
Abstract
The trigeminal nerve (Vn) is the largest cranial nerve and it supplies sensory fibres to all craniofacial structures. Sensory innervation of the craniofacial region is important in functional, psychological and emotional aspects, given the significance of the head as an organ in whole, of facial communication and of all specialised sense organs of the head such as the retina, olfactory epithelium, taste papillae, tooth pulp and cochlea, which are highly innervated by trigeminal fibres [1]. Trigeminal fibres are organised to warn the organism against changing environmental conditions, ranging from changes in environmental chemicals, temperature, injury or other external stimuli. The craniofacial region has a rich innervation and an extensive somatosensory representation in the CNS. These aspects make the Vn the most complex of the 12 cranial nerves. Mechanisms of nociception along the trigeminal nerve are of particular interest in headache conditions and orofacial pain [2].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1 Introduction
The trigeminal nerve (Vn) is the largest cranial nerve and it supplies sensory fibres to all craniofacial structures. Sensory innervation of the craniofacial region is important in functional, psychological and emotional aspects, given the significance of the head as an organ in whole, of facial communication and of all specialised sense organs of the head such as the retina, olfactory epithelium, taste papillae, tooth pulp and cochlea, which are highly innervated by trigeminal fibres [1]. Trigeminal fibres are organised to warn the organism against changing environmental conditions, ranging from changes in environmental chemicals, temperature, injury or other external stimuli. The craniofacial region has a rich innervation and an extensive somatosensory representation in the CNS. These aspects make the Vn the most complex of the 12 cranial nerves. Mechanisms of nociception along the trigeminal nerve are of particular interest in headache conditions and orofacial pain [2].
2 The Trigeminal Nerve
The trigeminal ganglion (TG) is the sensory ganglion of the trigeminal nerve and occupies the Meckel’s cavity (cavum Meckelii) in the dura mater covering the trigeminal impression near the apex of the petrous part of the temporal bone [3]. The TG consists of pseudounipolar primary sensory neurons (the dendrite of these neurons are located in the trigeminal nerve, the cell bodies are located in the trigeminal ganglion and the axons protrude through the sensory root and into the ventrolateral midpons) and is analogous to the dorsal root ganglia (DRG) of the spinal cord, which contain the cell bodies of incoming sensory fibres from the rest of the body [2, 4]. TG neurons have no synaptic interconnections with one another and they are surrounded by satellite glial cells (SGCs). However, the SGCs are connected with the TG neurons with gap junctions that intimately communicate between them [5].
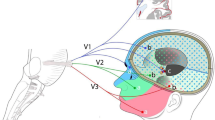
The TG gives rise to the trigeminal nerve (Vth cranial nerve), which is the largest of the cranial nerves. The trigeminal nerve trifurcates into ophthalmic (V1), maxillary (V2) and mandibular nerves (V3) distally from the trigeminal ganglion. The divisions of the trigeminal nerve exit the skull base through the superior orbital fissure for V1, through the foramen rotundum for V2, and through the foramen ovale for V3 [6]. The ophthalmic and maxillary nerves are purely sensory, whereas the mandibular nerve has both sensory and motor functions. These three branches converge on the TG from which a single large sensory root enters the brainstem at the level of the pons. Immediately adjacent to the sensory root, a smaller motor root emerges from the pons at the same level, and thus the trigeminal nerve is a mixed nerve containing both motor and sensory components [1, 3]. Motor fibres are distributed together with sensory fibres in branches of the mandibular nerve and supply the muscles of mastication and the tensor tympani and tensor veli palatine muscles.
The sensory fibres of the ophthalmic, maxillary and mandibular nerves have a diversity of arrangement of trigeminal endings in craniofacial tissues, supplying the cutaneous exteroceptors of the face, the retina, cochlea, the mucous membranes of the nasal and oral cavities, and a large portion of the intracranial dura mater and vessels [2, 7]. Early anatomical studies provided evidence for the meningeal representation in the trigeminal ganglion by using horseradish peroxidase histochemistry [7, 8] and more specifically using retrograde tracing with True Blue [9,10,11]. Most of the nociceptors around meningeal vessels were found to project mainly to the ophthalmic division of the ipsilateral trigeminal ganglion and to a minor degree to the maxillary and mandibular divisions [12, 13]. In addition the True Blue tracing revealed that the distribution was no strictly unilateral because some overlap existed for autonomic as well as sensory innervation [9, 14, 15]. The dermatomes of the three branches of the trigeminal nerve have relatively little overlap, unlike dermatomes in the rest of the body, which show considerable overlap. More specifically the three branches of the trigeminal nerve cover the following sensory areas [16]:
-
The ophthalmic nerve carries sensory information from the skin of the forehead, the upper eyelids and the nose ridge and the mucosa of the nasal septum and some paranasal sinuses.
-
The maxillary nerve innervates the skin of the middle facial area, the side of the nose and the lower eyelids, the maxillary dentition, the mucosa of the upper lip, the palate, the nasal conchae and the maxillary sinus.
-
The mandibular nerve innervates the skin of the lower facial area, the mandibular dentition, the mucosa of the lower lip, cheeks and floor of the mouth, part of the tongue and part of the external ear.
Within the TG the somata of neurons giving rise to the three branches of the trigeminal nerve are somatotopically organised. Somatotopic organisation is not only found within the ganglion but also in the brainstem distribution in the trigeminocervical complex. The cell bodies of the ophthalmic nerve are found medially in the ganglion, those of the mandibular nerve are grouped laterally, while in the middle of these two groups, the cell bodies of the maxillary nerve are grouped [17,18,19]. The proprioceptive fibres in the motor root of the trigeminal nerve have their cell bodies in the mesencephalic nucleus of the pons. The axons of these motor neurons run pass the trigeminal ganglion as an independent bundle without synapsing within it. The motor trigeminal nucleus is directly stimulated via the corticobulbar tract, originating from the contralateral cerebral cortex. Within the motor nucleus, there is also a large amount of somatotopy. Via efferent fibres the motor trigeminal nucleus receives proprioceptive information from the masticatory muscles, temporomandibular joint and periodontium.
3 The Primary Trigeminal Sensory Fibres
The trigeminal sensory fibres convey information regarding pain, temperature, touch and proprioception. The nociceptors are the sensory fibres that convey nociceptive information. The nociceptors run largely adjacent to the blood vessels and transmit nociceptive information mainly through Aδ- (thinly myelinated) and C- (unmyelinated) fibre types [20,21,22] although other types of primary afferents transmitting somatic sensations have also been characterised [23]. Recent work has shown that the C-fibres store CGRP while the Aδ-fibres contain CGRP receptor elements [24]. Similarly to somatic pain, the pain associated with trigeminal Aδ-fibres activation is characterised by an initial extremely sharp pain and is referred to as the “first” pain. The “second” pain is referred to the more prolonged and delayed feeling of dull ache or burning pain as a result of C-fibre activation. What makes trigeminal nerve unique is that it has ~100 times more dense C-fibres than any other nerve (Fig. 1.1).

In human TG, CGRP positive pearl-like fibres were observed (thin arrows). Thick arrow points at autofluorescent lipofuscin and arrowhead at a satellite glial cell
The peripheral terminal of the nociceptor is where noxious stimuli are detected and transduced into inward currents that, if sufficiently large, begin to drive action potentials along the axon to the CNS and set a train of events that ultimately lead to a conscious awareness of the noxious stimulus [25]. The sensory specificity of the nociceptor is established by expression of ion channels which respond with a high threshold only to particular features of the mechanical, thermal and chemical environment [25, 26]. The high threshold of these transducers differentiates nociceptors from sensory neurons that respond to innocuous stimuli by expressing transducers with low thresholds [25]. Such transducer channels are TRPV1-4, TRPM8, TRPA1, ASICs, P2X3, TREK, kainate receptors and 5-HT1B/1D receptors [27,28,29,30,31,32,33,34,35,36]. A number of studies have involved these channels in trigeminal nociception. Potentially, blockade of these transducers could act as an emerging treatment for trigeminal related disorders. 5-HT1B/1D receptors are the target of triptans, the migraine specific medications [37]. Single nucleotide polymorphisms in the TRPM8 gene have been repeatedly found to be significant in migraine genome-wide association studies [38,39,40,41,42].
Transmission of nociception occurs in response to calcium influx at the central terminal and releasing glutamate, as well as multiple neuropeptides and other signalling molecules that act as synaptic modulators which will activate post-synaptic receptors on second order neurons [23]. In response to inflammatory or noxious stimuli, trigeminal ganglia neurons release neuropeptides and other molecules that initiate and maintain neurogenic inflammation in the peripheral tissue that facilitate peripheral sensitisation of trigeminal nociceptors [43].
3.1 Neuropeptides of the Trigeminal Ganglion
Neurons of the TG express at different percentages several different neuropeptides, including calcitonin gene-related peptide (CGRP), substance P (SP), pituitary adenylate-cyclase-activating polypeptide (PACAP), neuropeptide Y (NPY), somatostatin, dynorphin, galanin, orexin, nociception, neurokinin A and neurokinin B, as well as nitric oxide synthase, inter alia [44]. Peripherally, upon their release, the majority of these neuropeptides are vasodilators, while centrally they are involved in signal transmission by acting as neuromodulators. TG neurons and their terminals also express many of the receptors for these neuropeptides. The actions of these neuropeptides have been extensively reviewed by Lazarov [44] and Goto and colleagues [45].
The expression of different neuropeptides in the TG, as well as the expression of their receptors, is altered following inflammation or injury, potentially to induce an autocrine-like reaction. For example, the expression of SP and its receptor neurokinin 1-receptor (NK1-R) increases following maxillary molar extraction [46], while CGRP is increased following induction of periodontium inflammation [47]. It has been suggested that the build-up of these vasodilatory neuropeptides at injury site may be related to the development of neurogenic inflammation, ectopic neural activity and to contribute to the development of neuropathic pain. Interestingly though, such upregulation is often beyond their nerve distribution. As TG neurons are anatomically isolated from one another and not synaptically interconnected, other means of interaction may exist between the three clusters of neurons giving rise to the three branches of the trigeminal nerve, as mentioned above related to the function of the SGCs [5]. One possibility is through SGCs and/or microglia/macrophage-like cells (MLCs), particularly with regard to interactions between the mandibular and maxillary neurons in the TG. SGCs initially become activated by receiving the signal from TG neurons. Following, SGCs activate adjacent SGCs or other TG neurons by release of neurotransmitters. Similar to the dorsal root ganglion, MLCs in the TG are activated by uptake of a transmitter from TG neurons or SGCs. This communication between neurons, SGCs, and MLCs is believed to contribute to the development of ectopic pain, hyperesthesia or peripheral sensitisation [45].
3.1.1 Calcitonin-Gene-Related Peptide
The α-CGRP isoform is expressed in about 50% of TG neurons and is a key neuropeptide involved in both neural and vascular responses [48,49,50]. CGRP is present in C-fibre neuronal cells [50,51,52]. About 30% of CGRP-positive TG neurons also co-express SP [53]. CGRP immunoreactive axons, derived from the ipsilateral ophthalmic division of the trigeminal nerve, are abundant on the walls of the rostral circulation of the major cerebral arteries in the circle of Willis, the rostral cerebral circulation, the dura mater and the eye [54,55,56]. Sensory terminals expressing CGRP have been also identified in the nasal mucosa, periodontium and gingivae [57,58,59]. Recent work has described in detail the distribution of CGRP and its receptor in the retina [60]. Some of these CGRP containing fibres originate in the TG, putatively involved in migraine attacks. CGRP acts mainly on the CLR/RAMP1 receptor, which is also found on trigeminal fibres [50] (Fig. 1.2).

RAMP1 immunoreactivity was exclusively found in satellite glial cells in human TG (arrowheads). Arrow points at lipofuscin
CGRP is released from large dense-core vesicles demonstrated in the human temporal artery [61] and the human middle meningeal artery [62]. CGRP is regulated by P/Q-type, N- and L-type voltage-dependent calcium channels, and it is [63] co-released with glutamate contained in separate vesicles [64]. Release from synaptic vehicles involves the SNAP-25 protein of the SNARE complex and hence, like glutamate release, it can be inhibited by botulinum toxin type A [65]. These complexes are found also in the TG [66]. Botox is currently an established preventive treatment in chronic migraine [67]. Its spontaneous basal release follows a circadian rhythm as it is increased at night [68].
CGRP is the most potent vasodilator when released peripherally, through direct activation of its receptor CLR/RAMP1 on smooth muscle cells [48, 69]. Its release from primary trigeminal afferents innervating blood vessels of the dura mater and the cerebral circulation is one of the main mechanisms of trigeminovascular activation [48], which is believed to be involved in the pathophysiology of primary headaches [70, 71]. CGRP can also induce vasodilation indirectly by activating endothelium CLR/RAMP1, resulting in a rise in cAMP [63, 72] and nitric oxide (NO) production [73]. Diffusion of NO into the smooth muscle cell activates guanylate cyclase inducing relaxation. CGRP as a vasodilator is involved in cardiovascular regulation and may have a protective role against ischaemia. CGRP is spontaneously released during acute blood pressure reflex for cardiovascular regulation, it antagonises sympathetic system-induced vascular resistance and appears to be protective against ischaemia and to reduce brain injury following a stroke [74, 75]. In human aneurysmal subarachnoid haemorrhage (aSAH) has been shown to counterbalance the blood induced vasoconstriction, hence reduced levels are seen in the perivascular nerves with an increase in vascular smooth muscle cells activity [76]. In human aSAH only CGRP was reduced after a fatal stroke [77] and infusion of CGRP could in vivo in patients reduce the vasospasm [78, 79].
CGRP and co-stored with SP are in the periphery involved in mediating axon-reflex mechanisms and an inflammation response [80,81,82]. CGRP application on the dura mater does not activate or sensitise the ascending trigeminal pathway [83, 84]. Application of CGRP in the temporomandibular joint (TMJ) in rats has been shown to increase expression of mitogen-activated protein kinases (MAPK) in trigeminal ganglia and of c-Fos neurons in the spinal trigeminal nucleus, as well as expression of glial fibrillary acidic protein (GFAP) in astrocytes and OX-42 in microglia [43]. Centrally, CGRP on its own has either no effect on spontaneous neuronal firing or a slow excitatory effect on non-nociceptive neurons [85, 86]. However it can facilitate SP- evoked firing [86, 87]. Intracerebral CGRP may locally induce increase in local cerebral blood flow (Edvinsson, unpublished).
A number of studies have also investigated the actions of CGRP on glutamate excitation given their co-release following stimulation of sensory fibres. It has been shown that CGRP can facilitate, inhibit or cause no changes to glutamate-evoked firing [86,87,88,89]. Interestingly, CGRP was shown to facilitate nociceptive-evoked firing on second order neurons and CGRP antagonists to inhibit nociceptive activity [86,87,88,89].
CGRP has been implicated in migraine pathophysiology as its levels were found to be elevated during a migraine attack in plasma, saliva and CSF samples from patients [70, 90,91,92]. Intravenous infusion of CGRP has been shown to trigger a migraine-like attack without aura in a proportion of sufferers [92], while CGRP antagonists had been used in clinical trials for the treatment of migraine [93,94,95,96]. CGRP antibodies and CGRP receptor antibodies have now been studied in clinical trials for the preventive treatment of frequent episodic and chronic migraine with promising results [97, 98]. These monoclonal antibodies are now approved by the FDA and the EMA. Triptans, 5-HT1B/D receptor agonists and migraine specific treatments, have been also shown to reduce CGRP plasma levels in migraine patients [99] and in cluster headache [100], but not in healthy subjects [101, 102].
Evidence for the importance of CGRP in migraine also comes from experimental animal models. Stimulation of the cat superior sagittal sinus led to increased release of CGRP and VIP levels while there was no change in SP or NPY [103]. When the dura mater was electrically stimulated in rats, it caused dilation of dural blood vessels [104], due to CGRP release from trigeminal sensory nerves that innervate the cranial blood vessels [48] since this effect was abolished by the rat CGRP receptor antagonist CGRP8–37 [104]. Significant attenuation of the neurogenic meningeal vasodilator response is similarly seen with triptans, such as sumatriptan [105]. Intravenous administration of CGRP also causes extracranial dural blood vessel dilation that is similarly abolished by CGRP8–37. CGRP-induced dilation however is not abolished by sumatriptan, indicating that it is likely the triptans act pre-junctionally to prevent CGRP release [106], rather than on the smooth muscles of the blood vessels [105]. In the TCC, CGRP receptor antagonists inhibited trigeminovascular neurons activated by L-glutamate, demonstrating a possible central site of action for CGRP receptor antagonists [88].
3.1.2 Substance P
Substance P (SP) is present in about 10–30% of TG neurons with nearly all fibres that store SP being unmyelinated, arising from small to medium-sized neurons [107,108,109]. All SP-containing TG cells are also immunopositive for CGRP [110, 111], and coexist with the excitatory neurotransmitter glutamate in primary afferents that respond to painful stimulation. SP-positive fibres innervate same structures as CGRP fibres [112,113,114,115]. Like CGRP it can be released both peripherally and centrally [116]. Interestingly, alterations in the expression of SP and its receptor NK1 have been implicated in pathogenesis of sudden perinatal death [117]. Besides SP, the other two peptides of the neurokinin family, Neurokin A and Neurokinin B, are also found in the TG system [118]. These are found in the same neurons as SP and functionally they appear to have slightly less vasodilatory effects.
Peripherally, SP is a potent vasodilator and, along with CGRP, is implicated in the development of neurogenic vasodilatation [119, 120]. Substance P-induced vasodilatation is mediated by the endothelial cell NK1 receptor [121], and depends on NO release [122]. In brain vessels it is in the same way; SP relaxes via an endothelium dependent mechanism and involves NO [48]. In contrast to CGRP, endogenous SP does not appear to contribute to the maintenance of peripheral vascular tone or systemic blood pressure [121]. SP is involved in the axon reflex-mediated vasodilatation and flare reaction, following application of heat or an injury.
Peripherally, SP appears to have an important role in the development of neurogenic inflammation, which is a local inflammatory response to certain types of infection or injury. SP can activate macrophages and mast cells to release inflammatory mediators such as interleukins, arachidonic acid compound, cytokines/chemokines and histamine. The release of these chemical mediators is crucial for inflammatory response [123]. This neuropeptide has been also implicated in the development of neuropathic orofacial pain [124, 125], and its levels, as well as of its receptor NK1-R, are upregulated in TG neurons following an injury [46] or inflammation [126]. Several lines of physiological evidence also indicate that SP has excitatory effects and depolarises TG neurons [127]. SP can release histamine from mast cells of the dura mater in both animal and human material [128].
Centrally, SP is seen in lamina I–II of the C1–2 and in the TNC. Its release on second order neurons following nociceptive peripheral stimulation promotes hyperexcitability and increased sensitivity to pain, by recruiting inflammatory cytokines and inducing glial activation [129]. SP excites second order neurons and facilitates glutamatergic neuronal firing, as well as nociceptive-evoked firing. A number of studies suggest its involvement in the development of central sensitisation by amplifying glutamatergic responses [130]. Studies in NK1 knockout demonstrated its importance in the development of the characteristic “wind up” amplification of second order neurons firing following nociceptive stimulation [131]. NK1 knockout mice also demonstrated attenuated nociceptive behaviour and reductions of Fos-positive neurons in spinal relay centres, indicating a reduction in nociceptive input to the spinal cord [131].
Although SP exists within the trigeminal system, this peptide does not appear to play an important role in the development of nociception. Its direct involvement in facial pain in man has not been explored, although it has been suggested to be involved in trigeminal neuralgia [132]. Based on the ability of SP to be involved in neurogenic inflammation several companies made selective NK-1 antagonists which were studied in migraine, however they all failed to demonstrate any efficacy as an acute or preventive treatment [133,134,135]. Therefore at the end of 1999 this line of research was abandoned because of futility.
3.1.3 PACAP
PACAP was first isolated from rat hypothalamus [136] and shares two-thirds sequence homology with the N-terminal domain of VIP. It occurs in two isoforms: PACAP-27 and PACAP-38 with 27 or 38 amino acids, respectively. Its actions are mediated mainly by the receptor PAR1, which is a member of the family of seven-transmembrane G protein-coupled receptors [41,42,43, 49, 137, 138]. PACAP is also a weak agonist of the two known receptors for VIP termed VPAC1 and VPAC2. Within the TG, PACAP is found in a subpopulation of small- to medium-size TG neurons, which in addition store CGRP [49, 139] (Fig. 1.3). Other structures relevant to the pathogenesis of migraine, such as in trigeminal afferents in the dura mater, the cerebral vessels, the trigeminocervical complex, brainstem nuclei, as well as the sphenopalatine and otic ganglia, also express PACAP [92,93,94]. Its levels appear to be elevated in the trigeminal system following an injury of the trigeminal nerve [140]. However, it is worth pointing out that PACAP together with VIP and NOS are main players in the parasympathetic system in the head. Thus, all these three molecules are very abundant in the otic and sphenopalatine ganglia [141]. Hence, when we analyse the release of these molecules in conjunction it is not always clear from where the peptides originate.

PACAP38 immunoreactivity was observed in many neuronal cell soma (thin arrow) and satellite glial cells (thick arrowhead). In addition, neurons that were not PACAP immunoreactive were also observed (thin arrowhead). Thick arrow points at lipofuscin
PACAP has a broad spectrum of biological effects. In the periphery, PACAP is a potent vasodilator [142]. Application of PACAP in TG neuronal cultures induces neurite growth, a PAC1 mediated effect [143]. PACAP appears to have a small stimulating effect on trigeminal ganglion neurons as shown by slow increase in intracellular free calcium concentration after PACAP1-38 administration on cultured TG neurons [144]. In a recent preclinical study it was found that both VIP and PACAP similarly cause transient vasodilation of meningeal arteries, yet only PACAP was able to trigger a delayed sensitisation of second order trigeminocervical neurons [145, 146]. Stimulation of the trigeminal ganglion was shown to increase PACAP expression in the trigeminal nucleus caudalis, a phenomenon blocked by glutamate receptor antagonists [147].
In migraine patients, elevated levels of plasma PACAP-38 were revealed in the ictal migraine period but not during interictal phase in migraineurs [148,149,150]. Additionally, intravenous infusion of PACAP-38, but not VIP, was shown to trigger migraine-like headaches in migraine patients [151,152,153]. PACAP levels have been also found to be increased in cluster headache patients [154]. Currently, a novel molecule, AMG 301, is a PAC1 receptor selective monoclonal antibody which has been developed for the prevention of migraine, potentially by inhibition of trigeminal autonomic signalling. A phase IIa randomised double-blind placebo controlled study that aims to evaluate the efficacy and safety of AMG 301 in migraine prevention is currently underway.
4 Trigeminal Nerve and Blood Vessel Relation
4.1 Blood Supply of the Trigeminal Nerve
The trigeminal sensory and motor nerve roots exit the brainstem at the anterolateral aspect of the pons. At this level, the arterial supply is provided by a vascular network around the trigeminal nerve root, formed by 2–6 trigeminal arteries [155, 156]. The parent vessels of the vessels supplying the trigeminal nerve include the superior cerebellar artery, the posterolateral, superolateral and inferolateral arteries; and the anterior inferior cerebellar artery, all branches of the basilar artery.
Blood vessel compression of the trigeminal nerve is a common cause of trigeminal neuralgia, an extremely severe facial pain disorder. The superior cerebellar artery is responsible for most (60–90%) cases of neurovascular compression, while the anteroinferior cerebellar artery and basilar artery may cause trigeminal nerve compression [157, 158]. Trigeminal nerve compression is thought to induce discharges along nociceptive fibres that could be responsible for the development of trigeminal neuralgia [159]. Histopathologic studies have revealed focal axonal degeneration and demyelination in postoperative specimens collected from patients with TN due to neurovascular compression [160,161,162,163]. The reference-standard treatment for refractive TN caused by neurovascular compression is microvascular decompression [164].
4.2 Perivascular Nerve Fibres Innervating the Cranial Circulation
As discussed above, nearly all neuropeptides released in the periphery by trigeminal fibres have vasodilatory properties. Vasomotor control in the areas innervated by the trigeminal nerve is also the result of a balance between the sympathetic and parasympathetic fibres innervating the cranial circulation [165].
4.2.1 Sympathetic System
Sympathetic nerve fibres arising from the superior cervical ganglion in the thoracic spinal cord supply the cranial vasculature with neuropeptide Y (NPY), noradrenaline (NA) and ATP [70, 166, 167]. NPY and NA both cause vasoconstriction of the cerebral circulation and are secreted at rest; they are therefore thought to provide a tonic vasoconstriction. All these molecules have been studied extensively in human cerebral, middle meningeal and temporal arteries, and they produce vasoconstriction of different magnitude. In some vessels NPY can act as a potentiator of constriction induced by other agents [62, 166].
4.2.2 Parasympathetic System
Parasympathetic nerve fibres arising from the sphenopalatine and otic ganglia as well as the carotid miniganglia [166] supply the cranial vasculature with VIP, peptide histidine isoleucine (PHI), acetylcholinesterase (AChE), peptide histidine methionine 27 (PHM), PACAP as well as other VIP-related peptides [168]. The parasympathetic innervation of the cranial circulation is a vasodilatory system, with VIP, acetylcholine and PHM all being potent vasodilators in human cranial arteries. It is believed to become activated during attacks in trigeminal autonomic cephalalgias (TACs).
5 Cytoarchitecture of the Trigeminocervical Complex and Somatotopical Organisation
The sensory pseudounipolar neurons of the trigeminal and upper cervical ganglia that innervate the pain producing cranial structures project centrally and terminate on second order neurons in the trigeminocervical complex (TCC), which extends from the rostral pons to the upper cervical spinal cord levels and consists of a complex of subnuclei divided into the principal trigeminal nuclei (Vp), at which a major part of the trigeminal nerve terminates, and the spinal trigeminal nucleus (Vsp) [169]. The Vsp is further divided into three subnuclei; the nucleus oralis (Vo), interpolaris (Vi) and caudalis (Vc) arranged in a rostrocaudal manner [170, 171]. The TCC extends from the trigeminal nucleus caudalis to the segments of C2–C3 in the rat, cat and monkey [172]. The total complex of trigeminal nuclei that includes the mesencephalic nucleus where the cell bodies of the trigeminal motor neurons lie is known as the trigeminal brainstem nuclear complex.
The subnucleus caudalis (Vc), also known as the trigeminal nucleus caudalis (TNC) or medullary dorsal horn (MDH) extends from the obex to the cervical spinal cord and is analogous to the dorsal horn of the spinal cord [173]. It is composed of separate layers similar in appearance to the spinal cord dorsal horn with the outermost layer, the subnucleus marginalis corresponding to lamina I. Ventral to this lies the subnucleus gelatinosus (lamina II), and the subnucleus magnocellularis which corresponds to laminae III and IV.
The TCC is organised somatotopically, with the three trigeminal divisions being represented in a sequence from ventrolateral to dorsomedial direction [174].
-
Mandibular afferents are mainly represented on the dorsal part of each subnucleus.
-
Ophthalmic afferents terminate ventral in the trigeminal subnuclei or on the ventrolateral aspect of the TNC [175].
-
Maxillary afferents terminate between the mandibular and ophthalmic representations in the trigeminal subnuclei.
Given the strong somatotopic organisation of the trigeminal system both in the TG and in the trigeminal brainstem nuclear complex, the differential diagnosis of lesions causing trigeminal neuropathy can be quite varied and is best examined by location along the trigeminal pathway [6].
Primary afferent trigeminal sensory fibres converge on second order neurons in laminae I–VI, which constitute the dorsal horn, and on second order neurons in the TNC. According to their responses when activated by different stimuli, these second order neurons have been classified into three categories [23] and all three have been identified in the TCC [176]:
-
1.
Nociceptive-specific (NS) neurons are silent at rest and become activated in response to high intensity, noxious stimuli and receive inputs from Aδ- and C-fibres.
-
2.
Non-nociceptive low-threshold (LT) neurons that respond to innocuous stimulation only.
-
3.
Wide-dynamic range (WDR) neurons exhibit a dynamic response over a broad stimulus range eliciting an incremental response to both innocuous and noxious stimuli. WDR neurons also receive considerable convergent inputs from extracranial cutaneous and intracranial visceral structures and may respond to C-, Aδ- and Aβ- fibres.
The Vp mainly receives touch and pressure impulses from the entire oral area, whereas the Vsp receives information on pain, temperature and pressure from the entire trigeminal area. An organisation pattern of cutaneous, primary afferent inputs to the dorsal horn of the spinal cord has been suggested with C-fibres projecting in lamina I, outer lamina II and laminae VI and X, Aδ-fibres terminating in lamina I, outer lamina II and laminae III-V, and Aβ-fibres terminating in laminae II(inner)-VI and X. This is however not a strict organisation, as it does not quantitatively differentiate between various laminae as concerns primary afferent input, and the cell types (NS, WDR or LT) are qualitatively rather than quantitatively represented [23, 176].
Earlier studies have shown that the spinal trigeminal nucleus, especially the TNC, has an important role in the mediation of pain and temperature sensations from the head and facial regions [177, 178]. Sensory inputs from the dural blood vessels (such as the superior sagittal sinus and the middle meningeal artery), synapse on second order neurons in the TCC and nociceptive electrical and mechanical stimulation of the superior sagittal sinus result in Fos expression in this nuclei complex [179,180,181]. The sensory central projections of the trigeminal fibres innervating superficial temporal artery and the superior sagittal sinus in rats terminate in the TNC, the trigeminal nucleus interpolaris and the dorsal horn in the segment C1–C3 [182, 183]. Stimulation of the superior sagittal sinus or certain other dural components increases neuronal activity in the TNC [21, 22, 184, 185] and most of these also have facial receptive fields located in the ophthalmic division [184]. Electrical stimulation of the superior sagittal sinus causes increased metabolic activity and blood flow in the TNC and in C1 and C2 of the spinal dorsal horn [186]. CGRP-like immunoreactivity, which represents CGRP carrying afferents from the trigeminal ganglion [187], is abundant in the TCC and stimulation of the trigeminal ganglion causes increased release of CGRP and SP [188].
Clinical correlates, that indicate an important role of the brainstem in migraine, come from imaging studies, which showed activation of the pons and brainstem during migraine attacks [189], and this activation is migraine specific [190]. Both experimental and clinical evidence suggest that abnormal neuronal modulation at the level of the brainstem is clearly implicated in migraine pathophysiology [191].
5.1 Convergence of Trigeminal and Cervical Fibres in the Trigeminocervical Complex
An important aspect of trigeminal nociception is the convergence of trigeminal fibres and of cervical fibres arising from cervical DRGs on second order neurons in the TCC, particularly in the C1 and C2 regions [22, 192, 193]. The greater occipital nerve (GON) arises from fibres of the dorsal primary ramus of the C2 nerve and to a lesser extent fibres from the C3 nerve. It supplies the medial portion of the posterior scalp as far anterior as the vertex. The lesser occipital nerve arises from the ventral primary rami of C2 and C3 nerves. The lesser occipital nerve divides into cutaneous branches that innervate the lateral portion of the posterior scalp and the cranial surface of the pinna of the ear. The GON projects centrally, mainly in C2 and C3 spinal levels, but also to C1 and Vsp and has a somatotopic organisation [192, 194, 195]. As early as in 1961, Kerr and Olafson [196] showed that stimulation of trigeminal and occipital fibres can modulate the same second order neurons. It is estimated that about 40% of second order neurons receiving trigeminal inputs also receive occipital fibres. Stimulation of occipital fibres appears to excite, inhibit second order neurons or even to induce brief bursts followed by inhibition, inhibition followed by rebound or sensitisation [22, 193, 196].
From a clinical point of view the finding of convergence on second order neurons is of considerable interest as a possible mechanism for the diffuse spread of headache to occipital regions often seen in migraine and of the headache in the trigeminal territory that often accompanies occipital neuralgia as a form of referred pain, a similar functional relationship to the convergence of somatic and visceral afferent fibres seen in the dorsal horn of lower spinal levels. From a therapeutic point of view, GON block is often used as a preventive treatment in migraine and cluster headache [197,198,199]. Weiner and Reed [200] first reported a series of cases of intractable occipital neuralgia responding to occipital nerve stimulation (ONS). ONS has been since used as a treatment of chronic migraine [201,202,203] and chronic cluster headache [204,205,206] with mixed results.
6 Trigemino-Autonomic Reflex: Relevance to Trigeminal Autonomic Cephalalgias
The trigeminal-autonomic reflex refers to the anatomical and physiological relationship of the afferent trigeminal nerve and the efferent pathway that arises in the superior salivatory nucleus (SSN) [207, 208]. The trigemino-autonomic reflex has been implicated in the pathophysiology of trigeminal autonomic cephalalgias (TACs), which consist of cluster headache, paroxysmal hemicrania and SUNCT/SUNA (Short-lasting Unilateral Neuralgiform headache attacks with Conjunctival injection and Tearing/cranial Autonomic features), and by definition they have cranial autonomic symptoms, either parasympathetic activation: lacrimation, conjunctival injection, nasal symptoms, aural symptoms, peri-orbital swelling; or sympatholytic manifestations: ptosis, miosis. These symptoms are also seen in some migraine patients and even manifest in the premonitory phase [209, 210]. Trigeminal, sympathetic and parasympathetic fibres are well known to innervate cranial structures and to control together vessel dilatation [10, 14, 211, 212].
As discussed above, trigeminal fibres project to the trigeminocervical complex and second order neurons give rise to the trigeminothalamic pathway. Part of the trigeminothalamic projections synapse in the SSN [213, 214]; these appear to involve mainly projections from second order neurons that receive inputs from the ophthalmic division of the Vn [215]. Fibres from the SSN pass via the facial nerve (VII cranial nerve) to synapse post-ganglionic parasympathetic neurons in the ganglion (SPG). SPG neurons express the vasoactive neuropeptides VIP and PACAP, as well as nitric oxide synthase [216,217,218] and they also drive cerebral vasodilation following activation of the SPG or the facial nerve [219, 220]. Interestingly, increased levels of VIP and PACAP [100, 154], potentially of SPG origin, have been found in cluster headache patients, along with increased levels of CGRP which is thought to be of trigeminal origin [221, 222]. This SSN-SPG pathway is modulated from the hypothalamus [223,224,225]. Deep brain stimulation in the hypothalamic region has been used as a treatment in refractory chronic cluster headache with good results [226], while currently SPG stimulation is used as an abortive treatment of episodic cluster attacks [227, 228]. Which system becomes first activated in TACs is not yet clear, however, the autonomic dysregulation as seen in these primary headache disorders might be due to hypothalamic disturbances [229, 230], given the consistent hypothalamic activation seen in brain imaging in cluster attacks [231,232,233], as well as the efficacy of DBS in the hypothalamic area [226]. Other theories that include nociceptive trigeminal discharges driving autonomic activation as a secondary phenomenon, or perivascular oedema due to trigeminal-parasympathetic over activity that also compromise sympathetic fibres have been discussed [215, 225].
7 Ascending and Descending Pathways of the Trigeminal System
7.1 Ascending Pathways
The majority of the secondary neurons in the TCC decussate at the level of the medulla and travel up the brainstem through the ventral trigeminal tract, which ascends in close relationship with the contralateral medial lemniscus, and carry sensory information from the face and the meninges to higher brain areas. Their role is not only to facilitate the perception and detection of noxious stimuli, but also to communicate with cognitive circuits which control mood associated with pain, the attention to and memory of pain as well as the tolerance of pain [234].
The trigeminothalamic tract (also called the quintothalamic tract) transmits information from the trigeminocervical complex and synapse mainly at the ventral posteromedial nucleus (VPM) of the contralateral thalamus. Ipsilateral projections have been reported in some species, associated with the spinothalamic tract carrying sensory information from the body [235]. In addition to the sensory thalamus, neurons from the trigeminocervical complex also project to a number of diencephalic and brainstem areas involved in the regulation of autonomic, endocrine, affective and motor functions. Of them, the trigeminohypothalamic tract is not well studied, however, it appears to be formed by neurons located bilaterally in the TCC and their axons synapse mainly to the lateral preoptic, anterior, lateral, perifornical and caudal hypothalamic nuclei [236]. Of interest TCC neurons projecting to the hypothalamus appear to be nociceptive specific [236]. Although the structures that receive axons from the TCC have not been well studied, it is believed to act as the mediators of activation of descending efferent projections to the TCC that modulate nociceptive information within this relay centre.
7.2 Glutamate in the Ascending Trigeminovascular and Trigeminothalamic Pathways
Glutamate is the excitatory neurotransmitter that drives activation of the ascending trigeminovascular and trigeminothalamic pathways [237]. Trigeminal pain-relay structures, including the trigeminal ganglion, TCC and thalamus, contain glutamate-positive neurons [238, 239]. Glutamate activates neurons in the trigeminal nucleus caudalis [240] by acting both on ionotropic and metabotropic GluRs [241], and it is involved in signaling from trigeminothalamic tract and hypothalamic pathways and corticothalamic afferents [242]. In vivo studies using microdialysis and blood flow measurements demonstrated increased levels of glutamate in the TCC during and post stimulation of dural structures and following noxious stimulation along the trigeminal nerve [243,244,245]. Glutamate is released from trigeminal ganglion neurons along with CGRP by a calcium channels depended mechanism [64]. It has been further demonstrated that the majority of glutamatergic neurons in the trigeminal ganglia carry 5-HT1B/D/F receptors, which could possibly modulate glutamate release [246]. Glutamate plays additionally a crucial role in the transmission of nociceptive information in the sensory thalamus. Extracellular levels of glutamate, measured by microdialysis, are increased in the rat VPM following experimentally produced pain [247] and it triggers post-synaptic excitatory potentials by activating multiple GluRs [248].
The presence of glutamate in the transmission of sensory information implicates the involvement of GluRs that modulate glutamate responses, in key CNS areas involved in migraine pathophysiology. Each of the ionotropic and metabotropic GluRs has been identified in the superficial laminae of the trigeminal nucleus caudalis [241] and the sensory thalamus among other pain related areas of the rat brain [249]. Expression of NMDA, kainate and mGluRs messenger RNA has been found in trigeminal ganglion neurons [250,251,252].
Migraineurs have elevated levels of glutamate [253, 254] and glutamine [255] in the cerebrospinal fluid (CSF) compared to controls, suggesting an excess of neuroexcitatory amino acids in the CNS. A correlation between the glutamate levels and the mean headache scores has been reported [253], suggesting a persistent neuronal hyperexcitability that becomes heightened during an attack in migraineurs. In support of this hypothesis is the finding that migraineurs exhibit cutaneous allodynia during an attack, and thus exhibit signs of the development of central sensitisation [256]. Central sensitisation following peripheral sensory stimulation involves glutamate release and in some part is glutamate receptor activation mediated [257]. Evidence from animals support that increased glutamate levels parallel changes in sensory thresholds of facial receptive fields, as recorded from secondary neurons in the TCC [245]. This further supports the involvement of glutamate in the development of cutaneous allodynia and central sensitization, as seen in migraine patients.
7.3 Descending Pathways
Anti-nociceptive and modulatory networks are only discussed briefly here and the readers can find more extensive reviews on descending efferent connections to the TCC in [258,259,260].
The TCC has been shown to receive efferent monoaminergic, enkephalinergic, dopaminergic and other peptidergic projections from brainstem, pons and midbrain areas [259]. These include the nucleus raphe magnus and the reticular formation that project to the outer laminae of the TCC [261], the dopaminergic hypothalamic nucleus A11 [262], and the periaqueductal gray [263].
Brain imaging studies in migraine patients showed increased perfusion in the rostral brainstem and cingulate cortex during spontaneous and triggered migraine attacks. The increased perfusion was further shown to even after pharmacological intervention with headache relief. This gave rise to the theory that brainstem activation is more than a simple reactive response to pain [189, 190, 264, 265]. The area suspected to be involved in this increased perfusion studies is the PAG. The PAG is an anti-nociceptive modulatory structure as shown in many animal models of pain [263]. High resolution magnetic resonance imaging (MRI) of the PAG has identified a possible impairment of iron homeostasis, which can be indicative of a neuronal dysfunction in both migraine with and without aura [266]. The brain stem activation seen during migraine attacks is thought to be specific to migraine pathophysiology, as it is not seen in experimentally induced or atypical facial pain [267, 268], acute cluster headache [269] and short-lasting, unilateral, neuralgiform headache attacks with conjunctival injection and tearing (SUNCT) [270].
References
Crossman AR, Neary D. Cranial nerves and cranial nerve nuclei. In: Crossman AR, Neary D, editors. Neuroanatomy. Manchester: Elsevier; 2000. p. 103–16.
Shankland WE. The trigeminal nerve. Part I: an over-view. Cranio. 2000;18(4):238–48.
Schaltenbrand G, Walker AE. Anatomy of the brainstem. In: Stereotaxy of the human brain. New York: Thieme Medical Publishers; 1997. p. 36–59.
Tandrup T. Are the neurons in the dorsal root ganglion pseudounipolar? A comparison of the number of neurons and number of myelinated and unmyelinated fibres in the dorsal root. J Comp Neurol. 1995;357(3):341–7.
Hanani M. Satellite glial cells: more than just ‘rings around the neuron’. Neuron Glia Biol. 2010;6(1):1–2.
Go JL, Kim PE, Zee CS. The trigeminal nerve. Semin Ultrasound CT MR. 2001;22(6):502–20.
Shankland WE. The trigeminal nerve. Part II: the ophthalmic division. Cranio. 2001;19(1):8–12.
Messlinger K, Dostrovsky JO, Strassman AM. Anatomy and physiology of head pain. In: Olesen J, et al., editors. The headaches. Philadelphia: Lippincott Williams & Wilkins; 2006. p. 95–109.
Uddman R, Hara H, Edvinsson L. Neuronal pathways to the rat middle meningeal artery revealed by retrograde tracing and immunocytochemistry. J Auton Nerv Syst. 1989;26(1):69–75.
Uddman R, Edvinsson L. Neuropeptides in the cerebral circulation. Cerebrovasc Brain Metab Rev. 1989;1(3):230–52.
Edvinsson L, Hara H, Uddman R. Retrograde tracing of nerve fibers to the rat middle cerebral artery with true blue: colocalization with different peptides. J Cereb Blood Flow Metab. 1989;9(2):212–8.
Mayberg MR, Zervas NT, Moskowitz MA. Trigeminal projections to supratentorial pial and dural blood vessels in cats demonstrated by horseradish peroxidase histochemistry. J Comp Neurol. 1984;223(1):46–56.
Steiger HJ, Meakin CJ. The meningeal representation in the trigeminal ganglion--an experimental study in the cat. Headache. 1984;24(6):305–9.
Uddman R, Edvinsson L, Hara H. Axonal tracing of autonomic nerve fibers to the superficial temporal artery in the rat. Cell Tissue Res. 1989;256(3):559–65.
Hara H, et al. Acetylcholine and vasoactive intestinal peptide in cerebral blood vessels: effect of extirpation of the sphenopalatine ganglion. J Cereb Blood Flow Metab. 1989;9(2):204–11.
David OJ. Anatomy of the trigeminal nerve. Rev Fac Odontol Univ Nac (Cordoba). 1977;9(2):95–121.
Arvidsson J, Pfaller K, Gmeiner S. The ganglionic origins and central projections of primary sensory neurons innervating the upper and lower lips in the rat. Somatosens Mot Res. 1992;9(3):199–209.
Aigner M, et al. Somatotopic organization of primary afferent perikarya of the guinea-pig extraocular muscles in the trigeminal ganglion: a post-mortem DiI-tracing study. Exp Eye Res. 2000;70(4):411–8.
Marfurt CF. The somatotopic organization of the cat trigeminal ganglion as determined by the horseradish peroxidase technique. Anat Rec. 1981;201(1):105–18.
Schepelmann K, et al. Response properties of trigeminal brain stem neurons with input from dura mater encephali in the rat. Neuroscience. 1999;90(2):543–54.
Strassman A, et al. Response of brainstem trigeminal neurons to electrical stimulation of the dura. Brain Res. 1986;379(2):242–50.
Bartsch T, Goadsby PJ. Increased responses in trigeminocervical nociceptive neurons to cervical input after stimulation of the dura mater. Brain. 2003;126(Pt 8):1801–13.
Millan MJ. The induction of pain: an integrative review. Prog Neurobiol. 1999;57(1):1–164.
Eftekhari S, et al. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain. 2013;14(11):1289–303.
Woolf CJ, Ma Q. Nociceptors--noxious stimulus detectors. Neuron. 2007;55(3):353–64.
Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. 2006;68:619–47.
Sousa-Valente J, et al. Transient receptor potential ion channels in primary sensory neurons as targets for novel analgesics. Br J Pharmacol. 2014;171(10):2508–27.
Dussor G, et al. Targeting TRP channels for novel migraine therapeutics. ACS Chem Neurosci. 2014;5(11):1085–96.
Bron R, et al. Piezo2 expression in corneal afferent neurons. J Comp Neurol. 2014;522(13):2967–79.
Huang D, et al. Expression of the transient receptor potential channels TRPV1, TRPA1 and TRPM8 in mouse trigeminal primary afferent neurons innervating the dura. Mol Pain. 2012;8:66.
Marone IM, et al. TRPA1/NOX in the soma of trigeminal ganglion neurons mediates migraine-related pain of glyceryl trinitrate in mice. Brain. 2018;141(8):2312–28.
Kim YS, et al. Expression of transient receptor potential ankyrin 1 (TRPA1) in the rat trigeminal sensory afferents and spinal dorsal horn. J Comp Neurol. 2010;518(5):687–98.
Yamamoto Y, Hatakeyama T, Taniguchi K. Immunohistochemical colocalization of TREK-1, TREK-2 and TRAAK with TRP channels in the trigeminal ganglion cells. Neurosci Lett. 2009;454(2):129–33.
Holland PR, et al. Acid-sensing ion channel 1: a novel therapeutic target for migraine with aura. Ann Neurol. 2012;72(4):559–63.
Fu H, et al. Acid-sensing ion channels in trigeminal ganglion neurons innervating the orofacial region contribute to orofacial inflammatory pain. Clin Exp Pharmacol Physiol. 2016;43(2):193–202.
Goadsby PJ, Classey JD. Evidence for serotonin (5-HT)1B, 5-HT1D and 5-HT1F receptor inhibitory effects on trigeminal neurons with craniovascular input. Neuroscience. 2003;122(2):491–8.
Ferrari MD, et al. Oral triptans (serotonin 5-HT(1B/1D) agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet. 2001;358(9294):1668–75.
Chasman DI, et al. Selectivity in genetic association with sub-classified migraine in women. PLoS Genet. 2014;10(5):e1004366.
Chasman DI, et al. Genome-wide association study reveals three susceptibility loci for common migraine in the general population. Nat Genet. 2011;43(7):695–8.
Esserlind AL, et al. Replication and meta-analysis of common variants identifies a genome-wide significant locus in migraine. Eur J Neurol. 2013;20(5):765–72.
Freilinger T, et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat Genet. 2012;44(7):777–82.
Ligthart L, et al. Meta-analysis of genome-wide association for migraine in six population-based European cohorts. Eur J Hum Genet. 2011;19(8):901–7.
Cady RJ, et al. Calcitonin gene-related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain. 2011;7:94.
Lazarov NE. Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog Neurobiol. 2002;66(1):19–59.
Goto T, et al. Neuropeptides and ATP signaling in the trigeminal ganglion. Jpn Dent Sci Rev. 2017;53(4):117–24.
Gunjigake KK, et al. Correlation between the appearance of neuropeptides in the rat trigeminal ganglion and reinnervation of the healing root socket after tooth extraction. Acta Histochem Cytochem. 2006;39(3):69–77.
Kimberly CL, Byers MR. Inflammation of rat molar pulp and periodontium causes increased calcitonin gene-related peptide and axonal sprouting. Anat Rec. 1988;222(3):289–300.
Uddman R, et al. Innervation of the feline cerebral vasculature by nerve fibers containing calcitonin gene-related peptide: trigeminal origin and co-existence with substance P. Neurosci Lett. 1985;62(1):131–6.
Tajti J, et al. Messenger molecules and receptor mRNA in the human trigeminal ganglion. J Auton Nerv Syst. 1999;76(2–3):176–83.
Eftekhari S, et al. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. Neuroscience. 2010;169(2):683–96.
Lawson SN, Crepps B, Perl ER. Calcitonin gene-related peptide immunoreactivity and afferent receptive properties of dorsal root ganglion neurones in guinea-pigs. J Physiol. 2002;540(Pt 3):989–1002.
Lawson SN. Phenotype and function of somatic primary afferent nociceptive neurones with C-, Adelta- or Aalpha/beta-Fibres. Exp Physiol. 2002;87(2):239–44.
Reuss S, Riemann R, Vollrath L. Substance P- and calcitonin gene-related peptide-like immunoreactive neurons in the rat trigeminal ganglion--with special reference to meningeal and pineal innervation. Acta Histochem. 1992;92(1):104–9.
Wahlestedt C, et al. Calcitonin gene-related peptide in the eye: release by sensory nerve stimulation and effects associated with neurogenic inflammation. Regul Pept. 1986;16(2):107–15.
Tsai SH, et al. Cerebral arterial innervation by nerve fibers containing calcitonin gene-related peptide (CGRP): I. distribution and origin of CGRP perivascular innervation in the rat. J Comp Neurol. 1988;271(3):435–44.
Edvinsson L. The Trigeminovascular pathway: role of CGRP and CGRP receptors in migraine. Headache. 2017;57(Suppl 2):47–55.
Silverman JD, Kruger L. An interpretation of dental innervation based upon the pattern of calcitonin gene-related peptide (CGRP)-immunoreactive thin sensory axons. Somatosens Res. 1987;5(2):157–75.
Luthman J, et al. Immunohistochemical studies of the neurochemical markers, CGRP, enkephalin, galanin, gamma-MSH, NPY, PHI, proctolin, PTH, somatostatin, SP, VIP, tyrosine hydroxylase and neurofilament in nerves and cells of the human attached gingiva. Arch Oral Biol. 1988;33(3):149–58.
Takeda N, et al. Neurogenic inflammation in nasal allergy: histochemical and pharmacological studies in guinea pigs. A review. Acta Otolaryngol Suppl. 1993;501:21–4.
Blixt FW, et al. Distribution of CGRP and its receptor components CLR and RAMP1 in the rat retina. Exp Eye Res. 2017;161:124–31.
Jansen I, et al. Localization and effects of neuropeptide Y, vasoactive intestinal polypeptide, substance P, and calcitonin gene-related peptide in human temporal arteries. Ann Neurol. 1986;20(4):496–501.
Jansen I, et al. Distribution and effects of neuropeptide Y, vasoactive intestinal peptide, substance P, and calcitonin gene-related peptide in human middle meningeal arteries: comparison with cerebral and temporal arteries. Peptides. 1992;13(3):527–36.
Edvinsson L, et al. Perivascular peptides relax cerebral arteries concomitant with stimulation of cyclic adenosine monophosphate accumulation or release of an endothelium-derived relaxing factor in the cat. Neurosci Lett. 1985;58(2):213–7.
Xiao Y, Richter JA, Hurley JH. Release of glutamate and CGRP from trigeminal ganglion neurons: role of calcium channels and 5-HT1 receptor signaling. Mol Pain. 2008;4:12.
Meng J, et al. Synaptobrevin I mediates exocytosis of CGRP from sensory neurons and inhibition by botulinum toxins reflects their anti-nociceptive potential. J Cell Sci. 2007;120(Pt 16):2864–74.
Frederiksen SD, et al. Expression of pituitary adenylate cyclase-activating peptide, calcitonin gene-related peptide and headache targets in the trigeminal ganglia of rats and humans. Neuroscience. 2018;393:319–32.
Andreou AP, et al. Prospective real-world analysis of OnabotulinumtoxinA in chronic migraine post-National Institute for Health and Care Excellence UK technology appraisal. Eur J Neurol. 2018;25(8):1069–e83.
Trasforini G, et al. Circadian profile of plasma calcitonin gene-related peptide in healthy man. J Clin Endocrinol Metab. 1991;73(5):945–51.
Brain SD, et al. Calcitonin gene-related peptide is a potent vasodilator. Nature. 1985;313(5997):54–6.
Edvinsson L, Goadsby PJ. Neuropeptides in the cerebral circulation: relevance to headache. Cephalalgia. 1995;15(4):272–6.
Edvinsson L, Uddman R. Adrenergic, cholinergic and peptidergic nerve fibres in dura mater--involvement in headache? Cephalalgia. 1981;1(4):175–9.
Edvinsson L, et al. Calcitonin gene-related peptide and cerebral blood vessels: distribution and vasomotor effects. J Cereb Blood Flow Metab. 1987;7(6):720–8.
Bull HA, et al. Neuropeptides induce release of nitric oxide from human dermal microvascular endothelial cells. J Invest Dermatol. 1996;106(4):655–60.
Chai W, et al. The role of calcitonin gene-related peptide (CGRP) in ischemic preconditioning in isolated rat hearts. Eur J Pharmacol. 2006;531(1–3):246–53.
MaassenVanDenBrink A, et al. Wiping out CGRP: potential cardiovascular risks. Trends Pharmacol Sci. 2016;37(9):779–88.
Edvinsson L, et al. Involvement of perivascular sensory fibers in the pathophysiology of cerebral vasospasm following subarachnoid hemorrhage. J Cereb Blood Flow Metab. 1990;10(5):602–7.
Edvinsson L, et al. Reduced levels of calcitonin gene-related peptide-like immunoreactivity in human brain vessels after subarachnoid haemorrhage. Neurosci Lett. 1991;121(1–2):151–4.
Juul R, et al. Calcitonin gene-related peptide-LI in subarachnoid haemorrhage in man. Signs of activation of the trigemino-cerebrovascular system? Br J Neurosurg. 1990;4(3):171–9.
Juul R, et al. Alterations in perivascular dilatory neuropeptides (CGRP, SP, VIP) in the external jugular vein and in the cerebrospinal fluid following subarachnoid haemorrhage in man. Acta Neurochir. 1995;132(1–3):32–41.
Couture R, Cuello AC. Studies on the trigeminal antidromic vasodilatation and plasma extravasation in the rat. J Physiol. 1984;346:273–85.
Foreman JC. Substance P and calcitonin gene-related peptide: effects on mast cells and in human skin. Int Arch Allergy Appl Immunol. 1987;82(3–4):366–71.
Foreman JC. Peptides and neurogenic inflammation. Br Med Bull. 1987;43(2):386–400.
Covasala O, et al. Calcitonin gene-related peptide receptors in rat trigeminal ganglion do not control spinal trigeminal activity. J Neurophysiol. 2012;108(2):431–40.
Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;58(5):698–705.
Miletic V, Tan H. Iontophoretic application of calcitonin gene-related peptide produces a slow and prolonged excitation of neurons in the cat lumbar dorsal horn. Brain Res. 1988;446(1):169–72.
Biella G, et al. Facilitatory role of calcitonin gene-related peptide (CGRP) on excitation induced by substance P (SP) and noxious stimuli in rat spinal dorsal horn neurons. An iontophoretic study in vivo. Brain Res. 1991;559(2):352–6.
Leem JW, et al. Effects of iontophoretically applied substance P, calcitonin gene-related peptide on excitability of dorsal horn neurones in rats. Yonsei Med J. 2001;42(1):74–83.
Storer RJ, Akerman S, Goadsby PJ. Calcitonin gene-related peptide (CGRP) modulates nociceptive trigeminovascular transmission in the cat. Br J Pharmacol. 2004;142(7):1171–81.
Yu Y, Lundeberg T, Yu LC. Role of calcitonin gene-related peptide and its antagonist on the evoked discharge frequency of wide dynamic range neurons in the dorsal horn of the spinal cord in rats. Regul Pept. 2002;103(1):23–7.
Bellamy JL, Cady RK, Durham PL. Salivary levels of CGRP and VIP in rhinosinusitis and migraine patients. Headache. 2006;46(1):24–33.
Juhasz G, et al. NO-induced migraine attack: strong increase in plasma calcitonin gene-related peptide (CGRP) concentration and negative correlation with platelet serotonin release. Pain. 2003;106(3):461–70.
Hansen JM, et al. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. Cephalalgia. 2010;30(10):1179–86.
Voss T, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36(9):887–98.
Yao G, et al. Therapeutic effects and safety of olcegepant and telcagepant for migraine: a meta-analysis. Neural Regen Res. 2013;8(10):938–47.
Bramanti P, et al. Ictal and interictal hypoactivation of the occipital cortex in migraine with aura. A neuroimaging and electrophysiological study. Funct Neurol. 2005;20(4):169–71.
Nichols PL, et al. Potent oxadiazole CGRP receptor antagonists for the potential treatment of migraine. Bioorg Med Chem Lett. 2010;20(4):1368–72.
Israel H, Neeb L, Reuter U. CGRP monoclonal antibodies for the preventative treatment of migraine. Curr Pain Headache Rep. 2018;22(5):38.
Edvinsson L. CGRP receptor antagonists and antibodies against CGRP and its receptor in migraine treatment. Br J Clin Pharmacol. 2015;80(2):193–9.
Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33(1):48–56.
Goadsby PJ, Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies. Brain. 1994;117(Pt 3):427–34.
Juhasz G, et al. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia. 2005;25(3):179–83.
Hansen JM, et al. Sumatriptan does not change calcitonin gene-related peptide in the cephalic and extracephalic circulation in healthy volunteers. J Headache Pain. 2009;10(2):85–91.
Zagami AS, Goadsby PJ, Edvinsson L. Stimulation of the superior sagittal sinus in the cat causes release of vasoactive peptides. Neuropeptides. 1990;16(2):69–75.
Williamson DJ, et al. Intravital microscope studies on the effects of neurokinin agonists and calcitonin gene-related peptide on dural vessel diameter in the anaesthetized rat. Cephalalgia. 1997;17(4):518–24.
Williamson DJ, et al. Sumatriptan inhibits neurogenic vasodilation of dural blood vessels in the anaesthetized rat--intravital microscope studies. Cephalalgia. 1997;17(4):525–31.
Goadsby PJ, Edvinsson L. Joint 1994 Wolff award presentation. Peripheral and central trigeminovascular activation in cat is blocked by the serotonin (5HT)-1D receptor agonist 311C90. Headache. 1994;34(7):394–9.
Bae JY, et al. Quantitative analysis of afferents expressing substance P, calcitonin gene-related peptide, isolectin B4, neurofilament 200, and Peripherin in the sensory root of the rat trigeminal ganglion. J Comp Neurol. 2015;523(1):126–38.
Del Fiacco M, et al. Substance P-like immunoreactivity in the human trigeminal ganglion. Neurosci Lett. 1990;110(1–2):16–21.
Lehtosalo JI, et al. Substance P-like immunoreactivity in the trigeminal ganglion. A fluorescence, light and electron microscope study. Histochemistry. 1984;80(5):421–7.
Skofitsch G, Jacobowitz DM. Calcitonin gene-related peptide coexists with substance P in capsaicin sensitive neurons and sensory ganglia of the rat. Peptides. 1985;6(4):747–54.
Lee Y, et al. Coexistence of calcitonin gene-related peptide and substance P-like peptide in single cells of the trigeminal ganglion of the rat: immunohistochemical analysis. Brain Res. 1985;330(1):194–6.
Liu-Chen LY, Han DH, Moskowitz MA. Pia arachnoid contains substance P originating from trigeminal neurons. Neuroscience. 1983;9(4):803–8.
Lehtosalo JI. Substance P-like immunoreactive trigeminal ganglion cells supplying the cornea. Histochemistry. 1984;80(3):273–6.
Mingomataj E, et al. Trigeminal nasal-specific neurons respond to nerve growth factor with substance-P biosynthesis. Clin Exp Allergy. 2008;38(7):1203–11.
Saito K, Liu-Chen LY, Moskowitz MA. Substance P-like immunoreactivity in rat forebrain leptomeninges and cerebral vessels originates from the trigeminal but not sympathetic ganglia. Brain Res. 1987;403(1):66–71.
Cuello AC, Del Fiacco M, Paxinos G. The central and peripheral ends of the substance P-containing sensory neurones in the rat trigeminal system. Brain Res. 1978;152(3):499–500.
Mehboob R. Substance P/neurokinin 1 and trigeminal system: a possible link to the pathogenesis in sudden perinatal deaths. Front Neurol. 2017;8:82.
Edvinsson L, et al. Neurokinin A in cerebral vessels: characterization, localization and effects in vitro. Regul Pept. 1988;20(3):181–97.
McCarthy PW, Lawson SN. Cell type and conduction velocity of rat primary sensory neurons with substance P-like immunoreactivity. Neuroscience. 1989;28(3):745–53.
McCarthy PW, Lawson SN. Cell type and conduction velocity of rat primary sensory neurons with calcitonin gene-related peptide-like immunoreactivity. Neuroscience. 1990;34(3):623–32.
Newby DE, et al. Substance P-induced vasodilatation is mediated by the neurokinin type 1 receptor but does not contribute to basal vascular tone in man. Br J Clin Pharmacol. 1999;48(3):336–44.
Bossaller C, et al. In vivo measurement of endothelium-dependent vasodilation with substance P in man. Herz. 1992;17(5):284–90.
Nicoletti M, et al. Impact of neuropeptide substance P an inflammatory compound on arachidonic acid compound generation. Int J Immunopathol Pharmacol. 2012;25(4):849–57.
Bill A, et al. Substance P: release on trigeminal nerve stimulation, effects in the eye. Acta Physiol Scand. 1979;106(3):371–3.
Teodoro FC, et al. Peripheral substance P and neurokinin-1 receptors have a role in inflammatory and neuropathic orofacial pain models. Neuropeptides. 2013;47(3):199–206.
Lu X, et al. Substance P expression in the distal cerebrospinal fluid-contacting neurons and spinal trigeminal nucleus in formalin-induced the orofacial inflammatory pain in rats. Brain Res Bull. 2009;78(4–5):139–44.
Spigelman I, Puil E. Substance P actions on sensory neurons. Ann N Y Acad Sci. 1991;632:220–8.
Ottosson A, Edvinsson L. Release of histamine from dural mast cells by substance P and calcitonin gene-related peptide. Cephalalgia. 1997;17(3):166–74.
Guo W, et al. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J Neurosci. 2007;27(22):6006–18.
Caudle RM, et al. Central sensitization in the trigeminal nucleus caudalis produced by a conjugate of substance P and the A subunit of cholera toxin. J Pain. 2010;11(9):838–46.
De Felipe C, et al. Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature. 1998;392(6674):394–7.
Strittmatter M, et al. Substance P, somatostatin and monoaminergic transmitters in the cerebrospinal fluid of patients with chronic idiopathic trigeminal neuralgia. Schmerz. 1996;10(5):261–8.
Goldstein DJ, et al. Lanepitant an NK-1 antagonist in migraine prophylaxis. Cephalalgia. 1999;19:377.
Goldstein DJ, et al. Lanepitant, an NK-1 antagonist, in migraine prevention. Cephalalgia. 2001;21(2):102–6.
Goldstein DJ, et al. Ineffectiveness of neurokinin-1 antagonist in acute migraine: a crossover study. Cephalalgia. 1997;17(7):785–90.
Miyata A, et al. Isolation of a novel 38 residue-hypothalamic polypeptide which stimulates adenylate cyclase in pituitary cells. Biochem Biophys Res Commun. 1989;164(1):567–74.
Dickson L, Finlayson K. VPAC and PAC receptors: from ligands to function. Pharmacol Ther. 2009;121(3):294–316.
Moller K, et al. Pituitary adenylate cyclase activating peptide is a sensory neuropeptide: immunocytochemical and immunochemical evidence. Neuroscience. 1993;57(3):725–32.
Eftekhari S, et al. Localization of CGRP, CGRP receptor, PACAP and glutamate in trigeminal ganglion. Relation to the blood-brain barrier. Brain Res. 2015;1600:93–109.
Larsen JO, et al. Expression of pituitary adenylate cyclase-activating polypeptide (PACAP) in the mesencephalic trigeminal nucleus of the rat after transsection of the masseteric nerve. Brain Res Mol Brain Res. 1997;46(1–2):109–17.
Csati A, et al. Calcitonin gene-related peptide and its receptor components in the human sphenopalatine ganglion -- interaction with the sensory system. Brain Res. 2012;1435:29–39.
Bhatt DK, et al. PACAP-38 infusion causes sustained vasodilation of the middle meningeal artery in the rat: possible involvement of mast cells. Cephalalgia. 2014;34(11):877–86.
Nakajima E, et al. Pituitary adenylate cyclase-activating peptide induces neurite outgrowth in cultured monkey trigeminal ganglion cells: involvement of receptor PAC1. Mol Vis. 2013;19:174–83.
Saghy E, et al. Stimulatory effect of pituitary adenylate cyclase-activating polypeptide 6-38, M65 and vasoactive intestinal polypeptide 6-28 on trigeminal sensory neurons. Neuroscience. 2015;308:144–56.
Markovics A, et al. Pituitary adenylate cyclase-activating polypeptide plays a key role in nitroglycerol-induced trigeminovascular activation in mice. Neurobiol Dis. 2012;45(1):633–44.
Akerman S, Goadsby PJ. Neuronal PAC1 receptors mediate delayed activation and sensitization of trigeminocervical neurons: relevance to migraine. Sci Transl Med. 2015;7(308):308ra157.
Kortesi T, et al. Kynurenic acid inhibits the electrical stimulation induced elevated pituitary adenylate cyclase-activating polypeptide expression in the TNC. Front Neurol. 2017;8:745.
Gazerani P, Cairns BE. New insight in migraine pathogenesis: vasoactive intestinal peptide (VIP) and pituitary adenylate cyclase-activating polypeptide (PACAP) in the circulation after sumatriptan. Scand J Pain. 2013;4(4):208–10.
Cernuda-Morollon E, et al. No change in interictal PACAP levels in peripheral blood in women with chronic migraine. Headache. 2016;56(9):1448–54.
Zagami AS, Edvinsson L, Goadsby PJ. Pituitary adenylate cyclase activating polypeptide and migraine. Ann Clin Transl Neurol. 2014;1(12):1036–40.
Guo S, et al. Part I: pituitary adenylate cyclase-activating polypeptide-38 induced migraine-like attacks in patients with and without familial aggregation of migraine. Cephalalgia. 2017;37(2):125–35.
Guo S, et al. Part II: biochemical changes after pituitary adenylate cyclase-activating polypeptide-38 infusion in migraine patients. Cephalalgia. 2017;37(2):136–47.
Hansen JM, et al. Vasoactive intestinal polypeptide evokes only a minimal headache in healthy volunteers. Cephalalgia. 2006;26(8):992–1003.
Tuka B, et al. Release of PACAP-38 in episodic cluster headache patients - an exploratory study. J Headache Pain. 2016;17(1):69.
Cetkovic M, et al. Vasculature and neurovascular relationships of the trigeminal nerve root. Acta Neurochir. 2011;153(5):1051–7; discussion 1057.
Krisht A, et al. The blood supply of the intracavernous cranial nerves: an anatomic study. Neurosurgery. 1994;34(2):275–9; discussion 279.
Love S, Coakham HB. Trigeminal neuralgia: pathology and pathogenesis. Brain. 2001;124(Pt 12):2347–60.
Adamczyk M, et al. Trigeminal nerve - artery contact in people without trigeminal neuralgia - MR study. Med Sci Monit. 2007;13(Suppl 1):38–43.
Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self-sustaining discharge in the trigeminal ganglion. Pain. 1994;56(2):127–38.
Sindou M, Howeidy T, Acevedo G. Anatomical observations during microvascular decompression for idiopathic trigeminal neuralgia (with correlations between topography of pain and site of the neurovascular conflict). Prospective study in a series of 579 patients. Acta Neurochir. 2002;144(1):1–12; discussion 12–3.
Hilton DA, et al. Pathological findings associated with trigeminal neuralgia caused by vascular compression. Neurosurgery. 1994;35(2):299–303; discussion 303.
Love S, Hilton DA, Coakham HB. Central demyelination of the Vth nerve root in trigeminal neuralgia associated with vascular compression. Brain Pathol. 1998;8(1):1–11; discussion 11–2.
Devor M, Govrin-Lippmann R, Rappaport ZH. Mechanism of trigeminal neuralgia: an ultrastructural analysis of trigeminal root specimens obtained during microvascular decompression surgery. J Neurosurg. 2002;96(3):532–43.
Brisman R, Khandji AG, Mooij RB. Trigeminal nerve-blood vessel relationship as revealed by high-resolution magnetic resonance imaging and its effect on pain relief after gamma knife radiosurgery for trigeminal neuralgia. Neurosurgery. 2002;50(6):1261–6; discussion 1266–7.
Edvinsson L, Uddman R. Neurobiology in primary headaches. Brain Res Brain Res Rev. 2005;48(3):438–56.
Edvinsson L, Goadsby PJ. Neuropeptises in headache. Eur J Neurol. 1998;5:329–41.
Edvinsson L, et al. Neuropeptide Y: cerebrovascular innervation and vasomotor effects in the cat. Neurosci Lett. 1983;43(1):79–84.
Olesen J, Jansen-Olesen I. Nitric oxide mechanisms in migraine. Pathol Biol (Paris). 2000;48(7):648–57.
Edvinsson L, et al. Cerebellar distribution of calcitonin gene-related peptide (CGRP) and its receptor components calcitonin receptor-like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) in rat. Mol Cell Neurosci. 2011;46(1):333–9.
Nieuwenhuys R, Voogd J, van Huijzen C. The human central nervous system- A synopsis and atlas. Berlin: Springer; 1988.
Wilkinson JL. Neuroanatomy for medical students. 3rd ed. Bristol: John Wright & Sons; 1986.
Bartsch T, Goadsby PJ. The trigeminocervical complex and migraine: current concepts and synthesis. Curr Pain Headache Rep. 2003;7(5):371–6.
Olszewski J. On the anatomical and functional organization of the spinal trigeminal nucleus. J Comp Neurol. 1950;92(3):401–13.
Strassman AM, Potrebic S, Maciewicz RJ. Anatomical properties of brainstem trigeminal neurons that respond to electrical stimulation of dural blood vessels. J Comp Neurol. 1994;346(3):349–65.
Hu JW, Dostrovsky JO, Sessle BJ. Functional properties of neurons in cat trigeminal subnucleus caudalis (medullary dorsal horn). I. Responses to oral-facial noxious and nonnoxious stimuli and projections to thalamus and subnucleus oralis. J Neurophysiol. 1981;45(2):173–92.
Hu JW. Response properties of nociceptive and non-nociceptive neurons in the rat’s trigeminal subnucleus caudalis (medullary dorsal horn) related to cutaneous and deep craniofacial afferent stimulation and modulation by diffuse noxious inhibitory controls. Pain. 1990;41(3):331–45.
Mosso JA, Kruger L. Receptor categories represented in spinal trigeminal nucleus caudalis. J Neurophysiol. 1973;36(3):472–88.
Dubner R, Bennett GJ. Spinal and trigeminal mechanisms of nociception. Annu Rev Neurosci. 1983;6:381–418.
Goadsby PJ, Hoskin KL. The distribution of trigeminovascular afferents in the nonhuman primate brain Macaca nemestrina: a c-fos immunocytochemical study. J Anat. 1997;190(Pt 3):367–75.
Hoskin KL, Kaube H, Goadsby PJ. Central activation of the trigeminovascular pathway in the cat is inhibited by dihydroergotamine. A c-Fos and electrophysiological study. Brain. 1996;119(Pt 1):249–56.
Kaube H, et al. Expression of c-Fos-like immunoreactivity in the caudal medulla and upper cervical spinal cord following stimulation of the superior sagittal sinus in the cat. Brain Res. 1993;629(1):95–102.
Liu Y, et al. Central projections of sensory innervation of the rat superficial temporal artery. Brain Res. 2003;966(1):126–33.
Liu Y, Broman J, Edvinsson L. Central projections of sensory innervation of the rat superior sagittal sinus. Neuroscience. 2004;129(2):431–7.
Davis KD, Dostrovsky JO. Activation of trigeminal brain-stem nociceptive neurons by dural artery stimulation. Pain. 1986;25(3):395–401.
Bolton S, O’Shaughnessy CT, Goadsby PJ. Properties of neurons in the trigeminal nucleus caudalis responding to noxious dural and facial stimulation. Brain Res. 2005;1046(1–2):122–9.
Goadsby PJ, Zagami AS. Stimulation of the superior sagittal sinus increases metabolic activity and blood flow in certain regions of the brainstem and upper cervical spinal cord of the cat. Brain. 1991;114(Pt 2):1001–11.
Henry MA, Nousek-Goebl NA, Westrum LE. Light and electron microscopic localization of calcitonin gene-related peptide immunoreactivity in lamina II of the feline trigeminal pars caudalis/medullary dorsal horn: a qualitative study. Synapse. 1993;13(2):99–107.
Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol. 1988;23(2):193–6.
Weiller C, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1(7):658–60.
Bahra A, et al. Brainstem activation specific to migraine headache. Lancet. 2001;357(9261):1016–7.
Goadsby PJ, Lipton RB, Ferrari MD. Migraine--current understanding and treatment. N Engl J Med. 2002;346(4):257–70.
Scheurer S, Gottschall J, Groh V. Afferent projections of the rat major occipital nerve studied by transganglionic transport of HRP. Anat Embryol (Berl). 1983;167(3):425–38.
Bartsch T, Goadsby PJ. Stimulation of the greater occipital nerve induces increased central excitability of dural afferent input. Brain. 2002;125(Pt 7):1496–509.
Bakker DA, Richmond FJ, Abrahams VC. Central projections from cat suboccipital muscles: a study using transganglionic transport of horseradish peroxidase. J Comp Neurol. 1984;228(3):409–21.
Neuhuber WL, Zenker W. Central distribution of cervical primary afferents in the rat, with emphasis on proprioceptive projections to vestibular, perihypoglossal, and upper thoracic spinal nuclei. J Comp Neurol. 1989;280(2):231–53.
Kerr FW, Olafson RA. Trigeminal and cervical volleys. Convergence on single units in the spinal gray at C-1 and C-2. Arch Neurol. 1961;5:171–8.
Elchami Z, et al. Effectiveness of occipital nerve block for the treatment of acute exacerbation of cluster headache. Headache. 2014;54:37.
Inan L, et al. Greater occipital nerve blocks with bupivacaine in the treatment of chronic migraine. Randomized, multicenter, double-blind, parallel, placebo-controlled study. Eur J Neurol. 2014;21:52.
Lambru G, et al. Greater occipital nerve blocks in chronic cluster headache: a prospective open-label study. Eur J Neurol. 2014;21(2):338–43.
Weiner RL, Reed KL. Peripheral neurostimulation for control of intractable occipital neuralgia. Neuromodulation. 1999;2(3):217–21.
Bermejo PE, del Pozo C. Long term results for occipital nerve stimulation in refractory chronic migraine. Cephalalgia. 2017;37:120–1.
Serra G, Marchioretto F. Occipital nerve stimulation for chronic migraine: a randomized trial. Pain Physician. 2012;15(3):245–53.
Goadsby PJ, et al. ONSTIM: occipital nerve stimulation for the treatment of chronic migraine. Eur J Neurol. 2005;12:198.
Guillamon EG, et al. Long-term outcome in occipital nerve stimulation in refractory chronic cluster headache. Cephalalgia. 2015;35:84.
Lainez MJA, et al. Efficacy and safety of occipital nerve stimulation for treatment of chronic cluster headache. Headache. 2008;48:S15.
Schwedt TJ, et al. Occipital nerve stimulation for chronic cluster headache and hemicrania continua: pain relief and persistence of autonomic features. Cephalalgia. 2006;26(8):1025–7.
Nakai M, et al. Parasympathetic cerebrovasodilator center of the facial nerve. Circ Res. 1993;72(2):470–5.
May A, et al. Correlation between structural and functional changes in brain in an idiopathic headache syndrome. Nat Med. 1999;5(7):836–8.
Obermann M, et al. Prevalence of trigeminal autonomic symptoms in migraine: a population-based study. Cephalalgia. 2007;27(6):504–9.
Karsan N, et al. The phenotype of premonitory symptoms and migraine headache triggered with nitroglycerin. J Neurol Neurosurg Psychiatr. 2016;87(12):e1.75.
Drummond PD, Lance JW. Pathological sweating and flushing accompanying the trigeminal lacrimal reflex in patients with cluster headache and in patients with a confirmed site of cervical sympathetic deficit. Evidence for parasympathetic cross-innervation. Brain. 1992;115(Pt 5):1429–45.
Edvinsson L, Uddman R, Juul R. Peptidergic innervation of the cerebral circulation. Role in subarachnoid hemorrhage in man. Neurosurg Rev. 1990;13(4):265–72.
Spencer SE, et al. CNS projections to the pterygopalatine parasympathetic preganglionic neurons in the rat: a retrograde transneuronal viral cell body labeling study. Brain Res. 1990;534(1–2):149–69.
Akerman S, et al. A translational in vivo model of trigeminal autonomic cephalalgias - therapeutic characterization with brainstem stimulation. J Headache Pain. 2013;14(S1):P36.
Frese A, Evers S, May A. Autonomic activation in experimental trigeminal pain. Cephalalgia. 2003;23(1):67–8.
Edvinsson L, et al. Innervation of the human cerebral circulation. J Auton Nerv Syst. 1994;49 Suppl:S91–6.
Goadsby PJ, Uddman R, Edvinsson L. Cerebral vasodilatation in the cat involves nitric oxide from parasympathetic nerves. Brain Res. 1996;707(1):110–8.
Steinberg A, et al. Expression of messenger molecules and receptors in rat and human sphenopalatine ganglion indicating therapeutic targets. J Headache Pain. 2016;17(1):78.
Goadsby PJ. Characteristics of facial nerve-elicited cerebral vasodilatation determined using laser Doppler flowmetry. Am J Phys. 1991;260(1 Pt 2):R255–62.
Goadsby PJ. Sphenopalatine ganglion stimulation increases regional cerebral blood flow independent of glucose utilization in the cat. Brain Res. 1990;506(1):145–8.
Fanciullacci M, et al. Increase in plasma calcitonin gene-related peptide from the extracerebral circulation during nitroglycerin-induced cluster headache attack. Pain. 1995;60(2):119–23.
Nicolodi M, Del Bianco E. Sensory neuropeptides (substance P, calcitonin gene-related peptide) and vasoactive intestinal polypeptide in human saliva: their pattern in migraine and cluster headache. Cephalalgia. 1990;10(1):39–50.
Malick A, Burstein R. Cells of origin of the trigeminohypothalamic tract in the rat. J Comp Neurol. 1998;400(1):125–44.
Malick A, Strassman RM, Burstein R. Trigeminohypothalamic and reticulohypothalamic tract neurons in the upper cervical spinal cord and caudal medulla of the rat. J Neurophysiol. 2000;84(4):2078–112.
May A. Cluster headache: pathogenesis, diagnosis, and management. Lancet. 2005;366(9488):843–55.
Leone M, et al. Hypothalamic deep brain stimulation for intractable chronic cluster headache: a 3-year follow-up. Neurol Sci. 2003;24(Suppl 2):S143–5.
Lainez MJ, et al. Sphenopalatine ganglion stimulation for the treatment of cluster headache. Ther Adv Neurol Disord. 2014;7(3):162–8.
Schoenen J, et al. Stimulation of the sphenopalatine ganglion (SPG) for cluster headache treatment. Pathway CH-1: a randomized, sham-controlled study. Cephalalgia. 2013;33(10):816–30.
Ferraro S, et al. Defective functional connectivity between posterior hypothalamus and regions of the diencephalic-mesencephalic junction in chronic cluster headache. Cephalalgia. 2018;38(13):1910–8.
Qiu E, et al. Abnormal brain functional connectivity of the hypothalamus in cluster headaches. PLoS One. 2013;8(2):e57896.
Sprenger T, et al. Specific hypothalamic activation during a spontaneous cluster headache attack. Neurology. 2004;62(3):516–7.
Goadsby PJ, May A. PET demonstration of hypothalamic activation in cluster headache. Neurology. 1999;52(7):1522.
May A, et al. Hypothalamic activation in cluster headache attacks. Lancet. 1998;352(9124):275–8.
Craig AD, et al. A thalamic nucleus specific for pain and temperature sensation. Nature. 1994;372(6508):770–3.
Gaze RM, Gordon G. The representation of cutaneous sense in the thalamus of the cat and monkey. Q J Exp Physiol Cogn Med Sci. 1954;39(4):279–304.
Burstein R, et al. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79(2):964–82.
Andreou AP, Goadsby PJ. Therapeutic potential of novel glutamate receptor antagonists in migraine. Expert Opin Investig Drugs. 2009;18(6):789–803.
Greenamyre JT, Young AB, Penney JB. Quantitative autoradiographic distribution of L-[3H]glutamate-binding sites in rat central nervous system. J Neurosci. 1984;4(8):2133–44.
Kai-Kai MA, Howe R. Glutamate-immunoreactivity in the trigeminal and dorsal root ganglia, and intraspinal neurons and fibres in the dorsal horn of the rat. Histochem J. 1991;23(4):171–9.
Hill RG, Salt TE. An ionophoretic study of the responses of rat caudal trigeminal nucleus neurones to non-noxious mechanical sensory stimuli. J Physiol. 1982;327:65–78.
Tallaksen-Greene SJ, et al. Excitatory amino acid binding sites in the trigeminal principal sensory and spinal trigeminal nuclei of the rat. Neurosci Lett. 1992;141(1):79–83.
Broman J, Ottersen OP. Cervicothalamic tract terminals are enriched in glutamate-like immunoreactivity: an electron microscopic double-labeling study in the cat. J Neurosci. 1992;12(1):204–21.
Goadsby PJ, Classey JD. Glutamatergic transmission in the trigeminal nucleus assessed with local blood flow. Brain Res. 2000;875(1–2):119–24.
Bereiter DA, Benetti AP. Excitatory amino release within spinal trigeminal nucleus after mustard oil injection into the temporomandibular joint region of the rat. Pain. 1996;67(2–3):451–9.
Oshinsky ML, Luo J. Neurochemistry of trigeminal activation in an animal model of migraine. Headache. 2006;46(Suppl 1):S39–44.
Ma QP. Co-localization of 5-HT(1B/1D/1F) receptors and glutamate in trigeminal ganglia in rats. Neuroreport. 2001;12(8):1589–91.
Silva E, et al. Extracellular glutamate, aspartate and arginine increase in the ventral posterolateral thalamic nucleus during nociceptive stimulation. Brain Res. 2001;923(1–2):45–9.
Salt TE. Glutamate receptor functions in sensory relay in the thalamus. Philos Trans R Soc Lond Ser B Biol Sci. 2002;357(1428):1759–66.
Halpain S, Wieczorek CM, Rainbow TC. Localization of L-glutamate receptors in rat brain by quantitative autoradiography. J Neurosci. 1984;4(9):2247–58.
Watanabe M, Mishina M, Inoue Y. Distinct gene expression of the N-methyl-D-aspartate receptor channel subunit in peripheral neurons of the mouse sensory ganglia and adrenal gland. Neurosci Lett. 1994;165(1–2):183–6.
Sahara Y, et al. Glutamate receptor subunits GluR5 and KA-2 are coexpressed in rat trigeminal ganglion neurons. J Neurosci. 1997;17(17):6611–20.
Tamaru Y, et al. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106(3):481–503.
Peres MF, et al. Cerebrospinal fluid glutamate levels in chronic migraine. Cephalalgia. 2004;24(9):735–9.
Martinez F, et al. Neuroexcitatory amino acid levels in plasma and cerebrospinal fluid during migraine attacks. Cephalalgia. 1993;13(2):89–93.
Rothrock JF, et al. Cerebrospinal fluid analyses in migraine patients and controls. Cephalalgia. 1995;15(6):489–93.
Burstein R, et al. An association between migraine and cutaneous allodynia. Ann Neurol. 2000;47(5):614–24.
Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89(2–3):107–10.
Messlinger K, Burstein R. Anatomy of the central nervous sytem pathways related to head pain. In: The headaches. Philadelphia: Lippincott Williams & Wilkins; 2000. p. 55–76.
Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219–45.
Akerman S, Holland PR, Goadsby PJ. Diencephalic and brainstem mechanisms in migraine. Nat Rev Neurosci. 2011;12(10):570–84.
Fields HL, et al. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J Neurosci. 1983;3(12):2545–52.
Bjorklund A, Skagerberg G. Evidence for a major spinal cord projection from the diencephalic A11 dopamine cell group in the rat using transmitter-specific fluorescent retrograde tracing. Brain Res. 1979;177(1):170–5.
Behbehani MM. Functional characteristics of the midbrain periaqueductal gray. Prog Neurobiol. 1995;46(6):575–605.
Afridi SK, et al. A positron emission tomographic study in spontaneous migraine. Arch Neurol. 2005;62(8):1270–5.
Afridi SK, et al. A PET study exploring the laterality of brainstem activation in migraine using glyceryl trinitrate. Brain. 2005;128(Pt 4):932–9.
Welch KM, et al. Periaqueductal gray matter dysfunction in migraine: cause or the burden of illness? Headache. 2001;41(7):629–37.
Derbyshire SW, et al. Cerebral responses to pain in patients with atypical facial pain measured by positron emission tomography. J Neurol Neurosurg Psychiatry. 1994;57(10):1166–72.
May A, et al. Experimental cranial pain elicited by capsaicin: a PET study. Pain. 1998;74(1):61–6.
May A, Goadsby PJ. Cluster headache: imaging and other developments. Curr Opin Neurol. 1998;11(3):199–203.
May A, et al. Functional magnetic resonance imaging in spontaneous attacks of SUNCT: short-lasting neuralgiform headache with conjunctival injection and tearing. Ann Neurol. 1999;46(5):791–4.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Andreou, A.P., Edvinsson, L. (2020). Trigeminal Mechanisms of Nociception. In: Lambru, G., Lanteri-Minet, M. (eds) Neuromodulation in Headache and Facial Pain Management. Headache. Springer, Cham. https://doi.org/10.1007/978-3-030-14121-9_1
Download citation
DOI: https://doi.org/10.1007/978-3-030-14121-9_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-14120-2
Online ISBN: 978-3-030-14121-9
eBook Packages: MedicineMedicine (R0)