
Abstract
Cardiovascular disease (CVD) is a class of diseases that encompasses heart diseases, brain vascular diseases and blood vessel diseases. CVDs and their risk factors are a leading cause of death and morbidity in the world [1–3]. According to World Health Organization estimates, CVDs are responsible for 151,377 million disability-adjusted life years (DALY). DALY is the number of years lost because of ill health, disability or premature death. Coronary heart disease (CHD) accounts for 41.35% (or 62,587 million) of these years, while cerebrovascular diseases account for another 30.78% (or 46,591 million) of these years [1].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


4.1 Introduction
Cardiovascular disease (CVD) is a class of diseases that encompasses heart diseases, brain vascular diseases and blood vessel diseases. CVDs and their risk factors are a leading cause of death and morbidity in the world [1,2,3]. According to World Health Organization estimates, CVDs are responsible for 151,377 million disability-adjusted life years (DALY). DALY is the number of years lost because of ill health, disability or premature death. Coronary heart disease (CHD) accounts for 41.35% (or 62,587 million) of these years, while cerebrovascular diseases account for another 30.78% (or 46,591 million) of these years [1].
CVD is subdivided into two major groups: (1) CVD due to atherosclerosis and (2) other CVDs. In atherosclerosis, fatty material and cholesterol are deposited inside blood vessels, making it harder for blood to efficiently supply oxygen and nutrients to cells. Such deposits are also known as atherosclerotic plaques. As a result, blood vessels become less pliable. The build-up of atherosclerotic plaques is referred to as coronary artery disease (CAD) (also known as CHD). Over time, the atherosclerotic plaques can rupture, triggering the formation of a blood clot that deprives blood flow. In acute cases, obstruction of the coronary artery to the heart will lead to a heart attack (i.e. myocardial infarction, MI) [1, 4, 5].
Epidemiologic studies have revealed a variety of risk factors for CAD that can be broadly subdivided into behavioural (e.g. sedentary lifestyle, smoking, unhealthy diet), metabolic (e.g. hypertension, diabetes, cholesterol) and other factors (e.g. age, gender, genetic disposition) [1, 5, 6]. The interplay between lifestyle and genetic risk factors is characteristic of complex or multifactorial diseases, such as CAD [5, 7]. This chapter focuses on disentangling the genetics of CAD.
4.2 The Role of Genetic Studies in Deciphering CAD Mechanisms
As a complex trait, the mode of CAD inheritance follows Fisher’s 1918 ‘infinitesimal model’ [8]. In this model, discrete and continuous traits are consistent if quantitative trait variation is caused by a combination of many segregating genes, each with a small (infinitesimal) effect on the trait. This mechanism leads to a normal distribution of genetic values and, together with normally distributed environmental effects, results in a normal distribution of phenotypes in the population. This theory implies that the genetic and non-genetic sources of variation can be estimated by quantifying the correlation between relatives, without any knowledge of specific genes that potentially affect the trait [9]. In 1938, the first familial CAD risk was described [10], followed by clinical observations in the 1950s and subsequent familial and twin studies that supported this theory [11]. For example, the Framingham Heart Study found that there was a 29% increased risk of CAD for an individual with a family history of CAD [12, 13]. In addition, the first large-scale prospective Swedish twin study (N = 21,004 twins) [14] identified that the risk of death from CHD was greater in monozygotic (MZ) twins than in dizygotic (DZ) twins in both men and women independent of CHD risk factors. A follow-up study showed that CHD heritability was 0.57 (95% CI, 0.45–0.69) and 0.38 (0.26–0.50) for male and female twins, respectively, with heritable effects most evident in younger individuals [15].
Such heritability estimates originate from family-based study designs. Briefly, family studies can be subdivided into three main groups: (1) single affected family member, (2) relative pairs and (3) extended families. Examples of study types with a single affected family member are case-control studies, trios (case and both parents) and case-only designs. However, the disadvantage of collecting single affected family members is that for complex diseases, multiple affected individuals are required to determine identity-by-descent (IBD) sharing. Therefore, relative pairs and/or extended family study designs are used. Examples of relative (affected and non-affected) pairs would be sib-pairs, twins or avuncular (e.g. aunt-nephew) pedigrees. Finally, extended family groups are large families with multiple affected individuals across many generations [16].
Heritability estimates are generally more precise using close relatives, whereas distant relatives are less precise and less biased [9]. Therefore, the use of twin (pedigree design) and full sibling (within-family design) data has been an important starting point for understanding CAD/MI genetics. In the proceeding paragraphs, we will briefly highlight a couple of studies that have paved the way for our understanding of CAD/MI genetics.
4.2.1 Twin Studies
Twin studies are a special case of pedigree studies consisting of six different types depending on the researcher’s aim [17]. The first evidence of a genetic basis of CAD/MI was provided from a ‘classical’ twin design [18]. This study design used the phenotypic resemblance of MZ (genetically identical as a result of the division of a single fertilised egg) and DZ (non-identical twins that are formed from the separate fertilisation of two eggs) twins to estimate the contribution of genetic and environmental variation to phenotypic variation. As MZ and DZ twin pairs are exposed to similar pre- and postnatal environmental factors, the genetic origin of a trait can be determined [18].
The advantage of twin studies for complex or multifactorial traits such as CAD is the distinct characteristics of a twin pair, i.e. twins are the same age and exhibit a higher degree of shared family environment (e.g. lifestyle) compared to sib-pair, thereby ‘controlling’ the influence of environmental risk factors into a study model and attributing phenotypic differences to twin genetics. Furthermore, errors caused by non-paternity (i.e. different fathers) are reduced or nullified in comparison to sib-pair studies [18]. As previously mentioned, the twin pairs used to investigate the genetic basis of CAD mortality [14, 15] were the first to highlight the utility of twin studies in understanding CAD/MI genetics. In 2001, Wienk et al. used a Danish twin study to report that heritability estimates of frailty in CHD were within the range of 0.53–0.58 for males and females, respectively [19]. Subsequently, in 2005, Wienk et al. reported heritability estimates of 0.45 for both sexes in an additive genetics-unique environment (also known as AE, where A represents the additive genetic factors and E the unique environmental factors) model without covariates [20]. The lower heritability estimates and discrepancies between these two Wienk et al. studies could be due to different sample selection methods and overall age differences between the cohorts, as the Danish twin cohort is much older than the Swedish twin cohort.
Over the last few decades, twin studies have emphasised the genetic component of numerous CAD/MI risk factors such as smoking [21], plasma lipids, lipoproteins, and apolipoproteins [22]. Although genetic methods and molecular technologies continue to evolve more ‘sophisticated’ study designs (e.g. such as improved genotype chips), twin studies remain an important resource considering the unique features of this type of study design. For example, the advantage of studying the effects of epigenetic factors through DNA methylation or histone modification between twins with different lifestyles may help elucidate the environmental effect of genome expression or within-pair epigenetic drift over time [23, 24]. These epigenetic factors may help to explain why most identical twins do not contract CHD and may die of different causes and hint towards a structural gene variant mechanism. For example, Gordon and colleagues [25] investigated a cohort of 250 mothers and their newborn twins focusing on two cell types: human umbilical vein endothelial cells and cord blood mononuclear cells. They found that birthweight—a known predisposing factor for cardiovascular disease—was associated with gene expression involved in cardiovascular function. Subsequent studies using twin data will enhance our understanding of CAD pathomechanisms.
4.2.2 Full Sibling Studies
Similar to twin studies, full sibling study designs have been seminal in the ongoing search for CAD-/MI-causing genes. Murabito and colleagues [26], using population-based offspring cohort data from the Framingham Heart Study (N = 2475), found that middle-aged adult siblings display an increased risk for CVD events with an odds ratio (OR) 1.55 (95% CI: 1.19–2.03). Moreover, the OR for sibling CVD risk (OR = 1.99, 95% CI: 1.32–3.00) exceeded that for the parental CVD (OR = 1.45, 95% CI: 1.02–2.05). This implies that sibling CVD prevalence conferred an increased risk of future CVD events beyond the established risk factors and parental CVD. Interestingly, a 2003 review by the same authors reported that on average, there is a two- to threefold increase in CAD risk in first-degree relatives of cases, and having two or more first-degree relatives with CAD is associated with a three- to sixfold increased risk in developing CAD [27].
4.2.3 Linkage-Based Family Studies
Another type of genetic study design that is used to map the chromosomal locations of genes is linkage analyses. Briefly, a family (or families) is genotyped with polymorphic markers that span their genome, and the genotyping data is then analysed. This technique results in a logarithm of odds (LOD) score for each marker. A significant LOD score (LOD ≥ 3.0) indicates that in the family there is co-segregation with the disease and is identified as linkage.
There are two different types of linkage analyses that are used to map the chromosomal locations of genes for CAD and MI: (1) a model-based linkage analysis using large families in which the inheritance pattern in the families is clearly defined and (2) a model-free analysis using hundreds of small nuclear families with at least two affected siblings in each family [28]. Each of these types of linkage analyses will be discussed in detail.
-
(1)
Model-based linkage analysis
Wang and colleagues [29] carried out a genome-wide linkage scan of a large Caucasian family (13 patients with CAD, of which nine were also affected with MI) that showed an autosomal dominant pattern of CAD/MI. The authors identified that there is a significant linkage score (LOD = 4.19) on chromosome 15q26.3 that contained approximately 93 genes. Of the known genes, myocyte enhancer factor 2 (MEF2A), which encodes a transcription factor, was a strong candidate for CAD/MI susceptibility due to its role in vasculogenesis and its potential role in controlling vascular morphogenesis [30, 31]. Subsequent in vitro studies showed that mice deficient in MEF2A because of a seven amino acid deletion had an effect on gene function [29]. A follow-up mutational study found 3 new mutations in exon 7 of MEF2A in 4 of the 207 independent CAD/MI patients [32]. However, follow-up efforts to re-sequence the coding sequence and splice sites of MEF2A in 300 patients with premature CAD failed to detect a MI-causing mutation or mutation co-segregation [30]. These negative findings were echoed in a later study of Iranian families [33] and in a separate Caucasian family with a history of CAD [34].
-
(2)
Model-free linkage analysis
Using this linkage analysis approach, Helgadottir et al. [35] observed a suggestive linkage on chromosome 13q12-13 that they successfully mapped to arachidonate 5-lipoxygenase-activating protein (ALOX5AP) gene encoding a 5-lipoxygenase-activating protein (FLAP). This gene was associated with a twofold increase in MI risk in 296 multiplex Icelandic families. Furthermore, they observed that the gain-of-function mutation was largely attributed to male carriers of the at-risk haplotype who also had the strongest associations with the ALOX5AP haplotype. However, they did not find an association between an at-risk haplotype called HapA and MI in a British cohort.
Some success with using linkage analysis to map chromosomal positions associated with CAD and MI has been reported in other studies. Three chromosomal positions have been mapped for CAD, namely, 2q21.1-22 [36], 3q13 [37] and Xq23-26 [36]. Two chromosomal positions have been mapped for MI, namely, 1p34-36 [38] and chromosome 14 (map position 123–130) [39]. These types of studies have been both a success and failure. In some circumstances, contradictory results may be due to the shortfall of analytical tools used. Referring back to the previously mentioned role of MEF2A variant in CAD susceptibility, two studies [29, 32] identified a 21 bp deletion of MEF2A. However, subsequent studies did not reach the same conclusion [33, 40]. In 2016, Xu et al. [41] conducted both exome and Sanger sequencing on a four-generation Chinese Han family with familial CAD and found a novel deletion in exon 11 of MEF2A that co-segregated with CAD/MI cases.
It is important to note that the downside of linkage analyses for complex traits such as CAD is that the effect sizes (or penetrance) of the individual causal variants may be too small to allow detection via co-segregation [42]. Therefore, the power to detect genes may be minimal [43] and mapping resolution may be low [44]. In this case, an alternative solution for gene identification may be to use unrelated individuals in hypothesis and hypothesis-free-based association studies.
4.2.4 Candidate Gene Studies
One strategy to identify risk variants associated with a particular disease is candidate gene studies. Briefly, these studies test whether selected genes are related to a disease based on prior knowledge about the gene function or pathophysiology of the disease. Succinctly, the following steps are used: (1) select candidate genes based on prior knowledge; (2) select the gene variant (also known as single nucleotide polymorphisms, SNPs) that is tagged by affecting gene regulation and/or its protein product; and then (3) confirm SNP association with a disease by detecting its occurrence in random cases versus controls [45].
Since the early 1990s, almost 5000 studies have analysed candidate genes in relation to CAD and MI with only 58% of variants showing consistent results in replication studies. Possible explanations for this disparity could be small study populations, false-positive associations and ethnic variations among studies [46]. Other reasons could be due to the inherent disadvantage of using candidate gene studies.
4.2.5 Genome-Wide Association Studies
There are three main disadvantages of the candidate gene approach: (1) the reliance on prior knowledge of the function of the studied gene(s), (2) the inherent bias towards choosing a candidate gene that is geared towards the researcher’s specific study or interest and (3) causative variants outside the region of study that may be missed [47]. Considering the limitations of candidate gene studies, in addition to the improvement of genotyping chip designs, their continuous lowering of costs and the development of improved statistical methods (e.g. imputation and haplotype tagging), there has been an increased popularity of genome-wide association studies (GWAS).
One such imperative technological breakthrough that aided in GWAS rapid success rate is the 2007 completion and public availability of the International HapMap (short for haplotype map) Project [48], which allowed the mapping of haplotype landscapes to SNPs in three continental populations. Several years later, in 2015, the 1000 Genome Project [49] took advantage of the development of sequencing technology and released freely available human genetic variation data based on low-coverage whole-genome sequencing that reached its pinnacle with a reference panel called 1000GP3. Recently, the Haplotype Reference Consortium (HRC) [50] combined all whole-genome sequencing data sets into a single haplotype reference panel to facilitate genotype imputation. Promisingly, the HRC reference panel has been used in the imputation stage of an endophenotype of glaucoma meta-analysis GWAS. This was shown to improve the concordance between assayed and imputed genotypes, markedly in cases of low-frequency variants. In turn, this technique significantly improved p-values, particularly for suggestive variants [51], thereby outperforming 1000GP-based imputation concordance and final p-value results.
Unlike linkage studies, GWAS use unrelated subjects to detect associations between genetic variants and disease/traits, making it easier to obtain large sample sizes. The foundation of GWAS is the ‘common disease, common variant’ (CDCV) hypothesis that was first put forward by Lander [52]. This hypothesis implies that common genetic variants in the population with low penetrance (by common we mean allelic variants present in more than 1–5% of the population [53]) contribute to the genetic susceptibility to common complex traits and disease [52].
Exploiting GWAS analyses has led to significant progress in understanding the genetics of CAD/MI. For CAD-GWAS studies, 2007 was an important year when three independent GWAS studies discovered an association of variants on chromosome 9p21.3 and CAD in European ancestry population [54,55,56] and multiple races [57,58,59,60,61], apart from African Americans [58]. This result may be consistent with the ‘Out Of Africa’ hypothesis which states that all present population groups of Homo sapiens have evolved from a primitive African population [62] (Fig. 4.1).

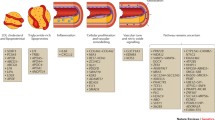
Milestones in cardiovascular genetics from 2007 and beyond
Over the past decade, GWAS for CAD/MI has seen much success, which was catapulted in 2007 with the discovery of 9p21, by independent research groups. By 2009, twelve other genetic risk variants were discovered through GWAS to be associated with CAD [63]. Subsequently in 2013, with a larger sample size, the CARDIoGRAMplusC4D Consortium reported 46 loci associated with CAD, both confirming previously published and finding new variants [64]. This was followed by the identification of ten additional new loci in 2015 [65]. Currently, in 2017 and 2018, CARDIoGRAMplusC4D data together with the UK Biobank [66] data have been proven to be a wealthy resource of genetic data reflected by the increase in new CAD-associated loci [67,68,69].
In spite of these new CAD loci findings, less focus has been given to the X- and Y-chromosomes with fewer in studies on the X-chromosome [70]. It is common knowledge that there exists sexual dimorphism regarding the incidence, prevalence, morbidity and mortality of CVD and/or MI with men having an increased risk compared to age-matched women [71,72,73,74,75]. This is due to two barriers: (a) markers on genotyping chips and (b) statistical methods [70]. These are acutely present when analysing the X-chromosome, due to its unique properties such as X-inactivation, and the presence of two X-chromosome copies in females compared to males [70, 75, 76]. There are publications [76, 77] that do offer possible solutions and recommendations for incorporating X-chromosome. However, the results have been mixed with a 2016 meta-analysis study [78] reporting no association of CAD and X-chromosome variants, compared to a recent 2017 American Heart Association/American Stroke Association (AHA/ASA) conference abstract [79] which found three novel CAD susceptibility loci on the X-chromosome. Much work remains for the inclusion of the sex chromosomes. Nevertheless, we can say that for the autosomal chromosomes, there are currently a total number of 163 loci associated with CAD.
Using the CAD/MI GWAS results thus far, we can tentatively say (a) the majority of common variants found show modest CAD risk increase; (b) most of the variants found are situated outside protein-coding regions; and (c) we have improved our understanding of CAD risk with the loci we have found so far [5] (Fig. 4.2).

Circos plot [80] with 163 risk loci identified by January 2018. Figure provided by Syed M. Ijlal Haider, Institute for Cardiogenetics
4.3 9p21 Locus and Its Role in CAD/MI
Carried by 75% of the global population (excluding black Africans), 9p21 risk alleles are associated with coronary atherosclerosis risk [81]. Moreover, a prior study [82] showed that 9p21 is significantly associated with the risk of first CHD events (1.19 hazard ratio of first event; 95% CI: 1.17–1.22) compared to subsequent CHD events (1.01 hazard ratio of first event; 95% CI: 0.97–1.06). These observations suggest that 9p21.3 stimulates coronary atherosclerosis (i.e. CAD) rather than MI [81, 83]. The 9p21.3 locus has been characterised as a ‘gene desert’ containing dispersed haplotype blocks [84]. It is thought that the CVD-associated region is adjacent to the last exons of a long non-coding RNA (lncRNA), specifically the antisense non-coding RNA in the INK4 locus (ANRIL; also known as CDKN2BAS) [85]. The closest protein-coding (candidate) genes include the cyclin-dependent kinase (CDK) inhibitors CDKN2A and CDKN2B [83]. Furthermore, Holdt et al. [86] reported that CDKN2A/B gene protein products (p16INK4a, p14ARF and p15INK4b) that are expressed in smooth muscle cell layers in both normal arteries and atherosclerotic plaques participate in atherosclerotic lesions. As covered in a review by Hannou and colleagues [83], several studies have failed to decipher the exact mechanism through which CDKN2A/B gene products work or which pathways are involved. Using unbiased genomic techniques based on chromosome conformation capture (3C) [87], Harismendy, O and colleagues [88] detected long-distance interactions between the enhancer interval containing the CAD locus and CDKN2A/B. Considering the effects of interactions across large distances, this observation tentatively (and excitingly) points to the possibility that 9p21.3 disease-associated SNPs interact and modify other distant genes. Very recently, Holdt and colleagues identified circANRIL as a prototype of a circRNA regulating ribosome biogenesis and conferring atheroprotection, thereby showing that circularisation of long non-coding RNAs may alter RNA function and in general might protect from human disease [89].
Broadly summarising, there are seven categories with their respective identified risk loci that underlie the pathways to CAD. Of the total number of loci (Fig. 4.3), 70% of loci are known to be involved in lipid metabolism (12%), blood pressure (7%), cell cycle and gene regulation (12%), vascular remodelling (12%), angiogenesis (9%), inflammation (10%) and nitric oxide signalling (9%). Unsurprisingly, due to the laborious and extensive experimental follow-up required, the vast majority (30%) of identified loci pathways have yet to be explained.

Selection of CAD risk genes and their proven and/or predicted involvement in pathways related to CAD and MI
4.4 Does Sample Size Matter to Identify CAD/MI Risk Loci?
To detect significant SNP contributions in GWAS, large numbers of cohort participants are required, ranging from the tens to hundreds of thousands of subjects. Large GWAS consortia are formed to reach such numbers, usually through meta-analysis of GWAS. For example, a large Coronary ARtery DIsease Genome-wide Replication and Meta-analysis (CARDIoGRAM) Consortium [90] (N cases = 22,233 and N controls = 64,762) was constituted and identified 13 new CAD loci by a meta-analysis of GWAS (sometimes abbreviated to meta-GWAS). In parallel, the Coronary Artery Disease (C4D) Genetics Consortium [91] (N total_cases = 15,420 and N total_controls = 15,062) found five newly associated CAD loci. In an effort to increase sample size, these two consortiums merged and are now known as the CARDIoGRAMplusC4D Consortium [64]. The merging, CARDIoGRAMplusC4D Consortium achieved a sample size of 63,746 CAD cases and 130,681 controls and identified an additional 15 loci to the already known CAD loci. Proceeding, the first interim 150,000 genotyped individuals from the UK Biobank have recently been made available [92] with the intention to release the remaining 350,000 participants in the near future. This would mean a publically available health resource of an unprecedented 500,000 individuals. Interestingly, this year, 3 studies [67, 68, 93] each found 13, 14 and 15 new loci associated with CAD, of which 7 loci overlapped between the 3 studies. Subtle differences in study design (e.g. phenome-wide association scan [67] vs. false discovery rate approach [93]) and phenotype definitions (e.g. CAD cases defined as multiple International Classification of Disease (ICD) 10 code [68] vs. subdivision of ICD10-coded CAD cases with/without angina [93]) could be a minor contributing reason as to why the loci overlap between studies is low; however, further studies are merited. These three studies exemplify the gain of a large sample size for GWASs within the last decade: from ≈60 common genetic variants to >95 total number of CAD-associated loci [94].
A larger sample size could help low-frequency variant studies to reach the required level of significance. A prior large-scale exome-wide study of more than 120,000 participants only had 80% power to detect an OR of ±2.0 for CAD-associated variants with a minor allele frequency (MAF) of 0.1% [95].
Even with relatively common genetic variants (MAF > 0.01%), large cohorts like the CARDIoGRAMplusC4D [64] and the recent UK Biobank could facilitate the identification of single variant statistical analyses for common variants across the exome and thereby reach exome-wide significance, such as in the case of atherosclerosis lesions in young participants [96].
4.5 Rare/Low-Frequency Variants of CAD: ‘Can “In-Betweeners” Explain the Missing Heritability?’
In classical genetics, narrow-sense heritability represents the joint distribution of allele frequency and effect sizes [97], meaning that dominant or epistatic effects are not considered.
In GWASs (and the CDCV hypothesis), the quantification of the proportion of additive genetic variance because of LD between the genotyped and imputed SNPs with the unknown causal variants (i.e. ‘SNP heritability’) implies that genetic variations can be tagged by common SNPs via LD [97]. It has been found that between one-third and two-thirds of the additive genetic variation in a population is tagged and is often referred to as the ‘missing heritability’ [42, 92, 97, 98]. This ‘missingness’ is clearly observed when comparing common variants (by common we mean MAF, >0.05%) of small effects (i.e. GWAS) and very rare variants (by rare we mean MAF ≤ 0.05%) with large effects (i.e. whole exome studies), even after controlling for other effects such as environment [92]. One early example was the haplotype association of a rare CAD variant and SLC2A-LPAL2-LPA gene cluster [99]. More recently, the Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators [95] investigated the effects of loss-of-function mutations in 72,868 CAD patients and 120,770 controls. They identified low-frequency loss-of-function missense variants in ANGPTL4 gene (that was also associated with a protection against CAD) and an association with increased CAD risk in low-frequency coding variants in SVEP1 gene.
Challenges remain in the search for rare CAD variants. For example, there are difficulties in detecting and replicating rare variants that are restricted to specific population groups. To overcome this issue, it has been suggested that combining summary GWAS association statistics by using ‘local SNP heritability’ could provide replication at the locus level rather than the SNP variant level [92]. Another issue for successful GWAS is the requirement for very large sample sizes. However, using large cohorts such as the UK Biobank would overcome this particular issue (Fig. 4.4).

Allelic spectrum of CAD ranging from private variants with monogenic effect on disease to common variants conferring only low disease risk (adapted from Manolio et al. 2009)
4.6 A Note: Missing Does Not Imply Absence
Considering that the current CAD-associated SNPs only explain 10.6% of CAD variability and 40% heritability, it is natural to ask whether the ‘missing’ heritability is actually absent. However, this is not the case with most complex traits that present missing heritability. As we mentioned above, other explanations (e.g. rare SNPs that present large effects) or new analytical approaches (e.g. Generalized Compound Double Heterozygosity [GCDH] that can detect genetic associations by a relaxed form of CH) help to explain this gap. Another proposed explanation for the missing variability and heritability would be that some common risk alleles might have (very) small effects that are too small to pass the traditional GWAS significance level. This leads to a question of how many of these common risk alleles with small effects would a study need to explain 100% of CAD heritability? During the peak of GWAS studies, Wray and colleagues [100] addressed this question and reported that causal risk variants can be estimated as a function of the disease heritability and prevalence in the scenario that all risk alleles have the same relative risk and frequency [100, 101]. Another enticing idea put forward to help explain CAD ‘missing heritability’ is that heritable risk for CAD and common complex diseases could be partly attributed to interactions among diseases and traits [102, 103]. Making the case for the pleiotropic effect among complex disease, a recent publication by Webb et al. [103] identified six new CAD loci. In addition, they showed that 47% of the loci were association with another disease/trait with several loci showing multiple associations.
4.7 Adding Biological Meaning to GWAS Findings
An inherent limitation of association studies, such as GWASs, is that they do not provide biological meaning of the casual variants that were tagged via genome-wide significant SNPs (tagSNPs). To translate GWAS results to biological function, post-GWAS analysis is necessary. Usually, post-GWAS starts with inexpensive in silico (bioinformatics) analysis of GWAS-found tagSNPs that is then followed up with more time and costly in vitro and/or in vivo studies. There is an increasing number of in silico tools and statistical techniques used to give meaning and prioritisation to the resulting associated loci that are subsequently investigated with experiments. In the following paragraphs, we will briefly cover some of the most popular or recent methods.
4.8 Mendelian Randomisation
Although randomised controlled trial (RCT) studies are the optimum way to establish the causal relationship between risk factors, exposure and disease of interest (in our case, CAD), sometimes this is not possible. An alternative is to use Mendelian randomisation (MR) analysis of GWAS data. The rationale is to use genetic variants as a proxy for a potentially modifiable exposure to identify causal effects for risk of CAD [104], thus making MR analysis resistant to confounding factors, an advantage over randomised control trials [105]. Other useful characteristics of MR studies are that multiple genetic variants can be used to increase power and investigate pleiotropic influence on the trait of interest, bidirectional MR studies can be used to determine the direction of causal effects in more complex networks, gene-by-environment interactions can be studied and epigenetic profiles can be used as an intermediate phenotype [104] (Fig. 4.5).

Mendelian randomization basic principles used to find causal pathways. Compliance to three MR assumptions regarding the genetic variant must be kept, namely, the genetic variant must be robustly associated with the exposure variable (shown by the thick blue arrow) and should not be directly related to the outcome or have any confounding factors (shown by the two pink lines)
One of the main utilities of MR results is their applicability in drug development. For example, MR studies of loss-of-function mutations in proprotein convertase subtilisin/kexin type 9 (PCSK9) have led to the development of several PCSK9 monoclonal antibodies. These antibodies are currently under study in four phase 3 trials to test whether such drugs reduce cardiovascular events. Various genetic studies have reported that gain- and loss-of-function mutations in PCSK9 increase low-density lipoprotein-cholesterol (LDL-C) concentration and premature atherosclerosis and reduce LDL-C with low rates of CHD, respectively. Succinctly, this is achieved via hepatocyte endocytosis where circulating LDL-C is cleared from the blood by the binding of LDL-C to low-density lipoprotein receptor (LDL-R) that is situated on the hepatocyte cell membrane. PCSK9 regulates LDL-R metabolism by binding to it, thus leading it to be destroyed by lysosomes and thereby decreasing its recirculation [106].
4.9 Next-Generation Sequencing (NGS) Approaches for CAD and MI
We would be remiss if we didn’t concisely highlight next-generation sequencing (NGS)—with whole-genome sequencing being the most widely used NGS technology [107]—and also MR, studies in providing strong CAD pathophysiological insight [108]. Following the lipoprotein example, Emdin et al. [109] used the NGS approach (via LPA gene sequencing) to show that one standard deviation (SD) genetically lowered lipoprotein(a) level is associated with a lower risk of 29% for CHD, of 31% for peripheral vascular disease, of 13% for stroke, of 17% for heart failure and of 37% for aortic stenosis.
4.10 Expression Quantitative Trait Loci (eQTL): CAD/MI
EQTLs are SNPs, previously found to be associated with the phenotype of interest (i.e. CAD), which have either a local effect (cis) from where the associated variant was found or distant effect (trans; e.g. more than 5 Mb away) from the associated variant [110, 111]. This method uses quantitative trait loci (QTLs) by looking at mRNA levels—the primary genome product—and correlating gene/protein/methylation level (i.e. intermediate molecular quantitative traits) with the genetic variants found [110, 112]. RNA samples from patients and healthy subjects are recruited and converted into microarray data creating a RNA expression data set. Publicly available multi-tissue databases usually accompany association studies and are used to add strong evidence that the associated SNP has a functional effect [113], in turn adding a targeted approach of candidate genes to follow up with experimental work. A variety of tissue disease-specific databases are available such as ENCODE [114], GTEx [115], Epigenome RoadMap [116] and STARNET [117], some of which have been used to focus on CAD-relevant tissues. For example, a study [118] using STARNET showed that cis- and trans-genes could act as a mechanism for multiple risk loci to contribute to cardiometabolic diseases (precursors of CAD) heritability (Fig. 4.6).

Flow chart describing GWAS and post-GWAS analyses
4.11 Network Analysis
There are a few tools [119,120,121,122] available that either prioritise variants found during association analysis or perform tissue enrichment analysis. An interesting study published last year [123] used various tissue-specific regulatory networks and protein-protein interaction networks that do not solely rely on a priori knowledge. They were able to detect well-implicated CAD genes in the prevalence of CAD and also unravelled novel key regulators (LUM, HGD, F2, ANXA3 and STAT3) for CAD. Recently, Vilne et al. demonstrated how hypercholesterolaemia can hinder mitochondrial activity during atherosclerosis progression and identified oestrogen-related receptor-α and its cofactors PGC1-α and PGC1-β as potential therapeutic targets to counteract these processes using a network approach [124].
4.12 The ‘Omics Era’
One of the fields that has seen an almost exponentially rapid rise in popularity due to lowered cost and high-throughput analysis has been the ‘-omics’ studies (namely, genomics, epigenomics, transcriptomics, proteomics, metabolomics and microbiomics), with a vital participatory role of cloud [125] and/or Web [126] computing that facilitates the handling of huge ‘-omics’ study data volumes. As we become more aware that (1) identified CAD loci only explain a small percentage of heritability; (2) common diseases, such as CAD, tend to occur because of gene regulation changes; and (3) similar genetic variants contribute to different final outcomes, it may come as no surprise that systems genetics evolved to integrate the various ‘-omics’ studies. In our case, this enabled the methodology to help explain the complexity of the underlying molecular patterns that are associated with CAD.
4.13 Systems Genetics Approaches in CAD/MI
As defined by Björkegren et al. [111], systems genetics uses molecular mechanisms to define disease-driving molecular processes that underlie GWAS, whole exome sequencing (WES) or whole-genome sequencing (WGS) and to integrate such processes with functional genomic data. One such example of associated SNPs exerting a tissue-dependent effect on gene expression is exemplified by Musunuru et al. [127]. They integrated eQTL and protein QTL (pQTL) information and found that a MI risk variant alters the expression of SORT1 gene in the liver (via a lipoprotein metabolism-regulated pathway) and not in the blood. This observation is dissimilar to prior GWASs that identified a strong association between 1p13 locus and plasma LDL-C levels in MI patients and, moreover, that this locus influenced the risk of MI by conferring changes to plasma LDL-C. Another recent study [128] used a systems genetics approach to integrate DNA genotypes and gene expression profiles from seven CAD-relevant tissues with CAD CARDIoGRAM GWAS information. With this analytical perspective, they showed that RNA-processing genes play a pivotal role in causing CAD and furthermore identify several strongly inherited, evolutionarily conserved, risk-enriched CAD genes that cause regulatory gene network changes across vascular and metabolic tissues.
4.14 Multi-omics Approaches
Compared to looking at an individualised -omics approach, multi-omics provides a greater understanding of the flow of information from the disease initiator to its functional consequence or interaction. Multifactorial diseases, such as CAD, prove to be extremely entangled, which may be a contributing factor as to the lack of multi-omics studies of CAD. However, a preprint study by Santolini et al. [129] used >100 genetically diverse mice strains to investigate a multi-omics approach to cardiac hypertrophy and heart failure. Interestingly, they developed a personalised strategy to investigate stressor-induced heart failure and identified 36-fold change genes that were enriched in human cardiac disease genes and hypertrophic pathways and were missed by the traditional population-wide differentially expressed gene method. Additionally, the genes that they found were linked to both upstream regulators and signalling networks, providing insight into cardiac hypertrophy severity and resistance. Finally, they validated Hes1 as a novel regulator of cardiac hypertrophy (Fig. 4.7).

Overview of the multi-omics approach. From a micro-level approach, we are able to consecutively combine information to form a picture at the ‘-ome’ level, which, when aggregated together, results in a phenome informational level. Furthermore, different informational pieces can affect between multiple -omics layers. SNPs, single nucleotide polymorphisms; CNVs, copy number variants; sRNA, small RNA; lncRNA, long non-coding RNA
4.15 Exciting Times Ahead
4.15.1 Reverse Genetics
An interesting topic that has appeared in the literature recently is the concept of ‘human knockouts’. It is based on the idea that the accumulation of rare homozygous mutations is most likely in highly consanguineous populations, as is the case with the Pakistan Risk of Myocardial Infarction Study (PROMIS) [130]. It is known that heterozygous deficiency of the APOC3 gene confers protection against CHD. PROMIS participants who were homozygous for APOC3 loss-of-function mutations were challenged with an oral fat load. These same individuals were then compared with family members lacking the mutation, and they showed significantly improved clearance of the usual post-prandial rise in plasma triglycerides from their circulation. This study highlights the potential and impact of reverse genetics and functional research and the relevance of drug targets prior to their costly development.
4.15.2 Leveraging Genomic Data to Identify Novel CAD/MI Drug Targets
As has been published [131] and commented [132], looking at these naturally occurring ‘human knockout’ population groups, researchers can directly (and non-invasively) see what would happen when a protein’s function or regulatory mechanism is completely removed thereafter derive subsequent dose-response curves in drug development, for example, if we follow the starting path of the GUCY1A3 and CCT7 genes. Prior GWAS studies [90, 133] had found a CAD/MI association with a common variant on chromosome 4q32.1 which overlapped with the GUCY1A3 gene. Subsequently in 2013, Erdmann and colleagues [134] studied two German families and unrelated MI/CAD cases and found two rare variants (both heterozygous mutations) in the GUCY1A3 and CCT7 genes. With confirmation through in vitro/in vivo experimental work, they evidenced that mutations in the soluble guanylyl cyclase-dependent nitric oxide signalling pathway could be linked to MI. Moving on to today, Kessler et al. [135] elucidated via human samples and cell lines (vascular smooth muscle cell migration and platelet function experiments) that GUCY1A3 affects the expression of soluble guanylyl cyclase, smooth muscle cell migration and platelet function. Further studies are merited to bring this pathway to its destination, which would hopefully be drug targets for individuals carrying the GUCY1A3 risk allele (Table 4.1).
4.16 Precision Medicine
GWAS data has been used as a stepping stone to identify risk genes and, with subsequent post-GWAS analysis strategies, may help to elucidate disease-associated pathways that would then be used in drug development or selection. This is the case with PCSK9 (mentioned previously). Although GWAS studies have been useful, it must be emphasised that these studies do not provide the necessary information required to stratify individuals according to severity, prognosis and responsiveness that is required for drug development and/or selection. To obtain such information, Morita and Komuro [143] suggested stratifying large-scale prospective studies according to clinically affected subphenotypes in patients with similar clinical presentations and then adding a second layer independent of the variant associated with the disease onset that would look for a variant associated (or driving pathways) with the subphenotype within the disease [143, 144]. One example of such stratification would be to discriminate between dyslipidemic patients with CAD and patients without CAD. In this example, a subpopulation analysis to identify genetic variants associated with CAD susceptibility could help in the selection of individuals susceptible to CAD who should receive proactive, perhaps intensive, cardiometabolic abnormality management to prevent CAD [143]. On a macro level, current pharmacogenomics research is following these lines. Contrary to the subphenotype method, some argue that in CAD patients, it is more useful to highlight the blend of genetic and environmental causal factors (or pathways) that underlie CAD patients in large-population-scale cohorts [144]. Such discussion can only promote this type of research approach and can be used as a thoroughfare towards a future of individualised precision medicine.
4.17 Closing Remarks
Indeed, much has happened since the journey towards unravelling the genetics underlying CAD/MI began. It will most likely be decades before we fully grasp the pathomechanisms that result in CAD/MI. However, considering the global burden that is CAD/MI, we owe it to ourselves to continue our efforts in unravelling the genetic basis of these diseases. Luckily, we have allies in the continued lowering of genetic sequencing costs, increased computational facilities and strategies (such as machine learning and data mining) that can handle the influx of large data and new research approaches. Exciting times lie ahead of us.
References
Mendis S, Puska P, Norrving B, editors. Global atlas on cardiovascular disease prevention and control. Geneva: World Health Organization (WHO) in collaboration with World Heart Foundation and the World Stroke Organization; 2011.
Nichols M, Townsend N, Scarborough P, Rayner M. Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J. 2014;35:2950–9.
Mozaffarian D, et al. Heart disease and stroke statistics—2016 update. Circulation. 2015;133:e38–e360.
Buja LM. Coronary artery disease: pathological anatomy and pathogenesis. In: Willerson JT, Holmes Jr DR, editors. Coronary artery disease. London: Springer; 2015.
Khera AV, Kathiresan S. Genetics of coronary artery disease: discovery, biology and clinical translation. Nat Rev Genet. 2017;18(6):331–44.
Ozaki K, Tanaka T. Molecular genetics of coronary artery disease. J Hum Genet. 2016;61:71–7.
Khera AV, et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–58.
Fisher RA. The correlation between relatives on the supposition of mendelian inheritance. Philos Trans R Soc Edinb. 1918;52:399–433.
Vinkhuyzen AAE, Wray NR, Yang J, Goddard ME, Visscher PM. Estimation and partitioning of heritability in human populations using whole genome analysis methods. Annu Rev Genet. 2013;47:75–95.
Müller C. Xanthomata, hypercholesterolemia, angina pectoris. Acta Med Scand. 1938;95(S89):75–84.
Gertler MM, Garn SM, White PD. Young candidates for coronary heart disease. J Am Med Assoc. 1951;147(7):621–5.
Schildkraut JM, Myers RH, Cupples LA, Kiely DK, Kannel WB. Coronary risk associated with age and sex of parental heart disease in the Framingham Study. Am J Cardiol. 1989;64(10):555–9.
Myers RH, Kiely DK, Cupples LA, Kannel WB. Parental history is independent risk factor for coronary artery disease: The Framingham Study. Am Heart J. 1990;120(4):963–9.
Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–6.
Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, de Faire U. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med. 2002;252:247–54.
Banerjee A. A review of family history of cardiovascular disease: risk factor and research tool. Int J Clin Pract. 2012;66(6):536–43.
Boomsma D, Busjahn A, Peltonen L. Classical twin studies and beyond. Nat Rev Genet. 2002;3:872–82.
Mangino M, Spector T. Understanding coronary artery disease using twin studies. Heart. 2013;99(6):373–5.
Wienke A, Holm N, Skytthe A, Yashin AI. The heritability of mortality due to heart disease: a correlated frailty model applied to Danish twins. Twin Res. 2001;4(4):266–74.
Wienke A, Herskind AM, Christensen K, Skytthe A, Yashin AI. The heritability of CHD mortality in Danish Twins after controlling for smoking and BMI. Twin Res Hum Genet. 2005;8(1):53–9.
Koopmans JR, Slutske WS, Heath AC. The genetics of smoking initiation and quantity smoked in Dutch adolescent and young adult twins. Behav Genet. 1999;29(6):383–93.
Snieder H, van Doornen LJP, Boomsma DI. The age dependency of gene expression for plasma lipids, lipoproteins, and apolipoproteins. Am J Hum Genet. 1997;60(3):638–50.
Tamoki AD, Tamoki DL, Molnar AA. Past, present and future of cardiovascular twin studies. Cor Vasa. 2014;56(6):e486–93.
Fraga MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102(30):10604–9.
Gordon L, et al. Expression discordance of monozygotic twins at birth: Effect of intrauterine environment and a possible mechanism for fetal programming. Epigenetics. 2011;6:579–92.
Murabito JM, et al. Sibling cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults. J Am Med Assoc. 2005;294(24):3117–23.
Scheuner MT. Genetic evaluation for coronary artery disease. Genet Med. 2003;5(4):269–85.
Wang Q. Advances in the genetic basis of coronary artery disease. Curr Atheroscler Rep. 2005;7(3):235–41.
Wang L, Fan C, Topol SE, Topol EJ, Wang Q. Mutation of MEF2A in an inherited disorder with features of coronary artery disease. Science. 2003;302(5650):1578–81.
Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineage during mouse embryogenesis. Development. 1994;120:1251–63.
Subramanian SV, Nadal-Ginard B. Early expression of the different isoforms of the myocyte enhancer factor-2 (MEF2) protein in myogenic as well as non-myogenic cell lineages during mouse embryogenesis. Mech Dev. 1996;57(1):103–12.
Bhagavatula MRK, et al. Transcription factor MEF2A mutations in patients with coronary artery disease. Hum Mol Genet. 2004;13(24):3181–8.
Inanloo Rahatloo K, Davaran S, Elahi E. Lack of association between the MEF2A gene and coronary artery disease in Iranian families. Iran J Basic Med Sci. 2013;16(8):950–4.
Lieb W, et al. Lack of association between the MEF2A gene and myocardial infarction. Circulation. 2008;117:185–91.
Helgadottir A, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36(3):233–9.
Pajukanta P, et al. Two loci on chromosome 2 and X for premature coronary heart disease identified in early- and late-settlement populations of Finland. Am J Hum Genet. 2000;67(6):1481–93.
Hauser ER, et al. A genome-wide scan for early-onset coronary artery disease in 438 families: the GENECARD study. Am J Hum Genet. 2004;75(3):436–47.
Wang Q, et al. Premature myocardial infarction novel susceptibility locus on chromosome 1P34-36 identified by genomewide linkage analysis. Am J Hum Genet. 2004;74:262–71.
Broeckel U, et al. A comprehensive linkage analysis for myocardial infarction and its related risk factors. Nat Genet. 2002;30(2):210–4.
Weng L, et al. Lack of MEF2A mutations in coronary artery disease. J Clin Invest. 2005;115(4):1016–20.
Xu D-L, et al. Novel 6-bp deletion in MEF2A linked to premature coronary artery disease in a large Chinese family. Mol Med Rep. 2016;14(1):649–54.
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24.
Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7.
Wang X, Prins BP, Siim S, Laan M, Snieder H. Beyond genome-wide association studies: new strategies for identifying genetic determinants of hypertension. Curr Hypertens Rep. 2011;13:442–51.
Patnala R, Clements J, Batra J. Candidate gene association studies: a comprehensive guide to useful in silico tools. BMC Genet. 2013;14:39.
Mayer B, Erdmann J, Schunkert H. Genetics and heritability of coronary artery disease and myocardial infarction. Clin Res Cardiol. 2007;96(1):1–7.
Zhu M, Zhao S. Candidate gene identification approach: progress and challenges. Int J Biol Sci. 2007;3:420–7.
International HapMap Consortium. The international HapMap project. Nature. 2003;426(6968):789–96.
The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Iglesias AI, et al. Haplotype reference consortium panel: practical implications of imputations with large reference panels. Hum Mutat. 2017;38(8):1025–32.
Lander ES. The new genomics: global views of biology. Science. 1996;274:536–9.
Manolio TA. Genomewide association studies and assessment for the risk of disease. N Engl J Med. 2010;363:166–1676.
Helgadottir A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316(5830):1491–3.
McPherson R, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007;316(5830):1488–91.
The Wellcome Trust Case Control Consortium. Genome-wide association study of 14 000 cases of seven common diseases and 3 000 shared controls. Nature. 2007;447:661–78.
Hinohara K, et al. Replication of the association between a chromosome 9p21 polymorphism and coronary artery disease in Japanese and Korean populations. J Hum Genet. 2008;53:357–9.
Assimes TL, et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum Mol Genet. 2008;17(15):2320–8.
Ding H, et al. 9p21 is a shared susceptibility locus strongly for coronary artery disease and weakly for ischemic stroke in Chinese Han population. Circ Cardiovasc Genet. 2009;2:338–46.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Pignataro P, et al. Association study between coronary artery disease and rs1333049 polymorphism at 9p21.3 locus in Italian population. J Cardiovasc Transl Res. 2017;10(5–6):455–8.
Tattersall I. Human origin: out of Africa. Proc Natl Acad Sci USA. 2009;106(38):16018–21.
Roberts R. A genetic basis for coronary artery disease. Trends Cardiovasc Med. 2015;25(3):171–8.
CARDIoGRAMplusC4D Consortium, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
the CARDIoGRAMplusC4D Consortium. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121.
Bycroft C, et al. Genome-wide genetic data on ~500,000 UK Biobank participants. bioRxiv. 2017.
Klarin D, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. 2017;49(9):1392–7.
Verweij N, Eppinga RN, Hagemeijer Y, van der Harst P. Identification of 15 novel risk loci for coronary artery disease and genetic risk of recurrent events, atrial fibrillation and heart failure. Sci Rep. 2017;7:2761.
van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433–43.
Editorial. Accounting for sex in the genome. Nat Med. 2017;23:1243.
Miller VM. Family matters: sexual dimorphism in cardiovascular disease. Lancet. 2012;379(9819):873–5.
Charchar FJ, et al. Inheritance of coronary artery disease in men: an analysis of the role of the Y chromosome. Lancet. 2012;379(9819):915–22.
Papakonstantinou NA, Stamou MI, Baikoussis NG, Goudevenos J, Apostolakis E. Sex differentiation with regard to coronary artery disease. J Cardiol. 2013;62(1):4–11.
Chiha J, Mitchell P, Gopinath B, Plant AJH, Kovoor P, Thiagalingam A. Gender differences in the severity and extent of coronary artery disease. IJC Heart Vasc. 2015;8:161–6.
Winham SJ, de Andrade M, Miller VM. Genetics of cardiovascular disease: Importance of sex and ethnicity. Atherosclerosis. 2015;241(1):219–28.
König IR, Loley C, Erdmann J, Ziegler A. How to include chromosome X in your genome-wide association study. Genet Epidemiol. 2014;38(2):97–103.
Gao F, et al. XWAS: a software toolset for genetic data analysis and association studies of the X chromosome. J Hered. 2015;106(5):666–71.
Loley C, et al. No association of coronary artery disease with X-chromosomal variants in comprehensive international meta-analysis. Sci Rep. 2016;6:35278.
Assimes TL, et al. Abstract 16167: A GWAS of EHR-Defined CAD Identifies Multiple Novel Loci Including the First 3 Loci on the X-Chromosome: The Million Veteran Program. Circulation. 2017;136(Suppl 1):A16167.
Krzywinski MI, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45.
Chen H-H, Almontashiri NAM, Antoine D, Stewart AFR. Functional genomics of the 9p21.3 locus for atherosclerosis: clarity or confusion? Curr Cardiol Rep. 2014;16(7):502.
Patel RS, et al. Genetic variants at chromosome 9p21 and risk of first versus subsequent coronary heart disease events. J Am Coll Cardiol. 2014;63(21):2234–45.
Hannou SA, Wouters K, Paumelle R, Staels B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? Trends Endocrinol Metab. 2015;26(4):176–84.
Paquette M, Chong M, Luna Saavedra YG, Pare G, Dufour R, Baass A. The 9p21.3 locus and cardiovascular risk in familial hypercholesterolemia. J Clin Lipidol. 2017;11:406–12.
Congrains A, et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis. 2012;220(2):449–55.
Holdt LM, Sass K, Gäbel G, Bergert H, Thiery J, Tuepser D. Expression of Chr9p21genes CDKN2B (p15INK4b), CDKN2A (p16INK4a, p14ARF) and MTAP in human atherosclerotic plaque. Atherosclerosis. 2011;214:264–70.
Nagano T, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502(7469):59–64.
Harismendy O, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011;470(7333):264–8.
Holdt LM, et al. Circular non-coding RNA ANRIL modulates ribosomal RNA maturation and atherosclerosis in humans. Nat Commun. 2016;7:12429.
Schunkert H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43(4):333–8.
The Coronary Artery Disease (C4D) Genetics Consortium. A genome-wide association study in Europeans and South Asians identifies five new loci for coronary artery disease. Nat Genet. 2011;43(4):339–44.
Haley CS. Ten years of the genomics of common diseases: ‘The end of the beginning’. Genome Biol. 2016;17(1):254.
Nelson CP, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49:1385–91.
van der Harst P, Verweij N. The identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433–43.
Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators. Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N Engl J Med. 2016;374:1134–44.
Hixson JE, et al. Whole exome sequencing to identify genetic variants associated with raised atherosclerotic lesions in young persons. Sci Rep. 2017;7(4091):2045–322.
Visscher PM, et al. 10 years of GWAS discover: biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22.
Manolio TA, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53.
Tregouet D-A, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41(3):283–5.
Wray NR, Goddard ME, Visscher PM. Prediction of individual genetic risk to disease from genome-wide association studies. Genome Res. 2007;17(10):1520–8.
Prins BP, Lagou V, Asselbergs FW, Snieder H, Fu J. Genetics of coronary artery disease: genome-wide association studies and beyond. Atherosclerosis. 2012;225:1–10.
Kovacic JC. Unraveling the complex genetics of coronary artery disease. J Am Coll Cardiol. 2017;69(7):837–40.
Webb TR, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69(7):823–36.
Burgess S, Timpson NJ, Ebrahim S, Davey Smith G. Mendelian randomization: where are we now and where are we going? Int J Epidemiol. 2015;44(2):379–88.
Jansen H, Lieb W, Schunkert H. Mendelian randomization for the identification of causal pathways in atherosclerotic vascular disease. Cardiovasc Drugs Ther. 2016;30(1):41–9.
Giugliano RP, Sabatine MS. Are PCSK9 inhibitors the next breakthrough in the cardiovascular field? J Am Coll Cardiol. 2015;65(24):2638–51.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–51.
Domenico G, Chiara P, Martinelli N, Corrocher R, Olivieri O. A decade of progress on the genetic basis of coronary artery disease. Practical insights for the internist. Eur J Intern Med. 2017;41:10–7.
Emdin CA, et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;68(25):2761–72.
Westra H-J, Franke L. From genome to function by studying eQTLs. Biochim Biophys Acta. 2014;1842(10):1896–902.
Björkegren JL, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: How important are they?: Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. 2015;65(8):830–45.
Jansen RC, Nap J-P. Genetical genomics: the added value from segregation. Trends Genet. 2001;17(7):388–91.
Hartiala J, Schwarzman WS, Gabbay J, Ghayalpour A, Bennett BJ, Allayee H. The genetic architecture of coronary artery disease: current knowledge and future opportunities. Curr Atheroscler Rep. 2017;19(2):6.
The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74.
Lonsdale J, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580–5.
Roadmap Epigenomics Consortium, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30.
Hägg S, et al. Multi-organ expression profiling uncovers a gene module in coronary artery disease involving transendothelial migration of leukocytes and LIM domain binding 2: The Stockholm Atherosclerosis Gene Expression (STAGE) Study. PLOS Genet. 2009;5(12):e1000754.
Franzén O, et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016;353(6301):827–30.
Tranchevent L-C, et al. Endeavour update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36:W377–84.
Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–11.
Pers TH, Dworzynski P, Thomas CE, Lage K, Brunak S. MetaRanker 2.0: a web server for prioritization of genetic variation data. Nucleic Acids Res. 2013;41:W104–8.
Pers TH, et al. Biological interpretation of genome-wide association studies using predicted gene functions. Nat Commun. 2014;6:5890.
Zhao Y, Chen J, Freudenberg JM, Meng Q, Rajpal DK, Yang X. Network-based identification and prioritization of key regulators of coronary artery disease loci. Arterioscler Thromb Vasc Biol. 2016;36:928–41.
Vilne B, et al. Network analysis reveals a causal role of mitochondrial gene activity in atherosclerotic lesion formation. Atherosclerosis. 2017;267:39–48.
Wall DP, Kudtarkar P, Fusaro VA, Pivovarov R, Patil P, Tonellato PJ. Cloud computing for comparative genomics. BMC Bioinformatics. 2010;11:259.
Pavlovich M. Computing in biotechnology: omics and beyond. Trends Biotechnol. 2017;35(6):479–80.
Musunuru K, et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–9.
Talukdar HA, et al. Cross-tissue regulatory gene networks in coronary artery disease. Cell Syst. 2016;2(3):196–208.
Santolini M, et al. A personalized, multi-omics approach identifies genes involved in cardiac hypertrophy and heart failure. bioRxiv. 2017.
Saleheen D, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544(7649):235–9.
Plenge RM, Scolnick EM, Altshuler D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 2013;12(8):581–94.
Mullard A. Calls grow to tap the gold mine of human genetic knockouts. Nat Rev Drug Discov. 2017;16(8):515–8.
Lu X, et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44(8):890–4.
Erdmann J, et al. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. 2013;504(7480):432–6.
Kessler T, et al. Functional characterization of the GUCY1A3 coronary artery disease risk locus. Circulation. 2017;136(5):476.
Kathiresan S. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. N Engl J Med. 2008;358(21):2299–300.
Inactivating Mutations in NPC1L1 and Protection from Coronary Heart Disease. N Engl J Med. 2014;371(22):2072–82.
The TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371(1):22–31.
Dewey FE, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374(12):1123–33.
Ajufo E, Rader DJ. New therapeutic approaches for familial hypercholesterolemia. Annu Rev Med. 2018;69(1):113–31.
Callaway E. Protective gene offers hope for next blockbuster heart drug. Nat News. 2016.
Deaton AM, et al. A rare missense variant in NR1H4 associates with lower cholesterol levels. Commun Biol. 2018;1(1):14.
Morita H, Komuro I. A strategy for genomic research on common cardiovascular diseases aiming at the realization of precision medicine. Circ Res. 2016;119:900–9003.
Khera AV, Sekar K. Is coronary atherosclerosis one disease or many? Setting realistic expectations for precision medicine. Circulation. 2017;135(11):1005–7.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
The authors have nothing to disclose.
Conflict of Interest
None declared.
Funding
This work was supported by a grant from the Fondation Leducq [PlaqOmics], the German Federal Ministry of Education and Research (BMBF) within the framework of ERA-NET on Cardiovascular Disease, Joint Transnational Call 2017 [ERA-CVD: ENDLESS]. This work was also supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC 22167-390884018.
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Erdmann, J., Muñoz Venegas, M.L. (2019). The Genetics of Coronary Heart Disease. In: Erdmann, J., Moretti, A. (eds) Genetic Causes of Cardiac Disease. Cardiac and Vascular Biology, vol 7. Springer, Cham. https://doi.org/10.1007/978-3-030-27371-2_4
Download citation
DOI: https://doi.org/10.1007/978-3-030-27371-2_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-27370-5
Online ISBN: 978-3-030-27371-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)