
Abstract
The emission of sulfur oxides can have harmful effects on the environment. Biodesulfurization of fossil fuels is attracting more and more attention because such a bioprocess is environmentally friendly. Some bacteria, like Rhodococcus, have been used or studied to upgrading the fossil fuels on sulfur content limitation with their gentle desulfurization and high desulfurizing competence, without lowering the calorific value of the fuel. Recent advances have demonstrated the desulfurization pathway called “4S” pathway, including four enzymes, and the molecular mechanism for biodesulfurization has also been described. In addition, genetic manipulations, such as co-expression of flavin reductases, promoter modification, increasing the expression of key enzymes, expressing of desulfurization enzymes in heterologous hosts, and rearranging the dsz gene cluster were also used to improve sulfur removal efficiencies. In this chapter, we summarize the mechanism of biodesulfurization in Rhodococcus.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
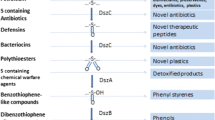


1 Introduction
Fossil fuels are humanity’s most important source of energy. Many of the benefits that we used in our way of life are due to fossil fuel use. There are three major fuels—coal, oil, and natural gas. Oil leads with a proportion to near 40% of the total consumption in the world, followed by coal (25%) and natural gas (22%). Almost all oil is consumed by burning, which causes pollution because of the chemical gases released.
Sulfur is the third most abundant element in crude oil and can vary from 0.05 to 10% of the composition. In addition to elemental sulfur, sulfate, sulfite, thiosulfate, and sulfide, more than 200 sulfur-containing organic compounds have been identified in crude oils. These include sulfides, thiols, thiophenes, substituted benzo- and dibenzothiophenes, benzonaphthothiophene, and many considerably more complex molecules (Monticello et al. 1985). The condensed thiophenes are the most common form in which sulfur is present (Kropp and Gerber 1998). Dibenzothiophene (DBT), benzothiophene (BT), and their substitutes are the major sulfur-containing aromatic compounds in fuels, accounting for up to 70% of the sulfur content (Fig. 1) (Kertesz 2001) because they have higher boiling points (more than 200 °C), and it is difficult to remove them from atmospheric tower outlet streams (e.g., middle distillates) (Kawatra and Eisele 2001; Shennan 1996). Benzothiophene (BT), non-β, single β, and di-β-substituted benzothiophenes (B.P.N219 °C) are the typical thiophenic compounds that are found up to 30% in diesel oils (McFarland et al. 1998).

Chemical structure of typical organic sulfur compounds in fossil fuel. The alkylated DBTs are different in type of substituents, number of substituents, and their bond position on benzene ring
The strict new regulations to lower sulfur content in fossil fuels require new economic and more efficient methods for desulfurization, especially for removing organic sulfur. The concentrations of BT and DBT in fossil fuels are prominently decreased by hydrodesulfurization (HDS) process (Monticello 1998), which has been commercially used for a long time. HDS has several disadvantages: (1) For refractory sulfur compounds, it requires higher temperature, pressure, and longer residence time; (2) it removes relatively simple sulfur compounds such as thiols, sulfides, and disulfides effectively. However, some complex aromatic sulfur-containing compounds such as DBTs, BTs, and polyaromatic sulfur heterocycles are resistant to HDS and form the most abundant organ sulfur compounds after HDS (Monticello 1998; Ma et al. 1994); (3) the cost of sulfur removal in industrial factories in HDS process is expensive, although HDS is considered to be a cost-effective method for fossil fuel desulfurization. Atlas et al. (2001) estimated the cost of lowering the sulfur content from 500 to 200 mg/kg to be approximately one cent per gallon. To reduce the sulfur content from 200 to 50 mg/kg, the desulfurization cost would be 4~5 times or higher.
Biodesulfurization (BDS) is a process that is being developed based on naturally occurring bacteria that can remove organically bound sulfur from diesel oil through their metabolism (Borgne and Quintero 2003). Intensive research has been conducted in BDS and has isolated many desulfurizing bacteria from many genera, such as Rhodococcus (Kilbane and Jackowski 1992; Izumi et al. 1994; Yu et al. 2006a, b; Ma et al. 2006a, b), Microbacterium (Li et al. 2005a), Gordonia (Rhee et al. 1998; Li et al. 2006a, b), Mycobacterium (Li et al. 2003, 2005b, 2007a, b; Chen et al. 2008), Pseudomonas (Gupta et al. 2005; Shan et al. 2003), and so on, of which Rhodococcus sp. is an important desulfurizing bacterium with a wide substrate range and deep desulfurizing activity. In this chapter, we introduce desulfurization by Rhodococcus.
2 Biodesulfurization Pathways in Rhodococcus
The genus Rhodococcus belongs to the phylum and class Actinobacteria, the order Actinomycetales, and the family Nocardiaceae. Rhodococci possess a variety of plasmids, which give them greater capability to remediating environment pollutions (Dosomer et al. 1988; Kayser 2002).
Most sulfur in fossil fuel can be removed easily by HDS. However, there is one type, known as refractory organic sulfur, which is very difficult to remove. The current methods that can remove the refractory part operate under extremely invasive conditions. They are very costly and produce considerable amounts of carbon dioxide. Microbial desulfurization is an environmentally friendly method that can remove sulfur from refractory organic compounds, such as DBTs and BTs, under ambient temperature and pressure without lowering the calorific value of the fuel. These features have been the reason to conduct extensive studies to develop methods by which desulfurization of refractory organic sulfur compounds under mild condition can be viable (Gupta et al. 2005; Soleimani and Bassi 2007).
2.1 DBT Biodesulfurization Pathway in Rhodococcus
Much effort has been put into the investigation of biological desulfurization systems using DBT or alkylated DBTs as model compounds. The pathway specifically cleaving the C–S bond during metabolic desulfurization has been termed the “4S” pathway (Fig. 2) (Gallagher et al. 1993; Kilbane 2006; Gray et al. 2003; Yan et al. 2000), because four different molecules are formed during DBT desulfurization.

“4S” pathway of microbial DBT desulfurization and involvement of relative enzymes and flavin reductase
The “4S” pathway for sulfur removal was first reported for Rhodococcus erythropolis IGTS8 in 1993 by Gallagher et al. (1993). Besides R. erythropolis IGTS8 (Kilbane and Jackowski 1992), other Rhodococcus that are also reported to follow this 4S pathway are R. erythropolis D1 (Izumi et al. 1994; Ohshiro et al. 1994), R. erythropolis DS-3 (Li et al. 2006a, b), Rhodococcus ECRD1 (Grossman et al. 1999), Rhodococcus B1 (Denis-Larose et al. 1997), Rhodococcus SY1 (Omori et al. 1992), Rhodococcus UM3 (Purdy et al. 1993), Rhodococcus sp. KT462, and R. erythropolis KA2-5-1 (Kobayashi et al. 2000). Among these, IGTS8 has been studied most extensively. R. erythropolis IGTS8 was isolated by Kilbane and Bielaga (1990) and was used by Energy Biosystems Corp. (EBC) for the development of their commercial microbial desulfurization plan. The strain IGTS8 is a Gram-positive rod-shaped bacterium approximately 0.5 μm long.
In the “4S” pathway, DBT is first converted to DBT sulfoxide (DBTO), then DBT sulfone (DBTO2), then 2′-hydroxybiphenyl 2-sulfinic acid (HBPS), and finally 2-hydroxybiphenyl (2-HBP), releasing sulfate into the medium. Isotopic labeling experiments have shown that the oxygen atom of the hydroxyl group of 2-HBP originates from molecular oxygen, implicating a role for an oxygenase or oxygenases in the pathway (Oldfield et al. 1997). BDS processing using this strain will be ideal. In a BDS process the end product, 2-HBP and its derivatives, would partition back into the oil, thus preserving the fuel value.
2.2 BT Biodesulfurization Pathway in Rhodococcus
In contrast to DBT-desulfurizing bacteria, little is known about bacteria that can desulfurize BT. BT predominates in gasoline. Oil contamination may impact the organisms that live in contaminated ecosystems because some of these compounds, such as benzothiophene derivatives, have been reported to be mutagenic and carcinogenic (Kropp and Fedorak 1998). Therefore the degradation pathway of BTH was also studied.
Based on the mass spectral data, two different pathways of desulfurization of sulfur from benzothiophene were proposed, and each end product has been identified (Fig. 3). In both of the pathways, BT is firstly converted to BT sulfoxide, then BT sulfone, and following by 2-(2′-hydroxyphenyl) ethan-1-al. The next desulfurization steps can proceed along two separated pathways. In the first pathway, the sulfinate group is removed with oxygenation of the molecule 2-(2′-hydroxyphenyl) ethan-1-al (Gilbert et al. 1998). This product is recovered as benzofuran due to dehydration under acidic extraction conditions. In the second pathway, the final product is o-hydroxystyrene, produced through desulfination of the molecule, which finally may oxygenate the carbon atom to dioxide carbon (Konishi et al. 2000). Most microorganisms can only desulfurize BT with one pathway, but Rhodococcus sp. JVH1 and Rhodococcus sp. WU-K2R have been reported to produce both end products from the desulfurization of benzothiophene (Kirimura et al. 2002).

Two possible degradation pathways of Benzothiophene (BT). (A) G. desulfuricans strain 213E (B) Paenibacillus sp. strain A11-2: (a) BT; (b) BT sulfoxide; (c) BT sulfone; (d) 2-(2′-hydroxyphenyl)ethen 1-sulfinate; (e) benzo[e][1,2]oxathiin S-oxide (sultine); (f) 2-(2′-hydroxyphenyl)ethan-1-al; (g) Benzofuran; (h) o-hydroxystyrene
3 Enzymes Involved in Specific Desulfurization
The enzymes involved in specific DBT-desulfurizing pathways have been purified and characterized; their optimum reacting conditions and activities have also been studied. Compared to DBT-desulfurizing enzymes, the enzymes in BT-desulfurizing pathway were rarely known.
3.1 Enzymes Involved in DBT Desulfurization of the 4S Pathway
The complete removal of sulfur from DBT through the 4S pathway requires four enzymes. DBT monooxygenase (DszC or DBT-MO) catalyzes the stepwise S-oxidation of DBT, first to DBTO and then to DBTO2. DBT-sulfone monooxygenase (DszA or DBTO2-MO) catalyzes the conversion of DBTO2 to HBPS. Both DBT-MO and DBTO2-MO are flavin-dependent and require a third enzyme (the flavin reductase, DszD) for activity. The fourth enzyme, HPBS desulfinase (DszB), catalyzes the desulfurization of HBPS to give HBP and sulfate, completing the reaction sequence (Gray et al. 1996; Ohshiro and Izumi 2000).
3.1.1 DBT-MO
The first enzyme catalyzes the conversion of DBT to DBT sulfone in a two-step process with DBT sulfoxide being the intermediate compound (DBT→DBTO→DBTO2). The presence of DBTO is difficult to detect because it is readily consumed. The first oxidation step (rate constant 0.06 min−1) is one-tenth of the rate of the second step (rate constant 0.5 min−1). Purified DBT-MO is shown to have a peak absorption at 281 nm. This enzyme shows homology to the acyl coenzyme A enzyme and is a homotetramer with a subunit molecular weight of 50 kDa, as reported by Gray et al. (1996). Ohshiro et al. (1994) isolated DBT-MO from R. erythropolis D-1 and reported it to be a homohexamer with a subunit molecular weight of 45 kDa. Its activity is maximum at a temperature of 40 °C and a pH of 8.0.
DBT-MO can act on the derivatives of DBT such as 4,6-dimethyl DBT, 2,8-dimethyl DBT, and 3,4-benzo-DBT, but it does not show any activity on carbazole, dibenzofuran, and fluorine; i.e., DBT atoms are substituted for sulfur atoms. Isotopic labeling studies indicated that the two oxygen atoms were derived from molecular oxygen. The DBT-MO from Rhodococcus, compared with other genera, has been shown to have a higher specific reaction rate for sparsely alkylated DBTs (Arensdorf et al. 2002).
3.1.2 DBTO2-MO
The DBTO2-MO enzyme is widely studied. It is a monooxygenase that oxidizes DBTO2 to HPBS. The enzyme isolated from R. erythropolis D-1, a thermophile, (Ohshiro et al. 1999) was found to have a molecular mass of 97 kDa and to consist of two subunits with identical masses of 50 kDa. The N-terminal amino acid sequence of the purified DBTO2-MO completely coincided with the deduced amino acid sequence for DBTO2-MO from R. erythropolis IGTS8 except for a methionine residue at the latter N-terminal. The optimal temperature and pH for DBTO2-MO activity are 35 °C and about 7.5.
Oldfield et al. (1997) found that DBTO2-MO from R. erythropolis IGTS8 catalyzed the conversion of dibenz[c,e][1,2]oxathiin 6,6-dioxide (sultone) to 2,2′- dihydroxybiphenyl (DHBP). Ohshiro et al. demonstrated that, by using DBTO2-MO from R. erythropolis D-1, sultone showed 54% activity as a substrate compared with DBT sulfone, and DHBP was formed as a product. In addition, dibenz[c,e][1,2] oxathiin 6-oxide (sultine) showed 23% activity and yielded DHBP as a product. However, DBTO2-MO did not act on DBT and HBPS. Although sultine was nonenzymatically hydrolyzed to form HBPS, it was also oxidized to sulfonic acid during shaking. It was thought that once sultone was nonenzymatically formed from sultine, it was immediately converted to DHBP by DBTO2-MO. DBTO2-MO may recognize the sulfone moiety within the structure of DBT sulfone and sultone.
Gray et al. (1996) demonstrated that 10 mM EDTA did not inhibit the activity of DBTO2-MO from R. erythropolis IGTS8. On the contrary, the activity of DBTO2-MO from R. erythropolis D-l was inhibited 50% by 1 mM EDTA. Moreover, 2,2′-bipyridine, 8-quinolinol, and the other metal-chelating reagents, such as Mn2+ and Ni2+, also inhibited the activity of the enzyme, suggesting that a metal might be involved in its activity. DBTO2-MO acted not only on DBT sulfone but also on dibenz[c,e][l,2]oxathiin 6,6-oxide and dibenz[c,e] (Ohshiro and Izumi 1999; Ohshiro et al. 1995), and oxathiin 6,6-dioxide. Dihydroxybiphenyl was formed from the latter two substrates.
3.1.3 HPBS Desulfinase
HPBS desulfinase is a novel enzyme, in that it can specifically cleave the carbon–sulfur bond of HBPSi to give 2-HBP and the sulfite ion without the aid of any other protein components or coenzymes. It has been demonstrated that the activity of HPBS desulfinase is the lowest among enzymes of desulfurization metabolism. It is the rate-limiting enzyme of 4S pathway. It is also the least studied enzyme since only a very small amount is produced.
It is a monomer with a subunit molecular weight of 40 kDa and shows enzyme activity over a wide temperature range (25–50 °C) with the optimum at 35 °C (Watkins et al. 2003). The working pH range for this enzyme is 6.0–7.5. Lee et al. (2004, 2006) elucidated the 3D structure of DszB, which was the first X-ray crystallographic study of enzymes involved in DBT desulfurization (Fig. 4). HPBS desulfinase does not require a metal cofactor for catalysis, and the inhibition by Zn2+ and Cu2+ is likely caused by interference of substrate binding or catalysis.

The overall structure of DszB. (a) ribbon model of DszB. Domain A and B are colored in light green and pink, respectively. Two crossover residues that define the domains are labeled. (b) Topology diagram of DszB. Helices and strands are colored in blue and red, respectively. Dotted green lines designate domain A and domain B. (c) Ribbon models of proteins structurally related to DszB. Ovotransferrin (left, PDB code 1NNT) and a sulfate-binding protein (right, PDB code 1SBP) are depicted in ribbon models. Two domains of each protein are colored in a similar fashion to (a). Substrate ferric carbonate and sulfate ions are depicted in space-filling models. (d) Stereo view of the active site. A glycerol molecule is depicted in space-filling model. Residues mentioned in the text are depicted as sticks
A Cys residue must be the catalytic center of DszB because SH reagents inhibited the enzyme activity. DszB has only one Cys residue, at position 27, and it was found that the C27S mutant enzyme lost its activity completely. Therefore, there is no doubt that this residue is the catalytic center.
Based on information about the 3D structure of DszB and a comparison of amino acid sequences between DszB and reported thermophilic and thermostable homologs (TdsB and BdsB), two amino acid residues, Tyr63 and Gln65, were selected as targets for mutagenesis to improve DszB. The promising mutant enzymes, which were replaced with these two residues by other amino acids, were purified and their properties examined. Among the wild-type and mutant enzymes, Y63F had higher catalytic activity but similar thermostability, and Q65H showed higher thermostability but less catalytic activity and affinity for the substrate. Furthermore, the double mutant enzyme Y63F-Q65H was purified and overcomes these drawbacks. This mutant enzyme had higher thermostability without loss of catalytic activity or affinity for the substrate.
Ohshiro et al. (2007) found that each mutation at positions 63 and 65 of DszB enhanced the maximum activity and thermal stability, respectively, and that the double mutation increased thermostability without losses in maximal activity or affinity for the substrate. For the purpose of developing microbial desulfurization as a practical process, it is necessary to improve DszB further by structural analysis of the mutant enzymes in the near future.
3.1.4 Flavin Reductase
The flavin reductases are associated with monooxygenases since monooxygenases cannot work in the absence of these reductases (DBT-MO and DBTO2-MO). The purified flavin reductase from the thermophilic strain R. erythropolis D-l contains no chromogenic cofactors and was found to have a molecular mass of 86 kDa with four identical 22 kDa subunits (Matsubara et al. 2001). The enzyme catalyzed NADH-dependent reduction of flavin mononucleotide (FMN).
For the flavin reductases from R. erythropolis D-l, flavin adenine dinucleotide was a poor substrate, and NADPH was inert. The enzyme did not catalyze the reduction of any nitroaromatic compound. The optimal temperature and optimal pH for enzyme activity were 35 °C and 6.0, respectively, and the enzyme retained 30% of its activity after heat treatment at 80 °C for 30 min. The N-terminal amino acid sequence of the purified flavin reductase was identical to that of the flavin reductase from R. erythropolis IGTS8.
Xi et al. (1997) studied the enhanced desulfurization activity of DszC and DszA under in vitro conditions by increasing the concentrations of flavin reductase, suggesting that the two are terminal oxygenases. The reaction rate with 1 unit/ml of flavin reductase was linear for 10–15 min, whereas it was linear for more than 20 min with a lower concentration.
The inhibition experiments revealed that the flavin reductase activity of R. erythropolis D-1 was inhibited by 7-hydroxycoumarin but not by other coumarin derivatives, including dicoumarol, which inhibited FRase I activity and was used for analysis of its crystal structure (Koike et al. 1998). FRase I was a flavoprotein possessing FMN as a prosthetic group. The flavin reductase of R. erythropolis D-1 has no flavin cofactor.
3.2 Enzymes Involved in BT-Desulfurizing Pathway
In contrast to DBT-desulfurizing enzymes, little is known about enzymes involved in the BT-desulfurizing pathway. At present, there are no related reports on BT-desulfurizing enzymes at home or abroad. The purified enzymes involved in BTH degradation would provide a detailed explanation for the degradation of BT.
4 Specific Desulfurizing Genes in Rhodococcus
In order to obtain better control over the machinery of specific sulfur removal, related research has been conducted on the molecular biology of this and similar strains since the metabolic identification of R. erythropolis IGTS8.
The primary genes involved in DBT metabolism, which are called both dsz and sox, have been cloned and are fairly well characterized. Although the sox (sulfur oxidation) designation was used first, the dsz (desulfurization) designation has generally been adopted.
The dsz genes are arranged in an operon-regulated system in a 4-kb conserved region of a 150 kb mega-plasmid, pSOX, in R. erythropolis IGTS8 (Oldfield et al. 1998) and a 100 kb plasmid in other strains. An insertion sequence (IS1166) was found to be associated with the dsz gene. It is a cluster of three genes (dszA, dszB, dszC) transcribed in the same direction, coding for three proteins Dsz A, Dsz B, and Dsz C, respectively (Piddington et al. 1995). The fourth gene, dszD, was on the chromosome of R. erythropolis IGTS8 rather than on pSOX with dszA, dszB, and dszC.
The termination codon of dszA and the initiation codon of dszB overlap (ATGA, Fig. 5), indicating that there may be translational coupling of these two genes. Between dszB and dszC, there was a 13-bp gap. Potential ribosome binding sites were also present upstream of each putative ATG initiation codon. The spacing and orientation of the three genes suggest that they are expressed as an operon, a suggestion that was also supported by the results of subclone analyses and promoter replacement analyses. Although expressed in the operon, Dsz B is present at concentrations severalfold less in the cytoplasm, as compared with Dsz A and Dsz C (Li et al. 1996).

Sequence of the 385-bp dsz promoter-containing fragment that starts immediately after the HindIII site. Nucleotides below the line indicate the positions of mutations, including the deletion of a C at 210. Boxed sequences are the protein-binding domain and the promoter region deduced from deletion analysis. The arrow indicates the G residue at position 11 in the 5′ end of the mRNA, and the nucleotides in bold are the putative −10 and −35 regions of the promoter. The HindIII site at the 5′ end and the SpeI site near the 3′ end were added for cloning purposes. Sequence numbering is for the native fragment, where 21 is the base preceding the A of the ATG initiation codon of dszA
These genes, when cloned into a non-desulfurized strain (called dsz−), confer the ability to desulfurize DBT to 2-HBP. The dsz operon was found on a large, 150 kb plasmid in R. erythropolis IGTS8 and on a 100 kb plasmid in other strains. An insertion sequence (IS1166) was found to be associated with the dsz gene.
To develop the biodesulfurization process, it was important to know under what conditions the desulfurization genes were expressed or repressed. Li et al. (1996) investigated the effect of various sulfur-containing compounds such as dimethyl sulfoxide (DMSO), cysteine, methionine, and sulfate on dsz gene expression. The results showed that desulfurization activity decreased when the concentration of cysteine, methionine, or sulfate in the media increased. In comparison, methionine caused the strongest repression in these substrates. When the concentration of these inhibitors reached more than 375 μM, desulfurization activity was strongly repressed. This repression was found to be due to the binding of a repressor protein next to the dsz promoter, which was located within the 385 bp region immediately upstream of dszA. Deletion analysis showed that the promoter was located to the region between −121 and −44. The S1 nuclease protection assay confirmed that the 5′ end of the dsz mRNA was the G at −46. A possible −35 promoter region with the sequence AAGTTTAA and a −10 region of GGGTGA are similar to those of Bacillus subtilis promoters that use the sigma factor σB (Li et al. 1996). The sequence at the transcription initiation site, TAG, is also the same as that of the dsz promoter, with two starting at the middle A and one starting at the G, as does the dsz promoter. The main difference between the two promoters is that the Bacillus promoters have a 14-bp spacer region between the −10 and −35 regions, where the dsz promoter is 23 bp.
The promoter region from −75 to −57 could be a potential case of dyad symmetry (Fig. 6). It is a strict inverted repeat sequence and could be part of an operator. An almost identical inverted repeat occurs within dszB from 1562 to 1578 and could be part of another operator.

Potential hairpin structure located in the dsz promoter. The hairpin is between −75 and −57 and has a free energy value of −15.4 kcal (1 cal = 4.184 J). Deletion of the G at −57 (indicated by the arrow), as in mutant R4, would reduce this to −13.2 kcal
Apart from promoter, the 385 bp fragment has at least three elements that affect Dsz activity (some overlapping the promoter region). The region from −263 to −244 proved to reduce dsz repression. However, deletion of the region did not affect repression or gene expression. The region from −144 to −121 could bind a protein such as an activator, and deletion of this region reduced gene expression, but not repression. The region between −98 and −57 may be a repressor-binding site (Li et al. 1996). It is possible that combinations of these mutations could further decrease repression.
5 Enhanced Biodesulfurization by Recombinant Bacteria
These specific bacteria can remove sulfur pollutants from petroleum and will reduce the amount of sulfur oxides released. However, genetic manipulations for the removal of harmful sulfur compounds from fossil fuels can be developed. In most times, engineered bacteria are required to remove more sulfur compounds with higher activities. Cultures with improved substrate ranges are also needed to better address the complicated mixture of chemicals present in petroleum.
5.1 Co-expression of Flavin Reductases
Since FMNH2 is essential for the activities of DszC and DszA, the overexpression of flavin reductase in Rhodococcus or in recombinant bacteria will enhance the activities of DszC and DszA.
Lei et al. (1997) found enhanced desulfurizing activities of purified DszC and DszA protein from R. erythropolis IGTS8 in vitro when activated with flavin reductase from Vibrio harveyi. Hirasawa et al. (2001) purified the flavin reductase DszD from R. erythropolis IGTS8, and the enzyme was overexpressed in Escherichia coli. The specific activity in crude extracts of the overexpressed strain was about 275-fold that of the wild-type strain.
Reichmuth et al. (1999) studied the desulfurization ability of an engineered E. coli DH10B strain that contained the plasmids pDSR2 and pDSR3. The plasmid pDSR2 contained a Vibrio harveyi NADH:FMN oxidoreductase gene, and pDSR3 encoded all of the three enzymes that converted DBT to HBP. In plasmid pDSR3 the native desulfurization control element, which located in the promoter, had been removed. Therefore, E. coli DH10B/pDSR3 could express its desulfurization trait even in the presence of sulfate ion or rich media such as LB. However, the oxidoreductase level proved to be insufficient for the overexpressed dszABC. Designing an operon that expresses the proper amount of FMN:NADH reductase to existed dszABC enzymes is crucial to reach an optimum desulfurization activity. Insufficient FMN:NADH reductase would make NADH the limiting step in DBT oxidation. On the other hand, a high concentration of FMNH2 will give rise to H2O2 formation, which would be lethal to cells (Gaudu et al. 1994; Galán et al. 2000).
In the search for the development of a method to provide the required amount of reduced flavin to DBT oxygenation, Galán et al. (2000) used hpaC, a flavin reductase from E. coli W, and connected it, in vitro, with a system of dszABC purified enzymes and an NADH source. They also used catalase in the desulfurization medium to minimize the probability of H2O2 formation, which might be produced by nonenzymatic reoxidation of FMNH2 under high oxygen concentrations. The addition of hpaC flavin reductase increased DBT desulfurization 7–10 times over 30 min. The enzyme HpaC flavin reductase and the oxidoreductase originated from IGTS8 were from the same subfamily of flavin:NAD(P)H reductases.
All the experiments confirm that the expression of an oxidoreductase with the dsz genes caused an increase in the rate of DBT removal.
5.2 Promoter Modification
The expression of dsz genes in most desulfurizing bacteria is repressed by sulfate, which is the product of biodesulfurization, through a repressor-binding site that may be in the promoter. So, looking for a new promoter that cannot be repressed by sulfate will be a new pathway to increase the desulfurizing rate.
Gallardo et al. (1997) subcloned the dsz cassette into the broad-host-range plasmid pVLT31 under the control of a hybrid promoter, Ptac, that has been shown to be functional in a wide range of bacteria. The resulting plasmid was transferred into Pseudomonas putida. The recombinant bacteria with dsz were shown to keep its desulfurization phenotype even in sulfate-containing media.
Several 16S ribosomal RNA promoters of mycobacteria have also been studied and found to be functionally constitutive (Ji et al. 1994). Matsui et al. (2002) reported a 16S ribosomal RNA promoter applied to the expression of dsz enzymes in Rhodococcus sp. strain T09. The putative Rhodococcus rrn promoter region was cloned from the Rhodococcus sp. strain T09, and the dibenzothiophene-desulfurizing gene, dsz, was expressed under the control of the putative rrn promoter in strain T09 using a Rhodococcus–E.coli shuttle vector. Strain T09 harboring the expression vector could desulfurize dibenzothiophene in the presence of inorganic sulfate, methionine, or cysteine, while the Dsz phenotype was completely repressed in recombinant cells carrying the gene under the control of the native dsz promoter under the same conditions.
At the same time, Noda et al. (2002) constructed a promoter probe transposon using a promoterless red-shifted green fluorescence protein gene (rsgfp). A 340 bp putative promoter element, kap1, was isolated from a recombinant strain, KA2-5-1, that had been shown to have high fluorescence intensity. The promoter element of kap1 was not repressed by 1 mM of sulfate, and it had about twofold greater activity than the rrn promoter from R. erythropolis. Kap1 stimulated cell growth with biodesulfurization activity without the repression of sulfate. In conclusion, kap1 is a convenient tool for improving biodesulfurization in Rhodococcus.
Otherwise, screening for recombinant bacteria that cannot be inhibited by sulfate is another substitutable method. Tanaka et al. (2002) isolated two mutants of the dibenzothiophene-desulfurizing R. erythropolis KA2-5-1 that express a high level of desulfurizing activity in the presence of sulfate using the transposome technique. The level of dibenzothiophene desulfurization by cell-free extracts prepared from mutants grown on sulfate was about fivefold higher than that by cell-free extracts from the wild type. Gene analysis of the mutants revealed that the same gene was disrupted and that the transposon-inserted gene in these strains was the gene for cystathionine β-synthase, cbs. The cbs mutants also expressed high levels of Dsz enzymes when methionine was used as the sole source of sulfur.
5.3 Increasing the Expression of Key Enzymes
The reaction catalyzed by DszC and DszB have been widely recognized as rate-limiting steps in the microbial desulfurization pathway. Several approaches have been performed by genetic engineering to improve desulfurizing enzyme activities, including those of DszC and DszB.
Coco et al. (2001) used random chimeragenesis on a transient template (RACHITT) to improve the activity of DszC by 20 times, and it must have increased the rate of the whole pathway.
As described previously, the rate of desulfurization is limited by the last enzyme in the pathway, DszB. In the native dsz operon, the ratio of mRNA of dszA, dszB, and dszC was 11:3.3:1, indicating that the translation levels of the desulfurization enzymes decreased according to their positions in the operon due to polar effects on dsz gene transcription; however, western blot analysis indicated that the expression level of dszB was far lower than that of dszC. These results suggest that the translation of dszB mRNA was not as efficient as dszA or dszB mRNA. Gene analysis revealed that the termination codon of dszA and the initiation codon of dszB overlapped, whereas there was a 13-bp gap between dszB and dszC. Potential ribosome binding sites were present upstream of each putative ATG initiation codon. In order to get a better, steady expression of DszB, Li et al. (2007a, b) removed the overlap structure by overlap polymerase chain reaction (PCR) and expressed the redesigned dsz operon in R. erythropolis without desulfurization activity, named R. erythropolis DR-2. Real-time PCR analysis confirmed that the transcription characteristics did not change in R. erythropolis DR-2 compared with R. erythropolis DR-1, which contains the original dsz operon. However, western blot analysis revealed that R. erythropolis DR-2 produced more DszB than R. erythropolis DR-1 did. The desulfurization activity of resting cells prepared from R. erythropolis DR-2 was about fivefold higher than that of R. erythropolis DR-1. That indicated that the enhanced expression level increased the metabolic rate of HBPS in the cells and contributed to the improved desulfurization rate of R. erythropolis DR-2.
To increase DszB production, Reichmuth et al. (2004) mutated the untranslated 5′ region of dszB using degenerate oligonucleotides. Because neither DszB activity nor the amount of DszB protein produced could be directly measured, it was difficult to determine the exact cause for the lack of HBP production. To clarify the results of our genetic manipulations, they chose to tag the production of the desulfurization transcripts and proteins by creating transcriptional and translational fusions with a fluorescent protein. This permitted a quick, straightforward, and direct determination of the amount of the desulfurization protein produced. This technique does not measure the activity of the proteins; however, activity screens could be used after protein production was optimized using fluorescent fusion tags. The protein used for those fusions was GFP. GFP has been widely used for the quantitative measurement of protein production and is known to be stable for a period of several days, allowing an integrative and quantitative measure of protein production (Albano et al. 1998; Cha et al. 2000). After screening only 96 mutants, several showed increased green fluorescence, and two showed increased DszB activity. When cotransformed with the full dszABC operon, the mutant dszB increased the rate of desulfurization ninefold relative to the native dszB.
R. erythropolis KA2-5-1 can desulfurize DBT into 2-HBP through the 4S pathway. Hirasawa et al. (2001) constructed an Escherichia coli-Rhodococcus shuttle vector, and the desulfurization gene cluster, dszABC, and the related reductase gene, dszD, were cloned from KA2-5-1, reintroduced into KA2-5-1, and efficiently expressed. The DBT desulfurization ability of the transformant carrying two dszABC and one dszD on the vector was about fourfold higher than that of the parent strain, and the transformant also showed improved desulfurization activity for light gas oil. Matsui et al. (2001) also enhanced the desulfurization rate by 3.3 times, by increasing the copy number of dsz genes.
5.4 The Expression of Desulfurization Enzymes in Heterologous Hosts
In fossil fuels, there are many kinds of compounds inhibiting on desulfurization process of Rhodococcus. Pseudomonas was found to be an ideal candidate for biodesulfurization because they are organic solvent tolerant and have a high growth rate. Pseudomonas sp. are among the best studied and most abundant microorganisms found in crude oil (Leahy and Colwell 1990), and a wide variety of genetic tools are now available for their molecular manipulation (Lorenzo and Timmis 1994). Furthermore, several biotechnological properties for the design of biocatalysts targeted to industrial biodesulfurization processes are present in Pseudomonas species. For example, while the solvent tolerance of Rhodococcus is the lowest reported (log P values from 6.0 to 7.0), that of the genus Pseudomonas (log P values from 3.1 to 3.4) is the highest known (Inoue and Horikoshi 1991), and several Pseudomonas strains that are highly resistant to heavy metals present in fossil fuels have been reported (Atlas 1994).
With the properties noted, dszABC genes from R. erythropolis XP were cloned into P. putida Idaho to construct a solvent-tolerant, desulfurizing P. putida A4. This strain, when contacted with sulfur refractory compounds dissolved in hydrocarbon solvent, maintained the same substrate desulfurization traits as observed in R. erythropolis XP. Resting cells of P. putida A4 could desulfurize 86% of DBT at 10% (v/v) p-xylene in 6 h. In the first 2 h, the desulfurization occurred with a rate of 1.29 mM DBT (gdw cell)−1 h−1. No DBT reduction was noticed when the experiment was repeated with R. erythropolis or P. putida Idaho under identical conditions (Tao et al. 2006).
In the development of engineered strains with potential industrial or environmental applications, a high degree of predictability in their performance and behavior is desirable. To achieve this goal, stable chromosomal insertion of the genes conferring the new trait is required. Therefore, Gallardo et al. (1997) constructed P. putida EGSOX, which carried dsz genes stably inserted into the chromosome of the host cell.
To improve the biodesulfurization process, it would be interesting to design a recombinant biocatalyst that combines the Dsz phenotype with another trait of potential interest, such as the production of biosurfactants. To accomplish this goal, Gallardo et al. (1997) also transferred dsz genes into P. aeruginosa PG201 (Ochsner et al. 1995), which cannot use DBT as the sole carbon and/or sulfur source and produces rhamnolipid biosurfactants. These are of increasing industrial relevance because of their applications in emulsification, wetting, phase separation, and viscosity reduction. The final recombinant bacteria were named P. aeruginosa EGSOX and carried dsz genes stably inserted into the chromosome of the host cell.
At 48 h of incubation, cultures of strain IGTS8 still contained DBT; however, this compound was exhausted by the two engineered Pseudomonas strains. P. aeruginosa EGSOX had the fastest metabolism of DBT, transforming 95% of the DBT at 24 h of incubation. Only 18% of the DBT was transformed by R. erythropolis IGTS8, and 40% was transformed by P. putida EGSOX. Remarkably, DBT depletion was concomitant with 2-HBP accumulation in all three strains, indicating that 2-HBP is a dead-end metabolite that cannot be further catabolized or used as a carbon source. These data demonstrated that the IGTS8-derived dsz cassette was efficiently expressed, allowing the elimination of sulfur with no loss of DBT carbon atoms, both in P. putida EGSOX and P. aeruginosa EGSOX. Moreover, in comparison with wild-type R. erythropolis IGTS8, the two recombinant biocatalysts showed enhanced biodesulfurization ability.
However, many Pseudomonas strains were unable to desulfurize DBT in the oil phase, and this will restrict their application in industry. Darzins et al. (1999) found that P. fluorescens with dszABC genes cannot desulfurize DBT in the oil phase; but the whole cell lysate with the cell wall removed can. The results showed a lack of DBT uptake ability from the oil phase to the inside of the recombinant Pseudomonas strains. Noda et al. (2003) transferred the dsz desulfurization gene cluster from R. erythropolis IGTS8 into the chromosome of P. aeruginosa NCIMB9571 using a transposon vector. All of the recombinant strains completely desulfurized 1 mM DBT in n-tetradecane (n-TD) except one, named PARM1. PARM1 was unable to desulfurize DBT in n-TD but was able to desulfurize it in water. The transposon tagging analysis indicated that the transposon is inserted into hcuA of the open reading frames hcuABC. The full-length hcuABC genes, when transformed into PARM1, achieved 87% recovery of the desulfurization activity of DBT in n-TD, but partial hcuABC genes achieved only 0–12%. These results indicated that DBT desulfurization in the oil phase by recombinant P. aeruginosa NCIMB9571 required the full-length hcuABC gene cluster. The hcuABC gene cluster is related to DBT uptake from the oil phase into the cell.
5.5 Rearranging the dsz Gene Cluster
As described before, the levels of transcription and translation of dszA, dszB, and dszC decreased according to the positions of the genes in the dsz operon. Furthermore, the translation of dszB was repressed by an overlapping structure in the dsz operon. In order to get better and steady expression of the Dsz enzymes and optimize the metabolic flux of DBT, the overlapped structure was removed, and the expression level of dszB was increased. The DBT desulfurization rate was 5 times faster than that of the native dsz operon (Li et al. 2007a, b), but this is still low in comparison to the requirements of a commercial process.
The rate of an enzyme catalytic reaction is determined by the catalytic activity, the quantity of the enzyme, and the substrate concentration. Higher levels of mRNA are the precondition for higher levels of the encoded protein. Therefore, rearranging these genes according to the catalytic capabilities of the enzymes and their reaction orders could not only balance the catalytic capabilities but also increase the substrate concentrations for the enzymes. Li et al. introduced a genetic rearrangement strategy for optimizing the metabolic pathway of DBT. By using recombinant PCR, the dsz operon of R. erythropolis DS-3 was rearranged according to the catalytic capabilities of the Dsz enzymes and their reaction orders in the 4S pathway (Fig. 7).

Rearrangement of the dsz operon by overlap PCR. Fragments of the 400-bp 5′ upstream segment (5′-U-S) and the 400-bp 3′ downstream segment (3′-D-S) of dszABC and the dszA and dszBC segments, including the overlap regions, were amplified by PCR, then the ligated 5′ upstream-dszBC segment (5′-U-S-dszBC) and the ligated dszA-3′ downstream segment (dszA-3′-D-S) were produced by overlap PCR via their overlap regions, and finally, the 5′ upstream-dszBC segment and the dszA-3′ downstream segment were linked together by overlap PCR via their overlap region to yield the reconstructed dsz operon. Black bars represent genes, and white bars represent overlap regions
The catalytic capabilities of the Dsz enzymes were approximately 25:1:5 (DszA:DszB:DszC). Hence, the dsz operon was rearranged according to the catalytic capabilities of the enzymes. The expression levels of dszB and dszC were improved by rearranging the order of the dsz genes to generate the operon dszBCA, which contained dszB, dszC, and dszA in tandem. After rearrangement, the ratio of dszA, dszB, and dszC mRNAs in the cells was changed, from 11:3.3:1 to 1:16:5. The desulfurization rate of the recombinant strain containing the rearranged dsz operon was 12 times faster than that of the native dsz operon. The maximum desulfurization rate was only about 26 μmol DBT/g DCW/h for the strain containing the native dsz operon. After removing the overlapped structure before the initiation codon of dszB, the rate was 120 μmol DBT/g DCW/h. The recombinant strain containing the rearranged dsz operon had the highest desulfurization rate, about 320 μmol DBT/g DCW/h. Therefore, the enhanced expression levels of DszC and DszB increased the desulfurization rate of the recombinant strain.
Feng et al. (2006) found that the function of the surfactant Tween 80 in the desulfurization was to decrease the product concentration associated with the cells, reducing product inhibition. The dsz genes of R. erythropolis DS-3 were also integrated into the chromosome of Bacillus subtilis ATCC 21332, which can secrete biosurfactant, yielding the recombinant strain B. subtilis M29, which has higher desulfurization efficiency than R. erythropolis DS-3 and showed no product inhibition (Ma et al. 2006a, b). It should be noted that the biosurfactant secreted from B. subtilis M29 significantly varied the interfacial tension of the supernatant. The biosurfactant therefore has an important function in the degradation of DBT.
6 Process Engineering for BDS with Rhodococcus
To apply the BDS process from laboratory to industrial level, it is necessary to improve the BDS process engineering. In this section, we summarized the main advances in process design for BDS with Rhodococcus strains from two parts: the application of immobilized cells during BDS process and the design of the novelty bioreactor for BDS.
6.1 The Application of Immobilized Cells
Though biodesulfurization with growing cells is a simple and widely accepted methods with different Rhodococcus strains, the application of immobilized cells during BDS process could help improve the desulfurization of DBT with a higher concentration, increase desulfurization efficiency, and enhance the separation of cells from the final products.
Rhodococcus spp. strains were immobilized for BDS process on silica (Si), alumina (Al), and sepiolite (Sep) or encapsulated in polymeric materials such as alginates with the advantages of biocompatibility and low-cost. However, the immobilization leads to the reduction mass transference of oil to the bacterial cells. One considerable solution to address this problem is the use of surfactants. The synthetic surfactants such as Tween 80, Span 80, and Triton X-100 have been confirmed to enhance biodesulfurization effectively. Derikvand and Etemadifar (2014) demonstrated that the addition of (Tween 80 and Span 80) increased the dissolved DBT concentration in the aqueous phase and facilitate its close to R. erythropolis R1 in the alginate beads and resulted in the increase of biodesulfurization. Taking into account the toxicity of synthetic surfactants, biological surfactants (biosurfactants) with the advantages of non-toxicity, biodegradation, and adaption to extreme pH and temperature were also studied. The biosurfactant from a marine bacteria strain was purified and added in the BDS system of R. erythropolis. The results showed that adding biosurfactants increased the higher BDS activity when compared with synthetic surfactant Tween 80. It may be due to the formation of micelles increases the solubilization of DBT (Dinamarca et al. 2014).
Generally, the bacterial surface was negatively charged which will lead to the adsorption with positively charged nano-ɣ Al2O3. The production of 2-HBP increased twofolds after 24 h in the Rhodococcus spp. strains alginate beads containing nano-ɣ Al2O3 when compared with the controls. The combination of nano-ɣ Al2O3 and alginate encapsulated cells could be an effective approach to enhance BDS process (Derikvand et al. 2014).
Hassan et al. (2013) synthesized the Fe3O4 magnetite nanoparticles, which could be magnetically separated from oil/water biphasic system conveniently. The magnetite nanoparticle–coated R. erythropolis FMF and R. erythropolis IGTS8 cells exhibited similar desulfurization activity with the free cells (67 ± 3 and 69 ± 4, respectively). Furthermore, the coated Fe3O4 nanoparticles facilitate the recovery of cells from the biodesulfurization systems and then increase the recycled times of the immobilized cells.
6.2 Bioreactor Design
A vertical rotating immobilized cell reactor (VRICR) was designed and investigated for its BDS activity with the R. erythropolis (Amin 2011). The maximum desulfurization rate was up to 167 mM 2-HBP/Kg/h, and 100% of sulfur could be removed from the model oil (dibenzothiophene in hexadecane) within a 120 h period. Another research studied the influence of bed lengths and support particle size on the desulfurization efficiency of immobilized R. rhodochrous cells in a catalytic bed reactor packed with silica. The results suggested that longer bed, lower substrate flow, and large particle size would be benefited for the desulfurization (Alejandro et al. 2014).
To increase the utilizing efficiency of bacteria cells for desulfurization, a new aqueous–organic two-layer partitioning and continuous process was designed. Different from the batch and fed-batch processes, the biphasic bioreactor showed the efficient biodesulfurization activity for a long period (Yang et al. 2007). In this bioreactor, the oil and the cells were kept in organic and aqueous phase, respectively. DBT transferred from the oil phase to the aqueous phase and desulfurized by the bacterial cells. And then the produced 2-HBP was washed out of the bioreactor to reduce the inhibition.
7 Future Perspectives
Our understanding of how microorganisms metabolize sulfur heterocyclic compounds in petroleum has increased rapidly. All the studies outlined above are significant steps to explore the biotechnological potential for developing an efficient biodesulfurization process. However, these technologies have not yet been available for large-scale applications. We still need a much better understanding of more aspects of this pathway to turn it into a commercial process earlier. Any progress that provides the possibility to remove sulfur in crude oil at higher temperature, with higher rate, or longer stability of desulfurization activity is considered to be a significant step toward industry level biodesulfurization. Microorganisms with a wider substrate range and higher substrate affinity in biphasic reaction containing toxic solvents or higher biodesulfurization activities could be engineered if the biocatalysts were to be used for petroleum treatment.
References
Albano C, Randers-Eichhorn L, Bentley W, Rao G (1998) Green fluorescent protein as a real time quantitative reporter of heterologous protein production. Biotechnol Prog 14:351–354
Alejandro DM, Orellana L, Aguirre J, Baeza P, Espinoza G, Canales C, Ojeda J (2014) Biodesulfurization of dibenzothiophene and gas oil using a bioreactor containing a catalytic bed with Rhodococcus rhodochrous immobilized on silica. Biotechnol Lett 36:1649–1652
Amin GA (2011) Integrated two-stage process for biodesulfurization of model oil by vertical rotating immobilized cell reactor with the bacterium Rhodococcus erythropolis. J Pet Environ Biotechnol 2:107–111
Arensdorf JJ, Loomis AK, DiGrazia PM, Monticello DJ, Pienkos PT (2002) Chemostat approach for the directed evolution of biodesulfurization gain-of-function mutants. Appl Environ Microbiol 68:691–698
Atlas RM (1994) Petroleum microbiology. Macmillan Publishing, New York
Atlas RM, Boron DJ, Deever WR, Johnson AR, McFarland BL, Meyer JA (2001) Method for removing organic sulfur from heterocyclic sulfur containing organic compounds. US patent number H1, p 986
Borgne S, Quintero R (2003) Review: biotechnological processes for the refining of petroleum. Fuel Process Technol 81:155–169
Cha H, Wu C, Valdes J, Rao G, Bentley W (2000) Observations of green fluorescent protein as a fusion partner in genetically engineered Escherichia coli: monitoring protein protein expression and solubility. Biotechnol Bioeng 67:565–574
Chen H, Zhang WJ, Chen JM, Cai YB, Li W (2008) Desulfurization of various organic sulfur compounds and the mixture of DBT + 4,6-DMDBT by Mycobacterium sp. ZD-19. Bioresour Technol 99:3630–3634
Coco WM, Levinson WE, Crist MJ, Hektor HJ, Darzins A, Pienkos PT, Squires CH, Monticello DJ (2001) DNA shuffling method for generating highly recombined genes and evolved enzymes. Nat Biotechnol 19:354–359
Darzins A, Xi L, Childs JD, Monticello DJ, Squires CH (1999) DSZ gene expression in pseudomonas hosts. US Patent No 5952208
Denis-Larose C, Labbe D, Bergeron H, Jones AM, Greer CW, al-Hawari J, Grossman MJ, Sankey BM, Lau PC (1997) Conservation of plasmid-encoded dibenzothiophene desulfurization genes in several Rhodococci. Appl Environ Microbiol 63:2915–2919
Derikvand P, Etemadifar Z (2014) Improvement of biodesulfurization rate of alginate immobilized Rhodococcus erythropolis R1. Jundishapur J Microbiol 7:e9123
Derikvand P, Etemadifar Z, Biria D (2014) Taguchi optimization of dibenzothiophene biodesulfurization by Rhodococcus erythropolis, R1 immobilized cells in a biphasic system. Int Biodeter Biodegr 86:343–348
Dinamarca MA, Rojas A, Baeza P, Espinoza G, Ibacache-Quiroga C, Ojeda J (2014) Optimizing the biodesulfurization of gas oil by adding surfactants to immobilized cell systems. Fuel 116:237–241
Dosomer JP, Dhaese P, Montagu MV (1988) Conjugative transfer of cadmium resistance plasmids in Rhodococcus fascians strains. J Bacteriol 170:2401–2405
Feng J, Zeng Y, Ma C, Cai X, Zhang Q, Tong M, Yu B, Xu P (2006) The surfactant tween 80 enhances biodesulfurization. Appl Environ Microbiol 72:7390–7393
Galán B, Díaz E, García JL (2000) Enhancing desulfurization by engineering a flavin reductase-encoding gene cassette in recombinant biocatalyst. Environ Microbiol 2:687–694
Gallagher JR, Olson ES, Stanley DC (1993) Microbial desulphurization of dibenzothiophene: a sulfur-specific pathway. FEMS Microbiol Lett 107:31–36
Gallardo ME, Ferrandez A, De Lorenzo V, Garcia JL, Diaz E (1997) Designing recombinant Pseudomonas strains to enhance biodesulfurization. J Bacteriol 179:7156–7160
Gaudu P, Touati D, Niviere V, Fontecave M (1994) The NAD(P)H: flavin oxidoreductase from Escherichia coli as a source of superoxide radicals. J Biol Chem 269:8182–8185
Gilbert SC, Morton J, Buchanan S, Oldfield C, McRoberts A (1998) Isolation of a unique benzothiophene-desulphurizing bacterium, Gordona sp. strain 213E (NCIMB 40816), and characterization of the desulphurization pathway. Microbiology 144:2545–2553
Gray KA, Pogrebinsky OS, Mrachko GT, Xi L, Monticello DJ, Squires CH (1996) Molecular mechanisms of biocatalytic desulfurization of fossil fuels. Nat Biotechnol 14:1705–1709
Gray KA, Mrachkoyz GT, Squiresy CH (2003) Biodesulfurization of fossil fuels. Curr Opin Microbiol 6:229–235
Grossman MJ, Lee MK, Prince RC, Garrett KK, George GN, Pickering IJ (1999) Microbial desulfurization of a crude oil middle-distillate fraction: analysis of the extent of sulfur removal and the effect of removal on remaining sulfur. Appl Environ Microbiol 65:181–188
Gupta N, Roychoudhury PK, Deb JK (2005) Biotechnology of desulfurization of diesel: prospects and challenges. Appl Microbiol Biotechnol 66:356–366
Hassan B, Jamshid R, Hossein M, Behnam R, Ayyoob A (2013) Desulfurization activity and reusability of magnetite nanoparticle-coated Rhodococcus erythropolis FMF and R. erythropolis IGTS8 bacterial cells. Biotechnol Appl Biochem 60:323–329
Hirasawa K, Ishii Y, Kobayashi M, Koizumi K, Maruhashi K (2001) Improvement of desulfurization activity in Rhodococcus erythropolis KA2-5-1 by genetic engineering. Biosci Biotechnol Biochem 65:239–246
Inoue A, Horikoshi K (1991) Estimation of solvent-tolerance of bacteria by the solvent parameter log P. J Ferment Bioeng 71:194–196
Izumi Y, Ohshiro T, Ogino H, Hine Y, Shinao M (1994) Selective desulphurisation of dibenzothiophene by R. erythropolis D-1. Appl Environ Microbiol 60:223–226
Ji YE, Colston MJ, Cox RA (1994) The ribosomal RNA (rrn) operons of fast-growing mycobacteria: primary and secondary structures and their relation to rrn operons of pathogenic slowgrowers. Microbiology 140:2829–2840
Kawatra SK, Eisele TC (2001) Coal desulfurization, high-efficiency preparation methods. Taylor & Francis, New York
Kayser KJ (2002) Molecular biological characterization and enhancement of the biodesulfurization (DSZ) pathway. PhD thesis. Illinois Institute of Technology, 106 pp
Kertesz L (2001) Building a scientific foundation for prevention. Healthplan 42:44–47
Kilbane JJ (2006) Microbial biocatalyst developments to upgrade fossil fuels. Curr Opin Biotechnol 17:305–314
Kilbane JJ, Bielaga BA (1990) Toward sulfur-free fuels. Chem Tech 20:747–751
Kilbane JJ, Jackowski K (1992) Biodesulphurisation of watersoluble coal-derived material by Rhodococcus rhodochrous IGTS8. Biotechnol Bioeng 40:1107–1114
Kirimura K, Furuya T, Sato R, Ishii Y, Kino K, Usami S (2002) Biodesulfurization of naphthothiophene and benzothiophene through selective cleavage of carbon-sulfur bonds by Rhodococcus sp. strain WU-K2R. Appl Environ Microbiol 68:3867–3872
Kobayashi M, Onaka T, Ishii Y, Konishi J, Takaki M, Okada H, Ohta Y, Koizumi K, Suzuki M (2000) Desulfurization of alkylated forms of both dibenzothiophene and benzothiophene by single bacterial strain. FEMS Microbiol Lett 187:123–126
Koike H, Sasaki H, Kobori T, Zenno S, Saigo K, Murphy MEP, Adman ET, Tanokura M (1998) 1.8Å crystal structure of the major NAD-(P)H:FMN oxidoreductase of a bioluminescent bacterium, Vibrio fischeri: overall structure, cofactor and substrate- analog binding, and comparison with related flavoproteins. J Mol Biol 280:259–273
Konishi J, Onaka T, Ishii Y, Suzuki M (2000) Demonstration of the carbon-sulfur bond-targeted desulfurization of benzothiophene by thermophile Paenibacillus sp. strain A11-2 capable of desulfurizing dibenzothiophene. FEMS Microbiol Lett 187:151–154
Kropp KG, Fedorak PM (1998) A review of the occurrence, toxicity, and biodegradation of condensed thiophenes found in petroleum. Can J Microbiol 44:605–622
Kropp P, Gerber WD (1998) Prediction of migraine attacks using a slow cortical potential, the contingent negative variation. Neurosci Lett 257:73–76
Leahy JG, Colwell RR (1990) Microbial degradation of hydrocarbons in the environment. Microbiol Rev 54:305–315
Lee WC, Ohshiro T, Matsubara T, Izumi Y, Tanokura M (2004) Crystallization and preliminary X-ray analyses of desulfurization enzyme DszB and its C27S mutant complexed with biphenyl-2-sulfinic acid. Acta Crystallogr D Biol Crystallogr 60:1636–1638
Lee WC, Ohshiro T, Matsubara T, Izumi Y, Tanokura M (2006) Crystal structure and desulfurization mechanism of 2′-hydroxybiphenyl-2- sulfinic acid desulfinase. J Boil Chem 281:32534–32539
Lei X, Squires CH, Monticello DJ, Child D (1997) A flavin reductase stimulates DszA and DszC proteins of Rhodococcus erythropolis IGTS8 in vitro. Biochem Biophys Res Commun 230:73–75
Li ZM, Squires CH, Monticello DJ, Childs JD (1996) Genetic analysis of the dsz promoter and associated regulatory region of Rhodococcus erythropolis IGTS8. J Bacteriol 178:6409–6418
Li FL, Xu P, Ma CQ, Luo LL, Wang XS (2003) Deep desulfurization of hydrodesulfurization treated diesel oil by a facultative thermophilic bacterium Mycobacterium sp. X7B. FEMS Microbiol Lett 223:301–307
Li W, Zhang Y, Wang MD, Shi Y (2005a) Biodesulfurization of dibenzothiophene and other organic sulfur compounds by a newly isolated Microbacterium strain ZD-M2. FEMS Microbiol Lett 247:45–50
Li FL, Xu P, Feng JH, Meng L, Zheng Y, Luo LL, Ma CQ (2005b) Microbial desulfurization of gasoline in a Mycobacterium goodii X7B immobilized-cell system. Appl Environ Microbiol 71:276–281
Li GQ, Ma T, Li JH, Li H, Liu RL (2006a) Co-expression of Rhodococcus sp. DS-3 dszABC and dszD gene with incompatible plasmids in Escherichia coli. Acta Microbiol Sin 46:275–279
Li W, Wang MD, Chen H, Chen JM, Shi Y (2006b) Biodesulfurization of dibenzothiophene by growing cells of Gordonia sp. in batch cultures. Biotechnol Lett 28:1175–1179
Li GQ, Ma T, Li SS, Li H, Liang FL, Liu RL (2007a) Improvement of dibenzothiophene desulfurization activity by removing the gene overlap in the dsz operon. Biosci Biotechnol Biochem 71:849–854
Li FL, Zhang ZZ, Feng JH, Cai XF, Xu P (2007b) Biodesulfurization of DBT in tetradecane and crude oil by a facultative thermophilic bacterium Mycobacterium goodii X7B. J Biotechnol 127:222–228
Lorenzo V, Timmis KN (1994) Analysis and construction of stable phenotypes in Gram-negative bacteria with Tn5 and Tn10-derived minitransposons. Methods Enzymol 235:386–405
Ma X, Sakanishi K, Mochida I (1994) Hydrodesulfurization reactivities of various sulfur compounds in diesel fuel. Ind Eng Chem Res 33:218–222
Ma CQ, Feng JH, Zeng YY, Cai XF, Sun BP, Zhang ZB, Blankespoor HD, Xu P (2006a) Methods for the preparation of a biodesulfurization biocatalyst using Rhodococcus sp. Chemosphere 65:165–169
Ma T, Li GQ, Li J, Liang FL, Liu RL (2006b) Desulfurization of dibenzothiophene by Bacillus subtilis recombinants carrying dszABC and dszD genes. Biotechnol Lett 28:1095–1100
Matsubara T, Ohshiro T, Nishina Y, Izumi Y (2001) Purification, characterization, and overexpression of flavin reduced involved in dibenzothiophene desulfurization by Rhodococcus erythropolis D-1. Appl Environ Microbiol 67:1179–1184
Matsui T, Hirasawa K, Koizumi KI, Maruhashi K, Kurane R (2001) Optimization of the copy number of dibenzothiophene desulfurizing genes to increase the desulfurization activity of recombinant Rhodococcus sp. Biotechnol Lett 23:1715–1718
Matsui T, Noda K, Tanaka Y, Maruhashi K, Kurane R (2002) Recombinant Rhodococcus sp. strain T09 can desulfurize DBT in the presence of inorganic sulfate. Curr Microbiol 45:240–244
McFarland BL, Boron DJ, Deever W, Meyer JA, Johnson AR, Atlas RM (1998) Biocatalytic sulfur removal from fuels: applicability for producing low sulfur gasoline. Crit Rev Microbiol 24:99–147
Monticello DJ (1998) Riding the fossil fuel biodesulfurization wave. Chem Tech 28:38–45
Monticello DJ, Bakker D, Finnerty WR (1985) Plasmid-mediated degradation of dibenzothiophene by Pseudomonas species. Appl Environ Microbiol 49:756–760
Noda K, Kimiko W, Kenji M (2002) Cloning of a rhodococcal promoter using a transposon for dibenthiophene biodesulfurization. Biotechnol Letters 24:1875–1882
Noda K, Watanabe K, Maruhashi K (2003) Isolation of the Pseudomonas aeruginosa gene affecting uptake of dibenzothiophene in n-tetradecane. J Biosci Bioeng 95:504–511
Ochsner UA, Reiser J, Fiechter A, Witholt B (1995) Production of Pseudomonas aeruginosa rhamnolipid biosurfactants in heterologous hosts. Appl Environ Microbiol 61:3503–3506
Ohshiro T, Izumi Y (1999) Microbial desulfurization of organic sulfur compounds in petroleum. Biosci Biotechnol Biochem 63:l–9
Ohshiro T, Izumi Y (2000) Purification, characterization and crystallization of enzymes for dibenzothiophene desulfurization. Bioseparation 9:185–188
Ohshiro T, Hine Y, Izumi Y (1994) Enzymatic desulfurization of dibenzothiophene by a cell-free system of Rhodococcus erythropolis D-1. FEMS Microbiol Lett 118:341–344
Ohshiro T, Hirata T, Izumi Y (1995) Microbial desulfurization of dibenzothiophene in the presence of hydrocarbon. Appl Microbiol Biotechnol 44:249–252
Ohshiro T, Kojima T, Torii K, Kawasoe H, Izumi Y (1999) Purification and characterization of dibenzothiophene (DBT) sulfone monooxygenase, an enzyme involved in DBT desulfurization, from Rhodococcus erythropolis D-l. J Biosci Bioeng 88:610–616
Ohshiro T, Ohkita R, Takikawa T, Manabe M, Lee WC, Tanokura M, Izumi Y (2007) Improvement of 2′-Hydroxybiphenyl-2-sulfinate Desulfinase, an Enzyme Involved in the Dibenzothiophene Desulfurization Pathway, from Rhodococcus erythropolis KA2-5-1 by Site-Directed Mutagenesis. Biosci Biotechnol Biochem 71:2815–2821
Oldfield C, Pogrebinsky O, Simmonds J, Olson ES, Kulpa CF (1997) Elucidation of the metabolic pathway for dibenzothiophene desulfurization by Rhodococcus sp. IGTS8 (ATCC 53968). Microbiology 143:2961–2973
Oldfield C, Wood NT, Gilbert SC, Murray FD, Faure FR (1998) Desulfurization of benzothiophene by actinomycete organisms belonging to the genus Rhodococcus, and related taxa. Antonie Van Leeuwenhoek 74:119–132
Omori T, Monna L, Saiki Y, Kodama T (1992) Desulfurization of dibenzothiophene by Corynebacterium sp. Strain SY1. Appl Environ Microbiol 58:911–915
Piddington CS, Kovacevich BR, Rambosek J (1995) Sequence and molecular characterization of a DNA region encoding the dibenzothiophene desulfurization operon of Rhodococcus sp. strain IGTS8. Appl Environ Microbiol 61:468–475
Purdy RF, Lepo JE, Ward B (1993) Biodesulfurization of organicsulfur compounds. Curr Microbiol 27:219–222
Reichmuth DS, Hittle JL, Blanch HW, Keasling JD (1999) Biodesulfurization of dibenzothiophene in Escherichia coli is enhanced by expression of Vibrio harveyi oxidoreductase gene. Biotechnol Bioeng 67:72–79
Reichmuth DS, Blanch HW, Keasling JD (2004) Dibenzothiophene biodesulfurization pathway improvement using diagnostic GFP fusions. Biotechnol Bioeng 88:94–99
Rhee SK, Chang JH, Chan YK, Chang HN (1998) Desulfurization of dibenzothiophene and diesel oils by a newly isolated Gordona strain, CYKS1. Appl Environ Microbiol 64:2327–2331
Shan GB, Xing JM, Luo MF, Liu HZ, Chen JY (2003) Immobilization of Pseudomonas delafieldii with magnetic polyvinyl alcohol beads and its application in biodesulfurization. Biotechnol Lett 25:1977–1981
Shennan JL (1996) Microbial attack on sulfur-containing hydrocarbons, implications for the biodesulphurization of oils and coals. J Chem Technol Biotechnol 67:109–123
Soleimani M, Bassi A (2007) Biodesulfurization of refractory organic sulfur compounds in fossil fuels. Biotechnol Adv 25:570–596
Tanaka Y, Matsui T, Konishi J, Maruhashi K, Kurane R (2002) Biodesulfurization of benzothiophene and dibenzothiophene by a newly isolated Rhodococcus strain. Appl Microbiol Biotechnol 59:325–328
Tao F, Yu B, Xu P, Ma CQ (2006) Biodesulfurization inbiophasic systems containing organic solvents. Appl Environ Microbiol 72:4604–4609
Watkins LM, Rodriguez R, Schneider D, Broderick R, Cruz M, Chambers R, Ruckman E, Cody M, Mrachko GT (2003) Purification and characterization of the aromatic desulfinase, 2-(2′-hydroxyphenyl) benzenesulfinate desulfinase. Arch Biochem Biophys 415:14–23
Xi L, Squires CH, Monticello DJ, Childs JD (1997) A flavin reductase stimulates DszA and DszC proteins of Rhodococcus erythropolis IGTS8 in vitro. Biochem Biophys Res Commun 230:73–75
Yan H, Kishimoto M, Omasa T, Katakura Y, Suga K, Okumura K, Yoshikawa O (2000) Increase in desulfurization activity of Rhodococcus erythropolis KA2-5-1 using ethanol feeding. J Biosci Bioeng 89:361–366
Yang J, Hu Y, Zhao D, Wang S, Lau PCK, Marison IW (2007) Two-layer continuous-process design for the biodesulfurization of diesel oils under bacterial growth conditions. Biochem Eng J 37:212–218
Yu B, Xu P, Shi Q, Ma C (2006a) Deep desulfurization of diesel oil and crude oils by a newly isolated Rhodococcus erythropolis strain. Appl Environ Microbiol 72:54–58
Yu B, Ma CQ, Zhou WJ, Wang Y, Cai XF, Tao F, Zhang Q, Tong MY, Qu JY, Xu P (2006b) Microbial desulfurization of gasoline by free whole-cells of Rhodococcus erythropolis XP. FEMS Microbiol Lett 258:284–289
Acknowledgements
We gratefully acknowledge the support for our research from the Natural Science Foundation of Tianjin, China (grant numbers 05YFJMJC00700, 09JCZDJC03200). We also thank the publisher of the American Society of Microbiology and the American Society for Biochemistry and Molecular Biology to offer kindly help with useful pictures.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Li, S., Ma, T. (2019). The Desulfurization Pathway in Rhodococcus. In: Alvarez, H. (eds) Biology of Rhodococcus. Microbiology Monographs, vol 16. Springer, Cham. https://doi.org/10.1007/978-3-030-11461-9_8
Download citation
DOI: https://doi.org/10.1007/978-3-030-11461-9_8
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-11460-2
Online ISBN: 978-3-030-11461-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)