
Abstract
All of the small number of studies conducted during the second half of last century to investigate the pharmacokinetics of polymyxins in animals used microbiological methods to quantify the compounds in biological fluids. Those methods generally lacked the accuracy and precision required for such investigations and, in the case of studies involving administration of colistin methanesulfonate (CMS), ongoing conversion to colistin during microbiological incubation of collected samples artifactually elevated the measured concentration of colistin. The pharmacokinetic studies reviewed in this chapter involved use of more accurate, precise and specific methods for the measurement of the relevant compounds in biological matrices. The studies have been conducted in a number of pre-clinical animal species following administration via various routes (e.g. intravenous, intrapulmonary), and have provided important insights into not only the global pharmacokinetics as viewed from plasma but also the tissue distribution and handling by key organs particularly the kidneys.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Colistimethate
- Colistin
- Polymyxin B
- Animals
- Global pharmacokinetics
- Tissue distribution
- Mechanisms involved in renal elimination
Polymyxin derivatives used in clinical practice correspond to complex mixtures of structurally related but distinct chemical entities obtained by fermentation. This raises a number of issues such as purity or differences in composition between brands and even between batches. But because of that, expressing polymyxin doses or concentrations is quite complex and may become confusing [1], in particular in the case of polymyxin E or colistin , the latter administered as a prodrug. Initial pharmacokinetic (PK) studies on colistin methansulfonate (CMS), colistin or polymyxin B in animals were conducted between 1970 and 2000 in various species (rabbits, dogs, ewes, calves) [2,3,4,5,6,7,8] using microbiological assays (i.e. bioassays) for measuring concentrations. However, as discussed in Chap. 6, these techniques are non-specific and cannot distinguish between CMS and colistin or between colistin and co-administered antibiotics cross-reacting with the selected test strain [9]. Therefore, only PK studies in animals conducted with chromatographic assays, including high performance liquid chromatography (HPLC) coupled with fluorimetric detection [10], tandem mass spectrometry (LC-MS/MS) [11] or ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS) [12] will be reviewed. Furthermore, polymyxins are occasionally administered orally for local decontamination, but since their oral absorption is virtually negligible, this specific situation will not be covered in this review.
7.1 Pharmacokinetics of Colistin after Parenteral Administration
The first pharmacokinetic study of colistin using a chromatographic assay was published by Li et al. in 2003 [13]. Colistin sulfate was administered to rats as a single 1 mg/kg intravenous (IV) bolus dose and a specific and sensitive HPLC assay with fluorimetric detection after derivatization, previously developed and validated by the same group, was used [10]. Colistin clearance (CL) and volume of distribution at steady state (Vss) were respectively estimated at 5.2 ± 0.4 mL/min/kg and 0.50 ± 0.06 L/kg. This low Vss value indicates a limited extravascular distribution, in agreement with physico-chemical characteristics of colistin including a large molecular weight and the presence of positive charges (5 amine functions with a pKa close to 10) at physiological pH. In this study, protein binding of colistin was determined in spiked plasma by equilibrium dialysis at three concentrations (4, 8 and 12 mg/L) leading to an average unbound fraction (fu) equal to 43–45% that was independent of the concentration. Noticeably, colistin A was more extensively bound (mean fu of 36%) than colistin B (mean fu of 52%) [13]. The initially reported elimination half-life of colistin (t1/2 = 74.6 ± 13.2 min) [13] was confirmed a few years later by Marchand et al. (t1/2 = 75.4 ± 14.1 min) [14] after subcutaneous administration of colistin 1.5 mg/kg, although estimates of clearance and volume (CL = 8.5 ± 1.0 mL/min/kg, Vss = 0.94 ± 0.25 L/kg) in this new study [14] were somewhat higher (up to two fold for Vss) than previously reported; it should be noted that the later study involved subcutaneous administration of colistin [14]. The virtually similar t1/2 estimate between studies suggests that colistin disposition is not rate limited by its absorption after subcutaneous administration.
Urine samples were also collected in the Li et al. study, and only 0.2% of the colistin dose was recovered unchanged in urine [13], with a corresponding renal clearance (CLr = 0.010 ± 0.008 mL/min/kg) much lower than renal clearance by glomerular filtration; estimated at 2.3 mL/min/kg under the assumption that colistin unbound fraction was equal to 0.44 [13] and glomerular filtration rate (GFR) was 5.2 mL/min/kg in rats [15]. This observation suggested an extensive tubular reabsorption of colistin, which was then confirmed by this group using an in vitro isolated perfused rat kidney model [16]. The extensive renal tubular reabsorption of colistin would be expected to enhance its accumulation in kidney tissue, which may have implications for its renal toxicity [16]. Active transport systems such as organic cation transporters (mainly OCTN1) and polypeptide transporter 2 (PEPT2) were proposed to be involved in the reabsorption of colistin [16, 17]. Because of this extensive tubular reabsorption, colistin elimination is mostly extra-renal. Yet mechanisms responsible for colistin elimination are mostly unknown and no degradation products have been identified. Furthermore in vitro degradation studies at 37 °C demonstrate that the rate of colistin disappearance is virtually similar in homogenates from various tissues such as liver, kidney, muscle or brain, but also not much different than in plasma or phosphate buffer at pH 7.4, suggesting that enzymes may not be involved in colistin elimination [18].
7.2 Pharmacokinetics of Colistin Methanesulfonate (CMS) and Colistin after Parenteral Administration of CMS
7.2.1 Pharmacokinetics of CMS and Colistin in Rats
The first pharmacokinetic study conducted after intravenous administration of CMS, the inactive prodrug of colistin used in clinical practice, in rats, was also conducted by Li et al. [19] at a dose of 15 mg/kg of CMS corresponding to ~6.3 mg/kg of colistin base activity (CBA) [1]. CMS and colistin were assayed by HPLC [20]. Noticeably as for any other chromatographic assays, CMS concentrations were not measured directly, but obtained by difference between colistin concentrations measured after and before CMS hydrolysis in plasma samples. Therefore, partially sulfomethylated CMS derivatives, that may be present within samples, cannot not be distinguished from CMS. Accordingly the authors clearly stated that estimated CMS pharmacokinetic parameters may only be considered as hybrid parameters for CMS [19]. With this limitation in mind, CMS volume of distribution (Vss) was estimated at 0.30 ± 0.06 L/kg; that is about 30% lower than that of colistin and therefore still close to the extracellular fluid volume [15]. Total CMS clearance was found equal to 11.7 ± 1.8 mL/min/kg which is about twice that of colistin [13]. Urinary recovery experiments showed that 61% ± 14% of the dose administered was recovered, mostly as unchanged CMS (2/3) and as colistin (1/3) [19]. However considering the much lower urinary recovery of colistin (0.2%) previously observed after its direct administration [13], it was concluded that post-excretion hydrolysis of CMS in urine was probably mainly responsible for this high recovery of colistin in urine after CMS administration. Consequently, a more reliable estimate of CMS renal clearance was obtained by assuming that the sum of CMS and colistin amounts recovered in urine was actually excreted as CMS. This CMS renal clearance estimate was equal to 7.2 ± 2.2 mL/min/kg, which is slightly higher than GFR in rats (5.2 mL/min/kg) [15]. Li et al. did not measure the plasma protein binding of CMS, but even if the unbound fraction was one (i.e. no binding in plasma) the relative magnitude of the renal clearance of CMS and of GFR is consistent with net tubular secretion [19]. However, it should again be remembered that CMS parameters correspond to hybrid values that are difficult to interpret. It was also possible during this study [19] to estimate that only 6.8% of the CMS dose was converted systemically into colistin in rats, and another interesting observation was that the elimination half-life of formed colistin (55.7 ± 19.3 min) was about twice that of CMS (23.6 ± 3.9 min) indicating that the disposition of formed colistin is not rate-limited by its formation.
A few years after this initial study, a dose-ranging pharmacokinetic study was conducted in rats by Marchand et al. [21], using the largest possible range of CMS doses (5–120 mg/kg of CMS base corresponding to ~2.1 to ~50 mg/kg of CBA), considering the limit of quantification of the LC-MS/MS analytical assay (0.078 μg/mL) and drug toxicity. No trend of non-linearity was observed and pharmacokinetic parameter values were consistent with those previously published by Li et al. [19], in particular when the dose administered was the same (15 mg/kg of CMS base corresponding to ~6.3 mg/kg of CBA). It was estimated that on average 10.2% of the intravenous CMS dose was converted systemically into colistin. This fm estimate is slightly higher than that previously reported (6.8%) [19] but still relatively low. Therefore, these two studies allow concluding, that at least in rats, only a small fraction of the intravenous dose of CMS is eventually converted systemically into colistin.
Interestingly also, the unusual colistin concentration versus time profiles in rats, compared with others species (discussed below), with almost instantaneous plasma peak concentrations and a slower decay of colistin compared with CMS over time, initially observed by Li et al. [19], were confirmed during the study by Marchand et al. [21] as illustrated in Fig. 7.1. A more recent pharmacokinetic study in rats by He et al. comparing four brands of CMS coming from various countries: Thailand (Atlantic Laboratories), United Kingdom (Forest Laboratories) and USA (X-GEN Pharmaceuticals and Paddock Laboratories) [22], may have provided an explanation for this unexpected behavior. Chemical analysis of the different brands was also performed and similar composition was observed for all brands by elementary analysis. However, chromatographic profiles of CMS showed several peaks with the Altantic CMS chromatographic profile distinct from the three other brands. This is consistent with CMS being a mixture of different fully and partially sulfomethylated derivatives that may vary between brands [19]. Following IV administration of these four brands at a dose of 28.1 mg/kg of CMS corresponding to ~11.7 mg/kg of CBA [22], CMS plasma concentrations versus time profiles (Fig. 7.2a) and CMS pharmacokinetic parameters were generally consistent with those previously described [19, 21]. Colistin elimination half-life was again longer than that of CMS, whatever the brand, confirming that colistin disposition is not limited by its formation. However, plasma concentration versus time profiles of formed colistin in the study by He et al. were different from those previously reported by Li et al. [19] and Marchand et al. [14, 21], and colistin peak concentration was considerably delayed to reach a peak after about 60 min on average (Fig. 7.2b). Similar to the results of He et al., a delay in attainment of peak plasma concentration of formed colistin following IV administration of CMS (Link Pharmaceuticals) in rats was observed by Yapa et al. [23].

Mean ± SD total plasma concentration versus time profiles of CMS and colistin in rats after a single 15 mg/kg IV dose of CMS (~6.3 mg/kg CBA) obtained by (a) Li et al., using CMS from Sigma (St Louis, USA) in 5 rats [19] or (b) Marchand et al. using CMS from Sanofi Aventis, (Paris, France) in 6 rats [21], by permission of Oxford University Press

Mean ± SD plasma concentration–time profiles of (a) CMS and (b) formed colistin in rats (n = 4) obtained by He et al., 2013, after IV administration of 28.1 mg/kg of CMS (~11.7 mg/kg CBA) from various brands, [22] by permission of Oxford University Press
Therefore the high initial concentrations of colistin after CMS injection observed by Li et al. [19] and Marchand et al. [14, 21] could be explained by the fact that in these initial studies, the administered CMS solutions may have contained a small fraction of partially sulfomethylated CMS derivatives, rapidly converted into colistin after administration. However, as discussed in the next section, this unexpected behavior was observed in rats but not in other species after IV administration of the same Sanofi-Aventis CMS brand.
Another PK study in rats was conducted to investigate the central nervous system (CNS) distribution of colistin in vivo and by in situ brain perfusion [24]. Brain-to-plasma ratios ranged between 2.1 and 3.7% and were not enhanced by co-administration of P-glycoprotein (P-gp) inhibitors (PSC833 or GF12918). Intraperitoneal injection of lipopolysaccharides to induce inflammation significantly increased brain AUC by two to threefold. In conclusion, blood brain barrier transport of colistin is negligible under healthy conditions but enhanced during systemic inflammation as might be observed in infected patients.
Bouchene et al. recently reported development of a whole-body physiologically-based pharmacokinetic (WBPBPK) model to characterize CMS and colistin distribution in various tissues of rats [25]. In order to describe the disposition of CMS and colistin, the work involved a combination of in vitro, in silico and in vivo data to construct the model. A key aspect of the study was the experimental determination of the tissue-to-plasma partition coefficient for CMS and colistin across 10 different tissues/organs, against which the model predictions were compared. Notably, the experimentally determined kidney-to-plasma partition coefficients for CMS (5.45) and colistin (19.7) were substantially higher than for any of the other tissues. The accumulation of CMS and colistin in kidney tissue is undoubtedly a key factor in the development of nephrotoxicity after administration of CMS. With appropriate validation studies, the WBPBPK model holds promise for inter-species extrapolations using species-specific physiological parameters [25].
7.2.2 Pharmacokinetics of CMS and Colistin in Various Animal Species
In a recent study, epithelial lining fluid (ELF) and systemic pharmacokinetics of CMS and colistin were determined in sheep after IV and pulmonary administration of both CMS (sodium) and colistin (sulfate) at respective doses of 4–8 mg/kg (corresponding to ~1.7–3.4 mg/kg CBA) and 2–3 mg/kg [26]. Concerning systemic pharmacokinetics, the maximal concentration of formed colistin was obtained at 3.13 ± 0.55 h after intravenous administration of CMS. CMS and colistin clearances were 2.29 ± 0.03 L/h (0.95 mL/min/kg) and 1.32 ± 0.23 L/h (0.55 mL/min/kg) which are at least 10 times lower than corresponding clearances in rats [13, 14, 19] and 2.5 times lower than in healthy volunteers for CMS (2.6 ± 0.3 mL/min/kg) [27]. Contrasting with previous results observed in rats and humans where colistin disposition was rate-limited by its own elimination, [19, 27] terminal half-life of formed colistin was not longer than that of CMS. The fm estimate in sheep (17.4%) is slightly higher than that previously reported in rats (6.8 and 10.2%) [19, 21] but still relatively low.
Other investigations have been conducted by our group in various species including mice (n = 36), rabbits (n = 3), baboons (n = 3) and pigs (n = 2) with the objective of developing a WBPBPK model to characterize CMS and colistin distribution in various tissues and eventually to allow between-species comparisons and extrapolations [18]. Animals received a single dose of CMS (Sanofi Aventis, Paris, France) as follows: 15 mg/kg of CMS base (~6.3 mg/kg CBA) administered subcutaneously to mice and intravenously to rabbits, on average 2.5 mg/kg of CMS base (~1.0 mg/kg CBA) infused intravenously over 10 min in baboons, and 149.5 mg of CMS base (160 mg of sodium salt) CMS per pig corresponding to ~67 mg CBA administered as a 1 h intravenous infusion. Multiple sampling of blood was conducted in all species except mice for which 4 animals were used at each of the 9 selected time points. Furthermore, colistin was also directly administered to baboons by subcutaneous 10 min infusion at doses ranging between 0.379 mg/kg and 0.485 mg/kg (of colistin sulfate), in order to estimate the fraction of the CMS dose converted into colistin in that particular species. Plasma concentrations of CMS and colistin were assayed by LC-MS/MS [11] and pharmacokinetic parameters were determined by a non-compartmental approach (WinNonLin version 6.2, Pharsight Corporation, Mountain View, California, USA). Data previously obtained in rats [14, 21] but also in healthy human volunteers [27], using the same CMS brand and analytical assay, can be used for comparison. It should be noted that different CMS batches were used, which may have an effect on concentration versus time profiles of CMS and, in particular, formed colistin. Furthermore, not the same route of administration was used for every species. Plasma concentration versus time profiles in these various species are presented in Fig. 7.3. Corresponding pharmacokinetic parameters are listed in Table 7.1 after correction of volume and clearance terms by body weight.

Mean ± SD plasma concentration–time profiles of CMS (closed circles and full line) and formed colistin (open circles and dotted line) in: mice (n = 4 per time point) after subcutaneous administration of CMS base 15 mg/kg (~6.3 mg/kg CBA); rats (n = 6) after IV bolus administration of CMS base 15 mg/kg (~6.3 mg/kg CBA) [21]; rabbits (n = 3) after IV bolus administration of CMS base 15 mg/kg (~6.3 mg/kg CBA); pigs (n = 2) after 1 h infusion of 149.5 mg of CMS base (~62.3 mg CBA); baboons (n = 3) after 10 min infusion of on average 2.5 mg/kg of CMS base (~1.0 mg/kg CBA), and in healthy volunteers (n = 12) after 1 h infusion of 1MIU of CMS equivalent to 80 mg of sodium CMS (~33 mg CBA)
In humans, baboons, pigs and rabbits, CMS plasma concentrations decayed more rapidly with time than colistin (Fig. 7.3) and accordingly, CMS half-lives were shorter (Table 7.1) [18]. These profiles indicate that in these species the disposition of formed colistin is rate limited by its own elimination and not by its formation. By contrast, CMS and colistin plasma concentrations decayed in parallel with time in mice, which is typical of a formation rate limited process. However, these data must be interpreted carefully since CMS was administered subcutaneously in mice. With the exception of mice, CMS Vss varied between approximately 130 and 330 mL/kg (Table 7.1), close to the volume of extracellular fluid (ECF) [15]. Moreover, these values are in reasonable agreement with the Vss for CMS estimated in healthy volunteers (196 mL/kg) [27]. CMS CL values in rabbits, pigs and baboons were virtually identical and close to the value estimated in humans (2.6 mL/min/kg) [27] but CL estimates for CMS in rats and mice were respectively about 5 and 10 times higher than in human volunteers.
Recently, Viel et al. reported details of a WBPBPK model to characterize the disposition of CMS and colistin in pigs, especially in regard to the renal handling of these compounds [28]. A number of different pharmacokinetic studies were conducted; specifically to elucidate the plasma and kidney pharmacokinetics after single IV and IM administration of CMS as well as after repeated IM administration, and also investigations of tissue-to-plasma partition coefficients and plasma protein binding of CMS. The experimental data were subjected to WBPBPK modelling. Key findings of the experimental and modelling studies were: extensive accumulation of colistin in kidney, followed by slow elimination from that organ; very substantial contribution of tubular secretion in the renal elimination of CMS, with some conversion to colistin within tubular cells; extensive tubular reabsorption of colistin; some degradation of colistin within tubular cells; and, a plasma unbound fraction of approximately 0.4 [28].
Colistin PK-PD has been investigated in vivo using mice after direct subcutaneous administration of colistin sulfate to demonstrate that the AUC to MIC ratio corrected for plasma protein binding (fAUC/MIC) is the relevant PK-PD index to predict colistin efficacy against Pseudomonas aeruginosa and Acinetobacter baumannii [29]. Those studies were preceded by single-dose studies at 10, 20 and 40 mg/kg to elucidate the pharmacokinetics of colistin in infected neutropenic mice. The plasma protein binding in these animals was determined using both ultracentrifugation and rapid equilibrium dialysis, with very careful attention to minimize non-specific binding to equipment used in the measurements. Over the dose range examined, the pharmacokinetics of colistin was nonlinear; the apparent clearance decreased with increasing dose while the half-life increased. The percentage of colistin bound in plasma of infected neutropenic mice was independent of plasma concentration over a wide range (~2–50 mg/L). The average plasma unbound fraction of colistin over this range by the two methods of determination was 0.084; this was very much lower than the value of ~0.5 found for critically-ill patients and healthy humans [29]. PK differences observed between rodents and non-rodents, such as differences in plasma protein binding of colistin, should be considered before extrapolating efficacy results from rodents to humans after treatment by CMS.
7.3 Pharmacokinetics of CMS and Colistin after Nebulization
7.3.1 Pharmacokinetics of CMS and Colistin after Nebulization in Rodents
CMS nebulization seems very appealing for the treatment of pulmonary infections. However because CMS is an inactive prodrug, it was interesting and important to characterize its pre-systemic conversion into colistin after nebulization. More specifically, it was important to estimate how much of the CMS dose is converted to colistin pre-systemically (in the lungs) and how much of the dose is directly absorbed. This question was addressed for the first time in rats and data were published in 2010 [14]. A schema for the pharmacokinetic behavior of CMS and colistin after CMS intra-tracheal administration in rats is presented in Fig. 7.4.

Schema of CMS disposition after nebulization: FCMS, lung corresponds to the fraction of the CMS dose absorbed systematically; fm,syst, is the fraction of CMS converted into colistin within the systemic circulation; fm,lung is the fraction of the CMS dose converted into colistin pre-systematically and Fcoli,lung is the fraction of colistin which is then absorbed into the systemic circulation [14]
In order to estimate the various pharmacokinetic parameters appearing on Fig. 7.4, CMS (Sanofi-Aventis) was administered to healthy rats at a dose equal to 15 mg/kg in base (~6.3 mg CBA), either by intra-tracheal nebulization using a microsprayer aerosolizer, model IA-1B (PennCentury INC., Pennsylvania, USA) (n = 5) or intravenous administration (n = 6) [14]. In order to complete the PK analysis, it was necessary to include another group of rats (n = 6) administered directly with colistin sulfate. The subcutaneous route was selected at a dose of 1.5 mg/kg. Blood samples were collected for concentration measurements in plasma, and broncho-alveolar lavage (BAL) was conducted in extra rats (n = 14) after intra-tracheal administration for determination of CMS and colistin in ELF, after correction by urea concentration. CMS and colistin were assayed by LC-MS/MS in plasma and BAL as well as urea in BAL. A non-compartmental pharmacokinetic analysis was conducted [14]. Using the Penn Century aerosolizer, most of the nebulized dose of CMS was eventually absorbed either directly or after pre-systemic conversion into colistin. It is important to realize that in clinical practice only a small fraction of the aerosolized dose is most likely capable of reaching the absorption/infection site, which precludes direct data extrapolation from rat to human. CMS maximum plasma concentrations were much lower after nebulization than after IV administration and the peak was delayed (Fig. 7.5b). Colistin plasma concentration versus time profiles observed after CMS intravenous administration and nebulization were distinct, with a delayed peak and sustained concentrations with time after CMS nebulization. However, plasma colistin concentrations were in the same order of magnitude independent of the route of administration (Fig. 7.5). It was estimated that after intra-tracheal administration with the PennCentury system, 70% of the CMS dose was directly absorbed to reach the systemic circulation and that 39% was converted pre-systematically and absorbed as colistin. These estimates reflected the experimental error and it was considered that 2/3 of the CMS was directly absorbed and 1/3 converted pre-systemically into colistin. The fraction of CMS converted into colistin after CMS intravenous administration in rats was estimated at 12.5% in the current study [14], compared with 10.2% in the previous dose-ranging study conducted by our group [21] and 6.8% in the pioneer study by Li et al. [19]. As a consequence of the relatively important CMS pre-systemic conversion in the lung, colistin systemic exposure (AUC) was about 4 times greater after CMS nebulization than after IV administration in this experimental animal model [14]. However although a significant fraction of CMS was converted into colistin pre-systemically, ELF concentrations of the active moiety were much lower (about 10 times lower at 30 and 120 min post-nebulization) than corresponding CMS concentrations, suggesting that colistin formation rate limits its absorption. In other words, colistin is rapidly absorbed after being formed within ELF, explaining that its concentrations remain always low compared with those of CMS. No modeling to better characterize these rate-limiting steps was conducted during this study.

Mean ± SD total plasma concentration-versus-time profiles of CMS and colistin after (a) IV administration of 15 mg/kg of CMS base (~6.3 mg/kg CBA) (n = 6), or (b) intra-tracheal nebulization of 15 mg/kg of CMS base (~6.3 mg/kg CBA) (n = 5). (Adapted from Marchand et al., 2010 [14])
Interestingly, these questions have been addressed in more detail by Yapa et al. [23]. In this study, CMS was provided by Link Pharmaceutical Ltd. (Auckland, New Zealand). CMS and colistin were administered to rats intra-tracheally using a 2.5 cm polyethylene tube inserted via the mouth to the tracheal carina at various doses, respectively 14 or 28 mg/kg for CMS (~5.8 and ~11.6 mg/kg CBA) and 0.41, 0.62, 0.99 and 1.49 mg/kg for colistin. They were also administered intravenously at either 14, 28 or 56 mg/kg for CMS (~5.8, ~11.6 or ~23.2 mg/kg CBA) and 0.21, 0.41 or 0.62 mg/kg for colistin. CMS and colistin concentrations were measured in plasma and BAL. Concentration versus time profiles of CMS and colistin in plasma and concentrations in ELF at predetermined sampling times after intra-tracheal administration of CMS compare favorably with those obtained by Marchand et al. at the same dose [14]. However, using the Link Pharmaceutical Ltd. CMS brand, colistin plasma concentration-time profiles after CMS IV administration, showed a delayed colistin peak as observed by He et al. [22] but not Marchand et al. [14] and Li et al. [19]. CMS and colistin concentrations in plasma and ELF were analyzed with a simultaneous population pharmacokinetic model including multi-compartments to describe a relatively complex colistin disposition within lung after intra-tracheal administration [23]. The fraction of the dose exposed to the lung after intra-tracheal administration was 40.9% for CMS and 48.5% for colistin, lower than previously described by Marchand et al. [14]. However, Yapa et al. estimated that on average 22.6% of the CMS dose was converted into colistin in ELF compared with only 3% in the systemic circulation [23], while Marchand et al. obtained respectively 39% and 10–12% for these same parameters [14, 21]. One potential explanation for the extensive CMS conversion in lungs after intra-tracheal administration, is its reduced availability for renal clearance [23]. One of the major findings in the Yapa et al. study was that intra-tracheal administration of CMS achieved much higher and sustained exposure of colistin in lungs than was possible with IV administration [23].
More recently, Gontijo et al. observed that after direct intra-tracheal administration of colistin (0.35 mg/kg), average ELF to plasma AUC ratio was equal to 1214 [30] and that colistin exposure in lung was much higher after nebulization than IV administration, confirming observations by Yapa et al. [23]. A complex absorption pattern of colistin after nebulization was again observed by Gontijo et al., but the best PK model to describe the data incorporated non-linear transfer which was further challenged by increasing the dose [30].
A recent study in neutropenic infected mice compared ELF and plasma pharmacokinetics of colistin after intra-tracheal nebulization at two doses (2.64 mg/kg and 5.28 mg/kg) versus intravenous administration (2.64 mg/kg) [31]. Whereas plasma concentrations of colistin were similar after intravenous administration and nebulization, concentrations in ELF were significantly higher than plasma concentrations after nebulization with corresponding maximum concentrations equal to 169 and 5.72 mg/L (dose of 2.64 mg/kg).
7.3.2 Pharmacokinetics of CMS and Colistin after Nebulization in Other Species
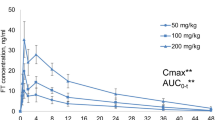
A pharmacokinetic study after CMS nebulization has been conducted in pigs [32]. In particular the aerosol delivery system used in pigs is currently used in patients and is expected to deliver much less of the dose to the pulmonary alveoli than when using the Penn Century system or direct intra-tracheal administration in rats. Furthermore, this study was conducted in piglets infected with an experimental model of pneumonia induced by P. aeruginosa whereas healthy rats were used previously [14, 23]. Twelve ventilated, infected piglets were included in the study, 6 received an IV infusion of CMS (3.2 mg/kg (~1.3 mg CBA) over 30 min every 8 h) and the remaining 6 received 8 mg/kg of CMS (~3.3 mg/kg CBA) over 30 min, every 12 h, by nebulization using a vibrating plate nebulizer (Aeronen Pro®, Aerogen Ltd., Galway; Ireland) [32]. Lung tissue samples were respectively obtained 1 h after the 4th intravenous infusion and the 3rd nebulization. CMS and colistin concentrations were measured in plasma and lung by HPLC [33]. After CMS IV infusions, colistin could not be detected in lung tissue whereas after aerosol delivery a median peak of colistin was estimated at 2.8 μg/g within lung tissue. The absence of colistin in lung tissue after IV administration of CMS was correlated with a lack of antimicrobial efficacy. After 24 h of treatment, 67% of pulmonary segments had bacterial counts <102 CFU/g following nebulization and 28% after IV administration. Therefore, these data suggest that CMS nebulization in patients could be of better value than intravenous dosing for the treatment of pulmonary infections. However measuring antibiotic concentrations in whole tissue homogenates is not recommended for reasons previously discussed [34]. ELF concentrations are probably more relevant to characterize the intrapulmonary distribution of antibiotics and predict antimicrobial efficacy against extra-cellular pathogens. Interestingly dose-normalized colistin plasma peak concentrations (Cmax) and area under concentration-time curve (AUC) were respectively 6 and 2.7 times lower after nebulization than after IV administration of CMS, suggesting that systemic side effects could be lower after CMS nebulization than IV administration [32].
Two different devices used in clinical practice, Eflow rapid® and Pari LC star® were compared by scintigraphy after nebulization of CMS at a dose of 1 MIU (corresponding approximately to 33 mg of CBA) in baboons [35]. A higher aerosol distribution into lungs was observed by imaging when nebulization was performed with a Pari LC® Star than with an Eflow Rapid® nebulizer. Accordingly, ELF concentrations simulated by PK modelling from measured plasma concentrations have confirmed a higher ELF CMS and colistin exposure after nebulization through the Pari LC® Star system than Eflow Rapid® nebulizer [35].
As partially presented above, the most recent study performed by Landersdorfer et al., has documented ELF and plasma pharmacokinetics of CMS and colistin in sheep after IV and pulmonary administration of both molecules [26]. CMS and colistin were not quantifiable in broncho-alveolar lavage fluid following intravenous CMS administration. CMS and formed colistin were not quantifiable in plasma after endotracheal nebulization of CMS at a dose of 2.6 mg/kg CBA (~6.2 mg/kg CMS sodium) whereas very high concentrations of both molecules were observed in ELF (between 1147 ± 710 mg/L and 63 ± 34 mg/L at 1 h and 24 h for CMS and between 400 ± 243 mg/L and 184 ± 190 mg/L at the same times for colistin). The therapeutic availability and the drug targeting index which characterize the targeting advantage in terms of exposure to CMS or formed colistin after nebulization were higher than 1, consequently, and in accordance with studies performed in rats [23, 26], a targeting advantage of pulmonary administration compared to intravenous administration was observed in sheep.
Pharmacokinetics of Polymyxin B in Animals
Polymyxin B and colistin are both old antibiotics used in the treatment of multidrug-resistant Gram-negative infections but not with the same clinical availability in various parts of the world. Europe and Australia have access to only colistin whereas in some other countries (e.g. United States, Brazil), both drugs are available. They have similar chemical structures with only one amino acid different in the ring structure [36]. However, a major difference in formulation exists since colistin is administered as an inactive prodrug (CMS), whereas polymyxin B is administered directly as its sulfate salt, which is active. Differences in terms of antibacterial concentration achievable both in plasma and in urine, in terms of toxicity but also in terms of pharmacokinetics are observable with these two molecules [36].
Due to its administration as an active form, the polymyxin B pharmacokinetics is simpler than that of colistin. The first recent modern pharmacokinetic study of polymyxin B in animals was performed in rats and relied on the measurement of four major components: polymyxin B1, isoleucine-polymyxin B1, polymyxin B2 and polymyxin B3, after IV administration of a single bolus dose equal to 4 mg/kg [37]. Since no major pharmacokinetic difference was observed between these four components, polymyxin B1, which is the most abundant component, was selected as the representative entity to describe the pharmacokinetics of polymyxin B. Polymyxin B1 pharmacokinetics was described by a one-compartment model and characterized by a clearance of 1.65 ± 0.62 mL/min and a volume of distribution of 198.1 ± 44.12 mL. Consequently the mean half-life value was estimated at 1.46 ± 0.39 h. Similar to colistin, the renal excretion of polymyxin B was negligible, with less than 1–5% of the dose recovered unchanged in urine collected up to 48 h [37, 38]. Consequently, renal insufficiency did not appear to have a significant impact on polymyxin B elimination in rats, in accordance with previous observations in patients with renal insufficiency [39, 40]. However, accumulation of polymyxin B in kidney tissue was observed, with concentrations 30 times higher in tissue than in serum in rats [41], and proximal tubular cells in the renal cortex and outer stripe of outer medulla seemed to be the main region of polymyxin accumulation [38]. Similar accumulation in renal proximal tubular cells was also observed in mice [42]. In rats, the accumulation of polymyxin B in renal tubular cells appeared to involve at least in part the transporter megalin that mediates uptake of substrates from tubular urine, and the accumulation was correlated with the onset of polymyxin B-induced nephrotoxicity [43]. Polymyxin B elimination may also occur by biliary excretion since the four major compounds of polymyxin B (B1, B2, B3 and isoleucine-polymyxin B) were detected in bile 4 hours after intravenous administration of 3 mg/kg in rats [38]. Polymyxin B concentrations were similar in muscle, heart, liver and in serum, but low concentrations were observed in brain [38]; however, whole tissue homogenate concentrations used in this study should be considered with great caution [34]. Polymyxin distribution studies in lung are limited and divergent results were found between this present study in rats which evaluated lung tissue homogenate concentrations [38] and a previous study in mice using ELF concentrations [12].
7.4 Pharmacokinetics of Novel Polymyxin-Like Compounds in Animals
Polymyxin B and colistin are used to treat serious infections caused by mutlidrug-resistant Gram-negative bacterial strains . However, both compounds are nephrotoxic which can restrict their use [44, 45]. Novel synthetic polymyxin-like antibiotics (NAB) which are potentially less toxic have been developed by Bachem AG (Budendorf, Switzerland) [46]. Among various compounds, NAB 739 and 740 present an antimicrobial activity that compares favorably with that of polymyxin B, although always slightly lower against most of the stains (Escherichia coli and Klebiella pneumoniae). Another derivative, NAB 7061, is not efficient by itself but demonstrates a strong synergism with clarithromycin and rifampicin [46, 47]. These NAB compounds present only 3 positive charges at physiological pH, compared with 5 for polymyxin B and colistin. Reducing the number of charges seems to reduce by a factor 6 to 7 the affinity of NAB compounds for isolated rat kidney brush border membrane [46] and consequently their potential renal toxicity compared with polymyxin B and colistin [48, 49]. The pharmacokinetics of NAB 739 and NAB 7061 was investigated in rats following IV bolus injection at a dose of 1 mg/kg [50]. The mean half-lives of NAB 739 and 7061 (respectively 69.0 ± 21.9 and 66.2 ± 12.3 min) were close to that of colistin (74.6 ± 13.2 min) [13] or polymyxin B (87.6 ± 23.4 min for polymyxin B1 and 79.8 ± 16.2 min for a polymyxin B2 and B3 mixture) [37]. The estimated volume of distribution of NAB 739 (222 ± 20.5 mL/kg) was slightly lower than that of NAB 7061 (339 ± 96 mL/kg), colistin (496 ± 60 mL/kg) [13] or polymyxin B (close to 800 mL/kg) [37]. Total clearance of NAB 739 (2.63 ± 0.54 mL/min/kg) was also lower than for other polymyxins (5.22 ± 0.4 mL/min/kg for colistin and between 6.5 and 7 mL/min/kg for polymyxin B). Although higher than those of colistin or polymyxin B, urinary recoveries were still relatively low with about 20% of the dose recovered in urine for NAB 739 and 7% for NAB 7061, with corresponding renal clearances estimated at 0.53 ± 0.30 mL/min/kg and 0.28 ± 0.16 mL/min/kg. These renal clearance values are approximately 30–50 times higher than those of colistin or polymyxin B which in fact constitutes the major pharmacokinetic specificity of these NAB compounds.
7.5 Conclusions
Although polymyxin antibiotics have been commercialized more than 50 years ago, reliable PK studies based on chromatographic assays have only been conducted recently. As a consequence, polymyxin PK has been investigated almost simultaneously in animals and humans. This is a rather unusual situation since traditionally animal studies are called pre-clinical studies as they are conducted first and provide valuable information before conducting studies in humans. As a result of research conducted over the last decade or so important data concerning the effect of CMS dose, brand or route of administration on colistin PK, have been obtained in animals. Other important questions such as non-renal elimination mechanisms remain to be addressed in animals. PB-PK approaches that could be of potential value to predict the effect of infection and of major pathophysiological alterations observed in critically ill patients on polymyxin disposition will be developed and validated in humans before being extrapolated to patients. Last but not least, because of the absence of major between-species differences in polymyxin PK, animal models of infection may be used to better characterize polymyxin PK-PD administered alone or in combination, which remains a major issue to improve treatment efficacy, reduce toxicity and delay mutant selection.
References
Nation RL et al (2014) Consistent global approach on reporting of colistin doses to promote safe and effective use. Clin Infect Dis 58(1):139–141
Schwartz BS et al (1959) Microbiological and pharmacological studies of colistin sulfate and sodium colistinmethanesulfonate. Antibiot Annu 7:41–60
al-Khayyat AA, Aronson AL (1973) Pharmacologic and toxicologic studies with the polymyxins. II. Comparative pharmnacologic studies of the sulfate and methanesulfonate salts of polymyxin B and colistin in dogs. Chemotherapy 19(2):82–97
Ziv G, Sulman FG (1973) Passage of polymyxins from serum into milk in ewes. Am J Vet Res 34(3):317–322
Ziv G, Schultze WD (1982) Pharmacokinetics of polymyxin B administered via the bovine mammary gland. J Vet Pharmacol Ther 5(2):123–129
Ziv G, Nouws JF, van Ginneken CA (1982) The pharmacokinetics and tissue levels of polymyxin B, colistin and gentamicin in calves. J Vet Pharmacol Ther 5(1):45–58
Renard L, Sanders P, Laurentie M (1991) Pharmacokinetics of colistin sulfate administered by intravenous and intramuscular routes in the calf. Ann Rech Vet 22(4):387–394
Lin B, Zhang C, Xiao X (2005) Toxicity, bioavailability and pharmacokinetics of a newly formulated colistin sulfate solution. J Vet Pharmacol Ther 28(4):349–354
Li J et al (2005) Evaluation of colistin as an agent against multi-resistant Gram-negative bacteria. Int J Antimicrob Agents 25(1):11–25
Li J et al (2001) A simple method for the assay of colistin in human plasma, using pre-column derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and reversed-phase high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 761(2):167–175
Gobin P et al (2010) Assay of colistin and colistin methanesulfonate in plasma and urine by liquid chromatography tandem mass spectrometry (LC-MS/MS). Antimicrob Agents Chemother 54:1941–1948
He J et al (2013) A validated ultra-performance liquid chromatography-tandem mass spectrometry method for the quantification of polymyxin B in mouse serum and epithelial lining fluid: application to pharmacokinetic studies. J Antimicrob Chemother 68(5):1104–1110
Li J et al (2003) Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob Agents Chemother 47(5):1766–1770
Marchand S et al (2010) Aerosol therapy with colistin methanesulfonate: a biopharmaceutical issue illustrated in rats. Antimicrob Agents Chemother 54(9):3702–3707
Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10(7):1093–1095
Ma Z et al (2009) Renal disposition of colistin in the isolated perfused rat kidney. Antimicrob Agents Chemother 53(7):2857–2864
Lu X et al (2016) Human oligopeptide transporter 2 (PEPT2) mediates cellular uptake of polymyxins. J Antimicrob Chemother 71(2):403–412
Bouchene S et al (2013) Comparison of colistin and colistimethate sodium (CMS) model-predicted whole-body distribution with measured tissue:plasma concentrations ratios in rats. In: 53rd interscience conference on antimicrobial agents and chemotherapy, Denver, CO
Li J et al (2004) Pharmacokinetics of colistin methanesulphonate and colistin in rats following an intravenous dose of colistin methanesulphonate. J Antimicrob Chemother 53(5):837–840
Li J et al (2002) Simple method for assaying colistin methanesulfonate in plasma and urine using high-performance liquid chromatography. Antimicrob Agents Chemother 46(10):3304–3307
Marchand S et al (2010) Dose-ranging pharmacokinetics of colistin methanesulphonate (CMS) and colistin in rats following single intravenous CMS doses. J Antimicrob Chemother 65(8):1753–1758
He H et al (2013) Pharmacokinetics of four different brands of colistimethate and formed colistin in rats. J Antimicrob Chemother 68(10):2311–2317
Yapa SWS et al (2013) Population pharmacokinetics of colistin methanesulfonate in rats: achieving sustained lung concentrations of colistin for targeting respiratory infections. Antimicrob Agents Chemother 57(10):5087–5095
Jin L et al (2011) Impact of p-glycoprotein inhibition and lipopolysaccharide administration on blood-brain barrier transport of colistin in mice. Antimicrob Agents Chemother 55(2):502–507
Bouchene S et al (2018) A whole-body physiologically based pharmacokinetic model for Colistin and Colistin Methanesulfonate in rat. Basic Clin Pharmacol Toxicol 123(4):407–422
Landersdorfer CB et al (2017) Substantial targeting advantage achieved by pulmonary administration of colistin methanesulfonate in a large-animal model. Antimicrob Agents Chemother 61(1):e01934-16
Couet W et al (2011) Pharmacokinetics of colistin and colistimethate sodium after a single 80-mg intravenous dose of CMS in young healthy volunteers. Clin Pharmacol Ther 89(6):875–879
Viel A et al (2018) A population WB-PBPK model of colistin and its prodrug CMS in pigs: focus on the renal distribution and excretion. Pharm Res 35(5):92
Cheah SE et al (2015) New pharmacokinetic/pharmacodynamic studies of systemically administered colistin against Pseudomonas aeruginosa and ACINETOBACTER baumannii in mouse thigh and lung infection models: smaller response in lung infection. J Antimicrob Chemother 70(12):3291–3297
Gontijo AV et al (2014) Biopharmaceutical characterization of nebulized antimicrobial agents in rats. 2. Colistin. Antimicrob Agents Chemother 58(7):3950–3956
Lin YW et al (2016) Pharmacokinetics/pharmacodynamics of pulmonary delivery of colistin against Pseudomonas aeruginosa in a mouse lung infection model. Antimicrob Agents Chemother 61(3):e02025-16
Lu Q et al (2010) Nebulized and intravenous colistin in experimental pneumonia caused by Pseudomonas aeruginosa. Intensive Care Med 36(7):1147–1155
Le Brun PP, de Graaf AI, Vinks AA (2000) High-performance liquid chromatographic method for the determination of colistin in serum. Ther Drug Monit 22(5):589–593
Mouton JW et al (2008) Tissue concentrations: do we ever learn? J Antimicrob Chemother 61(2):235–237
Marchand S et al (2015) Pharmacokinetics of Colistin Methansulphonate (CMS) and Colistin after CMS Nebulisation in Baboon Monkeys. Pharm Res 32(10):3403–3414
Nation RL, Velkov T, Li J (2014) Colistin and polymyxin B: peas in a pod, or chalk and cheese? Clin Infect Dis 59(1):88–94
Abdelraouf K et al (2012) Pharmacokinetics and renal disposition of polymyxin B in an animal model. Antimicrob Agents Chemother 56(11):5724–5727
Manchandani P et al (2016) Characterization of polymyxin B biodistribution and disposition in an animal model. Antimicrob Agents Chemother 60(2):1029–1034
Kwa AL et al (2011) Pharmacokinetics of polymyxin B in a patient with renal insufficiency: a case report. Clin Infect Dis 52(10):1280–1281
Zavascki AP et al (2008) Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis 47(10):1298–1304
Abdelraouf K et al (2012) Characterization of polymyxin B-induced nephrotoxicity: implications for dosing regimen design. Antimicrob Agents Chemother 56(9):4625–4629
Yun B et al (2015) Imaging the distribution of polymyxins in the kidney. J Antimicrob Chemother 70(3):827–829
Manchandani P et al (2017) Role of renal drug exposure in polymyxin B-induced nephrotoxicity. Antimicrob Agents Chemother 61(4):e02391-16
Oliveira MS et al (2009) Polymyxin B and colistimethate are comparable as to efficacy and renal toxicity. Diagn Microbiol Infect Dis 65(4):431–434
Akajagbor DS et al (2013) Higher incidence of acute kidney injury with intravenous colistimethate sodium compared with polymyxin B in critically ill patients at a tertiary care medical center. Clin Infect Dis 57(9):1300–1303
Vaara M et al (2008) Novel polymyxin derivatives carrying only three positive charges are effective antibacterial agents. Antimicrob Agents Chemother 52(9):3229–3236
Vaara M et al (2010) Susceptibility of carbapenemase-producing strains of Klebsiella pneumoniae and Escherichia coli to the direct antibacterial activity of NAB739 and to the synergistic activity of NAB7061 with rifampicin and clarithromycin. J Antimicrob Chemother 65(5):942–945
Mingeot-Leclercq MP et al (2012) Novel polymyxin derivatives are less cytotoxic than polymyxin B to renal proximal tubular cells. Peptides 35(2):248–252
Vaara M, Vaara T (2013) The novel polymyxin derivative NAB739 is remarkably less cytotoxic than polymyxin B and colistin to human kidney proximal tubular cells. Int J Antimicrob Agents 41(3):292–293
Ali FE et al (2009) Pharmacokinetics of novel antimicrobial cationic peptides NAB 7061 and NAB 739 in rats following intravenous administration. J Antimicrob Chemother 64(5):1067–1070
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Marchand, S., Grégoire, N., Couet, W. (2019). Pharmacokinetics of Polymyxins in Animals. In: Li, J., Nation, R., Kaye, K. (eds) Polymyxin Antibiotics: From Laboratory Bench to Bedside. Advances in Experimental Medicine and Biology, vol 1145. Springer, Cham. https://doi.org/10.1007/978-3-030-16373-0_7
Download citation
DOI: https://doi.org/10.1007/978-3-030-16373-0_7
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-16371-6
Online ISBN: 978-3-030-16373-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)