drugs.
The drug combination of tadalafil (PDE5i) + finasteride (5ARI) was significantly more efficacious in reducing LUTS/BPH (IPSS) than finasteride alone after 6 months; IIEF-EF improved only in the tadalafil arm by 4.7 points, whereas IIEF-EF remained unchanged with finasteride monotherapy [129]. Treatment satisfaction at week 26 was significantly greater with tadalafil/finasteride combination therapy compared to finasteride monotherapy for the total treatment satisfaction scale score and satisfaction with efficacy subscore [130].
Adverse Event Profile of PDE5i
All PDE5i may cause headache, flushing, dizziness, dyspepsia, nasal congestion, myalgia, hypotension, syncope, tinnitus or conjunctivitis [62]. PDE3 hydrolyzes cAMP and is mainly found in cardiomyocytes but also in the corpora cavernosa. PDE5i can cross-react with PDE3 and increase the level of cAMP in the heart, thereby increasing the heart rate with a positive inotropic effect. In this respect, PDE5i can cause myocardial infarction in patients with coronary heart disease, especially in those using nitrates or potassium channel openers. The probability of developing priapism or AUR during PDE5i therapy is very low [62]. PDE5i can also cross-react with PDE6 in the cones and rods of retina. Inhibition of PDE6 may cause visual adverse effects, such as chromatopsia or blurred vision, as reported by patients using sildenafil or udenafil [131]. A systematic review and meta-analysis demonstrated that adverse events during PDE5i treatment are generally mild [132]. However, the frequencies of these adverse events vary between different PDE5i. Tadalafil typically causes headache, back pain, dizziness and dyspepsia in 2–3% of patients [121].
PDE5i are contraindicated in patients using nitrates or the potassium channel opener nicorandil due to additional vasodilatation, which might cause arterial hypotension, myocardial ischemia in patients with coronary artery disease, or cerebrovascular strokes [62, 133]. Additionally, all PDE5i should not be used in patients who use immediate-release doxazosin or terazosin, have unstable angina pectoris, recently had a myocardial infarction (previous 3 months) or stroke (previous 6 months), have myocardial insufficiency NYHA >2, arterial hypotension, poorly controlled blood pressure, significant hepatic or renal insufficiency, or if non-arteritic anterior ischemic optic neuropathy (NAION) with sudden loss of vision is known or has appeared during previous use of PDE5i. Sildenafil and vardenafil are also contraindicated in patients with retinitis pigmentosa. Caution is advised when PDE5i are used together with other drugs which are metabolized by the same hepatic elimination pathway (CYP3A4), because these drugs can cause an increased serum concentration of PDE5i.
Androgens
The development of the prostate in the embryogenic period, prostate growth during puberty and prostate enlargement in elderly men with BPH are only possible with testicular androgens. The prostate is only rudimentary developed and remains free of BPH tissue in men with congenital hypopituitarism, congenital androgen-receptor defect, autosomal-recessive 5α-reductase deficiency (male pseudo-hermaphroditism) or castration before puberty. The prostate and seminal vesicales in these men are small or even missing, and prostate biopsies only show fibromuscular stroma without epithelial cells or glands. PSA is usually not detectable in the prostate, ejaculate or serum in the genetic diseases or are low in patients with early castration.
Testosterone is the predominant androgen in men. The active metabolite of testosterone is dihydrotestosterone (DHT) which results from 5α-reduction in the cytoplasm of the prostatic epithelial cells (Fig. 8.6). DHT has a stronger affinity to the nuclear bound androgen receptor and a 4- to 5-times higher androgenic potency than testosterone [135]. DHT accounts for approximately 90% of prostatic androgens. 5α-reductase, a NADPH-dependent enzyme, converts testosterone to DHT and exists in two isoforms [134, 136]:
-
5α-Reductase type 1 is encoded on chromosome 5 and has only minor expression and activity in the prostate but predominant activity in extra-prostatic tissues, such as skin and liver.
-
5α-Reductase type 2 is encoded on chromosome 2, is predominantly expressed and active in the stromal and basal cells of the prostate, and also exists in hair follicles as well as in cells of the seminal vesicles. The conversion of testosterone to DHT by 5α-reductase type 2 is 25-times faster than by 5α-reductase type 1.

The active metabolite of testosterone is dihydrotestosterone which results from 5α-reduction inside the prostatic epithelial cell. The dihydrotestosterone-androgen receptor complex initiates gene transcription resulting in protein and growth hormone synthesis as well as cell proliferation. 5α-Reductase type 2 inhibitors (5ARIs; dutasteride or finasteride) inhibit the conversion from testosterone to dihydrotestosterone and initiate apoptosis [134]
After synthesis of DHT, the active androgen binds to the androgen receptor on the nuclear membrane and migrates into the cell nucleus (together with a transportation complex), where the DHT-androgen receptor complex stimulates gene transcription which results in protein and growth hormone synthesis (e.g. PSA, epidermal growth factor [EGF] and vascular endothelial growth factor [VEGF]) as well as cell proliferation. Testosterone and DHT are important in the physiology of the muscle, fatty tissue, liver, bone, central nervous system as well as reproductive and sexual functions.
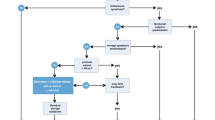
Hormonal and especially anti-androgenic therapies have a long tradition in the treatment of prostatic diseases. It is known since the end of the nineteenth century that androgen withdrawal by castration leads to shrinkage of the prostate [137, 138]. Molecular studies in men with androgen deprivation therapy (ADT) demonstrated that epithelial cells of the prostate induce a genetic mechanism which is characterized by enzymatic splitting of the DNA and irreversible cell death (apoptosis), depletion of epithelial cells and decrease in prostate volume. ADT is possible with bilateral surgical orchiectomy, synthetic anti-androgens, LHRH-agonists or -antagonists, estrogens or anti-estrogens. ADT is used for the treatment of advanced prostate cancer. Different drugs interfere in the gonadotropin-testosterone axis and initiate apoptosis of hormone-sensitive prostate cancer cells (Fig. 8.7). ADT results in shrinkage of the prostate but, however, also decrease of the serum testosterone concentration. Therefore, severe and bothersome adverse events resulting from androgen deficiency appear frequently and are only acceptable during the treatment of advanced prostate cancer. Libido loss typically emerges during treatment with LHRH-agonists or -antagonists, steroidal anti-androgens and estrogens; hot flushes and painful nipples typically appear during treatment with anti-androgens and estrogens; cardiovascular events (e.g. myocardial infarction) are frequently seen during treatment with estrogens or LHRH-agonists, and gastro-intestinal complaints or liver toxicity are observed during treatment with non-steroidal anti-androgens.

Androgen deprivation therapy for the treatment of advanced prostate cancer. Different drugs or drug classes interfere with the gonadotropin-androgen axis to inhibit growth of androgen-sensitive prostate cancer cells (modified according to Raja et al. [139]). 5ARI 5α-reductase inhibitor, ACTH adrenocorticotropic hormone, DHT dihydrotestosterone, T/AR testosterone/androgen receptor, LH luteinizing hormone, LH-RH luteinizing hormone-releasing hormone, NSAA non-steroidal antiandrogen (e.g. flutamide, nilutamide, bicalutamide, apalutamide, enzalutamide), SAA steroidal antiandrogen (e.g. cyproterone acetate)
Only local blockade of the DHT synthesis inside prostatic cells by selective 5α-reductase inhibitors (5ARIs) is a viable treatment option of LUTS/BPH. Thus, selective inhibition of intracellular 5α-reductase can prevent systemic (anti-)hormonal adverse events. Although testosterone concentrations in the serum and prostate even increase up to 10% during 5ARI therapy [140], the elevated concentrations are functionally irrelevant because testosterone is 5-times less potent in the prostate than DHT. Serum gonadotropin concentrations during 5ARI treatment remain unchanged which validates the hypothesis that the negative feedback on the secretion of LHRH in the hypothalamus and LH in the hypophysis is regulated by testosterone and not DHT [141]. Normal serum testosterone concentration is responsible for the preservation of libido and sexual function of patients treated with 5ARIs [142].
5α-Reductase Inhibitors (5ARIs)
Two 5ARIs are commercially available for the treatment of LUTS/BPH: dutasteride and finasteride. The pharmacokinetic profiles of the 5ARIs are listed in Table 8.3. Additionally, finasteride in a lower dose (1 mg once daily) has been licensed for the treatment of hair loss (androgenetic alopecia); dutasteride has only been licensed for this indication in Japan and South Korea but not in North America or Europe, as of 2018. Finasteride inhibits only 5α-reductase type 2, whereas dutasteride inhibits 5α-reductase types 1 and 2 with similar potency (dual 5α-reductase inhibitor). Continuous long-term treatment with 5ARIs reduces serum DHT concentration by approximately 70% with finasteride and 95% with dutasteride [143]. However, intra-prostatic DHT concentrations are reduced to a similar level (85–90%) by both 5ARIs. Therefore, the clinical role of dual 5α-reductase inhibition remains unclear. Both 5α-reductase inhibitors are metabolized in the liver and excreted into the feces. The elimination half-time is longer for dutasteride (3–5 weeks) than finasteride (6–8 h); consequently, serum DHT concentration is still reduced after >16 weeks after cessation of dutasteride treatment, whereas serum DHT concentration returned to normal within 4 weeks after finasteride use [144].
Clinical Efficacy of 5ARIs
5ARIs primarily reduce prostate volume by apoptosis of prostatic epithelial cells. Significant differences of prostate volumes in patients treated with 5ARIs compared to placebo were already seen as early as 1 month of treatment with dutasteride or finasteride. The transition, central and peripheral zones shrink equally [145]. Prostate volume reduction (cm3) is more pronounced in patients with greater volumes at baseline but volume reduction as percentage seems to be identical in patients with small, medium and large prostates [62]. Prostate volume continuously decreases within the first year of treatment with 5ARIs but, thereafter, remains more or less stable. After continuous 5ARI treatment >1 year, the prostate has lost ~18–28% of its original volume. Prostate volume reduction with 5ARIs is similar to the reduction seen with ADT (castration level) [146].
Clinically relevant effects secondary to prostate volume reduction and relative to placebo are only seen after minimum treatment duration of at least 6–12 months. After treatment for 2–4 years, 5ARIs reduce LUTS (IPSS) by approximately 15–30% and increase Qmax of free uroflowmetry by approximately 1.5–2.0 mL/s in patients with LUTS/BPE [62]. LUTS reduction by finasteride depends on initial prostate volume and may not be more efficacious than placebo in patients with prostates <40 cm3 [147]. Placebo effects appeared more frequently and were more pronounced in patients with prostate volumes <40 cm3 [148]. Similar meta-analyses have not been conducted with dutasteride but a significant IPSS reduction and Qmax increase were also documented with prostate volumes between 30 and 40 cm3 [149, 150]. Indirect comparison between individual studies and one direct comparative trial indicate that dutasteride and finasteride are equally effective in the treatment of LUTS/BPH [150,151,152,153].
Comparative studies with α1-blockers demonstrated that 5ARIs reduce symptoms more slowly and, for finasteride, less effectively [81, 154,155,156]. A long-term trial with dutasteride in symptomatic men with prostate volumes >30 cm3 (CombAT trial, mean prostate volume 55 cm3) showed that 5ARIs reduced LUTS at least as much or even more effectively than the α1-blocker tamsulosin [75, 157]. The greater the prostate volume (or the proxy parameter serum PSA-concentration) at baseline, the faster and more pronounced the symptomatic benefit of 5ARIs. IPSS reduction was significantly greater in men with a baseline prostate volume of ≥58 cm3 (PSA ≥4.4 ng/mL) at treatment month 15 or later compared to lower prostate volumes or PSA concentrations. Patient age (<65 years vs. ≥65 years) had no influence on the efficacy or tolerability of 5ARIs [158], but a prostate median lobe protruding into the bladder had a significant impact on efficacy [159]. In the latter study, patients with a comfortably full bladder were investigated by transabdominal ultrasound and the distance between bladder base and tip of the prostate median lobe (i.e. intravesical prostatic protrusion, IPP) was measured. 82 men had IPP <10 mm (IPP grades 1 and 2) and 29 men IPP ≥10 mm (IPP grade 3) [160]. After treatment for 26 months with dutasteride, significant improvements concerning LUTS (IPSS), Qmax, post-void residual and serum PSA concentration were only seen in the group with IPP grades 1–2. Patients with IPP grade 3 even significantly deteriorated (IPSS and Qmax). It was concluded that patients with a prominent prostate median lobe are poor candidates for 5ARI therapy.
AUR occurs in 18.3–35.9 symptomatic men/1000 patient-years [76]. Risk factors for AUR (MTOPS study data) are age (>62 years), PSA (>1.6 ng/mL), prostate volume (>31 cm3), Qmax (<10.6 mL/s), IPSS (>17 points) and post-void residual urine (>39 mL) [81]. Identical risk factors but slightly different threshold values for AUR were determined in the US-American Olmsted County Study and the German Herne-LUTS Study [161, 162]. Lately, the protrusion of the prostate median lobe into the bladder has been identified as another risk factor for AUR; patients with IPP ≥10 mm had a cumulative incidence rate for AUR or need for surgery during 3-year follow-up of 71.5% compared to 9.9% in patients with IPP <10 mm [159]. 5ARIs but not α1-blockers are able to reduce the long-term (>1 year) risk of AUR or need for surgery [74, 81, 163, 164]. Prevention of disease progression by 5ARIs is already detectable after 4 months and also in men with prostate volumes considerably smaller than 40 cm3 [75, 157]. The precise mechanism of action of 5ARIs in reducing disease progression is still unknown but it is most likely attributable to reduction of bladder outlet resistance due to shrinkage of the prostate and widening the prostatic urethra [62]. Accordingly, open-label computer-urodynamic trials demonstrated relevant reductions of voiding parameters in patients with long-term finasteride treatment (≥3 years) [165, 166]. The relative risk reduction with 5ARIs compared to placebo is approximately 50–67% for AUR and 30–64% for prostate surgery [74, 167, 168]. Therefore, 5ARIs should be used if prevention of disease progression is intended.
Combination therapy of α1-blockers together with 5ARI aims to couple the beneficial effects of both drug classes, i.e. fast symptom reduction within days or weeks with the α1-blocker and prevention of disease progression with the 5ARI. Initial studies with follow-up periods between 6 and 12 months consistently demonstrated that the α1-blocker was superior to finasteride in symptom reduction, whereas the combination treatment was not superior to the α1-blocker alone. In studies with a placebo arm, the α1-blocker was consistently more effective than placebo, whereas finasteride was not [154,155,156]. However, long-term combination therapy, as shown for finasteride + doxazosin in the 4-year MTOPS trial and dutasteride + tamsulosin in the 4 year CombAT trial, is superior to either monotherapy with regards to reduction of LUTS/BPH (IPSS), increase of Qmax and reduction of the risk of disease progression [75, 81, 157]. CombAT showed that combination treatment was superior to either monotherapy with regards to improvement of LUTS/BPH and Qmax starting from month 9 and superior to the α1-blocker with regards to the reduction of the risk of AUR as well as need for surgery after month 8 [75]. 5ARIs alone reduced prostate volume as effectively as combination treatment (−20 to −27%). Combination therapy was superior to monotherapy in both the MTOPS and CombAT trials in preventing overall clinical progression, as defined by IPSS increase ≥4 points, AUR, urinary tract infection, incontinence, or an increase in serum creatinine >50% compared to baseline values. For combination therapy in the MTOPS vs. CombAT trials, the following relative risk reductions were observed [62]:
-
Overall risk of disease progression: −66% vs. −44%
-
Symptomatic progression: −64% vs. −41%
-
AUR: −81% vs. −68%
-
urinary incontinence: −65% vs. −26%
-
BPH-related surgery: −67% vs. −71%
Nevertheless, monotherapy with 5ARIs reduced the risk of AUR and prostate-related surgery as effectively as combination therapy (differences not significant), although the prevention was more pronounced with combination therapy. These two long-term trials indicated that α1-blocker +5ARI combination treatment should be initiated in patients with moderate-to-severe LUTS, baseline characteristics of disease progression and when long-term treatment (>12 months) is intended [62].
Short-term use of 5ARIs (4 weeks) before scheduled prostate operations (e.g. TURP) can reduce microvessel density of prostatic tissue and, therefore, can decrease total blood loss, blood loss per gram resected prostate tissue, blood transfusions and concentration of vascular endothelial growth factor (VEGF, see above). A meta-analysis of 17 RCTs with a total of 1489 patients confirmed these beneficial effects for finasteride [169]. However, short-term use of finasteride was not able to not reduce the operation time, prostate volume or resected gland weight. A single center study recently confirmed these results for dutasteride [170].
5ARIs reduce serum PSA concentration by approximately 50% after 6–12 months of treatment [151]. Nevertheless, serum PSA-concentration can still be used for prostate cancer screening if the measurement results are multiplied by 2 [171]. The calculated values of patients with 5ARIs have a similar sensitivity (66%) and specificity (82%) during long-term treatment compared to patients without 5ARIs treatment [172]. However, the speed of serum PSA reduction varies between individual patients and, therefore, the false-positive rate of increased PSA concentration (>4 ng/mL) is slightly higher during the first year of treatment (35%) compared to the time afterwards (25%).
Adverse Event Profile of 5ARIs
The most relevant adverse events of 5ARIs are related to sexual function and include reduced libido, erectile dysfunction and, less frequently, ejaculation disorders, such as retrograde ejaculation, ejaculation failure, or decreased semen volume [75, 81, 151]. The incidence of sexual dysfunction and other adverse events was low in the individual trials and even decreased with trial duration. At least four meta-analyses have been published to evaluate sexual dysfunction associated with 5ARI treatment for LUTS/BPH or androgenetic alopecia [173,174,175,176]. The relative risks for sexual dysfunction in general and erectile dysfunction or decreased libido in particular were significantly increased for 5ARI users. The risk was higher in men with LUTS/BPH than with alopecia, most probably due to the lower 5ARI doses used in alopecia patients [176]. Ejaculation disorders were significantly more common with 5ARIs than with placebo (odds ratio 2.73), and both dutasteride (odds ratio 2.81) and finasteride (odds ratio 2.70) increased this risk [175]. Combination treatment of 5ARI + α1-blocker significantly increased the risk of ejaculation disorders compared to α1-blocker alone (odds ratio 3.75) or 5ARI alone (odds ratio 2.76). Meta-analyses calculated a significantly increased risk for hypoactive sexual desire (odds ratio 1.54) and erectile dysfunction (odds ratio 1.47) in patients using 5ARIs [173]. Some patients even reported persisting erectile dysfunction and diminished libido for more than 10 years after cessation of finasteride treatment (post-finasteride syndrome). A causal relationship between 5ARI treatment and prolonged sexual dysfunction is possible but not well understood. EMA and FDA warnings have been published on their homepages accordingly.
Other adverse events are gynecomastia (breast enlargement with breast or nipple tenderness in approximately 1–2% of patients), depression (with suicidal thoughts) and anxiety disorders. The psychiatric adverse events are a matter of EMA and FDA warnings and currently under closer investigation.
References
Berry SJ, Coffey DS, Walsh PC, Ewing LL. The development of human benign prostatic hyperplasia with age. J Urol. 1984;132:474–9.
McNeal JE. The prostate and prostatic urethra: a morphologic synthesis. J Urol. 1972;107:1008–16.
McNeal JE, Bostwick DG. Anatomy of the prostatic urethra. JAMA. 1984;251:890–1.
McNeal JE. Regional morphology and pathology of the prostate. Am J Clin Pathol. 1968;49:347–57.
McNeal JE. The zonal anatomy of the prostate. Prostate. 1981;2:35–49.
Roehrborn CG, McConnell JD. Etiology, pathophysilogy, epidemiology, and natural history of benign prostatic hyperplasia. In: Walsh PC, Retik AB, Vaughan ED, Wein AJ, editors. Campbell’s urology. 8th ed. Philadelphia: Saunders; 2002. p. 1297–336.
Villers A, Terris MK, McNeal JE, Stamey TA. Ultrasound anatomy of the prostate: the normal gland and anatomical variations. J Urol. 1990;143:732–8.
Hricak H, Dooms GC, McNeal JE, et al. MR imaging of the prostate gland: normal anatomy. AJR Am J Roentgenol. 1987;148:51–8.
Sommer FG, McNeal JE, Carrol CL. MR depiction of zonal anatomy of the prostate at 1.5 T. J Comput Assist Tomogr. 1986;10:983–9.
Reese JH, McNeal JE, Redwine EA, Stamey TA, Freiha FS. Tissue type plasminogen activator as a marker for functional zones, within the human prostate gland. Prostate. 1988;12:47–53.
Reese JH, McNeal JE, Redwine EA, Samloff IM, Stamey TA. Differential distribution of pepsinogen II between the zones of the human prostate and the seminal vesicle. J Urol. 1996;136:1148–52.
McNeal JE. Origin and development of carcinoma in the prostate. Cancer. 1969;23:24–34.
McNeal JE. Origin and evolution of benign prostatic enlargement. Invest Urol. 1978;15:340–5.
Abrams P. New words for old: lower urinary tract symptoms for “prostatism”. BMJ. 1994;308(6934):929–30.
D’Ancona C, Haylen B, Oelke M, et al. Standardisation Steering Committee ICS and the ICS Working Group on Terminology for Male Lower Urinary Tract & Pelvic Floor Symptoms and Dysfunction. The International Continence Society (ICS) report on the terminology for adult male lower urinary tract and pelvic floor symptoms and dysfunction. Neurourol Urodyn. 2019;38:433–77.
Frick J, Aulitzky W. Physiology of the prostate Infection. 1991;19(Suppl. 3):S115–8.
Grayhack JT, Lee C, Oliver L, Schaeffer AJ, Wendel EF. Biochemical profiles of prostatic fluid from normal and diseased prostate glands. Prostate. 1980;1:227–37.
Issa MM, Fenter TC, Black L, et al. An assessment of the diagnosed prevalence of diseases in men 50 years of age or older. Am J Manag Care. 2006;12:S83–9.
de Groat WC, Booth AM, Yoshimura N. In: Maggi CA, editor.. The autonomic nervous system. Nervous control of the urogenital system Neurophysiology of micturition and it’s modification in animal models of human disease. London: Harwood Academic Publishers; 1993. p. 227–89.
Lefkowitz RJ, Hoffmann BB, Taylor P. Neurohumoral transmission: the autonomic and somatic motor nervous system. In: Goodman AG, Wall TH, Nies AS, Taylor P, editors. Goodman and Gillmans pharmacological basis of therapeutics. New York: Pergamon Press; 1990. p. 84–121.
Oelke M, Höfner K, Berges RR, Jonas U. Pharmacological treatment of the benign prostatic syndrome (symptomatic BPH) using alpha1-adrenoceptor antagonists Basic principles and clinical results. Urologe A. 2002;41:425–41.
Langer SZ. History and nomenclature of alpha1-adrenoceptors. Eur Urol. 1999;36(Suppl. 1):2–6.
Schwinn DA. Novel role for alpha1-adrenergic receptor subtypes in lower urinary tract symptoms. BJU Int. 2000;86(Suppl. 2):11–20.
Learmonth JR. A contribution to the neurophysiology of the urinary bladder in man. Brain. 1931;54:147–76.
Caine M, Raz S, Zeigler M. Adrenergic and cholenergic receptors in the human prostate, prostatic capsule and bladder neck. Br J Urol. 1975;47:193–202.
Chapple CR, Aubry ML, James S, et al. Characterisation of human prostatic adrenoceptors using pharmacology receptor binding and localisation. Br J Urol. 1989;63:487–96.
Hedlund H, Andersson KE, Larsson B. Alpha-adrenoceptors and muscarinic receptors in the isolated human prostate. J Urol. 1985;134:1291–8.
Kobayashi S, Tang R, Shapiro E, Lepor H. Characterization and localization of prostatic alpha 1 adrenoceptors using radioligand receptor binding on slide-mounted tissue section. J Urol. 1993;150:2002–6.
Lepor H, Shapiro E. Characterization of alpha1 adrenergic receptors in human benign prostatic hyperplasia. J Urol. 1984;132:1226–9.
Shapiro E, Lepor H. Alpha 2 adrenergic receptors in hyperplastic human prostate: identification and characterization using [3H] rauwolscine. J Urol. 1986;135:1038–42.
Drescher P, Eckert RE, Sparwasser C, Madsen PO. Alpha-1 receptor mediated smooth muscle regulation in benign prostatic hyperplasia. Scand J Urol Nephrol Suppl. 1994;157:33–40.
Gup DI, Shapiro E, Baumann M, Lepor H. Autonomic receptors in human prostate adenomas. J Urol. 1990;143:179–85.
Hieble JP, Caine M, Zalaznik E. In vitro characterization of the alpha-adrenozeptors in human prostate. Eur J Pharmacol. 1985;107:111–7.
Kitada S, Kumazawa J. Pharmacological characteristics of smooth muscle in benign prostatic hyperplasia and normal prostatic tissue. J Urol. 1987;138:158–60.
Lepor H, Tang R, Shapiro E. The alpha-adrenoceptor subtype mediating the tension of human prostatic smooth muscle. Prostate. 1993;22:301–7.
Kawabe K, Moriyama N, Hamada K, Ishima T. Density and localization of alpha-1-adrenoceptors in hypertrophied prostate. J Urol. 1990;143:592–5.
Yamada S, Ashizawa N, Ushijima H, et al. Alpha-1 adrenoceptors in human prostate: characterization and alteration in benign prostatic hypertrophy. J Pharmacol Exp Ther. 1987;242:326–30.
Bylund DB, Eikenberg DC, Hieble JP, et al. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev. 1994;46:121–36.
Graham RM, Perez DM, Hwa J, Piascik MT. Alpha 1-adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ Res. 1996;78:737–49.
Michel MC, Kenny B, Schwinn DA. Classification of alpha1-adrenoceptor subtypes. Naunyn Schmiedeberg Arch Pharmacol. 1995;352:1–10.
Forray C, Bard JA, Wetzel JM, et al. The alpha 1-adrenergic receptor that mediates smooth muscle contraction in human prostate has the pharmacological properties of the cloned human alpha 1c subtype. Mol Pharmacol. 1994;45:703–8.
Hieble JP, Bylund DB, Clarke DE, et al. International Union of Pharmacology. X. Recommondation for nomenclature of alpha 1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–70.
Michel MC, Grubbel B, Taguchi K, et al. Drugs for treatment of benign prostatic hyperplasia: affinity comparison at cloned alpha 1-adrenoceptor subtypes and in human prostate. J Auton Pharmacol. 1996;16:21–8.
Nasu K, Moriyama N, Kawabe K, et al. Quantification and distribution of 1-adrenoceptor subtype mRNAs in human prostate: comparison of benign hypertrophied tissue and non-hypertrophied tissue. Br J Pharmacol. 1996;119:797–803.
Hatano A, Takahashi H, Tamaki M, et al. Pharmacological evidence of distinct alpha 1-adrenoceptor subtypes mediating the contraction of human prostatic urethra and peripherial artery. Br J Pharmacol. 1994;113:723–8.
Kohno Y, Saito H, Takita M, Kigoshi S, Muramatsu I. Heterogeneity of alpha 1-adrenoceptor subtypes involved in adrenergic contractions of dog blood vessels. Br J Pharmacol. 1994;112:1167–73.
Andersen K. alpha1-adrenoceptors and bladder function. Eur Urol. 1999;36(Suppl 1):96–102.
Kaplan PE, Nanninga JB. Reduction of bladder contractility after alpha-adrenergic blockade and after ganglionic blockade. Acta Neurol Scand. 1979;59:172–7.
Price D. Potential mechanisms of action of superselective alpha(1)-adrenoceptor antagonists. Eur Urol. 2001;40(Suppl. 4):5–11.
Shapiro E, Hartanto V, Lepor H. Quantifying the smooth muscle content of the prostate using double-immunoenzymatic staining and color assisted image analysis. J Urol. 1992;147:1167–70.
Appell RA, England HR, Hussell AR, McGuire EJ. The effects of epidural anesthesia on the urethral closure pressure profile in patients with prostatic enlargement. J Urol. 1980;124:410–1.
Donker PJ, Ivanovici F, Noach EL. Analyses of the urethral pressure profile by means of electromyography and the administration of drugs. Br J Urol. 1972;44:180–93.
Furuya S, Kumamoto Y, Yokoyama E, et al. Alpha-adrenergic activity and urethral pressure in prostatic zone in benign prostatic hypertrophy. J Urol. 1982;128:836–9.
Fusco F, Palmieri A, Ficarra V, et al. α1-Blockers improve benign prostatic obstruction in men with lower urinary tract symptoms: a systematic review and meta-analysis of urodynamic studies. Eur Urol. 2016;69:1091–101.
Caine M, Perlberg S, Meretyk S. A placebo-controlled double-blind study of the effect of phenoxybenzamine in benign prostatic obstruction. Br J Urol. 1978;50:551–4.
Caine M, Perlberg S, Shapiro A. Phenoxybenzamine for benign prostatic obstruction. Review of 200 cases. Urology. 1981;17:542–6.
Milroy E. Clinical overview of prazosin in the treatment of prostatic obstruction. Urol Int. 1990;45(Suppl 1):1–3.
Elliott WJ, Black HR. Treatment of hypertension in the elderly. Am J Geriatr Cardiol. 2002;11:11–20.
Lasagna L. Diuretics vs alpha-blockers for treatment of hypertension: lessions from ALLHAT. Antihyperensive and lipid-lowering treatment to prevent heart attack trial. JAMA. 2000;283:2013–4.
Miller JL. Doxazosin dropped from ALLHAT study. Am J Health Syst Pharm. 2000;57:718.
Poulter N, Williams B. Doxazosin for the management of hypertension: implications of the findings of the ALLHAT trial. Am J Hypertens. 2001;14:1170–2.
Oelke M, Bachmann A, Descazeaud A, et al. EAU guidelines on the treatment and follow-up of non-neurogenic male lower urinary tract symptoms, including benign prostatic obstruction. Eur Urol. 2013;64:118–40.
Michel MC, Chapple CR. Comparison of the cardiovascular effects of tamsulosin oral controlled absorption system (OCAS) and alfuzosin prolonged release (XL). Eur Urol. 2006;49:501–8.
Stevens HN, Speakman M. Behaviour and transit of tamsulosin oral controlled absorption system in the gastrointestinal tract. Curr Med Res Opin. 2006;22:2323–8.
van Kerrebroeck PE. The efficacy and safety of a new once-a-day formulation of an alpha-blocker. Eur Urol. 2001;39(Suppl. 6):19–26.
Thom S, Poulter N, Field J, et al. UMPIRE Collaborative Group. Effects of a fixed-dose combination strategy on adherence and risk factors in patients with or at high risk of CVD: the UMPIRE randomized clinical trial. JAMA. 2013;310:918–29.
Narayan P, Tewari A. Overview of alpha-blocker therapy for benign prostatic hyperplasia. Urology. 1998;51(4A Suppl):38–45.
Michel MC, Mehlburger L, Bressel HU, et al. Comparison of tamsulosin efficacy in subgroups of patients with lower urinary tract symptoms. Prostate Cancer Prostatic Dis. 1998;1:332–5.
Debruyne FM. Alpha blockers: are all created equal? Urology. 2000;56(5 Suppl 1):20–2.
Djavan B, Chapple C, Milani S, et al. State of the art on the efficacy and tolerability of alpha1-adrenoceptor antagonists in patients with lower urinary tract symptoms suggestive of benign prostatic hyperplasia. Urology. 2004;64:1081–8.
Yuan JQ, Mao C, Wong SY-S, et al. Comparative effectiveness and safety of monodrug therapies for lower urinary tract symptoms associated with benign prostatic hyperplasia: a network meta-analysis. Medicine (Baltimore). 2015;94:e974.
Becopoulos T, Mitropoulos D, Christofis I. Influence of prostate size on terazosin efficacy. Int J Urol. 1997;4:358–61.
Lepor H, Nieder A, Feser J, O’Connell C, Dixon C. Effect of terazosin on prostatism in men with normal and abnormal peak urinary flow rates. Urology. 1997;49:476–80.
McConnell JD, Bruskewitz R, Walsh P, et al. The effect of finasteride on the risk of acute urinary retention and the need for surgical treatment among men with benign prostatic hyperplasia. N Engl J Med. 1998;338:557–63.
Roehrborn CG, Siami P, Barkin J, et al. The effects of combination therapy with dutasteride and tamsulosin on clinical outcomes in men with symptomatic benign prostatic hyperplasia: 4-Year results from the CombAT study. Eur Urol. 2010;57:123–31.
Oelke M, Speakman MJ, Desgrandchamps F, Mamoulakis C. Acute urinary retention rates in the general male population and in adult men with lower urinary tract symptoms participating in pharmacotherapy trials: a literature review. Urology. 2015;86:654–65.
Fisher E, Subramonian K. Omar MI. The role of alpha blockers prior to removal of urethral catheter for acute urinary retention in men. Cochrane Database Syst Rev. 2014;10:CD006744.
Bechara A, Romano S, Casabé A, et al. Comparative efficacy assessment of tamsulosin vs. tamsulosin plus tadalafil in the treatment of LUTS/BPH. Pilot study. J Sex Med. 2008;5:2170–8.
Gacci M, Vittori G, Tosi N, et al. A randomized, placebo-controlled study to assess safety and efficacy of vardenafil 10 mg and tamsulosin 0.4 mg vs. tamsulosin 0.4 mg alone in the treatment of lower urinary tract symptoms secondary to benign prostatic hyperplasia. J Sex Med. 2012;9:1624–33.
Kaplan SA, Gonzalez RR, Te AE. Combination of alfuzosin and sildenafil is superior to monotherapy in treating lower urinary tract symptoms and erectile dysfunction. Eur Urol. 2007;51:1717–23.
McConnell JD, Roehrborn CG, Bautista O, et al. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N Engl J Med. 2003;349:2387–98.
Kaplan SA, Roehrborn CG, Rovner ES, et al. Tolterodine and tamsulosin for treatment of men with lower urinary tract symptoms and overactive bladder. JAMA. 2006;296:2319–28.
Kerrebroeck v, Haab F, Angulo J, et al. Efficacy and safety of solifenacin plus tamsulosin OCAS™ in men with voiding and storage LUTS: results from a phase 2, dose-finding study (SATURN). Eur Urol. 2013;64:398–407.
Ichihara K, Masumori N, Fukuta F, et al. A randomized controlled study of the efficacy of tamsulosin monotherapy and its combination with mirabegron for overactive bladder induced by benign prostatic obstruction. J Urol. 2015;2015(193):921–6.
Oelke M, Gericke A, Michel MC. Cardiovascular and ocular safety of α1-adrenoceptor antagonists in the treatment of male lower urinary tract symptoms. Expt Opin Drug Saf. 2014;13:1187–97.
Barendrecht MM, Koopmans RP, de la Rosette JJ, et al. Treatment for lower urinary tract symptoms suggestive of benign prostatic hyperplasia: the cardiovascular system. BJU Int. 2005;95(Suppl. 4):19–28.
Nickel JC, Sander S, Moon TD. A meta-analysis of the vascular-related safety profile and efficacy of a-adrenergic blockers for symptoms related to benign prostatic hyperplasia. Int J Clin Pract. 2008;62:1547–59.
Chapple CR, Montorsi F, Tammela TLJ, et al. Silodosin therapy for lower urinary tract symptoms in men with suspected benign prostatic hyperplasia: results of an international, randomized, double-blind, placebo- and active-controlled clinical trial performed in Europe. Eur Urol. 2011;59:342–52.
Choi WS, Cho MC, Lee JW, et al. Efficacy and safety of silodosin in the treatment of lower urinary tract symptoms in elderly men taking antihypertensive medications. Prostate Int. 2017;5:113–8.
Chang DF, Campbell JR. Intraoperative floppy iris syndrome associated with tamsulosin. J Cataract Refract Surg. 2005;31:664–73.
Chatziralli IP, Sergentanis TN. Risk factors for intraoperative floppy iris syndrome: a meta-analysis. Opthalmology. 2011;118:730–5.
van Dijk MM, de la Rosette JJ, Michel MC. Effects of a1-adrenoceptor antagonists on male sexual function. Drugs. 2006;66:287–301.
Roehrborn CG, Kaplan SA, Lepor H, Volinn W. Symptomatic and urodynamic responses in patients with reduced or no seminal emission during silodosin treatment for LUTS and BPH. Prostate Cancer Prostatic Dis. 2011;14:143–8.
Jung JH, Kim J, MacDonald R, et al. Silodosin for the treatment of lower urinary tract symptoms in men with benign prostatic hyperplasia. Cochrane Database Syst Rev. 2017;(11):CD012615.
Kawabe K, Yoshida M, Homma Y, Silodosin Clinical Study Group. Silodosin, a new a1A-adrenoceptorselective antagonist for treating benign prostatic hyperplasia: a results of a phase III randomized, placebo-controlled, double-blind study in Japanese men. BJU Int. 2006;98:1019–24.
Duan Y, Grady JJ, Albertsen PC, Helen Wu Z. Tamsulosin and the risk of dementia in older men with benign prostatic hyperplasia. Pharmacoepidemiol Drug Saf. 2018;27(3):340–8.
Tae BS, Jeon BJ, Choi H, et al. Alpha-blocker and risk of dementia in patients with benign prostate hyperplasia: a nationwide population-based study using the National Health Insurance Service database. J Urol. 2019. https://doi.org/10.1097/JU.0000000000000209 [Epub ahead of print].
Anderson KE, Ückert S, Stief C, Hedlund P. Phospodiesterases (PDEs) and PDE inhibitors for treatment of LUTS. Neurourol Urodyn. 2007;26:928–33.
Ückert S, Oelke M. Phosphodiesterase (PDE) inhibitors in the treatment of lower urinary tract dysfunction. Br J Clin Pharmacol. 2011;72:197–204.
Ghofrani HA, Voswinckel R, Reichenberger F, et al. Differences in hemodynamic and oxygenation responses to three different phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension: a randomized prospective study. J Am Coll Cardiol. 2004;44:1488–96.
Lincoln TM. Cyclic GMP and phosphodiesterase 5 inhibitor therapies: what’s on the horizon? Mol Pharmacol. 2004;66:11–3.
Kedia GT, Ückert S, Jonas U, et al. The nitric oxide pathway in the human prostate: clinical implications in men with lower urinary tract symptoms. World J Urol. 2008;26:603–9.
Andersson KE, Persson K. Nitric oxide synthase and the lower urinary tract: possible implications for physiology and pathophysiology. Scand J Urol Nephrol Suppl. 1995;175:43–53.
Conti M, Jin SL. The molecular biology of cyclic nucleotide phosphodiesterases. Prog Nucleic Acid Res Mol Biol. 1999;63:1–38.
Ückert S, Oelke M, Stief CG, et al. Immunohistochemical distribution of cAMP- and cGMP phosphodiesterase (PDE) isoenzymes in the human prostate. Eur Urol. 2006;49:740–5.
Ückert S, Küthe A, Jonas U, Stief CG. Characterization and functional relevance of cyclic nucleotide phosphodiesterase isoenzymes of the human prostate. J Urol. 2001;166:2484–90.
Ückert S, Sormes M, Kedia GT, et al. Effects of phosphodiesterase inhibitors on tension induced by norepinephrine and accumulation of cyclic nucleotides in isolated human prostatic tissue. Urology. 2008;71:526–30.
Berger AP, Deibl M, Leonhartsberger N, Bektic J, et al. Vascular damage as a risk factor for benign prostatic hyperplasia and erectile dysfunction. BJU Int. 2005;96:1073–8.
Berger AP, Horninger W, Bektic J, et al. Vascular resistance in the prostate evaluated by colour Doppler ultrasonography: is benign prostatic hyperplasia a vascular disease? BJU. Int 2006. 2006;98:587–90.
Berger AP, Bartsch G, Deibl M, et al. Atherosclerosis as a risk factor for benign prostatic hyperplasia. BJU Int. 2006;98:1038–42.
Kim NN. Phosphodiesterase type 5 inhibitors: a biochemical and clinical correlation survey. Int J Impot Res. 2003;15(Suppl 5):S13–9.
Ückert S, Kuczyk MA, Oelke M. Phosphodiesterase inhibitors in clinical urology. Expert Rev Clin Pharmacol. 2013;6:323–232.
Lue TF. Erectile dysfunction. N Engl J Med. 2000;342:1802–13.
Mulhall JP, Guhring P, Parker M, et al. Assessment of the impact of sildenafil citrate on lower urinary tract symptoms in men with erectile dysfunction. J Sex Med. 2006;3:662–7.
Sairam K, Kulinskaya E, McNicholas TA, et al. Sildenafil influences lower urinary tract symptoms. BJU Int. 2002;90:836–9.
McVary KT, Monnig W, Camps JL Jr, et al. Sildenafil citrate improves erectile function and urinary symptoms in men with erectile dysfunction and lower urinary tract symptoms associated with benign prostatic hyperplasia: a randomized, double-blind trial. J Urol. 2007;177:1071–7.
McVary KT, Roehrborn CG, Kaminetsky JC, et al. Tadalafil relieves lower urinary tract symptoms secondary to benign prostatic hyperplasia. J Urol. 2007;177:1401–7.
Gacci M, Corona G, Salvi M, et al. A systematic review and meta-analysis on the use of phosphodiesterase 5 inhibitors alone or in combination with α-blockers for lower urinary tract symptoms due to benign prostatic hyperplasia. Eur Urol. 2012;61:994–1003.
McVary KT, Peterson A, Donatucci C, et al. Differential impact of storage and voiding symptoms on quality-of-life during therapy for LUTS associated with BPH: structural equation modeling using an integrated randomized controlled trial tadalafil database. J Urol. 2016;196:824–30.
Pattanaik S, Mavuduru RS, Panda A, et al. Phosphodiesterase inhibitors for lower urinary tract symptoms consistent with benign prostatic hyperplasia. Cochrane Database Syst Rev. 2018;(11):CD010060.
Oelke M, Giuliano F, Mirone V, et al. Monotherapy with tadalafil or tamsulosin similarly improved lower urinary tract symptoms suggestive of benign prostatic hyperplasia in an international, randomised, parallel, placebo-controlled clinical trial. Eur Urol. 2012;61:917–25.
Oelke M, Giuliano F, Baygani SK, Melby T, Sontag A. Treatment satisfaction with tadalafil or tamsulosin versus placebo in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia: results from a randomized, placebo-controlled study. BJU Int. 2014;114:568–75.
Oelke M, Shinghal R, Sontag A, Baygani SK, Donatucci CF. Time to onset of clinically meaningful improvement with tadalafil 5 mg once daily in the treatment of men with lower urinary tract symptoms secondary to benign prostatic hyperplasia: analysis of data pooled from 4 pivotal, double-blind, placebo-controlled studies. J Urol. 2015;193:1581–9.
Donatucci CF, Brock GB, Goldfischer ER, et al. Tadalafil administered once daily for lower urinary tract symptoms secondary to benign prostatic hyperplasia: a 1-year, open-label extension study. BJU Int. 2011;107:1110–6.
Guiliano F, Oelke M, Jungwirth A, et al. Tadalafil once daily improves ejaculatory function, erectile function and sexual satisfaction in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia (LUTS/BPH) and erectile dysfunction (ED): results from a randomized, placebo- and tamsulosin-controlled 12-week double-blind study. J Sex Med. 2013;10:857–65.
Roehrborn CG, Chapple C, Oelke M, et al. Effects of tadalafil once-daily on maximum urinary flow rate in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia. J Urol. 2014;191:1045–50.
Oelke M, Wagg A, Takita Y, Büttner H, Viktrup L. Efficacy and safety of tadalafil 5 mg once daily in the treatment of lower urinary tract symptoms associated with benign prostatic hyperplasia in men aged 75 years or older: integrated analyses of pooled data from multinational, randomized, placebo-controlled clinical studies. BJU Int. 2017;119:793–803.
Vlachopoulos C, Oelke M, Maggi M, et al. Impact of cardiovascular risk factors and related comorbid conditions on the treatment response to tadalafil once-daily in men with lower urinary tract symptoms suggestive of benign prostatic hyperplasia: an integrated analysis of four randomized, double-blind, placebo-controlled clinical trials. Int J Clin Pract. 2015;69:1496–507.
Casabé A, Roehrborn CG, Da Pozzo LF, et al. Efficacy and safety of the coadministration of tadalafil once daily with finasteride for 6 months in men with lower urinary tract symptoms and prostatic enlargement secondary to benign prostatic hyperplasia. J Urol. 2014;191:727–33.
Roehrborn CG, Casabé A, Glina S, et al. Treatment satisfaction and clinically meaningful symptom improvement in men with lower urinary tract symptoms and prostatic enlargement secondary to benign prostatic hyperplasia: Secondary results from a 6-month, randomized, double-blind study comparing finasteride plus tadalafil with finasteride plus placebo. Int J Urol. 2015;22:582–7.
Cho MC, Paick JS. Udenafil for the treatment of erectile dysfunction. Ther Clin Risk Manag. 2014;10:341–54.
Yuan J, Zhang R, Yang Z, et al. Comparative effectiveness and safety of oral phosphodiesterase type 5 inhibitors for erectile dysfunction: a systematic review and network meta-analysis. Eur Urol. 2013;63:902–12.
Wright PJ. Comparison of phosphodiesterase type 5 (PDE5) inhibitors. Int J Clin Pract. 2006;60:967–75.
Russell DW, Wilson JD. Steroid 5 alpha-reductase: two genes/two enzymes. Annu Rev Biochem. 1994;63:25–61.
Andriole G, Bruchovsky N, Chung LW, et al. Dihydrotestosterone and the prostate: the scientific rationale for 5α-reductase inhibitors in the treatment of benign prostatic hyperplasia. J Urol. 2004;172(4. Pt 1):1399–403.
Makridakis NM, di Salle E, Reichardt JK. Biochemical and pharmacogenetic dissection of human 5 alpha-reductase type II. Pharmacogenetics. 2000;10:407–13.
Cabot A. The question of castration for enlarged prostate. Am Surg. 1896;26:265–85.
White J. The results of double castration in hypertrophy of the prostate. Am Surg. 1895;25:1–59.
Raja A, Hori S, Armitage JN. Hormonal manipulation of lower urinary tract symptoms secondary to benign prostatic obstruction. Indian J Urol. 2014;30:189–93.
Habib FK, Ross M, Tate R, Chrisholm GD. Differential effect of finasteride on the tissue androgen concentrations in benign prostatic hyperplasia. Clin Endocrinol (Oxf). 1997;46:137–44.
Rittmaster RS, Lemay A, Zwicker H, et al. Effect of finasteride, a 5 alpha-reductase inhibitor, on serum gonadotropins in normal men. J Clin Endocrinol Metab. 1992;75:484–8.
Vermeulen A, Giagulli VA, De Schepper P, Buntinx A, Stoner E. Hormonal effects of an orally active 4-azasteroid inhibitor of 5 alpha-reductase in humans. Prostate. 1989;14:45–53.
Goldenberg L, So A, Fleshner N, et al. The role of 5-alpha reductase inhibitors in prostate pathophysiology: is there an additional advantage to inhibition of type 1 isoenzyme? Cand Urol Assoc J. 2009;3(3 Suppl 2):S109–14.
Clark RV, Hermann DJ, Cunningham GR, et al. Marked suppression of dihydrotestosterone in men with benign prostatic hyperplasia by dutasteride, a dual 5alpha-reductase inhibitor. J Clin Endocrinol Metab. 2004;89:2179–84.
Marks LS, Partin AW, Dorey FJ, et al. Long-term effects of finasteride on prostate tissue composition. Urology. 1999;53:574–80.
Schroeder FH, Westerhof M, Bosch RJ, Kurth KH. Benign prostatic hyperplasia treated by castration or the LH-RH analogue buserelin: a report on 6 cases. Eur Urol. 1986;12:318–21.
Boyle P, Gould AL, Roehrborn CG. Prostate volume predicts outcome of treatment of benign prostatic hyperplasia with finasteride: meta-analysis of randomized clinical trials. Urology. 1996;48:398–405.
Nickel JC. Placebo therapy of benign prostatic hyperplasia: a 25-month study. Canadian PROSPECT Study Group. Br J Urol. 1998;81:383–7.
Gittelman M, Ramsdell J, Young J, et al. Dutasteride improves objective and subjective disease measures in men with benign prostatic hyperplasia and modest or severe prostate enlargement. J Urol. 2006;2006(176):1045–50.
Roehrborn CG, Lukkarinen O, Mark S, et al. Long-term sustained improvement in symptoms of benign protatic hyperplasia with the dual 5α-reductase inhibitor dutasteride: results of 4-year studies. BJU Int. 2005;96:572–7.
Naslund MJ, Miner M. A review of the clinical efficacy and safety of 5α-reductase inhibitors for the enlarged prostate. Clin Ther. 2007;29:17–25.
Nickel JC, Gilling P, Tammela TL, et al. Comparison of dutasteride and finasteride for treating benign prostatic hyperplasia: the Enlarged Prostate International Comparator Study (EPICS). BJU Int. 2011;108:388–94.
Roehrborn CG, Boyle P, Nickel JC, Höfner K, Andriole G. Efficacy and safety of a dual inhibitor of 5-alpha-reductase types 1 and 2 (dutasteride) in men with benign prostatic hyperplasia. Urology. 2002;60:434–41.
Debruyne FM, Jardin A, Colloi D, et al. On behalf of the European ALFIN Study Group. Sustained-release alfuzosin, finasteride and the combination of both in the treatment of benign prostatic hyperplasia. Eur Urol. 1998;34:169–75.
Kirby R, Roehrborn CG, Boyle P, et al. Prospective European Doxazosin and Combination Therapy Study Investigators. Efficacy and tolerability of doxazosin and finasteride, alone or in combination, in treatment of symptomatic benign prostatic hyperplasia: the Prospective European Doxazosin and Combination Therapy (PREDICT) trial. Urology. 2003;61:119–26.
Lepor H, Williford WO, Barry MJ, et al. The efficacy of terazosin, finasteride, or both in benign prostatic hyperplasia. N Engl J Med. 1996;335:533–9.
Roehrborn CG, Siami P, Barkin J, et al. CombAT Study Group. The effects of dutasteride, tamsulosin and combination therapy on lower urinary tract symptoms in men with benign prostatic hyperplasia and prostatic enlargement: 2-year results from the CombAT study. J Urol. 2008;179:616–21.
Kaplan SA, Holtgewe HL, Bruskewitz R, et al. Comparison of the efficacy and safety of finasteride in older versus younger men with benign prostatic hyperplasia. Urology. 2001;57:1073–7.
Yoshida T, Kinoshita H, Yoshida K, et al. Intravesical prostatic protrusion as a predicting factor for the adverse clinical outcome in patients with symptomatic benign prostatic enlargement treated with dutasteride. Urology. 2016;91:154–7.
Chia SJ, Heng CT, Chan SP, Foo KT. Correlation of intravesical prostatic protrusion with bladder outlet obstruction. BJU Int. 2003;91:371–4.
Berges R. Epidemiology of benign prostatic syndrome. Associated risks and management data in German men over age 50. Urologe A. 2008;47:141–8.
Jacobsen SJ, Jacobson DJ, Girman CJ, et al. Natural history of prostatism: risk factors for acute urinary retention. J Urol. 1997;158:481–7.
Roehrborn CG. BPH progression: concept and key learning from MTOPS, ALTESS, COMBAT, and ALF-ONE. BJU Int. 2008;101(Suppl 3):17–21.
Roehrborn CG, Siami P, Barkin J, et al. CombAT Study Group. The influence of baseline parameters on changes in International Prostate Symptom Score with dutasteride, tamsulosin, and combination therapy among men with symptomatic benign prostatic hyperplasia and enlarged prostate: 2-year data from the CombAT Study. Eur Urol. 2009;55:461–71.
Kirby RS, Vale J, Bryan J, et al. Long-term urodynamic effects of finasteride in benign prostatic hyperplasia: a pilot study. Eur Urol. 1993;24:20–6.
Tammela TLJ, Kontturi MJ. Long-term effects of finasteride on invasive urodynamics and symptoms in the treatment of patients with bladder outflow obstruction due to benign prostatic hyperplasia. J Urol. 1995;154:1466–9.
Andersen JT, Ekman P, Wolf H, et al. Can finasteride reverse the progress of benign prostatic hyperplasia? A two-year placebo-controlled study. The Scandinavian BPH Study Group. Urology. 1995;46:631–7.
Andersen JT, Nickel JC, Marshall VR, Schulman CC, Boyle P. Finasteride significantly reduces acute urinary retention and need for surgery in patients with symptomatic benign prostatic hyperplasia. Urology. 1997;49:839–45.
Zhu YP, Dai B, Zhang HL, Shi GH, Ye DW. Impact of preoperative 5α-reductase inhibitors on perioperative blood loss in patients with benign prostatic hyperplasia: a meta-analysis of randomized controlled trials. BMC Urol. 2015;15:47.
Bansal A, Arora A. Transurethral resection of the prostate and bleeding: a prospective, randomized, double-blind placebo-controlled trial to see the efficacy of short-term use of finasteride and dutasteride on operative blood loss and prostatic microvessel density. J Endourol. 2017;31:910–7.
Guess HA, Gormley GJ, Stoner E, Oesterling JE. The effect of finasteride on prostate specific antigen: review of available data. J Urol. 1996;155:3–9.
Andriole GL, Guess HA, Epstein JI, et al. Treatment with finasteride preserves usefulness of prostate-specific antigen in the detection of prostate cancer: results of a randomized, double-blind, placebo-controlled clinical trial. PLESS Study Group. Proscar Long-term Efficacy and Safety Study. Urology. 1998;52:195–201.
Corona G, Tirabassi G, Santi D, et al. Sexual dysfunction in subjects treated with inhibitors of 5α-reductase for benign prostatic hyperplasia: a comprehensive review and meta-analysis. Andrology. 2017;5:671–8.
Favilla V, Russo GI, Privitera S, et al. Impact of combination therapy 5-alpha reductase inhibitors (5-ARI) plus alpha-blockers (AB) on erectile dysfunction and decrease of libido in patients with LUTS/BPH: a systematic review with meta-analysis. Aging Male. 19:175–81.
Gacci M, Ficarra V, Sebastianelli A, et al. Impact of medical treatments for male lower urinary tract symptoms due to benign prostatic hyperplasia on ejaculatory function: a systematic review and meta-analysis. J Sex Med. 2014;11:1554–66.
Liu L, Zhao S, Li F, et al. Effect of 5-reductase inhibitors on sexual function: a meta-analysis and systematic review of randomized controlled trials. J Sex Med. 2016;13:1297–310.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Oelke, M. (2020). Physiology and Pharmacology of the Prostate. In: Chapple, C., Steers, W., Evans, C. (eds) Urologic Principles and Practice. Springer Specialist Surgery Series. Springer, Cham. https://doi.org/10.1007/978-3-030-28599-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-28599-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-28598-2
Online ISBN: 978-3-030-28599-9
eBook Packages: MedicineMedicine (R0)