
Abstract
The increasing prevalence of multidrug-resistant Gram-negative bacteria worldwide has resulted in colistin, administered as its inactive prodrug colistin methanesulfonate (CMS), being increasingly used as a last-line therapy to treat infections caused by these pathogens. Developed well before contemporary drug development procedures, substantial improvements in the understanding of its chemistry, pharmacokinetics (PK), pharmacodynamics (PD) and PK/PD relationships have occurred over the last decade which have enabled substantial progress towards optimising its clinical use in different patient populations. This has resulted in the first scientifically based dosing algorithm for various categories of critically ill patients receiving CMS to generate a desired target steady-state plasma concentration of formed colistin. It has become clear that monotherapy with CMS is unlikely to generate plasma colistin concentrations that are reliably efficacious. With nephrotoxicity preventing simply increasing the dose of CMS, combination therapy may be required in order to maximise efficacy and minimise the emergence of resistance.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Colistin
- Colistin methanesulfonate
- Pharmacokinetics
- Pharmacodynamics
- Dose optimization
- P. aeruginosa
- A. baumannii
- K. pneumoniae
- Multidrug resistance
Introduction
The increasing prevalence of infections caused by multidrug-resistant (MDR) Gram-negative bacteria, especially Pseudomonas aeruginosa, Acinetobacter baumannii and Klebsiella pneumoniae, and the dearth of new antibiotics with activity against these pathogens reaching the clinic (Livermore 2004; Boucher et al. 2009; Opal and Calandra 2009; IDSA 2010; Spellberg et al. 2011), mean that physicians are needing to resort to use of the polymyxin class of antibiotics (Li et al. 2006b; Landman et al. 2008; Nation and Li 2009; Lim et al. 2010; Michalopoulos and Karatza 2010). These are ‘old’ antibiotics that were discovered in the late 1940s and then became available in the clinic about a decade later. While a number of polymyxins are known, only two (polymyxin B and E, the latter also known more commonly as colistin) are used clinically. Of the two polymyxins, colistin is most commonly used in the majority of places in the world and is the subject of this chapter.
Drug development and regulatory approval processes were much different in the 1950s than those existing today. As a result, colistin was never subjected to the scientific rigour required of modern pharmaceuticals before they become available for use in patients. It is not surprising, therefore, that there has been a major paucity of pharmacological and other scientific information that is needed to guide rational use of colistin in various categories of patients. Over the last decade or so, since the resurgence in its clinical use, there have been a small number of research groups conducting studies to establish the scientific basis for clinical utilisation of colistin. In essence this antibiotic, that is more than half a century old, has been subjected over the last few years to scientific investigation and evaluation that is akin to the drug development procedures required of newly discovered pharmaceuticals.
At the outset, it is important to indicate what will, and what will not, be reviewed in this chapter. The literature supporting, or otherwise, the clinical efficacy of colistin will not be examined. Not unexpectedly in view of the history surrounding colistin, there is a shortage of information relating to clinical efficacy. Most reports have been based upon retrospective studies and randomised controlled trials are conspicuous by their absence; those interested may consult other sources of information (Li et al. 2006b; Landman et al. 2008; Molina et al. 2009). Like all other drugs, use of colistin may be associated with adverse effects. The most worrying potential adverse effect of colistin is nephrotoxicity (Falagas and Kasiakou 2006; Hartzell et al. 2009; DeRyke et al. 2010; Kwon et al. 2010; Ko et al. 2011). Fortunately, if this adverse effect occurs, it is usually mild to moderate in nature and reversible upon discontinuation of colistin therapy (Falagas et al. 2005; Falagas and Kasiakou 2006; Betrosian et al. 2008; Pintado et al. 2008; Hartzell et al. 2009). Colistin-induced nephrotoxicity appears to involve accumulation of colistin in renal tubular cells mediated by transporters (Li et al. 2003c, 2004; Ma et al. 2009) and ensuing oxidative stress (Ozyilmaz et al. 2011; Yousef et al. 2011, 2012). Clinically used antioxidants, N-acetylcysteine, melatonin and ascorbic acid, have been shown in animal models to ameliorate colistin-induced nephrotoxicity (Ozyilmaz et al. 2011; Yousef et al. 2011, 2012) and they hold promise for application in patients. Nephrotoxicity will not be further reviewed here. This chapter will provide an overview of the current state of microbiological and pharmacological knowledge, especially in relation to the pharmacokinetic (PK) and pharmacodynamic (PD) properties of colistin, and the PK/PD driver of its antibacterial effect. Because an appreciation of the chemistry and related terminology (including that applied to the material used in the clinic) is essential to an understanding of the pharmacological and microbiological properties of colistin, there is no option but to briefly review these aspects.
Important Aspects of Chemistry
Colistin is a polypeptide antibiotic produced by Bacillus colistinus (Koyama et al. 1950). It comprises a heptapeptide ring with a tripeptide side chain to which is covalently linked a fatty acyl tail. It is a multicomponent antibiotic, with colistin A (polymyxin E1) and colistin B (polymyxin E2), which differ by only one carbon and two protons in the fatty acyl tail, being the two major components (Koyama et al. 1950; Orwa et al. 2000, 2001) (Fig. 14.1a). Because of the biological origin of this antibiotic, there is supplier-to-supplier and batch-to-batch variation in the proportion of colistin A and colistin B in commercial material (Decolin et al. 1997; Li et al. 2001a). At physiological pH, the primary amines in the α,γ-diaminobutyric acid (Dab) residues (pKa approximately 10) are ionised; thus, colistin is a cationic antimicrobial peptide. Because colistin contains both polar (by virtue of the ionised Dab residues) and hydrophobic regions (fatty acyl tail), the molecule is amphiphilic and thereby exhibits surface activity (Wallace et al. 2010). As discussed below, both the cationic Dab residues and the hydrophobic fatty acyl tail are important for the interaction of colistin with the outer membrane of Gram-negative bacteria, a key first step in the bactericidal action of this antibiotic. Colistin is available commercially in the form of its colistin methanesulfonate salt. Colistin methanesulfonate is not administered parenterally, but is used in some countries in topical pharmaceutical formulations.

Structures of (a) colistin A and B and (b) colistin A and B methanesulfonate. Fatty acid: 6-methyloctanoic acid for colistin A and 6-methylheptanoic acid for colistin B; Thr threonine, Leu leucine, Dab α,γ-diaminobutyric acid. α and γ indicate the respective amino group involved in the peptide linkage
Colistin methanesulfonate (CMS, also known as colistimethate; Fig. 14.1b) is the form of ‘colistin’ that is administered parenterally and by inhalation. The sodium salt of CMS, in lyophilized form, is present in parenteral (and inhalational) formulations. It is crucial to understand the relationship between CMS and colistin. CMS was developed during the 1950s because of concerns in early studies about the relatively high level of toxicity that was associated with parenteral administration of colistin methanesulfonate. CMS is prepared from colistin by reaction of the free γ-amino groups of the five Dab residues with formaldehyde followed by sodium bisulfite (Barnett et al. 1964; Beveridge and Martin 1967). CMS is an inactive prodrug (Bergen et al. 2006) and is converted in vivo to the active antibacterial entity, colistin (Li et al. 2003a, 2004, 2005b; Markou et al. 2008; Plachouras et al. 2009; Imberti et al. 2010; Marchand et al. 2010a; Couet et al. 2011; Garonzik et al. 2011; Mohamed et al. 2012). It is essential to understand that the conversion of CMS to colistin, a requirement for antibacterial activity in vivo, may also occur in vitro as CMS is not stable in aqueous environments (Li et al. 2003b; Wallace et al. 2008b, 2010). Thus, the conversion of CMS to colistin, via a number of partially sulfomethylated derivatives, has been demonstrated to occur not only in vivo (see below) but also in vitro in plasma, urine, buffer solutions and microbiological culture medium (Li et al. 2003b, 2004; Bergen et al. 2006). The conversion of CMS to colistin also occurs in the solutions for administration to patients that are produced by reconstitution of the lyophilized powder in pharmaceutical products (Wallace et al. 2008b); to minimise this in vitro conversion, such reconstitution should occur immediately prior to administration. An awareness that CMS converts to colistin in aqueous media highlights the importance in PK and PK/PD studies of ensuring that blood, plasma and other biological samples are processed and stored appropriately to avoid in vitro conversion occurring after collection of samples (Dudhani et al. 2010a). Such conversion would result in a spuriously high estimate of the colistin concentration present in the sample at the time of its collection. Moreover, an appreciation of the facile conversion of CMS to colistin in aqueous media clearly leads to the conclusion that colistin methanesulfonate, rather than CMS sodium, must be used in determination of minimum inhibitory concentration (MIC). The use of CMS in MIC determinations will lead to an ‘apparent’ MIC that will represent the activity of the progressively increasing amount of colistin liberated from the CMS during the course of the microbiological incubation (Bergen et al. 2006). The time course of liberation of colistin may vary from laboratory to laboratory dependent upon conditions employed, and this would be expected to lead to variability in MIC values for a given strain.
Antibacterial Properties
Spectrum of Activity
Colistin exhibits a narrow antibacterial spectrum of activity, mostly against common Gram-negative pathogens (Li et al. 2005a). Colistin retains excellent in vitro bactericidal activity against most common species of Gram-negative bacilli or coccobacilli including P. aeruginosa (Walkty et al. 2009; Cernohorska and Slavikova 2010; Gales et al. 2011), Acinetobacter spp. (Walkty et al. 2009; Yau et al. 2009; Gales et al. 2011; Queenan et al. 2012) and Klebsiella spp. (Walkty et al. 2009; Hawser 2010; Gales et al. 2011; Sader et al. 2011), the organisms against which it is most commonly used clinically. Activity against other Gram-negative bacterial species has been reviewed elsewhere (Falagas and Kasiakou 2005; Li et al. 2005a). Colistin has no significant activity against most Gram-positive bacteria (Schwartz et al. 1959; Finland et al. 1976a, b) or fungi (Hoeprich 1970).
Susceptibility Breakpoints
The breakpoints for colistin susceptibility are based on colistin methanesulfonate given that CMS is an inactive prodrug (Bergen et al. 2006), and different breakpoints have been employed by various organisations (Comite de l’ Antibiogramme de la Societe Francaise de Microbiologie (SFM) 2005; Andrews and Howe 2011; Clinical and Laboratory Standards Institute (CLSI) 2012; European Committee on Antimicrobial Susceptibility Testing (EUCAST) 2012). The Clinical and Laboratory Standards Institute (CLSI) susceptibility breakpoints are ≤2 mg/L for both P. aeruginosa and A. baumannii using the microbroth dilution method (Clinical and Laboratory Standards Institute (CLSI) 2012). Given the emerging clinical pharmacokinetic and pharmacodynamic data (see sections below), the appropriateness of these breakpoints within a clinical context remains to be determined. Worryingly, colistin heteroresistance, the presence of resistant subpopulations within an isolate that is susceptible based upon its MIC, has been observed in A. baumannii (Li et al. 2006c; Owen et al. 2007; Tan et al. 2007; Hawley et al. 2008; Yau et al. 2009), K. pneumoniae (Poudyal et al. 2008; Meletis et al. 2011) and P. aeruginosa (Bergen et al. 2011a). While colistin still retains excellent activity generally, resistance to colistin is increasing in several key species including P. aeruginosa (Johansen et al. 2008; Lee et al. 2011), A. baumannii (Ko et al. 2007; Al-Sweih et al. 2011), K. pneumoniae and other Enterobacteriaceae (Tan and Ng 2006; Kontopoulou et al. 2010; Suh et al. 2010; Toth et al. 2010; Bogdanovich et al. 2011; Marchaim et al. 2011; Mezzatesta et al. 2011), and S. maltophilia (Tan and Ng 2006).
Mechanisms of Activity and Resistance
When considering the mechanism of antimicrobial activity it must be remembered that the polymyxins are polycationic, amphiphilic peptides. As colistin and polymyxin B are structurally similar, differing by only one amino acid, they are believed to share the same mechanism of antibacterial action. The initial target of the polymyxins against Gram-negative bacteria is the lipopolysaccharide (LPS) component of the outer membrane, initiated by electrostatic attraction between the cationic polymyxin molecule and the anionic lipid A of LPS, thereby displacing divalent inorganic cations (Ca2+, Mg2+) that assist in stabilising the LPS leaflet (Hancock and Chapple 1999). Once electrostatically bound to LPS, the N-terminal fatty-acyl tail is inserted into the outer membrane in a process driven by hydrophobic interactions. The overall result is permeabilization of the outer membrane, allowing the polymyxin to access the periplasmic space and the cytoplasmic membrane; this is the so-called ‘self-promoted uptake’ mechanism (Hancock and Chapple 1999). Originally it was proposed that the polymyxin inserted into the cytoplasmic membrane forming conductance events leading to leakage of cell contents and cell death (Hancock et al. 1995). However, there is an increasing body of evidence that suggests the polymyxins exert their effects through an alternative mode of action or that they may in fact act upon multiple bacterial cell targets (Hancock and Rozek 2002; Brogden 2005; Hale and Hancock 2007). The exact mechanism(s) by which they ultimately kill bacterial cells is still unknown.
Given that the crucial first step in the action of polymyxins on Gram-negative bacterial cells is the electrostatic interaction between the positively charged polymyxins and the negatively charged LPS, it is not surprising that resistance to polymyxins often involves changes in LPS structure which decrease the negative charge on the cell surface and hence the electrostatic interactions with the peptide. Modifications to the lipid A and/or core of LPS typically mask phosphate groups with moieties such as aminoarabinose and phosphoethanolamine. Such modifications have been observed in P. aeruginosa (Moskowitz et al. 2004), K. pneumoniae (Helander et al. 1996) and other bacterial species (Morrison and Wenzel 1984; Breazeale et al. 2005; Winfield et al. 2005) and have been shown to increase resistance to polymyxins (Moskowitz et al. 2004; Breazeale et al. 2005; Lewis et al. 2009; Beceiro et al. 2011). Interestingly, it was recently shown that resistance in A. baumannii can be mediated by complete loss of LPS (Moffatt et al. 2010). In K. pneumoniae, the presence of capsule may also be important for polymyxin resistance (Helander et al. 1996; Llobet et al. 2008).
Inconsistent Labelling and Dose Regimens of Pharmaceutical Products
Unfortunately, different conventions are used in various parts of the world for labelling the content of CMS pharmaceutical products and in defining the recommended daily doses (Li et al. 2006a, b). In some parts of the world (e.g. Europe), CMS parenteral vials are labelled in international units (IU). In these countries there are usually three vial sizes containing 500,000 IU, 1 million IU and 2 million IU per vial, corresponding to approximately 40, 80 and 160 mg of CMS sodium per vial (since there are ~12,500 IU per mg of CMS sodium). In many countries in the world (e.g. USA, Canada, Australia), the parenteral product available is labelled in terms of ‘colistin base activity’. In these countries, one vial size only is available and this contains 150 mg of colistin base activity, which actually corresponds to ~400 mg CMS sodium.
Very unfortunately, the inconsistency does not end with labelling of the pharmaceutical products; it extends to the recommended daily doses in the respective product information. For those products labelled in international units, the typical recommended dose for a patient over 60 kg and with normal renal function is 1–2 million IU three times daily (Li et al. 2006b), equivalent to 240–480 mg CMS sodium per day. For those products labelled in terms of colistin base activity, the recommended doses are 2.5–5 mg/kg colistin base activity per day in 2–4 divided doses (Li et al. 2006b), which is equivalent to about 6.67–13.3 mg/kg of CMS sodium per day. Thus, for a patient with normal renal function and bodyweight of 60 kg, the recommended dose of such a product labelled in terms of colistin base activity (recommended dose of 400–800 mg CMS sodium per day) is almost double that of the products that are labelled in international units (recommended dose of 240–480 mg CMS sodium per day, see above). The origin of this major discrepancy in recommended doses between products appears to be lost in the sands of time. Because both regimens appear to be equally well tolerated and because of the need to ensure the maximum antibacterial effect in an era of increasing multidrug resistance and shortage of new antibiotics, the higher of the two recommended dosage regimens (i.e. 2.5–5 mg/kg colistin base activity per day, equivalent to 400–800 mg CMS sodium per day) would seem to be a wise choice; as discussed below, a reduction in the daily dose may be appropriate for patients with impaired renal function.
The inconsistent labelling convention and the discrepant recommended dosage regimens have the potential to cause much confusion; indeed, even those clinicians who are familiar with the use of CMS are often unaware of this problem. Tragically, the confusion surrounding CMS labelling recently resulted in the death of a patient in the USA (Institute for Safe Medication Practices (ISMP) 2011). In that case, the physician ordered the dose as mg of CMS rather than as colistin base activity, the usual method of expressing the dose in the USA. This went unrecognised by the pharmacist and nurses and resulted in the patient receiving doses ~2.7-fold higher than intended. The patient subsequently developed acute renal failure and other complications that resulted in their death. Calls a number of years ago for an international consensus (Li et al. 2006a) have not been actioned. Clinicians reading the international literature to inform their practice will need to remain vigilant in regard to interpretation of dosage regimens used in published studies.
Pharmacokinetics, Pharmacodynamics and Pharmacokinetic/Pharmacodynamic Relationships
Important Methodological Considerations for PK, PD and PK/PD Studies
At the outset, it is essential to comment upon the analytical methods that have been employed in PK studies over the years. Such studies conducted through the 1950s to the 1990s inclusive were undertaken using microbiological assays (Mackay and Kaye 1964; al-Khayyat and Aronson 1973); indeed, such assays have been used in some of the more recent studies (Aoki et al. 2009). These assays, when applied to biological samples containing both CMS and colistin formed in vivo, are not capable of differentiating between the colistin actually present in the sample at the time of its collection from a subject administered CMS and the colistin formed in vitro during the incubation period of the microbiological assay. Thus, the use of such assays is incapable of providing accurate information on the time course of plasma concentrations of the prodrug (CMS) and the active entity (colistin). The pharmacokinetic characteristics described within the current prescribing information for the various parenteral products was obtained using microbiological assays; thus it is unhelpful and does not provide a solid scientific basis for understanding the disposition of administered CMS and the colistin formed from it in vivo.
An accurate and complete understanding of the PK of CMS and formed colistin has only been possible over the last 5–10 years since the development of HPLC (Li et al. 2001a, 2002) and LC/MS/MS (Jansson et al. 2009; Gobin et al. 2010; Dotsikas et al. 2011) analytical methods for the separate quantification of CMS and colistin in biological samples. Here, a couple of important points should be made. Firstly, all of the current methods for HPLC or LC/MS/MS analysis of ‘CMS’ involve so-called ‘difference assays’. That is, the ‘CMS’ concentration in a biological sample is determined as the difference between the colistin concentration measured in a sample that has been carefully processed and stored to prevent in vitro conversion of CMS to colistin and the colistin concentration in another aliquot of the sample where the conversion of CMS to colistin is forced to occur in vitro. The ‘CMS’ concentration determined using this approach represents the concentration of CMS (i.e. the penta-sulfomethylated species) and the numerous partially sulfomethylated species that are intermediates in the conversion of CMS to colistin. This type of analytical approach has been necessary because it has not been possible to directly quantify CMS due to the complex chemical nature and composition of CMS. Secondly, it is essential to appreciate that very careful procedures must be employed in the handling and storage of biological samples to avoid the in vitro conversion of CMS to colistin. Such in vitro conversion would lead to an underestimation of the ‘CMS’ concentration and, more importantly, an overestimation of the colistin concentration in the biological sample. Thus, upon their collection, samples must be placed on ice, processed rapidly (e.g. blood samples centrifuged to obtain plasma/serum) and stored under conditions to minimise in vitro conversion of CMS to colistin prior to analysis. In regard to the latter, it has been demonstrated that storage of plasma samples at −20 °C is generally not acceptable, unless the samples are analysed within 1 month of collection (Dudhani et al. 2010a). Samples should be stored at −70 °C to −80 °C and even then the samples must be analysed within 4 months of collection to avoid substantial conversion of CMS into colistin and the degradation of both entities.
Overview of the Pharmacokinetics of CMS and Formed Colistin
The availability in the past decade or so of liquid chromatographic methods has enabled increased understanding of the relatively complex disposition of the inactive prodrug, CMS and the (active) colistin formed from it in the body. The intravenous route is the most common way in which CMS is administered, especially in critically ill patients with life-threatening infections caused by Gram-negative bacteria. For this reason, and also because PK data obtained from studies using this route are the most informative in regard to dispositional characteristics, the major focus here will be on studies conducted following intravenous administration. This section will provide an overview of preclinical PK studies; this is important because there are aspects of the overall PK of CMS and formed colistin that are only possible to reveal by undertaking studies involving separate administration of CMS and preformed colistin, which cannot be readily performed in humans.
The differences in chemistry between CMS and colistin (see section entitled ‘Important Aspects of Chemistry’) translate into differences in the PK of these entities. Li et al. was the first to apply HPLC methods capable of distinguishing between CMS and colistin to studies undertaken in rats administered either CMS (Li et al. 2004) or colistin (Li et al. 2003c). Those studies provided very useful information concerning the differences in disposition of CMS and the colistin formed from it in vivo. Following intravenous administration of CMS in rats, colistin appeared in plasma soon after administration of the prodrug (Li et al. 2004). The terminal half-life of formed colistin was approximately twice that of the administered CMS and was similar to the half-life of colistin administered directly (Li et al. 2003c). This indicated that the overall disposition of formed colistin following administration of CMS was rate limited by its elimination rather than its formation. The fundamental aspects of the overall disposition of CMS and formed colistin observed by Li et al. (2004) were subsequently confirmed by Marchand et al. using a wide range of CMS doses (5–120 mg/kg intravenously) in a rat PK study (Marchand et al. 2010b). In people with cystic fibrosis, the terminal half-life of formed colistin (251 ± 79 min; mean ± SD) has also been reported to be approximately twice that of the administered CMS (124 ± 52 min) (Li et al. 2003a). More recent studies conducted in critically ill patients indicated that the terminal half-life of formed colistin is substantially longer (up to ~18 h) than that of the CMS (~3 h) that was administered (Markou et al. 2008; Plachouras et al. 2009; Garonzik et al. 2011; Mohamed et al. 2012); it is also evident that the half-life of formed colistin in critically ill patients is longer than that in people with cystic fibrosis (Li et al. 2003a), which may relate to differences in renal function and other patient characteristics (see section ‘How Appropriate Are Current Dosage Regimens?’). From studies conducted to date, the PK of CMS and formed colistin appears to be linear following intravenous administration of CMS. In rats administered intravenous CMS across the range 5–120 mg/kg (which generated plasma concentrations of CMS and formed colistin that span those that are clinically relevant) linear relationships were observed between CMS and colistin areas under the plasma concentration–time curves (AUC) to infinity and CMS doses, as well as between CMS and colistin maximum plasma concentration (C max) values and CMS doses (Marchand et al. 2010b). Following direct administration of colistin subcutaneously across a range of colistin doses to infected neutropenic mice, there was evidence of non-linear PK (plasma colistin concentration increased to a greater extent than the increase in dose) (Dudhani et al. 2010b); however, this may have resulted from non-linearity in the tissue binding of colistin, including at the subcutaneous site of administration, thereby impacting the fraction of the dose available for absorption.
Studies performed several decades ago employing microbiological assays indicated that colistin binds extensively to tissues of many organs, whereas a lesser degree of tissue binding was apparent for CMS (Kunin and Bugg 1971; Craig and Kunin 1973; Ziv et al. 1982; Leroy et al. 1989). The studies with CMS must be interpreted cautiously due to the use of a microbiological assay, which is non-specific for CMS as the assay measures the concentration of active colistin generated from CMS in vivo as well as during the incubation period of the microbiological assay. Protein binding studies in plasma from a range of healthy (i.e. non-infected) animals indicated that colistin was 30–70 % plasma bound (Ziv and Sulman 1972; al-Khayyat and Aronson 1973; Li et al. 2003c). A recent study of colistin binding in plasma from infected neutropenic mice indicated that the binding of colistin was higher than that above (Dudhani et al. 2010b). It is apparent that colistin binds to both albumin and α 1-acid glycoprotein (Dudhani et al. 2009), the latter being an acute-phase reactant protein whose plasma concentration increases in a variety of stressful conditions, including infection (Voulgari et al. 1982; Morita and Yamaji 1995). The extent of plasma binding of colistin in infected patients may therefore be subject to variations in the concentrations of albumin, α 1-acid glycoprotein and any other proteins involved in its binding.
There are very substantial differences in the clearance pathways for CMS and colistin. Following intravenous administration of colistin in rats, substantially less than 1 % of the dose was recovered in urine in unchanged form (Li et al. 2003c); the renal clearance involved very extensive renal tubular reabsorption to an extent greater than that occurring for water indicating that the reabsorption of colistin must be a carrier-mediated process (Li et al. 2003c; Ma et al. 2009). The very minor role for renal clearance in the overall body clearance of colistin was also observed for polymyxin B (differing from colistin in just one amino acid) in patients (Zavascki et al. 2008). In marked contrast, CMS was shown to be predominantly renally cleared in rats with a component of tubular secretion (Li et al. 2004). The trafficking through renal tubular cells of CMS by secretion [with the possibility of intracellular generation of colistin (Li et al. 2004)] and of colistin by tubular reabsorption (Li et al. 2003c) may explain in part the propensity for nephrotoxicity following administration of CMS. Comparison of the dose-normalised AUC of formed colistin arising from administration of CMS in rats with that arising from direct administration of colistin allowed estimation of the fraction of the dose of CMS that was converted systemically to colistin (Li et al. 2004); this revealed that only a very small proportion (~7 %) of the administered dose of CMS was converted to colistin. A subsequent study in rats by Marchand et al. (Marchand et al. 2010b) confirmed many of the observations of Li et al. (Li et al. 2004). Similar to the findings in rats, Couet et al. recently demonstrated that in young healthy volunteers administered a single dose of one million IU of CMS (infused over 1 h), CMS was predominantly excreted in the urine (70 % on average as both CMS and colistin, the majority of the latter forming in the urinary tract) (Couet et al. 2011). The low in vivo fractional conversion of the prodrug, CMS, to the active form, colistin, arises because the conversion clearance of CMS to colistin is substantially lower than the renal clearance of CMS (i.e. the fractional conversion is dictated by the relative efficiencies of parallel pathways for elimination of CMS).
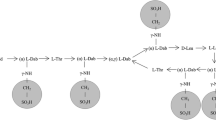
As a result of the understanding generated from these studies, the overall disposition of CMS and formed colistin has been summarised as shown in Fig. 14.2. The schema shown in Fig. 14.2 is consistent with the emerging data on the pharmacokinetics of CMS and formed colistin in humans, which is discussed below (see section ‘How Appropriate Are Current Dosage Regimens?’).

Schematic representation of the disposition of colistin methanesulfonate and the colistin generated from it in the body following administration of sodium colistin methanesulfonate. Modified after (Li et al. 2006b) with permission from Elsevier
Pharmacodynamics of Colistin
Although colistin is administered parenterally as CMS, it is important to recognise that antimicrobial activity results from formation of colistin, not from CMS or its partially sulfomethylated derivatives (Bergen et al. 2006). Thus, CMS should be considered an inactive prodrug of colistin. Most PD data on colistin have been generated using in vitro models. Time-kill studies with colistin (used as its sulfate salt) in both static and dynamic systems showed potent, concentration-dependent killing against P. aeruginosa (Eickhoff and Finland 1965; Li et al. 2001b; Gunderson et al. 2003; Bergen et al. 2008, 2010, 2011a, b; Bulitta et al. 2010; Lin et al. 2010), A. baumannii (Owen et al. 2007; Tan et al. 2007) and K. pneumoniae (Poudyal et al. 2008; Deris et al. 2012), including multidrug-resistant and colistin-heteroresistant strains. Initial killing is very rapid, with a large decrease in colony-forming units (cfu) per mL occurring as early as 5 min after exposure to colistin concentrations in the vicinity of the MIC and above. A modest post-antibiotic effect was found only at high colistin concentrations (Li et al. 2001b; Owen et al. 2007; Poudyal et al. 2008). Both the rate and extent of killing are markedly decreased at high compared to low inocula (Bulitta et al. 2010; Bergen et al. 2011a, b; Deris et al. 2012). Against a genetically characterised isolate of P. aeruginosa (PAO1), killing of the susceptible population by colistin was 23-fold slower at an inoculum of 109 cfu/mL and sixfold slower at 108 cfu/mL compared with 106 cfu/mL. Up to 32-fold higher colistin concentrations were required at the 109 compared with the 106 cfu/mL inoculum to achieve bactericidal activity (a ≥ 3-log10 cfu/mL decrease) (Bulitta et al. 2010). Thus, there is a potential need for higher colistin exposure or combination regimens to treat deep-seated, difficult-to-treat infections with high inocula.
A consistent finding of both in vitro and in vivo studies is regrowth with colistin monotherapy, even with concentrations well above those which can be safely achieved clinically. For example, two studies which utilised in vitro PD models (Gunderson et al. 2003; Bergen et al. 2010) reported regrowth of P. aeruginosa with colistin concentrations well above clinically achievable levels, the former with concentrations up to 200 mg/L, while similar regrowth has been reported in A. baumannii (Owen et al. 2007) and K. pneumoniae (Poudyal et al. 2008; Deris et al. 2012) with colistin concentrations up to 64 × MIC. Regrowth following colistin monotherapy has been shown to be the result of amplification of colistin-resistant subpopulations (Tan et al. 2007; Bergen et al. 2008, 2011a, b; Poudyal et al. 2008; Bulitta et al. 2010; Dudhani et al. 2010b; Deris et al. 2012). The difficulty of eradicating colistin-resistant subpopulations with colistin monotherapy, together with the potential for rapid amplification of colistin-resistant subpopulations, suggests caution with the use of colistin monotherapy and highlights the importance of investigating rational colistin combinations.
Which Pharmacokinetic/Pharmacodynamic Index Is Most Predictive of Efficacy?
Only recently have studies used a dose-fractionation design to investigate the relationship between the PK and PD of colistin, namely which PK/PD index [C max/MIC, AUC/MIC, or T>MIC (time for which concentrations exceed the MIC)] best correlates with colistin efficacy (Ketthireddy et al. 2007; Bergen et al. 2010; Dudhani et al. 2010b, c; Hengzhuang et al. 2012). Using an in vitro PK/PD model, Bergen et al. examined 37 different regimens involving various colistin C max and dosage frequencies (including intermittent dosing and continuous infusion regimens) against three strains of P. aeruginosa including a colistin-susceptible but MDR strain (Bergen et al. 2010); analysis was based upon unbound or free (ƒ) indices (i.e. ƒC max/MIC, ƒAUC/MIC, and ƒT>MIC). The overall killing effect was best correlated with ƒAUC/MIC (R 2 = 0.931); weaker correlations occurred for ƒC max/MIC (R 2 = 0.868) and ƒT>MIC (R 2 = 0.785) (Fig. 14.3). The magnitudes of ƒAUC/MIC required for 1- and 2-log10 reductions in the area under the cfu/mL curve relative to growth control were able to be determined.

Relationship between killing effect against three strains of P. aeruginosa as a function of three PK/PD indices: (a) ƒAUC/MIC, (b) ƒC max/MIC and (c) ƒT>MIC. ATCC 27853 (solid line and open circles), PAO1 (dashed line and solid triangles) and 19056 mucoid (dotted line and crosses). Each data point represents the result from a single treatment run. Lines represent model-generated fits. Reproduced from (Bergen et al. 2010) with permission from the American Society for Microbiology
In a conference abstract describing use of a neutropenic mouse thigh infection model, Ketthireddy et al. concluded that once-daily dosing of colistin was most effective against P. aeruginosa and that C max/MIC was likely the PK/PD index most predictive of efficacy; PK data, however, were not included in that study (Ketthireddy et al. 2007). Dudhani et al. employed neutropenic mouse thigh and lung infection models in dose-fractionation studies with colistin against three strains each of P. aeruginosa and A. baumannii which included MDR but colistin-susceptible and, for A. baumannii, colistin-heteroresistant strains (Dudhani et al. 2010b, c). In these studies, the time course of total (i.e. protein-bound plus unbound) and unbound plasma colistin concentrations were determined allowing the PK/PD analysis to be based upon unbound indices. The PK/PD index most predictive of the antibacterial effect against both P. aeruginosa and A. baumannii in both thigh and lung infection models was ƒAUC/MIC (see Fig. 14.4, for colistin against P. aeruginosa in murine lung infection model), in agreement with the results from dose-fractionation studies of colistin against P. aeruginosa in an in vitro PK/PD model (Bergen et al. 2010). That ƒAUC/MIC is the most predictive PK/PD index indicates that time-averaged exposure to colistin is more important than the achievement of high peak concentrations from the administration of larger, less frequent doses. For both P. aeruginosa and A. baumannii, ƒAUC/MIC targets required to achieve various magnitudes of kill were of the same order of magnitude in both the thigh and lung, although somewhat higher values were required to achieve maximal killing in the lung. Most recently, Hengzhuang et al. used a neutropenic murine lung biofilm infection model to determine the PK/PD indices for colistin most predictive of activity against planktonic and biofilm cells of a single strain of P. aeruginosa (Hengzhuang et al. 2012). The AUC/MIC was again the PK/PD index most closely correlated with bacterial killing of planktonic cells, whereas the AUC to minimal biofilm inhibitory concentration (MBIC) ratio (AUC/MBIC) was most predictive of killing for biofilm cells in the lung. In this study unbound colistin concentrations were not considered and AUC/MIC and AUC/MBIC values were those for total colistin. The AUC/MBIC targets identified to achieve various magnitudes of bacterial killing were substantially higher for biofilm infections than for planktonic cells. The observed differences in bacterial killing of planktonic and biofilm cells by Hengzhuang et al. (2012), as well as between thigh and lung infections by Dudhani et al. (2010b, c), suggest that dosage regimens may need to be altered depending upon the nature and/or site of infection.

Relationships for P. aeruginosa ATCC 27853 between the log10 CFU per lung at 24 h and the PK/PD indices (a) ƒAUC/MIC, (b) ƒC max/MIC and (c) ƒT>MIC. Each symbol represents the mean datum per mouse from two lungs. R 2 is the coefficient of determination. The dotted line represents the mean bacterial burden in lungs at the start of treatment. Reproduced from (Dudhani et al. 2010b, c) with permission from the American Society for Microbiology
Unfortunately, it is currently not possible to compare the ƒAUC/MIC targets identified from dose-fractionation studies in in vitro and animal infection models with the ƒAUC/MIC values achieved in infected patients receiving currently recommended CMS dosage regimens. Although, as discussed in the following section ‘How Appropriate Are Current Dosage Regimens?’, there is increasing information on the total plasma colistin concentrations occurring in CMS-treated patients, no information is currently available on unbound plasma concentrations. As such information is forthcoming it will be possible to not only assess the ability of current CMS dosage regimens to meet the above-mentioned ƒAUC/MIC targets but also to design optimised dosage regimens.
How Appropriate Are Current Dosage Regimens?
As previously discussed, colistin retains significant activity in vitro against many Gram-negative ‘superbugs’ and is often the only therapeutic option available to treat infections by these MDR Gram-negative pathogens (Bratu et al. 2005; Li et al. 2006b; Antoniadou et al. 2007; David and Gill 2008; Landman et al. 2008; Michalopoulos and Karatza 2010). With resistance to colistin beginning to emerge (Li et al. 2006b; Antoniadou et al. 2007; Ko et al. 2007; Johansen et al. 2008; Al-Sweih et al. 2011; Bogdanovich et al. 2011; Lee et al. 2011; Mezzatesta et al. 2011), it is imperative to administer CMS in regimens that maximise antibacterial activity and minimise resistance development, while also minimising the potential for adverse effects (e.g. nephrotoxicity). Unfortunately, a lack of information on the PK and PD of colistin and CMS has led to confusion regarding the ‘optimal’ dosing schedule (Nation and Li 2009). Current dosage regimens are primarily derived from manufacturers’ package inserts that were developed decades ago, before an understanding of modern PK/PD concepts. As discussed above (see section ‘Inconsistent Labelling and Dose Regimens of Pharmaceutical Products’), the daily dosage recommendations for parenterally administered CMS differ substantially among products that are used in various regions of the world; this causes confusion and potentially leads to a situation of sub-optimal use.
In the product package inserts, the recommended number of CMS doses per day is 2–4 for a person with normal renal function, with 2–3 doses daily being a common regimen (Bergen et al. 2008). Once-daily regimens have also been employed despite a lack of supporting PK/PD data (Gunderson et al. 2003; Rosenvinge et al. 2005), presumably to take advantage of the concentration-dependent activity of colistin that is evident in vitro. However, in an in vitro PK/PD model that simulated human dosing regimens incorporating higher doses of CMS administered once daily, there was greater emergence of resistance in P. aeruginosa than occurred with a thrice-daily regimen involving essentially the same total daily dose (Bergen et al. 2008). Furthermore, a study conducted in rats involving week-long multiple-dose regimens mimicking once- and twice-daily administration in humans of the same daily dose of CMS, to achieve clinically relevant plasma colistin concentrations, resulted in a greater range and severity of renal lesions with the once-daily dosing regimen (Wallace et al. 2008a). In vitro studies have shown that the toxic effects of colistin on mammalian cells is both concentration- and time-dependent (Lewis and Lewis 2004). Finally, colistin lacks a significant postantibiotic effect (Li et al. 2001b; Owen et al. 2007; Poudyal et al. 2008; Ozbek and Senturk 2010). Thus, higher doses administered less frequently may potentially increase both nephrotoxicity and resistance development, although this remains to be confirmed in patients.
Evidence is emerging that the PK of CMS and formed colistin differs across various patient groups, and that currently used dosage regimens of CMS are likely to generate sub-optimal exposure to colistin in many patients. Li et al. reported a study in which 12 people with cystic fibrosis (age range 13–39 years, body weight range 39–68 kg, all with normal serum creatinine) were administered intravenously 1–2 million international units (IU) of colistin methanesulfonate every eight hours (equivalent to 1.83–3.50 mg of colistin base activity/kg per day) (Li et al. 2003a). The plasma CMS and colistin concentrations across a dosage interval at steady state are shown in Fig. 14.5. The peak plasma concentration of colistin was typically found in the first blood sample collected following the CMS dose and the half-life of the formed colistin was ~4 h. The range of calculated plasma colistin C max at steady state was 1.2–3.1 mg/L while that for the minimum plasma concentration (C min) was 0.14–1.3 mg/L, and the range of AUC over 24 h for formed colistin was 16–53 mg h/L (Li et al. 2003a). Even without consideration of protein binding, plasma colistin concentrations in many cases failed to reach the CLSI breakpoint of 2 mg/L (Clinical and Laboratory Standards Institute (CLSI) 2012) defining susceptibility to colistin for P. aeruginosa and A. baumannii, with plasma concentrations falling rapidly below this level even when achieved. It is apparent that the CMS dosage regimens employed and the resulting exposure to plasma colistin in these patients was very likely to be sub-optimal, especially with CMS monotherapy.

Plasma concentrations of (a) colistin methanesulfonate and (b) colistin at steady state in 12 patients with cystic fibrosis following intravenous administration of colistin methanesulfonate 1–2 million IU every 8 h for at least 2 days. Reproduced from (Li et al. 2003a) with permission from Oxford University Press
It is increasingly obvious from studies undertaken in critically ill patients that the CMS dosage regimens used in many of these patients generate plasma colistin concentrations that are not likely to be very effective, especially when used as monotherapy (Li et al. 2005b; Markou et al. 2008; Plachouras et al. 2009; Fernandez et al. 2010; Imberti et al. 2010; Garonzik et al. 2011; Mohamed et al. 2012). Arguably, a 2005 report was the first to draw attention, based upon experimental data, to the lack of PK information for CMS and formed colistin in critically ill patients and to the lack of appropriate CMS dosage guidelines for these patients (Li et al. 2005b). Li et al. reported the disposition of CMS and formed colistin at steady state in a critically ill adult patient requiring CMS for treatment of an infection caused by MDR P. aeruginosa. The patient had multiple organ failure requiring continuous venovenous hemodiafiltration (CVVHDF). Intravenous CMS (equivalent to 150 mg colistin base activity every 48 h) was administered as last-line therapy; the regimen was based upon the product information which suggested that in patients with renal impairment the size of the dose should be essentially maintained and the dosing interval should be increased from the normal 8–12 h. The dose actually administered to this patient was also in accord with the suggestion made, without any supporting data whatsoever, in an influential report focussing upon antibiotic dosing in critically ill patients receiving continuous renal replacement therapy (Trotman et al. 2005). Li et al. demonstrated that both CMS and colistin were cleared by CVVHDF. Importantly, total plasma concentrations of formed colistin fell below the MIC for the infecting strain ~4 h after CMS dosing. Unfortunately, 12 days after commencing CMS therapy, the patient died. Clearly, dosage adjustment for CMS in CVVHDF patients should be much more modest than that used in this patient. Subsequent studies have confirmed both CMS and colistin are efficiently cleared by intermittent hemodialysis and continuous renal replacement therapy (either CVVHDF or continuous venovenous hemofiltration) (Marchand et al. 2010a; Garonzik et al. 2011).
Makou et al. reported plasma colistin concentrations across a CMS dosage interval at least 2 days after commencing therapy (Markou et al. 2008); all patients, who were adults, had creatinine clearance >46 mL/min and 13 of the 14 patients received 2.8 million IU CMS intravenously 8 or 12 hourly (corresponding to ~270 mg colistin base activity per day). The range of C max values for formed colistin was 1.15–5.14 mg/L while that for C min was 0.35–1.70 mg/L; the AUC over 24 h for formed colistin ranged from 12.8 to 60.0 mg h/L. The authors expressed concern about the low plasma concentrations of colistin achieved in these patients. It was not surprising, given that all patients had moderate to good renal function, that Makou et al. were not able to discern a relationship between plasma colistin C max or C min and creatinine clearance. Imberti et al. reported plasma colistin concentrations across a dosage interval at least 2 days after commencing therapy in 13 adult critically ill patients with ventilator-associated pneumonia caused by Gram-negative bacteria (Imberti et al. 2010). Each patient had a creatinine clearance of >96 mL/min and received CMS two million IU intravenously 8 hourly (equivalent to ~180 mg colistin base activity per day). There was no apparent relationship between plasma colistin C max (range 0.68–4.65 mg/L), C min (0.23–2.43 mg/L) or AUC over 24 h (8.9–75.2 mg h/L) and creatinine clearance; as with the study of Markou et al. (2008), failure to identify a relationship between colistin PK parameters and renal function is not at all surprising given that all patients had creatinine clearance values around 100 mL/min or greater. In the study of Imberti et al. bronchoalveolar lavage (BAL) was performed 2 h after administration of a CMS dose (Imberti et al. 2010). The authors did not concentrate the BAL fluid prior to analysis to increase the sensitivity of the assay, and the colistin concentration was below the limit of detection (0.05 mg/L). It is not possible to interpret this finding because of the extensive dilution of pulmonary epithelial lining fluid (ELF) that occurs during the BAL procedure. For example, if ~100-fold dilution occurs then even if the actual concentration of colistin in BAL fluid was 0.04 mg/L this would be equivalent to 4 mg/L in ELF.
Two studies by the same research group have made a useful contribution to the understanding of important facets of the disposition of CMS and formed colistin in adult critically ill patients (Plachouras et al. 2009; Mohamed et al. 2012). In the first study, the plasma colistin concentration–time profiles observed with an intravenous CMS regimen of 3 million IU 8 hourly (equivalent to ~270 mg colistin base activity per day) in 18 critically ill patients (creatinine clearance range of 41–126 mL/min), without administration of a loading dose, revealed that total plasma colistin concentrations remained well below the MIC breakpoints for the first few doses in the regimen (Plachouras et al. 2009). Indeed, the predicted plasma colistin C max from that study was 0.60 mg/L after the first dose, while plasma colistin concentrations ≥2 mg/L were not achieved until approximately 44 h after commencing therapy; the typical plasma colistin C max at steady state was estimated to be 2.3 mg/L. Even at steady state, the plasma concentrations of formed colistin were below the MIC breakpoint in many patients, without consideration of plasma protein binding. In a follow-up study, the same group reported clinical PK data on a further ten critically ill patients (creatinine clearance range of 24.9–214.3 mL/min; intravenous CMS maintenance doses of 1–3 million IU 8 hourly) (Mohamed et al. 2012). The PK data were analysed simultaneously with the data from the original study (Plachouras et al. 2009). Once again, steady-state plasma colistin concentrations were not achieved for 2–3 days, were low (the average colistin C max at steady-state was 2.3 mg/L) and a large fraction of the patients had plasma colistin concentrations below the MIC breakpoint of 2 mg/L (Mohamed et al. 2012). Importantly, delayed initiation of appropriate antimicrobial therapy is associated with increased mortality in critically ill patients (Kumar et al. 2006; Luna et al. 2006), and low colistin concentrations have been associated with the amplification of colistin-resistant subpopulations (Tan et al. 2007; Bergen et al. 2008, 2011a, b; Poudyal et al. 2008; Bulitta et al. 2010; Dudhani et al. 2010b). Mathematical modelling by Bulitta et al. predicted colistin regimens with a large colistin exposure during the first ~12 h may be beneficial, providing enough net killing such that the immune system may be able to eradicate any remaining colistin-resistant cells (Bulitta et al. 2010). Thus, it is evident from the data presented by Plachouras et al. and Mohamed et al. that therapy with CMS should commence with a loading dose, which was suggested by the authors (Plachouras et al. 2009; Mohamed et al. 2012). Because of the potential for nephrotoxicity, it is suggested that the loading dose for an adult should not exceed 300 mg colistin base activity, with the first maintenance dose administered 24 h later (Garonzik et al. 2011). Because there were only 28 patients in total in the two studies reported by Plachouras et al. and Mohamed et al., with all but one patient having a creatinine clearance of >41 mL/min, it was not possible for these investigators to identify any relationships between the CMS or colistin kinetics and renal function (Plachouras et al. 2009; Mohamed et al. 2012).
The impact of renal function in critically ill patients on the disposition of CMS and formed colistin is evident from the largest pharmacokinetic study to date involving 105 patients, 89 of whom had very diverse renal function (creatinine clearance 3–169 mL/min/1.73 m2) but were not receiving renal support, 12 who were receiving intermittent hemodialysis and 4 who were recipients of continuous renal replacement therapy (Garonzik et al. 2011). The plasma concentration-time profiles of CMS and formed colistin across a dosage interval at steady state in the 105 patients who were not receiving renal support are presented in Fig. 14.6. It is evident that within each patient there was generally little fluctuation in the plasma colistin concentrations across a dosage interval, consistent with a protracted half-life for formed colistin in these very sick patients. The CMS dosage regimens administered to these patients (median daily dose across the 105 patients was 200 mg colistin base activity; range 75–410 mg colistin base activity per day) achieved average steady-state plasma colistin concentrations of 0.48–9.38 mg/L (median, 2.36 mg/L; Fig. 14.6), corresponding to a range of AUC over 24 h of 11.5–225 mg h/L. That is, the ~5.5-fold range of CMS daily doses generated a ~20-fold range of exposure to colistin in plasma. The importance of renal function as a determinant of the plasma colistin concentrations achieved from a given daily dose of CMS can begin to be appreciated from the data presented in Fig. 14.7. Indeed, population PK modelling revealed that creatinine clearance was an important covariate for the clearance of CMS and the apparent clearance of colistin. That the clearance of CMS correlated with renal function is not surprising given that CMS is predominantly cleared by renal excretion. However, the fact that creatinine clearance was a covariate for the apparent clearance of formed colistin may, at first thought, seem rather surprising because colistin is mainly excreted by non-renal mechanisms (see section ‘Pharmacokinetics, Pharmacodynamics and Pharmacokinetic/Pharmacodynamic Relationships’). The explanation lies in the schema shown in Fig. 14.2. In patients with relatively normal renal function, only a very small fraction of an administered dose of CMS is converted to colistin because the renal clearance of CMS is substantially greater than the clearance for the formation of colistin from CMS. However, in patients with substantial reductions in kidney function, the renal clearance of CMS declines and consequently a greater fraction of the administered dose of CMS is converted to colistin. Thus, the apparent clearance of colistin correlates with creatinine clearance, leading to at least two important practical consequences. First, it is evident that in patients with moderate to good renal function, administration of a daily dose of colistin base activity at the upper limit of the current product-recommended dose range (300 mg colistin base activity per day) (Coly-Mycin 2005) was not able to generate plasma colistin concentrations that would be expected to be reliably efficacious (Fig. 14.6). The second practical consequence is that reduction of the daily dose of CMS is required as renal function declines, in patients who are not receiving renal support with either intermittent hemodialysis or continuous renal replacement therapy. In agreement with previous case reports concerning critically ill patients on intermittent hemodialysis Marchand et al. (2010a) or continuous renal replacement therapy (Li et al. 2005b), both CMS and colistin were shown to have relatively efficient extracorporeal clearance in the 12 and 4 patients, respectively, who were receiving these forms of renal support (Garonzik et al. 2011). A very important practical outcome of this study was the generation of the first scientifically based CMS-dosing algorithms for patients with a large range of renal function, including patients on intermittent hemodialysis or continuous renal replacement therapy; the algorithms allowed calculation of the CMS loading and maintenance doses required to achieve a desired target average steady-state plasma concentration for colistin. Overall, the observations from this study (Fig. 14.7) are a cause for significant concern, suggesting suboptimal exposure to formed colistin with current CMS dosage regimens, particularly when one or more of the following applies: (1) the patient has moderate to good renal function, in which case it is unlikely that even a CMS daily dose at the upper limit of the product-recommended dose range will generate plasma colistin exposure likely to be reliably efficacious; (2) MIC of the infecting strain is in the upper range (i.e. 2 mg/L) of the susceptibility region for colistin; and (3) the infection is associated with high bacterial numbers. Under these circumstances, the most appropriate approach is likely to be therapy with a rationally selected colistin combination regimen.

Steady-state plasma concentration-time profiles of (a) colistin methanesulfonate and (b) formed colistin in 105 critically ill patients (89 not on renal replacement, 12 on intermittent hemodialysis and 4 on continuous renal replacement therapy). The physician-selected daily dose ranged from 75 to 410 mg colistin base activity; the dosage intervals ranged from 8 to 24 h and hence the inter-dosing blood sampling interval spanned the same range. Reproduced from (Garonzik et al. 2011) with permission from the American Society for Microbiology

Relationship of (a) physician-selected daily dose of colistin base activity (CBA) and (b) the resultant average steady-state plasma colistin concentration with creatinine clearance in 105 critically ill patients. Reproduced from (Garonzik et al. 2011) with permission from the American Society for Microbiology
Conclusions
The last several years have seen significant advances in unravelling of key aspects of the pharmacokinetics and pharmacodynamics of colistin and the relationship between the pharmacokinetics and pharmacodynamics, resulting for the first time in the generation of scientifically based dosing algorithms for CMS. As both preclinical and clinical investigations continue there will be further steps towards understanding how to optimise the administration of colistin methanesulfonate. The future incorporation of human PK/PD data into dosing algorithms, including information on colistin plasma protein binding and endpoints such as clinical cure, bacteriological eradication and the development of resistance will be very important. In addition, studies examining colistin monotherapy versus combination therapy will facilitate further optimization of colistin therapy in the various categories of patients who now require this important last-line antibiotic against Gram-negative pathogens.
References
al-Khayyat AA, Aronson AL (1973) Pharmacologic and toxicologic studies with the polymyxins. II. Comparative pharmnacologic studies of the sulfate and methanesulfonate salts of polymyxin B and colistin in dogs. Chemotherapy 19:82–97
Al-Sweih NA, Al-Hubail MA, Rotimi VO (2011) Emergence of tigecycline and colistin resistance in acinetobacter species isolated from patients in Kuwait hospitals. J Chemother 23:13–16
Andrews JM, Howe RA (2011) BSAC standardized disc susceptibility testing method (version 10). J Antimicrob Chemother 66:2726–2757
Antoniadou A, Kontopidou F, Poulakou G et al (2007) Colistin-resistant isolates of Klebsiella pneumoniae emerging in intensive care unit patients: first report of a multiclonal cluster. J Antimicrob Chemother 59:786–790
Aoki N, Tateda K, Kikuchi Y et al (2009) Efficacy of colistin combination therapy in a mouse model of pneumonia caused by multidrug-resistant Pseudomonas aeruginosa. J Antimicrob Chemother 63:534–542
Barnett M, Bushby SR, Wilkinson S (1964) Sodium sulphomethyl derivatives of polymyxins. Br J Pharmacol 23:552–574
Beceiro A, Llobet E, Aranda J et al (2011) Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob Agents Chemother 55:3370–3379
Bergen PJ, Li J, Rayner CR et al (2006) Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob Agents Chemother 50:1953–1958
Bergen PJ, Li J, Nation RL et al (2008) Comparison of once-, twice- and thrice-daily dosing of colistin on antibacterial effect and emergence of resistance: studies with Pseudomonas aeruginosa in an in vitro pharmacodynamic model. J Antimicrob Chemother 61:636–642
Bergen PJ, Bulitta JB, Forrest A et al (2010) Pharmacokinetic/pharmacodynamic investigation of colistin against Pseudomonas aeruginosa using an in vitro model. Antimicrob Agents Chemother 54:3783–3789
Bergen PJ, Forrest A, Bulitta JB et al (2011a) Clinically relevant plasma concentrations of colistin in combination with imipenem enhance pharmacodynamic activity against multidrug-resistant pseudomonas aeruginosa at multiple inocula. Antimicrob Agents Chemother 55:5134–5142
Bergen PJ, Tsuji BT, Bulitta JB et al (2011b) Synergistic killing of multidrug-resistant Pseudomonas aeruginosa at multiple inocula by colistin combined with doripenem in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 55:5685–5695
Betrosian AP, Frantzeskaki F, Xanthaki A et al (2008) Efficacy and safety of high-dose ampicillin/sulbactam vs. colistin as monotherapy for the treatment of multidrug resistant Acinetobacter baumannii ventilator-associated pneumonia. J Infect 56:432–436
Beveridge EG, Martin AJ (1967) Sodium sulphomethyl derivatives of polymyxins. Br J Pharmacol Chemother 29:125–135
Bogdanovich T, Adams-Haduch JM, Tian GB et al (2011) Colistin-resistant, Klebsiella pneumoniae carbapenemase (KPC)-producing Klebsiella pneumoniae belonging to the international epidemic clone ST258. Clin Infect Dis 53:373–376
Boucher HW, Talbot GH, Bradley JS et al (2009) Bad bugs, no drugs: no ESKAPE! an update from the infectious diseases society of America. Clin Infect Dis 48:1–12
Bratu S, Landman D, Haag R et al (2005) Rapid spread of carbapenem-resistant Klebsiella pneumoniae in New York City: a new threat to our antibiotic armamentarium. Arch Intern Med 165:1430–1435
Breazeale SD, Ribeiro AA, McClerren AL et al (2005) A formyltransferase required for polymyxin resistance in Escherichia coli and the modification of lipid A with 4-amino-4-deoxy-L-arabinose. Identification and function oF UDP-4-deoxy-4-formamido-L-arabinose. J Biol Chem 280:14154–14167
Brogden KA (2005) Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250
Bulitta JB, Yang JC, Yohonn L et al (2010) Attenuation of colistin bactericidal activity by high inoculum of Pseudomonas aeruginosa characterized by a new mechanism-based population pharmacodynamic model. Antimicrob Agents Chemother 54:2051–2062
Cernohorska L, Slavikova P (2010) Antibiotic resistance and biofilm formation in Pseudomonas aeruginosa strains isolated from patients with urinary tract infections. Epidemiol Mikrobiol Imunol 59:154–157
Clinical and Laboratory Standards Institute (CLSI) (2012) Performance standards for antimicrobial susceptibility testing: twentieth informational supplement (M100-S20). CLSI, Wayne, PA
Coly-Mycin M Parenteral [package insert] (2005). Monarch Pharmaceuticals, Bristol, TN
Comite de l’ Antibiogramme de la Societe Francaise de Microbiologie (SFM) (2005). Recommandations du CSFM, Communique: edition de Janvier 2005. http://www.sfm.asso.fr/. Accessed 31 July 2012
Couet W, Gregoire N, Gobin P et al (2011) Pharmacokinetics of colistin and colistimethate sodium after a single 80-mg intravenous dose of CMS in young healthy volunteers. Clin Pharmacol Ther 89:875–879
Craig WA, Kunin CM (1973) Dynamics of binding and release of the polymyxin antibiotics by tissues. J Pharmacol Exp Ther 184:757–765
David MD, Gill MJ (2008) Potential for underdosing and emergence of resistance in Acinetobacter baumannii during treatment with colistin. J Antimicrob Chemother 61:962–964
Decolin D, Leroy P, Nicolas A et al (1997) Hyphenated liquid chromatographic method for the determination of colistin residues in bovine tissues. J Chromatogr Sci 35:557–564
Deris ZZ, Yu HH, Davis K et al (2012) Colistin and doripenem combination is synergistic against Klebsiella pneumoniae at multiple inocula and suppresses colistin resistance in an in vitro PK/PD model. Antimicrob Agents Chemother 56(10):5103–5112
DeRyke CA, Crawford AJ, Uddin N et al (2010) Colistin dosing and nephrotoxicity in a large community teaching hospital. Antimicrob Agents Chemother 54:4503–4505
Dotsikas Y, Markopoulou CK, Koundourellis JE et al (2011) Validation of a novel LC-MS/MS method for the quantitation of colistin A and B in human plasma. J Sep Sci 34:37–45
Dudhani RV, Li J, Nation RL (2009). Plasma binding of colistin involves multiple proteins and is concentration dependent: potential clinical implications (abstract A1-576, p41). In: Abstracts of the 49th annual interscience conference on antimicrobial agents and chemotherapy (ICAAC), American Society for Microbiology San Francisco, CA, 12–15 Sept 2009
Dudhani RV, Nation RL, Li J (2010a) Evaluating the stability of colistin and colistin methanesulphonate in human plasma under different conditions of storage. J Antimicrob Chemother 65:1412–1415
Dudhani RV, Turnidge JD, Coulthard K et al (2010b) Elucidation of the pharmacokinetic/pharmacodynamic determinant of colistin activity against Pseudomonas aeruginosa in murine thigh and lung infection models. Antimicrob Agents Chemother 54:1117–1124
Dudhani RV, Turnidge JD, Nation RL et al (2010c) fAUC/MIC is the most predictive pharmacokinetic/pharmacodynamic index of colistin against Acinetobacter baumannii in murine thigh and lung infection models. J Antimicrob Chemother 65:1984–1990
Eickhoff TC, Finland M (1965) Polymyxin B and colistin: in vitro activity against Pseudomonas aeruginosa. Am J Med Sci 249:172–174
European Committee on Antimicrobial Susceptibility Testing (EUCAST) (2012). Breakpoint tables for interpretation of MICs and zone diameters (Version 2.0, January 1, 2012). http://www.eucast.org/clinical_breakpoints/. Accessed 31 July 2012
Falagas ME, Kasiakou SK (2005) Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin Infect Dis 40:1333–1341
Falagas ME, Kasiakou SK (2006) Toxicity of polymyxins: a systematic review of the evidence from old and recent studies. Crit Care 10:R27
Falagas ME, Rizos M, Bliziotis IA et al (2005) Toxicity after prolonged (more than four weeks) administration of intravenous colistin. BMC Infect Dis 5:1
Fernandez L, Gooderham WJ, Bains M et al (2010) Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob Agents Chemother 54:3372–3382
Finland M, Garner C, Wilcox C et al (1976a) Susceptibility of beta-hemolytic streptococci to 65 antibacterial agents. Antimicrob Agents Chemother 9:11–19
Finland M, Garner C, Wilcox C et al (1976b) Susceptibility of pneumococci and Haemophilus influenzae to antibacterial agents. Antimicrob Agents Chemother 9:274–287
Gales AC, Jones RN, Sader HS (2011) Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006–09). J Antimicrob Chemother 66:2070–2074
Garonzik SM, Li J, Thamlikitkul V et al (2011) Population pharmacokinetics of colistin methanesulfonate and formed colistin in critically ill patients from a multicenter study provide dosing suggestions for various categories of patients. Antimicrob Agents Chemother 55:3284–3294
Gobin P, Lemaitre F, Marchand S et al (2010) Assay of colistin and colistin methanesulfonate in plasma and urine by liquid chromatography-tandem mass spectrometry. Antimicrob Agents Chemother 54:1941–1948
Gunderson BW, Ibrahim KH, Hovde LB et al (2003) Synergistic activity of colistin and ceftazidime against multiantibiotic-resistant Pseudomonas aeruginosa in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 47:905–909
Hale JD, Hancock RE (2007) Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev Anti Infect Ther 5:951–959
Hancock RE, Chapple DS (1999) Peptide antibiotics. Antimicrob Agents Chemother 43:1317–1323
Hancock RE, Rozek A (2002) Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol Lett 206:143–149
Hancock RE, Falla T, Brown M (1995) Cationic bactericidal peptides. Adv Microb Physiol 37:135–175
Hartzell JD, Neff R, Ake J et al (2009) Nephrotoxicity associated with intravenous colistin (colistimethate sodium) treatment at a tertiary care medical center. Clin Infect Dis 48:1724–1728
Hawley JS, Murray CK, Jorgensen JH (2008) Colistin heteroresistance in acinetobacter and its association with previous colistin therapy. Antimicrob Agents Chemother 52:351–352
Hawser SP (2010) Susceptibility of Klebsiella pneumoniae clinical isolates from 2007 to 2009 to colistin and comparator antibiotics. Int J Antimicrob Agents 36:383–384
Helander IM, Kato Y, Kilpelainen I et al (1996) Characterization of lipopolysaccharides of polymyxin-resistant and polymyxin-sensitive Klebsiella pneumoniae O3. Eur J Biochem 237:272–278
Hengzhuang W, Wu H, Ciofu O et al (2012) In vivo pharmacokinetics/pharmacodynamics of colistin and imipenem in Pseudomonas aeruginosa biofilm infection. Antimicrob Agents Chemother 56:2683–2690
Hoeprich PD (1970) The polymyxins. Med Clin North Am 54:1257–1265
Imberti R, Cusato M, Villani P et al (2010) Steady-state pharmacokinetics and BAL concentration of colistin in critically ill patients after IV colistin methanesulfonate administration. Chest 138:1333–1339
Infectious Disease Society of America (IDSA) (2010) The 10 x ’20 Initiative: pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin Infect Dis 50:1081–1083
Institute for Safe Medication Practices (ISMP) (2011) National Alert Network: Warning! Dosing confusion with colistmethate for injection. http://www.ashp.org/DocLibrary/Policy/PatientSafety/NANAlert-Colistimethatesodium.aspx. Accessed 1 Aug 2012
Jansson B, Karvanen M, Cars O et al (2009) Quantitative analysis of colistin A and colistin B in plasma and culture medium using a simple precipitation step followed by LC/MS/MS. J Pharm Biomed Anal 49:760–767
Johansen HK, Moskowitz SM, Ciofu O et al (2008) Spread of colistin resistant non-mucoid Pseudomonas aeruginosa among chronically infected Danish cystic fibrosis patients. J Cyst Fibros 7:391–397
Ketthireddy S, Lee DG, Murakami Y et al (2007) In vivo pharmacodynamics of colistin against Pseudomonas aeruginosa in thighs of neutropenic mice (abstract A-4, p1). In: Abstracts of the 47th interscience conference on antimicrobial agents and chemotherapy (ICAAC), American Society for Microbiology, Chicago, IL, 17–20 Sept 2007
Ko KS, Suh JY, Kwon KT et al (2007) High rates of resistance to colistin and polymyxin B in subgroups of Acinetobacter baumannii isolates from Korea. J Antimicrob Chemother 60:1163–1167
Ko H, Jeon M, Choo E et al (2011) Early acute kidney injury is a risk factor that predicts mortality in patients treated with colistin. Nephron Clin Pract 117:c284–c288
Kontopoulou K, Protonotariou E, Vasilakos K et al (2010) Hospital outbreak caused by Klebsiella pneumoniae producing KPC-2 beta-lactamase resistant to colistin. J Hosp Infect 76:70–73
Koyama Y, Kurosasa A, Tsuchiya A et al (1950) A new antibiotic ‘colistin’ produced by spore-forming soil bacteria. J Antibiot 3:457–458
Kumar A, Roberts D, Wood KE et al (2006) Duration of hypotension before initiation of effective antimicrobial therapy is the critical determinant of survival in human septic shock. Crit Care Med 34:1589–1596
Kunin CM, Bugg A (1971) Binding of polymyxin antibiotics to tissues: the major determinant of distribution and persistence in the body. J Infect Dis 124:394–400
Kwon JA, Lee JE, Huh W et al (2010) Predictors of acute kidney injury associated with intravenous colistin treatment. Int J Antimicrob Agents 35:473–477
Landman D, Georgescu C, Martin DA et al (2008) Polymyxins revisited. Clin Microbiol Rev 21:449–465
Lee JY, Song JH, Ko KS (2011) Identification of nonclonal Pseudomonas aeruginosa isolates with reduced colistin susceptibility in Korea. Microb Drug Resist 17:299–304
Leroy P, Decolin D, Nicolas S et al (1989) Residue determination of two co-administered antibacterial agents–cephalexin and colistin–in calf tissues using high-performance liquid chromatography and microbiological methods. J Pharm Biomed Anal 7:1837–1846
Lewis JR, Lewis SA (2004) Colistin interactions with the mammalian urothelium. Am J Physiol Cell Physiol 286:C913–C922
Lewis LA, Choudhury B, Balthazar JT et al (2009) Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect Immun 77:1112–1120
Li J, Milne RW, Nation RL et al (2001a) A simple method for the assay of colistin in human plasma, using pre-column derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and reversed-phase high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 761:167–175
Li J, Turnidge J, Milne R et al (2001b) In vitro pharmacodynamic properties of colistin and colistin methanesulfonate against Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother 45:781–785
Li J, Milne RW, Nation RL et al (2002) Simple method for assaying colistin methanesulfonate in plasma and urine using high-performance liquid chromatography. Antimicrob Agents Chemother 46:3304–3307
Li J, Coulthard K, Milne R et al (2003a) Steady-state pharmacokinetics of intravenous colistin methanesulphonate in patients with cystic fibrosis. J Antimicrob Chemother 52:987–992
Li J, Milne RW, Nation RL et al (2003b) Stability of colistin and colistin methanesulfonate in aqueous media and plasma as determined by high-performance liquid chromatography. Antimicrob Agents Chemother 47:1364–1370
Li J, Milne RW, Nation RL et al (2003c) Use of high-performance liquid chromatography to study the pharmacokinetics of colistin sulfate in rats following intravenous administration. Antimicrob Agents Chemother 47:1766–1770
Li J, Milne RW, Nation RL et al (2004) Pharmacokinetics of colistin methanesulphonate and colistin in rats following an intravenous dose of colistin methanesulphonate. J Antimicrob Chemother 53:837–840
Li J, Nation RL, Milne RW et al (2005a) Evaluation of colistin as an agent against multi-resistant Gram-negative bacteria. Int J Antimicrob Agents 25:11–25
Li J, Rayner CR, Nation RL et al (2005b) Pharmacokinetics of colistin methanesulfonate and colistin in a critically ill patient receiving continuous venovenous hemodiafiltration. Antimicrob Agents Chemother 49:4814–4815
Li J, Nation RL, Turnidge JD (2006a) Defining the dosage units for colistin methanesulfonate: urgent need for international harmonization. Antimicrob Agents Chemother 50(12):4231, author reply 4231–4232
Li J, Nation RL, Turnidge JD et al (2006b) Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis 6:589–601
Li J, Rayner CR, Nation RL et al (2006c) Heteroresistance to colistin in multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 50:2946–2950
Lim LM, Ly N, Anderson D et al (2010) Resurgence of colistin: a review of resistance, toxicity, pharmacodynamics, and dosing. Pharmacotherapy 30:1279–1291
Lin KH, Chuang YC, Lee SH et al (2010) In vitro synergistic antimicrobial effect of imipenem and colistin against an isolate of multidrug-resistant Enterobacter cloacae. J Microbiol Immunol Infect 43:317–322
Livermore DM (2004) The need for new antibiotics. Clin Microbiol Infect 10(Suppl 4):1–9
Llobet E, Tomas JM, Bengoechea JA (2008) Capsule polysaccharide is a bacterial decoy for antimicrobial peptides. Microbiology 154:3877–3886
Luna CM, Aruj P, Niederman MS et al (2006) Appropriateness and delay to initiate therapy in ventilator-associated pneumonia. Eur Respir J 27:158–164
Ma Z, Wang J, Nation RL et al (2009) Renal disposition of colistin in the isolated perfused rat kidney. Antimicrob Agents Chemother 53:2857–2864
Mackay DN, Kaye D (1964) Serum concentrations of colistin in patients with normal and impaired renal function. N Engl J Med 270:394–397
Marchaim D, Chopra T, Pogue JM et al (2011) Outbreak of colistin-resistant, carbapenem-resistant Klebsiella pneumoniae in metropolitan Detroit, Michigan. Antimicrob Agents Chemother 55:593–599
Marchand S, Frat JP, Petitpas F et al (2010a) Removal of colistin during intermittent haemodialysis in two critically ill patients. J Antimicrob Chemother 65:1836–1837
Marchand S, Lamarche I, Gobin P et al (2010b) Dose-ranging pharmacokinetics of colistin methanesulphonate (CMS) and colistin in rats following single intravenous CMS doses. J Antimicrob Chemother 65:1753–1758
Markou N, Markantonis SL, Dimitrakis E et al (2008) Colistin serum concentrations after intravenous administration in critically ill patients with serious multidrug-resistant, gram-negative bacilli infections: a prospective, open-label, uncontrolled study. Clin Ther 30:143–151
Meletis G, Tzampaz E, Sianou E et al (2011) Colistin heteroresistance in carbapenemase-producing Klebsiella pneumoniae. J Antimicrob Chemother 66:946–947
Mezzatesta ML, Gona F, Caio C et al (2011) Outbreak of KPC-3-producing, and colistin-resistant, Klebsiella pneumoniae infections in two Sicilian hospitals. Clin Microbiol Infect 17:1444–1447
Michalopoulos AS, Karatza DC (2010) Multidrug-resistant Gram-negative infections: the use of colistin. Expert Rev Anti Infect Ther 8:1009–1017
Moffatt JH, Harper M, Harrison P et al (2010) Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977
Mohamed AF, Karaiskos I, Plachouras D et al (2012) Application of a loading dose of colistin methanesulfonate in critically ill patients: population pharmacokinetics, protein binding, and prediction of bacterial kill. Antimicrob Agents Chemother 56:4241–4249
Molina J, Cordero E, Pachon J (2009) New information about the polymyxin/colistin class of antibiotics. Expert Opin Pharmacother 10:2811–2828
Morita K, Yamaji A (1995) Changes in the serum protein binding of vancomycin in patients with methicillin-resistant Staphylococcus aureus infection: the role of serum alpha 1-acid glycoprotein levels. Ther Drug Monit 17:107–112
Morrison AJ Jr, Wenzel RP (1984) Epidemiology of infections due to Pseudomonas aeruginosa. Rev Infect Dis 6(Suppl 3):S627–S642
Moskowitz SM, Ernst RK, Miller SI (2004) PmrAB, a two-component regulatory system of Pseudomonas aeruginosa that modulates resistance to cationic antimicrobial peptides and addition of aminoarabinose to lipid A. J Bacteriol 186:575–579
Nation RL, Li J (2009) Colistin in the 21st century. Curr Opin Infect Dis 22:535–543
Opal SM, Calandra T (2009) Antibiotic usage and resistance: gaining or losing ground on infections in critically ill patients? JAMA 302:2367–2368
Orwa JA, Van Gerven A, Roets E et al (2000) Development and validation of a liquid chromatography method for analysis of colistin sulphate. Chromatographia 51:433–436
Orwa JA, Govaerts C, Busson R et al (2001) Isolation and structural characterization of colistin components. J Antibiot (Tokyo) 54:595–599
Owen RJ, Li J, Nation RL et al (2007) In vitro pharmacodynamics of colistin against Acinetobacter baumannii clinical isolates. J Antimicrob Chemother 59:473–477
Ozbek B, Senturk A (2010) Postantibiotic effects of tigecycline, colistin sulfate, and levofloxacin alone or tigecycline-colistin sulfate and tigecycline-levofloxacin combinations against Acinetobacter baumannii. Chemotherapy 56:466–471
Ozyilmaz E, Ebinc FA, Derici U et al (2011) Could nephrotoxicity due to colistin be ameliorated with the use of N-acetylcysteine? Intensive Care Med 37:141–146
Pintado V, San Miguel LG, Grill F et al (2008) Intravenous colistin sulphomethate sodium for therapy of infections due to multidrug-resistant gram-negative bacteria. J Infect 56:185–190
Plachouras D, Karvanen M, Friberg LE et al (2009) Population pharmacokinetic analysis of colistin methanesulphonate and colistin after intravenous administration in critically ill patients with gram-negative bacterial infections. Antimicrob Agents Chemother 53:3430–3436
Poudyal A, Howden BP, Bell JM et al (2008) In vitro pharmacodynamics of colistin against multidrug-resistant Klebsiella pneumoniae. J Antimicrob Chemother 62:1311–1318
Queenan AM, Pillar CM, Deane J et al (2012) Multidrug resistance among Acinetobacter spp. in the USA and activity profile of key agents: results from CAPITAL Surveillance 2010. Diagn Microbiol Infect Dis 73:267–270
Rosenvinge A, Pressler T, Hoiby N (2005) Colistin intravenously is a safe and effective treatment of multidrug-resistant microorganisms in cystic fibrosis. J Cyst Fibros 4:S32
Sader HS, Farrell DJ, Jones RN (2011) Susceptibility of Klebsiella spp. to colistin and polymyxin B: results from the SENTRY Antimicrobial Surveillance Program (2006–2009). Int J Antimicrob Agents 37:174–175
Schwartz BS, Warren MR, Barkley FA et al (1959) Microbiological and pharmacological studies of colistin sulfate and sodium colistinmethanesulfonate. Antibiot Annu 7:41–60
Spellberg B, Blaser M, Guidos RJ et al (2011) Combating antimicrobial resistance: policy recommendations to save lives. Clin Infect Dis 52(Suppl 5):S397–S428
Suh JY, Son JS, Chung DR et al (2010) Nonclonal emergence of colistin-resistant Klebsiella pneumoniae isolates from blood samples in South Korea. Antimicrob Agents Chemother 54:560–562
Tan TY, Ng SY (2006) The in-vitro activity of colistin in gram-negative bacteria. Singapore Med J 47:621–624
Tan CH, Li J, Nation RL (2007) Activity of colistin against heteroresistant Acinetobacter baumannii and emergence of resistance in an in vitro pharmacokinetic/pharmacodynamic model. Antimicrob Agents Chemother 51:3413–3415
Toth A, Damjanova I, Puskas E et al (2010) Emergence of a colistin-resistant KPC-2-producing Klebsiella pneumoniae ST258 clone in Hungary. Eur J Clin Microbiol Infect Dis 29:765–769
Trotman RL, Williamson JC, Shoemaker DM et al (2005) Antibiotic dosing in critically ill adult patients receiving continuous renal replacement therapy. Clin Infect Dis 41:1159–1166
Voulgari F, Cummins P, Gardecki TI et al (1982) Serum levels of acute phase and cardiac proteins after myocardial infarction, surgery, and infection. Br Heart J 48:352–356
Walkty A, DeCorby M, Nichol K et al (2009) In vitro activity of colistin (polymyxin E) against 3,480 isolates of gram-negative bacilli obtained from patients in Canadian hospitals in the CANWARD study, 2007–2008. Antimicrob Agents Chemother 53:4924–4926
Wallace SJ, Li J, Nation RL et al (2008a) Subacute toxicity of colistin methanesulfonate in rats: comparison of various intravenous dosage regimens. Antimicrob Agents Chemother 52:1159–1161
Wallace SJ, Li J, Rayner CR et al (2008b) Stability of colistin methanesulfonate in pharmaceutical products and solutions for administration to patients. Antimicrob Agents Chemother 52:3047–3051
Wallace SJ, Li J, Nation RL et al (2010) Self-assembly behavior of colistin and its prodrug colistin methanesulfonate: implications for solution stability and solubilization. J Phys Chem B 114:4836–4840
Winfield MD, Latifi T, Groisman EA (2005) Transcriptional regulation of the 4-amino-4-deoxy-L-arabinose biosynthetic genes in Yersinia pestis. J Biol Chem 280:14765–14772
Yau W, Owen RJ, Poudyal A et al (2009) Colistin hetero-resistance in multidrug-resistant Acinetobacter baumannii clinical isolates from the Western Pacific region in the SENTRY antimicrobial surveillance programme. J Infect 58:138–144
Yousef JM, Chen G, Hill PA et al (2011) Melatonin attenuates colistin-induced nephrotoxicity in rats. Antimicrob Agents Chemother 55:4044–4049
Yousef JM, Chen G, Hill PA et al (2012) Ascorbic acid protects against the nephrotoxicity and apoptosis caused by colistin and affects its pharmacokinetics. J Antimicrob Chemother 67:452–459
Zavascki AP, Goldani LZ, Cao G et al (2008) Pharmacokinetics of intravenous polymyxin B in critically ill patients. Clin Infect Dis 47:1298–1304
Ziv G, Sulman FG (1972) Binding of antibiotics to bovine and ovine serum. Antimicrob Agents Chemother 2:206–213
Ziv G, Nouws JF, van Ginneken CA (1982) The pharmacokinetics and tissue levels of polymyxin B, colistin and gentamicin in calves. J Vet Pharmacol Ther 5:45–58
Acknowledgements
RLN and JL are supported by Award Number R01AI079330 and Award Number R01AI070896 from the National Institute of Allergy and Infectious Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health. JL is an Australian National Health and Medical Research Council Senior Research Fellow.
The authors do not have any financial, commercial or proprietary interest in any drug, device or equipment mentioned in this article.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Nation, R.L., Bergen, P.J., Li, J. (2014). Pharmacokinetics and Pharmacodynamics of Colistin. In: Vinks, A., Derendorf, H., Mouton, J. (eds) Fundamentals of Antimicrobial Pharmacokinetics and Pharmacodynamics. Springer, New York, NY. https://doi.org/10.1007/978-0-387-75613-4_14
Download citation
DOI: https://doi.org/10.1007/978-0-387-75613-4_14
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-0-387-75612-7
Online ISBN: 978-0-387-75613-4
eBook Packages: MedicineMedicine (R0)