
Abstract
Purpose
The objective was the development of a whole-body physiologically-based pharmacokinetic (WB-PBPK) model for colistin, and its prodrug colistimethate sodium (CMS), in pigs to explore their tissue distribution, especially in kidneys.
Methods
Plasma and tissue concentrations of CMS and colistin were measured after systemic administrations of different dosing regimens of CMS in pigs. The WB-PBPK model was developed based on these data according to a non-linear mixed effect approach and using NONMEM software. A detailed sub-model was implemented for kidneys to handle the complex disposition of CMS and colistin within this organ.
Results
The WB-PBPK model well captured the kinetic profiles of CMS and colistin in plasma. In kidneys, an accumulation and slow elimination of colistin were observed and well described by the model. Kidneys seemed to have a major role in the elimination processes, through tubular secretion of CMS and intracellular degradation of colistin. Lastly, to illustrate the usefulness of the PBPK model, an estimation of the withdrawal periods after veterinary use of CMS in pigs was made.
Conclusions
The WB-PBPK model gives an insight into the renal distribution and elimination of CMS and colistin in pigs; it may be further developed to explore the colistin induced-nephrotoxicity in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colistin is an old peptide antibiotic from the polymyxin family that is used in human and veterinary medicines. In food producing animals, colistin is widely used as colistin sulphate to treat bacterial digestive infections. The use of its pro-drug, the colistimethate sodium (CMS), is the most frequent in human medicine but CMS can also be found as animal treatment. In human, in many cases colistin has become the last resort antibiotic against multi-resistant bacteria (1). Colistin is the active moiety and is formed from CMS hydrolysis within the body (2). CMS is a mixture of methanesulfonated molecules, which are hydrolysed in colistin by loss of methanesulfonate groups (3). The structures of colistin and CMS are responsible for their complex absorption, distribution, metabolism, and excretion (ADME), which depend on poorly described biological mechanisms (see below). Because of the renewed interest for colistin, clarifying this complexity is nevertheless essential in order to improve dosing adjustments and to avoid toxic effects (4).
Colistin and CMS have a high molecular weight (1167 g/mol and 1632 g/mol, respectively) and are ionized (cationic and anionic, respectively) at physiological pH (5), implying a weak passage of cellular membranes and physiological barriers. Hence, their distributions are supposed to be mainly within the extracellular spaces (6). According to few studies in animals, colistin is also suspected to bind to tissues (7,8). Concerning elimination mechanisms, CMS is partially excreted unchanged in urine, as seen in healthy humans and rats (2,6). Conversely, colistin excretion in urine is very low due to a major tubular reabsorption after glomerular filtration (2), and colistin tends to accumulate in kidney tissue (9,10). Specifically, colistin mainly accumulates within cells of the proximal tubules (11), where this extensive reabsorption takes place (12,13). This is an active process for which carrier-mediated uptakes involving different renal transporters, like PEPT2 (14) or megalin (10), have been identified. Moreover, an accumulation of polymyxins in intracellular organelles (like mitochondria and endoplasmic reticulum) has been shown (12) which could be linked to cellular death-pathways (15) and nephrotoxic effects (15,16). Colistin metabolism has not been described, but considering its peptidic structure it probably involves hydrolysis mechanisms (1).
Thus, the disposition of CMS and colistin within kidneys is not fully understood but has great implication in clinical practice, due to the dose-limiting nephrotoxicity. Classical pharmacokinetic approaches could be used to describe plasma profiles of both compounds but are inefficient to handle tissue concentrations. Moreover, it is quite difficult to collect experimental data in humans, especially in tissues, for obvious ethical reasons. Physiologically-based pharmacokinetics (PBPK) modelling, based on animal experiments, is a pertinent approach for this task. In addition to their ability to describe tissue distributions, these models are useful to perform some extrapolations from animals to human (17). A previous whole-body PBPK (WB-PBPK) model has been developed for CMS and colistin from rat experiments (18). However, several assumptions were made, especially for the renal/urinary distribution of CMS and colistin, without experimental data in tissue to support them. One advantage of this PBPK model was the use of a non-linear mixed effect (NLME) modelling approach (19) in order to handle inter-individual variabilities. Here, we refined this WB-PBPK model using numerous experimental plasma, urinary and tissue data from pigs, with a special focus on kidney exposure to colistin. Pigs were chosen for their physiological proximity to human (20), facilitating future inter-specie extrapolations.
The description of colistin pharmacokinetics in whole body is also interesting in the veterinary field. Indeed, for food safety concern, maximal residue limits (MRL) for colistin have been defined in edible tissues originating from food producing animals (21), e.g. pigs. These MRLs ensure that consumers can eat products from animals treated with colistin, without risk for their health. Therefore, a period is necessary between the last administration and the production of foodstuffs in order that colistin concentrations decrease below the MRL. This time is regulatory defined as the withdrawal period (WP). Linear regression based-methods are traditionally used to estimate the WP (22). However, the development of population PBPK modelling for this purpose in on going, due to their ability to predict tissue concentrations (23).
The aim of this study was first the development of a population PBPK model in pigs for colistin and its prodrug, CMS, using a NMLE approach with a focus on the renal disposition of both compounds. As an application of this PBPK model, its predictive ability to describe plasma and tissue concentrations was then used for estimating withdrawal period of colistin in pigs, highlighting one of the advantages of this modelling approach.
Materials and Methods
Chemicals
CMS (Colymicine 1 MIU; Sanofi Aventis, Paris, France) was used for all experiments. It was freshly reconstituted in 0.9% NaCl prior to each administration. To avoid any spontaneous hydrolysis of CMS into colistin, reconstituted CMS solutions were kept at +4°C and administered within the first hour after reconstitution.
Animals
Forty-six (46) crossbred female swine (Duroc × Landrace × Large white) were purchased from INRA (Le Rheu, France) with no history of polymyxin treatments. The animals were housed in collective pens and acclimatized for one week under standard farming conditions before the experiments. They were between 12 and 14 weeks old with a body weight (BW) ranging from 45 to 55 kg at the beginning of the experiments.
Then pigs were housed by groups of two for those receiving repeated intramuscular (IM) administrations. Pigs carrying a venous catheter were kept alone in metabolism cages for a maximum of 4 days.
Animal killing was performed with electronarcosis, that induced instantaneous insensibility, following by bleeding with aorta section.
All experiments were conducted in accordance with the local ethical comity and were registered under the references 2905–2015112717486085 and 6528–201608251410563.
In Vivo Experiments
Catheter Implantation
For pigs receiving at least one intravenous (IV) administration of CMS, central venous catheters were implanted. These animals were firstly sedated with an IM injection of ketamine at 20 mg/kg (Imalgène, Mérial, Lyon, France) and xylazine at 2 mg/kg (Rompun, Bayer, Loos, France). Then, they were intubated and kept anesthetized by inhalation with isoflurane 2.5% (IsoFlo, Zoetis, Malakoff, France) during all the surgical procedure. An incision was performed on the neck under local anaesthesia with xylocaine (Xylovet, CEVA, Libourne, France). After dilaceration of superficial tissues and muscles, two catheters were implanted in the jugular vein, one for drug administration and one for blood sampling. After surgery, pigs rested 48 h alone in their box. Then, they were housed separately in metabolism cage in order to facilitate drug administration and blood/urine sampling.
Sampling
-
Blood sampling: for pigs harbouring a venous catheter, 1.5 mL of blood was taken at each sample. Catheter was then flushed with heparinised saline solution. Pigs without catheter were restrained by an operator using a snout rope while a second one was sampling blood with a vacuum tube from the external jugular vein. Immediately after sampling, plasma was chilled on ice bath, then separated by centrifugation (3000 g) at 4°C and kept in polypropylene tubes at −80°C until assay.
-
Urine sampling: spontaneous urination was gathered only from animals kept in metabolism cage; volume of urine was measured and then a 20 mL-sample was kept in polypropylene tubes at −80°C until assay.
-
Tissue sampling: each organ (except muscles, fat and skin) was entirely collected after killing, weighted and its volume measured by water displacement. A sample was taken from the area of the left gluteal muscle (including skin) for skin and muscles analysis. A piece of abdominal fat was taken for the adipose analysis. Then, tissues were cut into small pieces, rinsed with saline solution, put into polypropylene tubes and kept at −80°C until assay. Tissues were chilled within the first 30-min following the euthanasia of pigs. The possible hydrolysis of CMS during that period, and its impact on the estimations of partition coefficients (see experiment n°3 below), were considered as negligible.
Experimental Setup for PBPK Model Calibration
Different dosing regimens (doses and route of administration) of CMS were administered for model calibration (a brief description is given in Table I). Some pigs were used for several experiments (n°1, 2 and 3, see below): in that case the potential residual concentrations from previous administrations were considered for modelling.
-
Experiment N°1/Plasma and kidney PK after a single IV administration (10 pigs): a 1-h constant IV infusion of CMS at 125,000 UI/kg of BW (corresponding to 3.75 mg/kg CBA or 10 mg/kg of CMS base (24)) was administered via the central catheter. Blood samples (n = 12 per pig) were taken from 0.5 h to 32 h after the start of CMS administration. Urine samples were collected over two intervals (0-8 h and 8-24 h after CMS administration) for 6 pigs, and for the remaining 4 animals between 6 and 9 successive urine samples were collected depending on technician availability. Four pigs were sacrificed at 32 h and their kidneys were immediately removed and processed as described in sampling section.
-
Experiment N°2/Plasma PK after a single IM administration (6 pigs): CMS solution (125,000 UI/kg of BW) was administered as two injections of about 10 mL into gluteal muscle of each side. Blood samples (n = 12 per pig) were taken from 0.25 h to 24 h after the injection.
-
Experiment N°3/Tissue partition coefficients (6 pigs): A dosing regimen was elaborated to achieve steady state of CMS and colistin in order to estimate the tissue to plasma partition coefficients (Kp): pigs were firstly infused during 1 h with a loading dose of CMS at 75,000 UI/kg; then a break of 1.5 h was done to get sufficient in vivo hydrolysis of CMS into colistin; finally, 50,000 UI/kg of CMS was administered as a 4 h-infusion. Blood samples were taken during the 4 h-infusion to assess steady-state in plasma; at the end of the infusion pigs were sacrificed and their blood, lungs, brain, heart, abdominal fat, skin, gluteal muscle, duodenum, liver and kidneys were immediately removed and processed as described in sampling section.
-
Experiment N°4/Plasma and kidney PK during and after repeated IM administrations (15 pigs): repeated CMS administrations were performed to study colistin renal accumulation. Pigs were randomly divided into 5 groups of 3 individuals. They received two IM injections of CMS per day at 25,000 UI/kg with a day-delay of 9 h (and 15 h during night). One group received 2 administrations (1 day) and were slaughtered 15 h after last injection; one group received 6 administrations (3 days) and were slaughtered 15 h after last injection; last 3 groups received 14 administrations (7 days) and were sequentially slaughtered at 15, 39 and 63 h after last injection. IM administrations were given on the neck and on top of gluteal muscles with a side alternation at each injection. Blood samples were taken during the treatment period in a sparse sampling way (between 1 and 3 per pig), each animal being sampled every 48 h at most. At sacrifice, blood and kidneys were collected and processed as described in sampling section.
Experimental Setup for Model Validation
-
Experiment N°5/Tissue and plasma PK after IM injections of CMS following the recommended veterinary dose (20 pigs): repeated CMS administrations were performed over 3 days with two IM injections per day at 25,000 UI/kg with a day-delay of 9 h (and 15 h during night). Then, pigs were sacrificed by groups of 4 at 1 h, 3 h, 5.5 h, 7.5 h, 15 h after last administration and their blood and fat, muscles, kidney, liver, skin (edible tissues) were collected as described above.
Determination of the Unbound Fraction (fu) of CMS in Plasma
The plasma fu of colistin in pigs (40%) was obtained from the literature (25). As no value was retrieved for CMS in the literature, we determined fu_CMS by ultrafiltration. Briefly, CMS was added to blank plasma from pig at a theoretical concentration of 5 μg/mL and 0.5 μg/mL, then ultra-filtrated through a cellulose-membrane (Centrifree, Merck, Alsace, France) by centrifugation (3000 g) at 37°C during 30 min. A similar experiment was performed in buffer instead of plasma in order to take into account the loss due to CMS hydrolysis at 37°C in plasma (3), and the potential non-specific binding of CMS to the lab material (5). All filtrates were kept at −20°C before assay (less than 1 week). All these experiments were realized in triplicates.
Analytical Methods
Plasma and urinary CMS and colistin concentrations were assayed with a validated high performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS) method using polymyxin B as internal standard, as described elsewhere (26). With this analytical method, CMS determination is achieved in an indirect way: a separate aliquot of each sample was pre-treated with sulphuric acid at 0.5 M (for 1 h at room temperature) to hydrolyse CMS to colistin and the concentration of CMS was then determined by difference between the concentrations measured before and after the acid hydrolysis, accounting for the differences in molecular weights of CMS and colistin.
For tissues, this method was adapted. Briefly, standards and quality controls were prepared from blank tissues. A sample of 100 mg for each organ was taken and 980 μL of blank plasma was added before homogenization with T-18 Ultra-Turrax homogenizer (KA®-Werke GmbH & Co. KG, Germany). Then 20 μL of a colistin solution (diluted in a 50/50 mix plasma/water) was added to obtain standard curves. After vortexing and centrifugation (4000 rpm for 10 min), supernatants were assayed as described for plasma (26). For samples, the same procedure was realized but 20 μL of a 50/50 mix plasma/water solution was added instead of 20 μL of colistin solution. Calibration range of colistin was from 0.020 μg/mL to 10 μg/mL in plasma and 0.2 to 100 μg/g In tissues; the concentrations of quality controls were at 0.16, 0.63 and 3.80 μg/mL in plasma and 0.5,5 and 75 μg/g in tissues; the limit of quantification were 0.02 μg/mL for colistin in plasma; 0.2 μg/g for colistin in all tissues. For CMS, the LOQ was 0.15 μg/mL in plasma and 1 μg/g in tissues.
For the assay in kidney, the intracellular localization of colistin made imprecise the discrimination between colistin and CMS concentrations. Indeed, the lysis of cells occurred both during the crushing phase and the acidic phase used for CMS determination. Therefore, the concentrations measured in kidneys corresponded to the sum of CMS and colistin, expressed as colistin.
Development of the PBPK Model
Model Structure
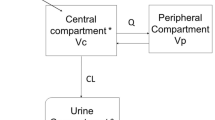
The PBPK model was based on a previous published model developed from rats, using plasma and various tissue concentrations (18). It is composed of 9 compartments corresponding to the main body organs (lungs, heart, liver, fat, skin, gastro-intestinal tract, brain, muscles, and kidneys), two blood compartments (arterial and venous) and one excretion compartment (urine) (Fig. 1a). Remaining body was lumped into a compartment named “rest of body”. Volumes of compartments and blood flows were fixed to physiological values reported in the literature (25,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42). These values depended on individual bodyweights and cardiac output (which also depends on bodyweight), respectively (Table II). The cardiac output was corrected by the haematocrit to get the total plasma flow. Because molecular weights of CMS and colistin are small with respect to passage across endothelial walls, it was expected that distribution within extracellular fluid was rapid (6) and a perfusion limited model was assumed for all organs except kidneys in which active intra-cellular transport occurs (see below).

A global diagram of the physiologically based pharmacokinetic (PBPK) model of CMS and colistin in swine. The whole body PBPK model is described (a), as well as the detailed mechanism in a generic tissue (except kidneys) (b), of the IM route (c) and of the deep compartments (d). During model development, supplementary components were added in the final model and are represented in blue (see Results). Kidney sub-compartments and urine are detailed in Fig. 2. All estimated parameters (in italic) are detailed in Table IV. Vtissue: tissue volume; Qtissue: blood flow; Kp_tissue: partition coefficient; IV: intravenous dose; IM: intramuscular dose.
Drug distribution in each tissue compartment (except kidney and bladder) was upon the dependency of Kps. CMS and colistin Kps were determined at the end of the perfusion established to reach the steady state for both drugs (experiment n°3) as follows (Eq. 1):
where Kp is the partition coefficient of the tissue, Ctis_ SS is the concentration measured at steady-state in the overall tissue, i.e. containing both extracellular and intracellular spaces; Cplas_ SS is the plasma concentration of CMS or colistin at steady-state.
The hydrolysis of CMS into colistin was assumed to take place in every compartment (including plasma), with a common hydrolysis constant (Khyd_CMS) (Fig. 1b). This assumption was supported by a previous experiment in rats, showing no significant differences of the hydrolysis rates between plasma and various tissue homogenates (18). This constant was estimated during model calibration. Thus, for each tissue an intrinsic hydrolysis clearance (CLhyd_CMS) was expressed as follows (Eq. 2):
Where V tissue is the volume of the corresponding tissue.
For colistin, the elimination occurs via mechanisms not yet described. Similar to CMS, an elimination process was defined based on a constant (Kdeg_COLI) common to all the organs (Fig. 1b) and estimated during model calibration. The intrinsic degradation clearance (CLdeg_COLI) was defined as (Eq. 3):
Where V tissue is the volume of the corresponding tissue.
Kidneys were divided into sub-compartments (Fig. 2) due to the particular distribution/elimination pathways of polymyxins within this organ. Renal vascular, extra-vascular and tubular intracellular spaces as well as luminal proximal tubular compartments were defined. CMS was eliminated in kidneys either by urine excretion or by hydrolysis into colistin. The latter one was assumed to take place in every sub-compartments of the kidney (according to a constant rate Khyd_CMS). Concerning the urine excretion of unchanged CMS, it was due either to glomerular filtration or potentially to secretion of CMS from the extra-vascular space to the tubular lumen (through tubular cells), as outlined for rats in a previous study (2). Therefore, a glomerular filtration clearance (CLGFR_CMS) was included in the structure of the base model, originating from kidney vascular space compartment and going into the proximal tubules one (43). CLGFR_CMS was calculated as the product of the glomerular filtration rate (GFR) and the fu_CMS in plasma (experimentally determined). The potential secretion of CMS was tested during model development.

Schematic representation of the renal sub-compartments in the PBPK model of CMS and colistin in swine. All estimated parameters (in italic) are detailed in Table IV. During model development, supplementary components were added in the final model and are represented in blue (see Results). QKID: renal blood flow; QURI: urinary flow; QTUB: tubular flow; CLGFR_CMS/CLGFR_COLI: filtration clearance of CMS and colistin.
Within kidney, colistin was also eliminated either by urine excretion or by metabolism (degradation). The urinary excretion corresponded to the colistin filtrated by the glomeruli (expressed by CLGFR_COLI) that was neither metabolized nor reabsorbed within the tubules. Indeed, colistin undergoes major tubular reabsorption: it was observed that more than 90% of excreted colistin was reabsorbed in rats (9). Colistin is then mainly located in proximal tubular cells as shown with in vitro studies with polymyxins (11,12,13). Therefore, a reabsorption clearance of colistin (CLreabs_COLI) was estimated in the model, originating from the tubular lumen into the intracellular sub-compartment. Colistin renal metabolism was assumed to take place in every sub-compartment (at constant rate corresponding to Kdeg_COLI). Due to the lack of data and the kidney physiology that is close between human and pig (20), the flow in the proximal tubule (Qtub) was fixed to the human value, i.e. about 67% of GFR (44). Bladder compartment was used as a transitory compartment receiving urine from tubular lumen and evacuating it, with an exit flow (Quri) determined in pigs kept in metabolism cage. This compartment was not considered as a vascularized tissue (no distribution).
For IM administrations, a compartment of depot was added (45). Two absorption rate constants originating from this depot compartment to the venous one were estimated, one for CMS (KIM_CMS) and one for colistin (KIM_COLI). Intrinsic hydrolysis of CMS and elimination of colistin were supposed to occur in this compartment such as in all other compartments (Fig. 1c).
The control file describing the PBPK model is presented in the supplementary files.
Model Calibration and Modelling Method
A NLME modelling approach was used for the estimation of unknown parameters, inter-individual variabilities (IIV) and residual variabilities (RV) (19). Model structure was modified if needed and model selection was based on the physiological plausibility (as described above) and a parsimonious approach. A decrease of the objective function value (OFV) of more than 3.84, corresponding to a 5% significance level, was considered as a significant improvement in model fit. The sampling importance resampling (SIR) method (46) was used to obtain the 95% confidence intervals (IC 95%) of the parameter estimates (5 iterations with 1000, 1000, 1000, 2000, 2000 samples and 200, 400, 500, 1000, 1000 resamples for each).
For IIV, a log-normal distribution of parameters was assumed. IIV was firstly estimated for each parameter separately and retained only if a significant decrease in OFV was observed without adding uncertainties in the estimation of fixed effects (forward selection process). In a second step, all IIV parameters for which the p-value was <0.05 in the univariate analysis were included in the model, and a stepwise backward selection was then performed, with a threshold of p-value of <0.05. For RV, additive and proportional error structure models were tested. Furthermore, for CMS and colistin concentrations determined in the same sample (see details of the analytical method), the L2 data item method was used in order to estimate potential correlation between their RV (47). The M3 method was used to handle data below the LOQ (BLOQ) (48).
The calibration step was performed with all experimental data described in section “Experimental setup for model calibration” and summarized in Table I. Concentrations were log-transformed before modelling. For urine data, CMS and colistin could not be discriminated due to the spontaneous hydrolysis of CMS into colistin after excretion (2,6). Thus, all observed concentrations were converted as CMS quantities (thanks to molecular ratio and measured urine volume) and pooled. Accordingly, urinary CMS and colistin model predictions were also pooled. Predictions in kidney corresponded to the amount of colistin and CMS in the different kidney sub-compartments (vascular, extra-vascular, intracellular and tubular lumen) divided by the sum of their respective volumes. Moreover, colistin and CMS concentrations in kidney were pooled for the analysis (cf. analytical methods).
The determination of CMS and colistin half-lives in plasma and kidneys was done by graphical identification of the different phases of decrease for predicted concentrations in log-scale, and then by calculating the time required for the typical concentrations to decrease by 50% in each phase. Total clearances of each compound were calculated as the dose of CMS, or as the total formed quantity of colistin, divided by the area under the curve of the plasma CMS, or colistin concentration, respectively. The renal clearances (CL R ) in this study accounted for the removal of compounds in kidney by different routes, i.e. secretion/excretion processes and hydrolysis of CMS or degradation of colistin. Thus, they were defined as the product of renal extraction ratio by the renal flow (Q kid ):
With C art : arterial concentration of CMS or colistin; Ckid_VA: concentrations of CMS or colistin in renal vascular compartment.
Model Evaluation
The performances of the PBPK model were tested in a 2-step approach. An internal validation was firstly done based on graphical and statistical criteria. Goodness-of-fit was assessed by plotting observed (DV) versus individual predictions (IPRED) and population predictions (PRED). Then Visual Predictive Checks (VPC) were generated, stratified by experimental designs and organs, with 1000 simulated replicates from the calibration dataset (Experiments n°1 to n°4, see Table I). The 5th and 95th percentile of the model predictions were plotted to check if 90% of the experimental data were included within this interval.
Then, an external validation was done with the independent dataset that was not used for calibration (Experiment n°5). This experiment followed the recommended veterinarian CMS doses. All parameters estimated during model calibration (fixed and random effects) were fixed. No RV was estimated for concentrations in tissues (except kidneys) during calibration because these tissue data were only used to determine the CMS and colistin Kps. Therefore, a common RV value between those estimated in plasma and kidney was chosen for the predictions of colistin concentrations in all other tissues. Concentrations of CMS and colistin within each organ were simulated with the final model and the predictive ability of the PBPK model was assessed by visualizing the distribution of the validation dataset within the 90% prediction intervals (10% of the data expected to be outside the interval). Only compartments involved in withdrawal period calculations (fat, skin, muscles, liver and kidney) and plasma were analysed.
A local sensitivity analysis was performed on the estimated structural parameters to assess their influence (associated to potential uncertainties) on model predictions. This analysis was performed only for kidney predictions, which was the main tissue of interest. The sensitivity analysis consisted in a ± 10% perturbation of each parameter estimate, all other parameters estimate being unchanged. The output considered for sensitivity was the time when the median model prediction for concentration in kidney felt below its MRL (0.20 μg/g).
Model Application: Withdrawal Period Estimation
To estimate withdrawal period, we generated a 98% prediction interval (i.e. 99% unilateral) (49) from 1000 simulations of the individual predicted profiles (without RV) of virtual pigs of 50 kg receiving the dosing scheme of CMS used in veterinary medicine. Then, the same approach was done with a virtual pig of 100 kg (which is close to the real slaughter weight). These simulations were performed with all structural parameters and their IIV (if present) estimated with the final model. Times for which the upper prediction limit felt below the MRL for each tissue intended for human consumption were determined. Then, the highest time from all of them was chosen as the final withdrawal period.
Software
The modelling was performed using NONMEM 7.4 (ICON Development Solutions, Ellicott City, Maryland, USA) with the first order conditional estimation method including eta-epsilon interaction (FOCE-I) and ADVAN 14. Perl speaks NONMEM (50) and Piraña (51) were used in order to facilitate the modelling work. All graphs were done using R software (version 3.4.1, www.R-project.org).
Results
Unbound Fraction of CMS in Plasma
We experimentally determined the fu_CMS by ultra-filtration at 37°C (Table III). About 28% of CMS was lost in buffer solution due to the CMS hydrolysis and to the non-specific binding to the tube. The measurement of colistin concentrations in the buffer samples at 37°C indicated that less than 6% of CMS was hydrolysed into colistin after 30 min (data not shown). Moreover, we had previously estimated in human and rat plasma samples spiked with CMS and kept for 30 min at 37°C, that about 8% of CMS was converted into colistin (in-house data). Therefore, the hydrolysis of CMS in plasma and buffer were quite low over the period of this experiment (30 min) and could be considered as negligible. Overall, by neglecting the degradation of CMS into colistin and assuming that the non-specific binding was similar during the ultrafiltration experiments in plasma and buffer, the average fu_CMS was estimated to be 37 ± 3% in pigs.
Model Structure and Calibration
The structure of the base model was developed in order to fit the experimental data. Colistin Kps were calculated as the ratios of concentrations measured at steady-state in tissue and plasma. However, for muscles, 3 concentrations over 6 were below the LOQ and were fixed at LOQ/2 for calculations of Kps. Results for Kps of colistin are reported in Table II. Concerning Kps of CMS, about 80% of tissue concentrations (except kidney) were under the LOQ (1 μg/g of tissue) at steady-state. Therefore, these Kps could not be measured experimentally and were estimated by the model (M3 method for data below the LOQ). However, estimation of one specific Kp for each organ was impossible and a Kp of CMS common for all tissues (Kpmix_CMS) as well as a common RV value for CMS concentrations in tissues were estimated (except for kidneys).
One additional compartment was added to each vascular compartment (arteries and veins) in order to fully describe the plasma colistin kinetic profile. These two compartments, referred as “deep compartments”, were volume-less and with two different estimated transfer constants (KDEEP_COLI and KDEEP_OUT_COLI) (Fig. 1d).
Structural modifications were needed for the kidney sub-model (permeability-limited model). Due to the protein binding of CMS, a secretion clearance of CMS (CLsec_CMS) from the extra-vascular compartment towards the tubular lumen through the intracellular compartment was added to explain the urinary excretion of CMS (Fig. 2). A rate of degradation of colistin (Kdeg_COLI) in kidneys different from that in other organs was estimated but did not improve significantly the fitting. Non-linear mechanisms for renal elimination of colistin were also tested, without significant improvement of the fitting. An intracellular binding compartment (volume less) significantly improved the fitting (OFV decrease of 20), with two different estimated “in and out” transfer constants (KON_COLI and KOFF_COLI). Because CMS and colistin could not be distinguished in urine, the fraction of colistin reabsorbed in proximal tubules could not be accurately estimated. Therefore, the proportion of colistin reabsorbed (driven by CLreabs_COLI) was estimated from data in human healthy volunteers (6) and fixed at 97.5%.
The parameter estimates of the PBPK model after model calibration are reported in Table IV. For each structural parameter, CI 95% were satisfying (the wider interval being for the colistin absorption constant from IM depot, KIM_COLI) highlighting the good precisions of the estimates. Overall, uncertainty for CMS parameters was lower than for colistin ones. Proportional residual errors were chosen as the best error models and the highest estimated RV was for kidney concentrations (57%). Two inter-individual variabilities, one associated to Kdeg_COLI (26.6%) and the other to CLSEC_CMS (43.5%), were estimated.
Model Evaluation
Model diagnostics showed acceptable goodness-of-fit plot for the final model (supplementary material, Fig. S1). The VPCs generated for the internal validation showed a good agreement between model median predictions and CMS and colistin concentrations measured in plasma, either after one IV of CMS (experiment n°1, Fig. 3a), one IM of CMS (experiment n°2, Fig. 3b) or after the dosing regimen implemented to achieve steady-state (experiment n°3, Fig. 4a). Median predictions of plasma concentrations were also reasonably well described after repeated IM administrations (experiment n°4, Fig. 4b), even if the median predictions were slightly below the measured peak of plasma concentrations for both compounds. However, distribution and elimination phases were well fitted. The wide prediction intervals, though, highlighted that variability could be overestimated. Overall, data BLOQ were well predicted by the model as shown on Fig. 3a, b. For the repeated IM injections, collected data were sparse so the fractions of data BLOQ were not presented due to graphical reasons. However, the only discrepancy was at 9 h just before the second IM injection because all observed colistin data were BLOQ in contrast to the model prediction (Fig. 4b).

Visual predictive checks of the PBPK model for CMS and colistin plasma concentrations after one IV (a) and one IM (b) of CMS, used for model calibration. Plasma data come from experiment n°1 for A (125,000 UI/kg of CMS after IV infusion over 1 h) and from experiment n°2 for B (125,000 UI/kg of CMS after IM injection). Blue dots represent the observed plasma concentrations; the grey areas represent the 90% prediction interval of the model, whereas the black solid line represents the median; the purple area represents the 95% confidence interval around the median; the horizontal dashed black lines represent the LOQ. In the smaller panels, blue areas represent the simulation-based 95% confidence intervals for the fraction of model simulated samples below the LOQ (BLOQ) at each time point, whereas the blue solid line represents the actual observed fraction of BLOQ samples.

Visual Predictive Checks of the PBPK model for CMS and colistin plasma concentrations after a dosing scheme to achieve steady-state (a) and repeated IM administrations (b) of CMS, used for model calibration. Plasma data come from experiment n°3 for A (75,000 UI/kg IV during 1 h; 1.5 h without administration; 50,000 UI/kg IV during 4 h) and from experiment n°4 for B (50,000 UI/kg of CMS divided in two IM injection per day). Blue dots represent the observed plasma concentrations; the grey areas represent the 90% prediction interval of the model, whereas the black solid line represents the median; the purple area represents the 95% confidence interval around the median; the horizontal dashed black lines represent the LOQ. No data below LOQ (BLOQ) were observed in A. For B, fractions of BLOQ are not represented due to the sparse sampling but they are discussed in the text.
Concerning kidney sub-model, cumulative urinary amounts after one IV administration of CMS were well predicted by the model (Fig. 5). For concentrations in kidney, the typical prediction captured well the renal accumulation (Fig. 6c), but there was a high variability between the different experiments, with some under-predictions (Fig. 6b) or over-predictions (Fig. 6a and late points of Fig. 6c). This variability was taken into account by the model thanks to the high estimated IIV and RV. However, the large 90% PI suggest that variability could be overestimated in kidneys.

Visual predictive checks of the PBPK model for cumulative urinary quantities concentrations after one IV of CMS, used for model calibration. Urinary data come from experiment n°1 (125,000 UI/kg of CMS after IV infusion over 1 h). Blue dots represent the observed plasma concentrations; the grey area represents the 90% prediction interval of the model, whereas the black solid line represents the median. The purple area represents the 95% confidence interval around the median.

Visual predictive checks of the PBPK model for total renal concentrations after one IV (a), the dosing scheme of infusions for steady-state (b) and repeated IM administrations (c) of CMS, used for model calibration. Kidney data come from experiment n°1 for A (125,000 UI/kg of CMS after IV infusion over 1 h); from experiment n°3 for B (75,000 UI/kg IV during 1 h; 1.5 h without administration; 50,000 UI/kg IV during 4 h, those pigs received one IV and one IM, 48 h and 24 h before t = 0 h, respectively) and from experiment n°4 for C (50,000 UI/kg of CMS divided in two IM injection per day). Blue dots represent the observed plasma concentrations; the grey areas represent the 90% prediction interval of the model, whereas the black solid line represents the median. The purple area represents the 95% confidence interval around the median. No data were below the LOQ (0.15 μg/g).
To assess the PBPK model predictive ability, an external validation was performed with an independent dataset that was not used during model calibration. Median plasma concentrations of both compounds were quite well fitted by the model typical predictions (Fig. 7). In kidneys, the elimination phase was in good match with observed data (Fig. 8) but the typical prediction under-estimated the maximal concentrations. For other tissue concentrations, most of the observed data were below the LOQ for CMS and colistin (Supplementary files, Fig. S2 and S3) and this was well predicted by the model. However, typical concentrations of CMS in muscles and of colistin in skin were slightly under-predicted. Overall, these results gave good confidence in the PBPK model predictive ability, even if there was a slight overestimation of the total variability.

VPC of the PBPK model for CMS (a) and colistin (b) plasma concentrations after 3 days of IM administrations of CMS, used for model validation. Observed data come from an independent experiment (n°5: 50,000 UI/kg of CMS divided in two IM injection per day during 3 days) that was not used for model calibration. Blue dots represent the observed plasma concentrations; the grey areas represent the 90% prediction interval of the model, whereas the black solid line represents the median; the purple area represents the 95% confidence interval around the median; the horizontal dashed black lines represent the LOQs. In the smaller panels, blue areas represent the simulation-based 95% confidence intervals for the fraction of model-simulated samples below the LOQ (BLOQ) at each time point, whereas the blue solid line represents the actual observed fraction of BLOQ samples.

VPC of the PBPK model for total renal concentrations after 3 days of IM administrations of CMS. Observed data come from an independent experiment (n°5: 50,000 UI/kg of CMS divided in two IM injection per day) that was not used for model calibration. Blue dots represent the observed plasma concentrations; highlighted with grey are the areas between the 5th and 95th percentiles of model simulations, whereas the black solid line represents the median. No data were below the LOQ (0.15 μg/g).
CMS and Colistin Pharmacokinetics in Plasma and Tissue
After IV and IM injections of CMS, plasma concentrations of CMS declined quickly and were under the LOQ 8 h post-administration (Fig. 3a, b). By contrast, colistin plasma concentrations declined slower and were still quantifiable more than 24 h after CMS administration. The estimated half-life (t1/2) of CMS was 1.2 h. For colistin, an initial (lasting from 0 to 10 h after dosing) half-life of distribution of 1.8 h was calculated, followed by a terminal half-live of 10.5 h. This terminal t1/2 for colistin was described in the model thanks to the additional deep compartments. The maximal concentrations of colistin were predicted to occur 1 h after the end of the IV infusion, or 2 h after IM injection. As shown by the median prediction in Fig. 4a, the plasma steady-state of CMS and colistin was achieved at the end of experiment n°3 for Kp determination, thanks to the use of a loading dose of CMS. Concerning drug elimination, total CMS clearance was estimated to 11.6 L/h for a virtual pig of 50 kg, whose 7.9 L/h was associated to renal clearance, which included the glomerular filtration, the tubular secretion and the CMS hydrolysis within kidneys. For colistin, the total clearance was found to be at 7.4 L/h whereas the renal clearance, which was mostly due to intracellular metabolism, accounted for 1.5 L/h (see below).
For tissues other than kidneys, all colistin Kp values were less than one (Table II) as well as the common estimated Kp for CMS (Table IV). In kidneys, the CMS concentrations were predicted to decrease quickly after an administration of CMS (Supplementary material, Fig. S4). Therefore, we can consider that at late time points after administration there was only colistin remaining in kidneys. Residual renal concentrations were still high 31 h after the single IV injection (7.0 ± 3.4 μg/g, Fig. 6a) and 63 h after the last IM injection (5.6 ± 1.9 μg/g, Fig. 6c); the terminal t1/2 of colistin in kidneys was estimated to be about 38 h. After twice daily IM administrations of CMS 25,000 UI/kg, colistin concentrations in kidney were 7.0 ± 1.3 μg/g after 1 day (2 doses), 14.7 ± 9.9 μg/g after 3 days (6 doses) and 24.6 ± 5.9 μg/g after 7 days (14 doses) (experiment n°4, Fig. 6c). The model predicts that steady-state in kidney should be almost reached after 5 days of treatment.
The relative disposition of CMS and colistin within the kidneys as predicted by the PBPK model is presented on Fig. 9. Typically, 68% of the initial dose is predicted to be excreted in urine as a mix of CMS and colistin, with 56% due to the net tubular secretion of CMS and only 12% due to the glomerular filtration of CMS. In urine, CMS accounted for more than 99% of the total quantities. Among the fraction of the CMS dose converted into colistin (32%), only 2% were converted into the kidney. Colistin extraction ratio in kidney would be 18%, with glomerular filtration of the unbound fraction in plasma, almost complete reabsorption (0.2% of formed colistin excreted in urine) and intracellular degradation in proximal tubules. Overall, kidneys would account for 20% of total colistin clearance.

CMS and colistin disposition within kidneys as given by the PBPK model. Each percentage represents the fraction of the initial dose (100%) involved in each process. fu_CMS/fu_COLI: unbound fraction of CMS/colistin; GFR: glomerular filtration rate.
For the other compartments, the evolution of the mass balance of CMS and colistin after one IV of CMS are represented in the supplementary materials (Fig. S5).
Model Application: Withdrawal Period Estimation
The withdrawal period was calculated from depletion in kidneys because colistin concentrations within this organ remained the longest above the LMR. The simulation of the PBPK model gave a WP of 23 days for a virtual pig of 50 kg (Fig. 10) and 25 days for a virtual pig of 100 kg (Supplementary material, Fig. S6). The sensitivity analysis revealed that KON_COLI,KOFF_COLI and Kdeg_COLI were the 3 parameters that particularly influenced the output, i.e. the time when the median model prediction of the renal concentration fell below the MRL. The most influential parameter was KOFF_COLI, because 10% variation of its value caused 8% to 9% variation of the output (Table V).

Withdrawal period estimation in a 50-kg pig. Model simulation in kidney after 3 consecutive days of CMS IM injections (50,000 UI/kg of CMS divided in two injections per day) for 1000 virtual pigs of 50 kg. The grey area includes the 1st and 99th percentiles of model simulations, whereas the black solid line represents the median; the horizontal dashed black line represents the kidney MRL for colistin (0.20 μg/g). WP: withdrawal period, rounded to the next whole day.
Discussion
A whole body PBPK model was developed for colistin and its pro-drug, CMS, with a nonlinear mixed effect modelling approach. This model could reasonably well predict the median CMS and colistin concentrations in plasma (Fig. 7) and tissues, especially for kidneys (Fig. 8). For pigs, the PK of CMS in plasma was monophasic, with a t1/2 of 1.2 h, whereas the PK of colistin was biphasic, with a distribution t1/2 of 1.8 h and a terminal t1/2 of 10.5 h (Fig. 3a, b). These half-lives in pigs were in good agreement with those in healthy volunteers (6) except that a monophasic elimination of colistin was described. However, this biphasic profile was also observed in sheep, another large-animal model (52). As colistin is known to non-specifically bind to biological and non-biological matrix (7,53,54), deep compartments linked to vascular ones were implemented in the PBPK model to fit the plasma concentrations of colistin at late time points. These compartments may reflect either a permeability-limited distribution of colistin in some organs, e.g. due to a weak intracellular penetration, or a high affinity binding to some extracellular component (e.g. red blood cells (55)), thus resulting in a slow release of colistin towards plasma. However, evidences of these mechanisms should be sought in experimental studies.
Concerning distribution, plasma unbound fraction of CMS was determined for the first time in pigs, thanks to an ultra-filtration method and by taking care of CMS degradation and potential adsorption to laboratory material. This value (37%) was close to that of colistin (40%) found in literature for pigs (25), highlighting a non-negligible protein binding. Colistin is known to bind to α-1-acid glycoprotein (at least for human) due to its cationic properties (56) but the mechanisms of CMS protein binding have not been investigated yet. Regarding tissue distribution, all Kp values (experimental ones for colistin and estimated ones for CMS) were lower than 1. This result reflected a poor distribution into tissues that could be in accordance with an extracellular distribution of CMS and colistin within organs. Our values were in good agreement with the experimental Kps determined in rats for colistin (18), suggesting that these values could be used for inter-species extrapolations.
Concerning the elimination, CMS total clearance was higher (0.23 L/h/kg) than the colistin one (0.15 L/h/kg) for a standard pig weighting 50 kg. These results compare favourably with previous results in pigs (57). By contrast, these clearances are greater than those reported in healthy volunteers for CMS and colistin (0.12 L/h/kg and 0.040 L/h/kg for a man weighting 73 kg, respectively) (6), which is in contradiction with the classical allometry scaling laws based on weight (58). Therefore, extrapolation of clearances between the two species may be challenging.
To our knowledge, this is the first time that an accumulation of colistin in kidneys was quantified over time after repeated CMS administrations. Indeed, after a twice-daily IM administration of CMS (50,000 UI/kg/day) for 7 days, colistin concentrations in kidney were more than 3-fold higher than after the first administration (Fig. 6c). This was related to the long t1/2 of colistin estimated in this organ (~38 h). A previous study in rats already attested that the concentrations of colistin were high in kidney after 7 days of treatment (65.7 fold higher than in plasma), but the renal accumulation was not investigated over time (59).
To go further into the underlying mechanisms of the renal disposition of CMS and colistin, we divided the kidney into physiological sub-compartments (Fig. 2) (43). The unbound fraction of CMS in plasma, estimated herein, implied a tubular secretion of CMS because glomerular filtration was insufficient to explain the amounts measured in urine (Fig. 2). According to our model, this tubular secretion was the major elimination pathway for CMS in kidney, 4 to 5-fold higher than glomerular filtration (Fig. 9). This net tubular secretion of CMS into urine was already supported by studies in rats (2) and suggested in humans (60), but as the unbound fraction of CMS in plasma was unknown, it remained hypothetic. The median proportion of the initial dose of CMS excreted in urine was predicted to be 68%, in accordance with the 60 to 70% of CMS dose recovered in urine in rats and humans (2,6).
Regarding colistin pharmacokinetics in kidneys, the tubular reabsorption is known to be career-mediated thanks to PEPT2 and megalin (10,14). In our model, the clearance of reabsorption from the tubular lumen (CLreabs_COLI) was fixed to a physiological value estimated from data in man (6), due to an identifiability problem. This reabsorption explained the colistin accumulation within tubular cells and the very small amount of colistin excreted in urine. Colistin might also undergo a tubular secretion, as for CMS, but because of the predominant reabsorption, this was not identifiable by our model.
Several hypotheses were considered in the model to describe the colistin accumulation in kidneys and its slow elimination in this organ. For instance, we tried to estimate a different colistin intrinsic constant of elimination (Kdeg_COLI) in kidney, or to estimate a release of colistin from the kidneys towards the systemic circulation. However, these hypotheses did not improve the fitting or parameters were not identifiable. Moreover, the latter assumption, besides the modelling results, was also in contradiction with the results of previous studies showing in vivo that when the reabsorption of colistin or polymyxin B in renal tubules was inhibited, the renal exposures was reduced considerably but the kinetic profiles in plasma remained unaltered (10,61). The fact that total clearance of polymyxin remained unchanged, whether the reabsorption was inhibited or not, suggested that polymyxin was eliminated within the kidney, either excreted in urine or metabolized, but did not go back to the systemic circulation. Finally, the use of a renal intracellular “binding” compartment, with a slow release, was the best choice for the goodness-of-fit and for physiological reasons. Indeed, the observed colocalization with cell organelles (12) and the known non-specific binding properties of polymyxins to cellular membranes (1) could support this assumption. Sensitivity analysis suggested that these intracellular binding parameters were the parameters that most influenced the kidney exposure to colistin (Table V). This kind of intracellular binding has already been presented in another PBPK model developed and validated for doxorubicin (belonging to an antitumor antibiotic family) (30). Furthermore, due to this intracellular accumulation, about 20% of the total formed colistin quantities were predicted to be metabolized within the kidneys, highlighting a major role of kidneys in colistin elimination. Further in vitro studies should be performed to investigate this intracellular binding and the intra-renal elimination.
These new renal data, about CMS tubular secretion and colistin accumulation over time, may be useful to explore the nephrotoxicity associated to the use of CMS and colistin. Indeed, in a recent meta-analysis, nephrotoxicity ranged from 24% to 74% in CMS-treated patients but most of the events were reversible (62). The duration therapy and the daily dose are risk factors of renal toxicity (1). In our study, the renal steady-state was quasi-achieved after 5 days of treatment (>120 h) and high renal concentrations were reached (>20 μg/g) (Fig. 6c). However, no clinical sign of renal insufficiency was observed in our animals and the creatinine concentrations in plasma stayed within the normal range over the whole treatment period (data not shown). Nevertheless, measurement of other biomarkers (like urinary creatinine or plasma cystatin C) might have been better to detect early signs of nephrotoxicity (63), in addition to histological analysis. According to modelling results, the intra-renal conversion of CMS into colistin was very minor compared to the colistin that was reabsorbed and accumulated inside tubular cells. The accumulation of polymyxin inside proximal tubular cells is supposed to be responsible for the nephrotoxic effects involving apoptosis and oxidative stress (15,16). However, the model slightly underestimated peak concentrations in kidney (Fig. 8), which may be of importance for nephrotoxic predictions. Of note, a high amount of CMS (56% of initial dose) transited inside tubular cell. This is important as CMS (and its numerous partially methanesulfonated derivatives) is invoked as a potential contributor to the observed nephrotoxic effects (60). Nevertheless, this model could be refined when new data will be available.
As a model application, we chose to estimate the withdrawal period (WP) after IM injections of CMS in pigs following the veterinarian recommended doses. WP is defined as the time after last administration for which 99% of animals have residual edible tissue concentrations below the MRL, in Europe. Kidney was the tissue of interest because of the accumulation and of the slow colistin depletion (Fig. 8), compared to all other edible tissues (Supplementary files, Fig. S2 and S3). Simulations from the PBPK model gave an estimated WP of 23 days for a 50-kg pig. Since our last experimental concentration was measured 3 days after last administration, further experimental data around the estimated WP would have been preferable to confirm it. Overall, the model prediction seemed reasonable as our estimated WP (23 days) was close to the official one given in the summary of product characteristics of the veterinary medicinal product (21 days). The renal accumulation observed in our repeated CMS injections experiment highlighted that an extra label use of CMS would probably need longer WP. No data of such use for colistin are currently recorded but the use of PBPK models in these extra label situations has already proved its interest (64). Furthermore, this PBPK model was developed with colistin and its prodrug CMS but it could be easily adapted to pigs injected directly with colistin, as possibly done in veterinary medicine (65).
To our knowledge, this is the first study using a NLME approach for a PBPK model related to withdrawal period calculation. The process of PBPK models development for food safety is well established as explained in a recent review of veterinary pharmacology (23). This method allows to predicting the time course of drug concentrations in any tissue of interest. Different doses and route of administrations can be used to develop a PBPK model as we did, enhancing its robustness of prediction (23). The classical statistical methodology for WP estimation uses inferences on a limited number of healthy animals whereas the real-life target is diseased animals. We also used healthy animals but the PBPK model can easily handle patho-physiological changes of parameters, like for a diseased animal, to see the effect on tissue drug concentrations. In addition, the use of NLME modelling brings many advantages. Noticeably, it gives estimation of population variabilities like inter-individual variability, which is discriminated from the unexplained but quantified residual variability. This is important as the WP calculation applies for a global “population of treated animals” and must include 99% of them. The prediction of WP was based on simulations taking into account IIV but not RV, because what is important is the actual concentration in tissue (which depends on IIV) and not the measured concentration (which depends also on RV). As there was only one tissue sample per animal (destructive sampling), IIV might be difficult to estimate and it is possible that RV, which was high for renal concentrations (57%), was inflated by unidentified IIV. This bias, resulting in an underestimation of the IIV, could result in an underestimation of the WP. On the other hand, VPCs indicated that there was a potential overestimation of the overall variability, without knowing if it was IIV or RV that was inflated: this bias would result in a contrary over-prediction of WP. This issue rises the necessity, for an accurate estimation of WP, of an accurate estimation of both IIV and RV. The NMLE approach is also efficient to handle sparse data (like in experiment n°4) and thus could limit the number of necessary animals. Furthermore, this method allows a sophisticated handling of data below the limit of quantification compared to the classic one. Indeed, in the latter approach, the rule is to omit or fix the data BLOQ at half of the LOQ but it could bias the results (22).
Lastly, it is necessary to highlight some analytical considerations. Due to the high instability of CMS, direct measure of its concentration was not possible (26). The indirect method used could not discriminate renal CMS and colistin concentrations, which were pooled. Therefore, the estimations of parameters in kidney might have been biased. Moreover, CMS is a mixture of many methanesulfonated derivatives carrying various number of methanesulfonate groups (3). It was not possible to determine the concentration of each component separately; therefore, all these derivatives were considered as being CMS. As previously explained, colistin (which has no methanesulfonate group) is widely reabsorbed, whereas CMS (which have 5 methanesulfonate groups) is not. Therefore, some partially methanesulfonated derivatives, i.e. considered as CMS, might also be reabsorbed. These compounds may have an ADME closer to colistin than to CMS. All these concerns highlight the need of further analytical developments allowing to discriminate CMS and colistin in kidneys and to quantify the various methanesulfonate derivatives.
Conclusion
To conclude, this PBPK model coupled with a NMLE approach gave new insight into the mechanistic pharmacokinetics of CMS and colistin, especially within kidneys. This may have implications to limit the colistin induced-nephrotoxicity in human medicine. We also used this model to estimate withdrawal period in pigs treated with CMS, highlighting the utility of such an approach in veterinary medicine. Furthermore, PBPK models are helpful to perform inter-species extrapolation (from animal to human), but also intra-specie extrapolation (from adult to children). Thus, this model could be useful to adapt CMS dosing-regimen in pediatric population, a sub-population which is less studied. Some works about this topic are ongoing in our team.
Acknowledgments and Disclosures
Alexis Viel was supported by a doctoral fellowship from the French National Institute of Health and Medical Research (Inserm) and the French Agency for Food, Environmental and Occupational Health & Safety (Anses).
Abbreviations
- ADME:
-
Absorption, distribution, metabolism, excretion
- BLOQ:
-
Below the limit of quantification
- BW:
-
Body weight
- CBA:
-
Colistin base activity
- CMS:
-
Colistimethate sodium
- DV:
-
Observed value
- fu:
-
Unbound fraction
- GFR:
-
Glomerular filtration rate
- GIT:
-
Gastro-intestinal tract
- HPLC-MS/MS:
-
High-performance liquid chromatography coupled with tandem mass spectrometry
- IIV:
-
Interindividual variability
- IM:
-
Intramuscular
- IPRED:
-
Individual prediction
- IV:
-
Intravenous
- LOQ:
-
Limit of quantification
- MRL:
-
Maximal residue limits
- NLME:
-
Nonlinear mixed effects
- OFV:
-
Objective function value
- PBPK:
-
Physiologically-based pharmacokinetic
- PK:
-
Pharmacokinetics
- PRED:
-
Population prediction
- RV:
-
Residual variability
- SIR:
-
Sampling importance resampling
- t1/2 :
-
Half-life
- VPC:
-
Visual predictive checks
- WB-PBPK:
-
Whole body physiologically-based pharmacokinetic
- WP:
-
Withdrawal period
References
Gregoire N, Aranzana-Climent V, Magreault S, Marchand S, Couet W. Clinical pharmacokinetics and pharmacodynamics of Colistin. Clin Pharmacokinet. 2017;56:1441–60.
Li J, Milne RW, Nation RL, Turnidge JD, Smeaton TC, Coulthard K. Pharmacokinetics of colistin methanesulphonate and colistin in rats following an intravenous dose of colistin methanesulphonate. J Antimicrob Chemother. 2004;53(5):837–40.
Li J, Milne RW, Nation RL, Turnidge JD, Coulthard K. Stability of Colistin and Colistin Methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob Agents Chemother. 2003;47(4):1364–70.
Forrest A, Garonzik SM, Thamlikitkul V, Giamarellos-Bourboulis EJ, Paterson DL, Li J, et al. Pharmacokinetic/toxicodynamic analysis of colistin-associated acute kidney injury in critically ill patients. Antimicrobial Agents and Chemotherapy. 2017:AAC. 01367–17.
Li J, Nation RL, Milne RW, Turnidge JD, Coulthard K. Evaluation of colistin as an agent against multi-resistant gram-negative bacteria. Int J Antimicrob Agents. 2005;25(1):11–25.
Couet W, Gregoire N, Gobin P, Saulnier P, Frasca D, Marchand S, et al. Pharmacokinetics of colistin and colistimethate sodium after a single 80-mg intravenous dose of CMS in young healthy volunteers. Clin Pharmacol Ther. 2011;89(6):875–9.
Craig WA, Kunin CM. Dynamics of binding and release of the polymyxin antibiotics by tissues. J Pharmacol Exp Ther. 1973;184(3):757–65.
Tomasi L, Giovannetti L, Rondolotti A, Rocca GD, Stracciari GL. Depletion of the residues of colistin and amoxicillin in turkeys following simultaneous subcutaneous administration. Vet Res Commun. 1996;20(2):175–82.
Ma Z, Wang J, Nation RL, Li J, Turnidge JD, Coulthard K, et al. Renal disposition of colistin in the isolated perfused rat kidney. Antimicrob Agents Chemother. 2009;53(7):2857–64.
Suzuki T, Yamaguchi H, Ogura J, Kobayashi M, Yamada T, Iseki K. Megalin contributes to kidney accumulation and nephrotoxicity of colistin. Antimicrob Agents Chemother. 2013;57(12):6319–24.
Yun B, Azad MA, Wang J, Nation RL, Thompson PE, Roberts KD, et al. Imaging the distribution of polymyxins in the kidney. J Antimicrob Chemother. 2015:70(3):827–9.
Yun B, Azad MA, Nowell CJ, Nation RL, Thompson PE, Roberts KD, et al. Cellular uptake and localization of polymyxins in renal tubular cells using rationally designed fluorescent probes. Antimicrob Agents Chemother. 2015;59(12):7489–96.
Azad MA, Roberts KD, Yu HH, Liu B, Schofield AV, James SA, et al. Significant accumulation of polymyxin in single renal tubular cells: a medicinal chemistry and triple correlative microscopy approach. Anal Chem. 2015;87(3):1590–5.
Lu X, Chan T, Xu C, Zhu L, Zhou QT, Roberts KD, et al. Human oligopeptide transporter 2 (PEPT2) mediates cellular uptake of polymyxins. J Antimicrob Chemother. 2016;71(2):403–12.
Dai C, Li J, Tang S, Li J, Xiao X. Colistin-induced nephrotoxicity in mice involves the mitochondrial, death receptor, and endoplasmic reticulum pathways. Antimicrob Agents Chemother. 2014;58(7):4075–85.
Azad MA, Akter J, Rogers K, Nation RL, Velkov T, Li J. Major pathways of polymyxin-induced apoptosis in rat kidney proximal tubular cells. Antimicrob Agents Chemother. 2015;59(4):2136–43.
Nestorov I. Whole-body physiologically based pharmacokinetic models. Expert Opin Drug Metab Toxicol. 2007;3(2):235–49.
Bouchene S, Marchand S, Couet W, Friberg LE, Gobin P, Lamarche I, et al. Comparison of Colistin and Colistimethate sodium (CMS) model-predicted whole-body distribution with measured tissue:plasma concentrations ratios in rats. 53rd interscience conference on antimicrobial agents and chemotherapy; Denver, Co 2013.
Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetic parameters II. Biexponential model and experimental pharmacokinetic data. J Pharmacokinet Pharmacodyn. 1981;9(5):635–51.
Swindle MM, Makin A, Herron AJ, Clubb FJ, Frazier KS. Swine as models in biomedical research and toxicology testing. Veterinary Pathology Online. 2012;49(2):344–56.
JECFA. Residue evaluation of certain veterinary drugs: joint FAO/WHO expert committee on food additives, 66th meeting 2006: Food & Agriculture org.; 2006.
EMA. Guideline on approach towards harmonisation of withdrawal periods. European Medicines Agency (EMA) - Committee for Medicinal Products for Veterinary Use (CVMP), 2016 EMA/CVMP/CHMP/231573/2016 Contract No.: EMA/CVMP/SWP/735325/2012.
Lin Z, Gehring R, Mochel J, Lavé T, Riviere J. Mathematical modeling and simulation in animal health–part II: principles, methods, applications, and value of physiologically based pharmacokinetic modeling in veterinary medicine and food safety assessment. J Vet Pharmacol Ther. 2016;39(5):421–38.
Nation RL, Li J, Cars O, Couet W, Dudley MN, Kaye KS, et al. Consistent global approach on reporting of Colistin doses to promote safe and effective use. Clin Infect Dis. 2014;58(1):139–41.
Rottbøll LAH, Friis C. Penetration of antimicrobials to pulmonary epithelial lining fluid and muscle and impact of drug physicochemical properties determined by microdialysis. J Pharmacol Toxicol Methods. 2016;78:58–65.
Gobin P, Lemaître F, Marchand S, Couet W, Olivier J-C. Assay of Colistin and Colistin Methanesulfonate in plasma and urine by liquid chromatography-tandem mass spectrometry. Antimicrob Agents Chemother. 2010;54(5):1941–8.
Buur JL, Baynes RE, Craigmill AL, Riviere JE. Development of a physiologic-based pharmacokinetic model for estimating sulfamethazine concentrations in swine and application to prediction of violative residues in edible tissues. Am J Vet Res. 2005;66(10):1686–93.
Chen K, Calibration SKY. Validation of a physiologically based model for soman intoxication in the rat, marmoset, Guinea pig and pig. J Appl Toxicol. 2012;32(9):673–86.
de Boer VC, Dihal AA, van der Woude H, Arts IC, Wolffram S, Alink GM, et al. Tissue distribution of quercetin in rats and pigs. J Nutr. 2005;135(7):1718–25.
Dubbelboer IR, Lilienberg E, Sjögren E, Lennernas H. A model-based approach to assessing the importance of intracellular binding sites in doxorubicin disposition. Mol Pharm. 2017;14(3):686–98.
Elowsson P, Carlsten J. Body composition of the 12-week-old pig studied by dissection. Lab Anim Sci. 1997;47(2):200–2.
Eskild-Jensen A, Jacobsen L, Christensen H, Frokiaer J, Jorgensen HS, Djurhuus JC, et al. Renal function outcome in unilateral hydronephrosis in newborn pigs. II. Function and volume of contralateral kidneys. J Urol. 2001;165(1):205–9.
Lødrup AB, Karstoft K, Dissing TH, Nyengaard JR, Pedersen M. The association between renal function and structural parameters: a pig study. BMC Nephrol. 2008;9:18.
Lundeen G, Manohar M, Parks C. Systemic distribution of blood flow in swine while awake and during 1.0 and 1.5 MAC isoflurane anesthesia with or without 50% nitrous oxide. Anesth Analg. 1983;62(5):499–512.
Rendas A, Branthwaite M, Reid L. Growth of pulmonary circulation in normal pig--structural analysis and cardiopulmonary function. J Appl Physiol. 1978;45(5):806–17.
Scotcher D, Jones C, Posada M, Rostami-Hodjegan A, Galetin A. Key to opening kidney for in vitro–in vivo extrapolation entrance in health and disease: part I: in vitro systems and physiological data. AAPS J. 2016;18(5):1067–81.
Suenderhauf C, Parrott N. A physiologically based pharmacokinetic model of the minipig: data compilation and model implementation. Pharm Res. 2013;30(1):1–15.
Ten have GAM, Bost MCF, Suyk-Wierts JCAW, van den Bogaard AEJM, Deutz NEP. Simultaneous measurement of metabolic flux in portally-drained viscera, liver, spleen, kidney and hindquarter in the conscious pig. Lab Anim. 1996;30(4):347–58.
Tranquilli WJ, Manohar M, Parks CM, Thurmon JC, Theodorakis MC, Systemic BGJ. Regional blood flow distribution in unanesthetized swine and swine anesthetized with halothane + nitrous oxide, halothane, or enflurane. Anesthesiology. 1982;56(5):369–79.
Upton RN. Organ weights and blood flows of sheep and pig for physiological pharmacokinetic modelling. J Pharmacol Toxicol Methods. 2008;58(3):198–205.
Vinegar A. Development of a physiologically based pharmacokinetic model for the anesthetics halothane, isoflurane, and desflurane in the pig (Sus scrofa). DTIC Document, 1999.
Drougas JG, Barnard SE, Wright JK, Sika M, Lopez RR, Stokes KA, et al. A model for the extended studies of hepatic hemodynamics and metabolism in swine. Lab Anim Sci. 1996;46(6):648–55.
Peters SA. Physiologically-based pharmacokinetic (PBPK) modeling and simulations: principles, methods, and applications in the pharmaceutical industry. New York: Wiley; 2012.
Scotcher D, Jones C, Rostami-Hodjegan A, Galetin A. Novel minimal physiologically-based model for the prediction of passive tubular reabsorption and renal excretion clearance. Eur J Pharm Sci. 2016;94:59–71.
Leavens T, Tell L, Clothier K, Griffith R, Baynes RE, Riviere J. Development of a physiologically based pharmacokinetic model to predict tulathromycin distribution in goats. J Vet Pharmacol Ther. 2012;35(2):121–31.
Dosne A-G, Bergstrand M, Harling K, Karlsson MO. Improving the estimation of parameter uncertainty distributions in nonlinear mixed effects models using sampling importance resampling. J Pharmacokinet Pharmacodyn. 2016;43(6):583–96.
Karlsson MO, Beal SL, Sheiner LB. Three new residual error models for population PK/PD analyses. J Pharmacokinet Pharmacodyn. 1995;23(6):651–72.
Beal SL. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001;28(5):481–504.
Chevance A, Jacques AM, Laurentie M, Sanders P, Henri J. The present and future of withdrawal period calculations for milk in the European Union: focus on heterogeneous, nonmonotonic data. J Vet Pharmacol Ther. 2017;40(3):218–30.
Lindbom L, Pihlgren P, Jonsson N. PsN-toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Prog Biomed. 2005;79(3):241–57.
Keizer RJ, Van Benten M, Beijnen JH, Schellens JH, Huitema AD. Pirana and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Prog Biomed. 2011;101(1):72–9.
Landersdorfer CB, Nguyen T-H, Lieu LT, Nguyen G, Bischof RJ, Meeusen EN, et al. Substantial targeting advantage achieved by pulmonary Administration of Colistin Methanesulfonate in a large-animal model. Antimicrob Agents Chemother. 2017;61(1):e01934–16.
Karvanen M, Malmberg C, Lagerback P, Friberg LE, Cars O. Colistin is extensively lost during standard in vitro experimental conditions. Antimicrob Agents Chemother. 2017;61(11):e00857–17.
Huang JX, Blaskovich MA, Pelingon R, Ramu S, Kavanagh A, Elliott AG, et al. Mucin binding reduces colistin antimicrobial activity. Antimicrob Agents Chemother. 2015;59(10):5925–31.
Hinderling PH. Red blood cells: a neglected compartment in pharmacokinetics and pharmacodynamics. Pharmacol Rev. 1997;49(3):279–95.
Azad MA, Huang JX, Cooper MA, Roberts KD, Thompson PE, Nation RL, et al. Structure–activity relationships for the binding of polymyxins with human α-1-acid glycoprotein. Biochem Pharmacol. 2012;84(3):278–91.
Bouchene S, Marchand S, Friberg LE, Björkman S, Couet W, Karlsson MO, editors. Whole body physiologically-based pharmacokinetic model for colistin and Colistimethate sodium (CMS) in six different species: mouse, rat, rabbit, baboon, pig and human. J Pharmacokinet Pharmacodyn 2013;40(S1):S115–S6
Holford NH, Anderson BJ. Allometric size: the scientific theory and extension to normal fat mass. Eur J Pharm Sci. 2017;109S:S59–S64.
Yousef JM, Chen G, Hill PA, Nation RL, Li J. Melatonin attenuates colistin-induced nephrotoxicity in rats. Antimicrob Agents Chemother. 2011;55(9):4044–9.
Zavascki AP, Nation RL. Nephrotoxicity of polymyxins: is there any difference between colistimethate and polymyxin B? Antimicrob Agents Chemother. 2017;61(3):e02319–16
Manchandani P, Zhou J, Babic JT, Ledesma KR, Truong LD, The TVH. Role of renal drug exposure in polymyxin B-induced nephrotoxicity. Antimicrob Agents Chemother. 2017;61(4):02391–16.
Vardakas KZ, Falagas ME. Colistin versus polymyxin B for the treatment of patients with multidrug-resistant gram-negative infections: a systematic review and meta-analysis. Int J Antimicrob Agents. 2017;49(2):233–8
Ordooei Javan A, Shokouhi S, Sahraei Z. A review on colistin nephrotoxicity. Eur J Clin Pharmacol. 2015;71(7):801–10.
Li M, Gehring R, Riviere JE, Development LZ. Application of a population physiologically based pharmacokinetic model for penicillin G in swine and cattle for food safety assessment. Food Chem Toxicol. 2017;107:74–87.
EMA. Updated advice on the use of colistin products in animals within the European Union: development of resistance and possible impact on human and animal health. European Medicines Agency (EMA), 2016 EMA/CVMP/CHMP/231573/2016.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Figure S1
Goodness-of-fit plots for model validation. Population predicted (PRED) versus observed concentrations or quantities (DV) in log-log scale (A) and linear scale (B). Individual predicted (PRED) versus observed concentrations or quantities (DV) in log-log scale (C) and linear scale (D). (GIF 70 kb)
Figure S2
Visual Predictive Checks of the PBPK model for colistin tissue data in liver (A), muscles (B), skin (C), fat (D), used for model validation. Observed data come from an independent experiment (n°5: 50,000 UI/kg of CMS divided in two IM injection per day during 3 days) that was not used for model calibration. Blue dots represent the observed tissue concentrations; highlighted with grey are the areas between the 5th and 95th percentiles of model simulations, whereas the black solid line represents the median; the purple area represents the 95% confidence interval around the median; the horizontal dashed black line represents the LOQ. In the lower panels, blue areas represent the simulation-based 95% confidence intervals for the fraction of data below the LOQ (BLOQ), whereas the blue solid line represents the actual observed fraction of BLOQ samples. (GIF 138 kb)
Figure S3
Visual Predictive Checks of the PBPK model for CMS tissue data in liver (A), muscles (B), skin (C), fat (D), used for model validation. Observed data come from an independent experiment (n°5: 50,000 UI/kg of CMS divided in two IM injection per day during 3 days) that was not used for model calibration. Blue dots represent the observed tissue concentrations; highlighted with grey are the areas between the 5th and 95th percentiles of model simulations, whereas the black solid line represents the median; the purple area represents the 95% confidence interval around the median; the horizontal dashed black line represents the limit of quantification. In the lower panels, blue areas represent the simulation-based 95% confidence intervals for the fraction of data below the LOQ (BLOQ), whereas the blue solid line represents the actual observed fraction of BLOQ samples. (GIF 94 kb)
Figure S4
Relative contribution of CMS and colistin in total kidney concentrations. CMS concentrations (green), colistin concentrations (red) and total concentrations in kidney after one IV of CMS (10 mg/kg) for a 50-kg pig. (GIF 23 kb)
Figure S5
Evolution of the mass balance predicted by the model after one IV of CMS, as expressed in relative quantities for CMS (A) and colistin (B) in each compartment. GIT: gastro-intestinal tract (GIF 73 kb)
Figure S6
Withdrawal period estimation in a 100-kg pig. Model simulation in kidney after 3 consecutive days of CMS IM injections (50,000 UI/kg of CMS divided in two injections per day) for 1000 virtual pigs of 100 kg. The grey area includes the 1st and 99th percentiles of model simulations, whereas the black solid line represents the median; the horizontal dashed black line represents the kidney MRL (0.20 μg/g). WP: withdrawal period, rounded to the next whole day (GIF 80 kb)
ESM 1
(TXT 28 kb)
Rights and permissions
About this article

Cite this article
Viel, A., Henri, J., Bouchène, S. et al. A Population WB-PBPK Model of Colistin and its Prodrug CMS in Pigs: Focus on the Renal Distribution and Excretion. Pharm Res 35, 92 (2018). https://doi.org/10.1007/s11095-018-2379-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11095-018-2379-4