
Abstract
Acquired immunodeficiency syndrome (AIDS) is a life-threatening disorder caused by infection of individuals with the human immunodeficiency virus (HIV). Entry of HIV-1 into target cells depends on the presence of two surface proteins on the cell membrane: CD4, which serves as the main receptor, and either CCR5 or CXCR4 as a co-receptor. A limited number of people harbor a genomic 32-bp deletion in the CCR5 gene (CCR5∆32), leading to expression of a truncated gene product that provides resistance to HIV-1 infection in individuals homozygous for this mutation. Moreover, allogeneic hematopoietic stem cell (HSC) transplantation with CCR5∆32 donor cells seems to confer HIV-1 resistance to the recipient as well. However, since Δ32 donors are scarce and allogeneic HSC transplantation is not exempt from risks, the development of gene editing tools to knockout CCR5 in the genome of autologous cells is highly warranted. Targeted gene editing can be accomplished with designer nucleases, which essentially are engineered restriction enzymes that can be designed to cleave DNA at specific sites. During repair of these breaks, the cellular repair pathway often introduces small mutations at the break site, which makes it possible to disrupt the ability of the targeted locus to express a functional protein, in this case CCR5. Here, we review the current promise and limitations of CCR5 gene editing with engineered nucleases, including factors affecting the efficiency of gene disruption and potential off-target effects.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Targeted gene editing
- Genome engineering
- CCR5 disruption
- HIV resistance
- HIV therapy
- Designer nucleases
- Zinc-finger nucleases
- TALEN
- CRISPR/Cas9
Introduction
As HIV research continues to identify novel druggable viral and host factors that promote virulence and latency, the long-term clinical management and survival of HIV-positive individuals has improved considerably. Combination therapies, like highly active antiretroviral therapy (HAART), continuously suppress HIV replication while attenuating the development of escape mutants. However, because HAART is unable to clear latent viral reservoirs [1, 2], patients require lifelong treatment, which not only is expensive but has been associated with multiple adverse side effects and the development of drug-induced diseases [3–5]. The sustained antiviral efficacy of these therapeutic regimens is also strongly influenced by the compliance of each patient, which remains a key factor in managing not only the HIV infection but also the development of any accompanying disease [6]. Ideally, a therapy aimed at eliminating both the replicating and latent viral populations would provide a long awaited cure.
HIV-1 fusion with the cell membrane and ensuing virus entry is an intricate process that requires the expression of both the CD4 transmembrane glycoprotein as well as an associated seven-pass G-protein coupled chemokine co-receptor, CCR5 or CXCR4 (Fig. 1), a receptor combination typically found on CD4+ T cells, macrophages and dendritic cells [7]. Virus attachment is mediated by gp120, a viral surface glycoprotein located in the lipid membrane of the HIV-1 virion. Initially gp120 binds to CD4, which then facilitates the sequential attachment of gp120 to either the CCR5 or CXCR4 co-receptors. R5-tropic viruses, most prominently detected during the early stages of HIV-1 infection, bind to the CCR5 co-receptor, whilst X4-tropic viruses bind to CXCR4. The subsequent conformational change of the viral envelope protein exposes the viral gp41 glycoprotein, which mediates fusion with the target cell membrane. The resulting formation of a transmembrane pore enables the delivery of the viral capsid, which initiates viral integration and replication. Whereas the majority of the population is susceptible to infection, a small percentage of individuals are protected from infection with particular HIV strains. This resistance to HIV infection has been linked to naturally occurring genetic variations, including polymorphisms within the locus encoding the CCR5 co-receptor [8–14]. As a consequence, rational design of novel therapeutic strategies has also focused on blocking viral entry with small molecule drugs or genetic engineering to generate HIV-resistant T cells.

Attachment and entry of R5-tropic HIV-1. (a) The HIV virion initially binds to target cells through interactions between the viral gp120 surface glycoprotein and the CD4 receptor. The CD4 receptor then draws the virion closer to the target cell, facilitating the interaction between the CCR5 co-receptor and gp120. This triggers a conformational change, allowing the gp41 glycoprotein to fuse to the cell membrane in order to create a transmembrane pore. The viral capsid, which contains the HIV RNA, integrase and reverse transcriptase, is then released into the target cell. (b) Initial binding of the HIV-1 virion occurs as described above; however, the CCR5Δ32 mutant form of this co-receptor is severely truncated and remains cytosolic, ultimately eliminating the gp120 binding site. As there is no co-receptor binding, the conformational change required to expose the gp41 protein is blocked, preventing viral fusion and entry
CCR5 as a Target for HIV Antiretroviral Therapy
CCR5 was first identified as the prominent co-receptor for R5-tropic viruses following the discovery that three chemokines, RANTES (CCL5), MIP-1α (CCL3), and MIP-1β (CCL4), impede HIV-1 binding [15]. Ever since, pharmaceutical companies have focused heavily on the development of HIV antiretroviral therapies based on entry and fusion inhibitors. One such drug, Maraviroc, binds to the transmembrane domains of CCR5, ultimately preventing viral attachment and fusion [16]. Based on successful clinical trials, Maraviroc has been approved for HIV-1 treatment in both Europe and the USA. However, as with most currently available HIV therapies, viral escape mutants have been isolated [17–19]. Furthermore, as Maraviroc binds to the CCR5 co-receptor, it is not effective against X4-tropic viral infections.
Moving away from traditional HAART therapy, the adoptive transfer of synergistic T cells and allogeneic stem cells has been investigated as potential curative treatments. Initially, the efficacy of synergistic or autologous transplantation of hematopoietic stem/progenitor cells (HSPCs) was described in HIV-positive patients that had developed lymphomas [20–23]. Whilst patients remained on antiviral therapy, the myeloablative conditioning required prior to transplantation facilitated the reconstitution of the T cell compartment. In 2007, Hoffmann and colleagues reported that the adoptive transfer of T cells between HIV-1 discordant twins resulted in improved CD4+ T cell counts [24]. The patients remain on antiretroviral therapy and required a total of 12 transfers to achieve a sustained expansion of CD4+ cells. Since myeloablative conditioning was not performed before adoptive transfers, these results suggest that HLA-matched T cells could help reprise aspects of the immune system, provided that HIV viral loads are continuously repressed. Nonetheless, neither approach is curative, as patients still require continuous antiretroviral therapy post-transplantation.
In contrast, an allogeneic HSPC transplantation from a donor homozygous for the CCR5Δ32 mutation has given rise to the first described permanent “cure” for HIV [25–27]. The CCR5Δ32 mutation was originally identified in a small group of people who, despite being repeatedly exposed to HIV, did not contract the disease [12, 13]. This 32 base pair deletion in the CCR5 gene induces a frameshift mutation and the resulting truncated protein does not support gp120 binding, ultimately preventing HIV-1 infection (Fig. 1). Although individuals who are homozygous for this mutation are resistant to R5-tropic HIV-1 infection, they remain susceptible to X4-tropic strains [28]. A number of studies showed that HIV-positive patients, who are heterozygous for the CCR5Δ32 mutation, have reduced disease progression and better overall prognosis than patients who are homozygous for the wild-type CCR5 gene [10, 11, 29, 30]. In 2009, Hütter and colleagues described the first curative allogeneic HSPC transplantation using an HLA-matched donor who was homozygous for the CCR5Δ32 mutation [26]. Timothy Brown (alternatively referred to as the “Berlin patient”), an HIV-positive patient on HAART therapy, received the initial HSPC transplant after developing acute myeloid leukemia (AML), which was refractory to induction and consolidation chemotherapy. As his AML relapsed, a second HSPC transplantation from the same homozygous CCR5Δ32 donor was performed. To date, the patient remains cancer-free and HIV negative in the absence of HAART [25–27], suggesting that homozygous CCR5Δ32 HSPC transplantation could be used to cure not only the blood-related malignancy but also HIV-1 infection. Although this presents an idealistic approach, the number of homozygous CCR5Δ32 donors is low, since only approximately 1 % of the Caucasian population has this HIV-1-resistant genotype [30, 31]. Accordingly, much research has focused on engineering homozygous CCR5Δ32-like mutations in patient-derived HSPCs and T cells using designer nucleases.
Gene Editing with Designer Nucleases
Designer nucleases are engineered enzymes that are comprised of a DNA binding domain, tailored to bind to a specific target sequence, and a DNA cleavage domain (Fig. 2). Binding of the engineered nuclease to a defined genomic target site results in the formation of a DNA double stranded break (DSB) which, in turn, elicits cellular DNA repair mechanisms that can be exploited to achieve targeted and permanent genetic modifications. Mammalian cells rely on two major DSB repair pathways: non-homologous end joining (NHEJ), which is active throughout the cell cycle, and homologous recombination (HR) based repair, which is restricted to the S/G2 phase. As compared to HR, NHEJ is an error-prone pathway, which can be harnessed to insert small insertion/deletion (indel) mutations at the DNA break in order to inactivate a target gene, such as CCR5. Conversely, HR relies on the genetic information contained in the sister chromatid for the accurate repair of a DSB. For gene editing, this pathway can be exploited by including a donor DNA template with specific sequence homology during the generation of nuclease-mediated DSBs [32, 33]. In this setting, the genetic information is transferred from the donor DNA to the target locus, thus allowing precise genomic modifications.

Designer nucleases to disrupt CCR5. (a) Schematic of the CCR5 protein localized to the cellular membrane. The dotted boxes indicate the corresponding regions of the genomic locus targeted by designer nucleases as well as the location of the ∆32 deletion. Three different designer nuclease platforms have been efficiently engineered to knock out CCR5 and the corresponding DNA target sites are indicated in green (RGN), light blue (TALEN), and orange (ZFN). The putative cleavage sites are indicated (black triangles) (b) Designer nucleases. RGENs are composed of the Cas9 nuclease and a guide RNA (gRNA) that directs the enzyme to the target site. The protospacer adjacent motive (PAM) required by the Cas9 enzyme to recognize and cleave the target site is indicated in red. The two nuclease domains within the Cas9 protein (RuvC and HNH) are highlighted. TALEN or ZFN monomers include a modular DNA binding domain that is engineered to recognize a specific DNA target sequence. Each TALE module specifically recognizes one nucleotide in the target subsite, while a ZF module binds to a nucleotide triplet. A short linker connects the respective DNA binding domain to the cleavage domain of the FokI restriction enzyme (light red), which cuts the DNA upon dimerization of the two monomers at the target site
Dimeric zinc-finger nucleases (ZFNs) have been traditionally used for genetic modifications [34]. The DNA binding domain is comprised of multiple zinc-finger modules, each recognizing three to four nucleotides in a sequence-specific manner. However, generating highly active ZFNs with novel specificities is challenging and cumbersome, as context-dependent interactions between individual modules within the zinc-finger array affect the overall binding efficiency [35]. In the last 15 years, ZFNs have been successfully used in basic research to study gene function [36–44] and to correct genetic defects underlying human disorders for therapeutic purposes [45–47] in preclinical settings. Their relatively small size has allowed ZFNs to be delivered using the most common viral and non-viral platforms as well as a direct protein delivery [48].
For therapeutic applications, a high specificity of the designer nuclease is of utmost importance, as off-target cleavage activity poses obvious concerns with regard to genotoxicity. Two studies assessing the genome-wide specificity of the CCR5-specific ZFN pair revealed a considerable level of non-specific off-target activity [49, 50]. In view of the complexity of generating highly specific ZFNs, the discovery of a novel modular DNA binding domain identified in transcription activator-like effectors (TALEs) of plant pathogens has provided new momentum to the genome engineering field. TALE-based nucleases (TALENs) can be easily customized to target any given sequence (Fig. 2) due to their simple recognition code in which a TALE module specifically recognizes one nucleotide [51–53]. When compared to an existing ZFN, some CCR5-specific TALENs showed similar activity but lower cytotoxicity [54, 55]. While more work needs to be invested to dissect the specificity signature of designer nucleases, initial results suggest that TALENs seem to harbor a rather high specificity [55–57]. TALENs have hence evolved as a valid alternative for the generation of transplantable HIV-resistant T cells. Unlike ZFNs, TALENs are relatively large proteins with a highly repetitive structure. While adenoviral vectors can be used to deliver single TALEN monomers, lentiviral vectors have failed to transfer intact TALEN encoding expression cassettes [58]. As a consequence, many labs have relied on in vitro transcribed mRNA or plasmid DNA to deliver the TALENs.
The newest addition to the toolbox for genome engineers is of bacterial origin as well. The clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 system is used by prokaryotes to defend themselves against invading DNA [59]. It consists of the Cas9 cleavage enzyme complexed to a guide RNA strand that directs the enzyme to a 20-nucleotide long target site [60, 61]. Exchanging a specific portion of the gRNA molecule allows researchers to redirect the Cas9 cleavage activity to a user-defined target sequence (Fig. 2). This versatile platform, also known as RNA-guided nuclease (RGN) technology, holds many advantages over both ZFNs and TALENs. The most obvious one is the simplicity to customize the enzyme to target any sequence of choice by simple molecular cloning techniques [62, 63]. Moreover, delivering the Cas9 protein with more than one gRNA molecule allows multiplexing, i.e., to target several sites simultaneously [64]. Although RGNs have been shown to target CCR5 efficiently [65, 66], concerns regarding their specificity have been raised [67–69]. On the other hand, further advances, such as Cas9 nickases [70], the use of truncated guide RNAs [71], and dimeric RNA-guided FokI nucleases [72], have shown promise to generate more specific RGNs.
Target Cells
Two potential cellular targets have been envisioned for a CCR5 disruption-based HIV therapy: CD4+ T cells, which are the mature lymphocytes infected by HIV, or CD34+ HSPCs, which would give rise to HIV-resistant T cells and macrophages.
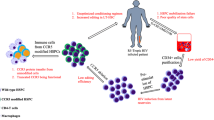
In the first scenario, patient derived CD4+ T cells will be collected by apheresis and modified ex vivo using designer nucleases [47]. Modified cells will then be amplified in vitro and subsequently reintroduced in the patient (Fig. 3). For the therapy to be effective, a large number of cells are required to retain proficient proliferative and effector functions. Consequently, patients enrolled in such trials should have a CD4+ T cell count above a set threshold that allows collection of enough CD4+ T cells to be genetically altered and subsequently expanded ex vivo. Transfer of the CCR5 modified T cells will at least temporarily restore T cell immunity of the patients. Discontinuation of antiretroviral medication would allow the virus to infect and replicate in susceptible cells. Over time, only cells devoid of CCR5 will be able to expand in presence of the virus. Once the modified pool of T cells is depleted as a result of cellular senescence, the transfer of modified T cells can be repeated. Of note, the CCR5 disrupted cells remain susceptible to CXCR4-tropic strains and discontinuing HAART could result in a flare of these X4-tropic strains. Importantly, however, a viral rebound was not observed in the “Berlin patient” although he was positive for CXCR4-tropic strains [26, 73]. Nonetheless, to overcome this potential limitation, a simultaneous disruption of CCR5 and CXCR4 has been reported in primary CD4+ T cells, and protection from both R5 and X4-tropic virus was verified in a mouse model [74].

Clinical application of modified T cells and CD34+ cells. After collection of cells by apheresis, CD4+ T cells or CD34+ hematopoietic stem cells are enriched and CCR5 disruption is accomplished by expression of designer nuclease. T cells are expanded ex vivo before adaptive transfer. In case of CD34+ cells, chemotherapy of AIDS lymphoma patients will assist the engraftment of the modified cells
The second approach is directed towards the targeting and manipulation of CD34+ HSPCs [45]. The main advantage of this strategy when compared to CD4+ T cell targeting is the ability of modified CD34+ cells to engraft and produce a long-lasting effect. HSPCs continuously differentiate in all the hematopoietic lineages, including T cells and macrophages that can be infected by HIV. The downside is that stem cells are difficult to manipulate and tend to lose their differentiation potential when cultured ex vivo. In addition, transplantation of HSPCs requires a mild preconditioning regimen to provide adequate space in the bone marrow for engraftment of the modified HSPCs. In this setting, leukopoiesis will occur from both modified and non-modified CD34+ cells, and the survival advantage in the presence of replicating HIV will occur on the level of CD4+ T cells and macrophages.
The advantages of the two approaches are apparent: since the genetic modification is performed in autologous cells, there is no need for HLA matching, which significantly decreases the risk of developing graft-versus-host-disease or graft rejection. Additionally, there is no need for post-transplantation immunosuppressive therapy. The patients will be provided with an autologous pool of HIV-resistant cells, which restores the immune system either transiently or permanently. An open question is whether active clearance of HIV reservoirs will occur in an autologous setting where the graft-versus-host effect is not present.
Applying Designer Nucleases for HIV Gene Therapy
Many HIV gene therapy trials based on the ex vivo modification of CD4+ T cells or HSPCs have used ribozymes, aptamers, and siRNAs [75]. Although none of these studies have reported clinical benefit in terms of decreased viral load or protection from HIV replication so far, they showed promising outcomes in terms of safety, long-term engraftment and survival of modified peripheral cells [76, 77], including maintenance of the genetic modification in mature myeloid and T cells [77, 78]. These positive aspects were the basis for the clinical trials aimed at disrupting the CCR5 co-receptor gene with designer nucleases. This strategy has a major advantage over conventional knockdown approaches using RNA interference, since it permits the generation of HIV-resistant cells after a single treatment. Indeed, when CCR5-specific ZFNs were delivered to primary human T cells by adenoviral transduction, a population of HIV-resistant T cells was observed in vivo 50 days after transplantation in a murine HIV infection model [47]. A similar approach was applied to human CD34+ HSPCs by nucleofection of DNA expression plasmids encoding CCR5-specific ZFNs. Following transplantation in a humanized HIV mouse model, CCR5 disrupted cells showed selective survival after challenge with HIV [45]. However, nucleofection of plasmid DNA into primary cells, and in particular into stem cells, can be associated with considerable cytotoxicity. This drawback has been recently overcome by delivering ZFNs in the form of in vitro transcribed mRNA [79].
Based on these preclinical accomplishments, the use of ZFNs as an HIV gene therapy for the generation of transplantable autologous HIV-resistant T cells has entered phase I/II clinical trials. The protocol was similar in all studies (Fig. 3): CD4+ T cells were isolated from HIV patients and transduced with an adenoviral vector expressing a ZFN pair targeted to CCR5. After ex vivo expansion, the cells were reinfused into the patients. In the first published study [80], 12 patients were recruited and received one infusion of 10 billion CD4 T cells. Six patients underwent a 12-week treatment interruption 4 weeks after infusion. The primary objective was the assessment of safety, while secondary objectives included the evaluation of increased CD4+ T cell counts, the trafficking of CCR5-modified cells to the gut mucosa, and a decrease in viral load. The modified CD4+ T cells engrafted and were detected in the patients up to 42 months after transfer. Moreover, modified cells were detected in all biopsies of the rectal mucosa, revealing successful trafficking. Treatment was prematurely discontinued and HAART reinitiated in two patients because of a rise in HIV RNA levels above the threshold. In four patients who completed the 12-week HAART interruption, a relative survival advantage of the modified cells was observed. The decrease in virus load correlated with the number of circulating cells carrying biallelic modifications at the CCR5 locus. Actually, the one patient with undetectable HIV load after treatment interruption was found to be heterozygous for the CCR5Δ32 allele. In summary, this first-in-human application of ZFN designer nucleases showed infusion of CCR5-modified T cells to be safe and well tolerated, and led to reduced virus loads in some patients. However, complete eradication of HIV could not be achieved, probably due to suboptimal engraftment and the low number of cells carrying a biallelic disruption. It will be interesting to learn what further safety evaluations involving a larger sample size and a long-term follow-up will reveal.
Based on these promising results, more studies have been initiated, including one which specifically enrolled ten patients heterozygous for CCR5Δ32 (NCT01044654). As expected, the biallelic modification frequency in the CCR5Δ32 cohort was doubled as compared to normal, and three out of eight subjects with high levels of engraftment had virus loads below detection limit up to 20 weeks following interruption of HAART (Sangamo Biosciences Inc., Richmond, CA: press release on Dec. 6, 2013). To improve engraftment and increase of CD4+ T cell counts, another study involving 12 patients has evaluated the use of escalating doses of cyclophosphamide (NCT01543152), a drug used for non-myeloablative lymphodepletion to enhance adoptive T cell transfer [81]. Conditioning with cyclophosphamide was reported to be safe and well tolerated, and a dose-dependent increase was observed for both normal and modified CD4+ T cells (Sangamo Biosciences Inc., Richmond, CA: press release on Dec. 6, 2013).
Since HIV can also use the CXCR4 co-receptor for viral entry, an alternative strategy for HIV treatment using CXCR4-specific ZFNs delivered by adenoviral vectors has been investigated [82]. However, while CCR5 disruption seems to be well tolerated by the immune system, the CXCR4 receptor plays an important role in immune regulation, especially in B cell development [83], and its disruption raises concerns of potential deleterious effects. ZFNs have not only been used to create HIV-resistant cells but novel strategies have also been developed to eradicate the provirus from infected cells [84]. While promising, this approach may be limited by the difficulties associated with targeting the integrated provirus, especially in rare cells like resting T cells or latently infected cells.
Concluding Remarks
The presented clinical results are encouraging and validate the CCR5 knockout strategy as an important development in fighting HIV infection. Furthermore, the data underline the number of T cells with biallelic CCR5 disruption to be a key factor for clinical success. On the other hand, off-target cleavage of designer nucleases is a major concern. This is especially true if applied in multipotent stem cells predestined to be transplanted in patients, as the potentially mutagenic events could prompt a malignant phenotype. Hence, specificity of engineered nucleases will be the second key factor required to pave the road for this new line of gene therapy into the clinic.
References
Shen L, Siliciano RF. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J Allergy Clin Immunol. 2008;122:22–8.
Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–7.
Blas-Garcia A, Apostolova N, Esplugues JV. Oxidative stress and mitochondrial impairment after treatment with anti-HIV drugs: clinical implications. Curr Pharm Des. 2011;17:4076–86.
Domingo P, Estrada V, Lopez-Aldeguer J, Villaroya F, Martinez E. Fat redistribution syndromes associated with HIV-1 infection and combination antiretroviral therapy. AIDS Rev. 2012;14:112–23.
Hawkins T. Understanding and managing the adverse effects of antiretroviral therapy. Antiviral Res. 2010;85:201–9.
Mills EJ, Lester R, Ford N. Adherence to antiretroviral therapy: supervision or support? Lancet Infect Dis. 2012;12:97–8.
Klasse PJ. The molecular basis of HIV entry. Cell Microbiol. 2012;14:1183–92.
Su B, Sun G, Lu D, Xiao J, Hu F, et al. Distribution of three HIV-1 resistance-conferring polymorphisms (SDF1-3'A, CCR2-641, and CCR5-delta32) in global populations. Eur J Hum Genet. 2000;8:975–9.
Martinson JJ, Hong L, Karanicolas R, Moore JP, Kostrikis LG. Global distribution of the CCR2-64I/CCR5-59653T HIV-1 disease-protective haplotype. AIDS. 2000;14:483–9.
Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–62.
Huang Y, Paxton WA, Wolinsky SM, Neumann AU, Zhang L, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–3.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86:367–77.
Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–5.
Kulkarni H, Marconi VC, Agan BK, McArthur C, Crawford G, et al. Role of CCL3L1-CCR5 genotypes in the epidemic spread of HIV-1 and evaluation of vaccine efficacy. PLoS One. 2008;3:e3671.
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, et al. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–5.
Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49:4721–32.
Westby M, Smith-Burchnell C, Mori J, Lewis M, Mosley M, et al. Reduced maximal inhibition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry. J Virol. 2007;81:2359–71.
Roche M, Jakobsen MR, Sterjovski J, Ellett A, Posta F, et al. HIV-1 escape from the CCR5 antagonist maraviroc associated with an altered and less-efficient mechanism of gp120-CCR5 engagement that attenuates macrophage tropism. J Virol. 2011;85:4330–42.
Ratcliff AN, Shi W, Arts EJ. HIV-1 resistance to maraviroc conferred by a CD4 binding site mutation in the envelope glycoprotein gp120. J Virol. 2012;87:923–34.
Fluri S, Ammann R, Luthy AR, Hirt A, Aebi C, et al. High-dose therapy and autologous stem cell transplantation for children with HIV-associated non-Hodgkin lymphoma. Pediatr Blood Cancer. 2007;49:984–7.
Krishnan A, Molina A, Zaia J, Nademanee A, Kogut N, et al. Autologous stem cell transplantation for HIV-associated lymphoma. Blood. 2001;98:3857–9.
Gabarre J, Azar N, Autran B, Katlama C, Leblond V. High-dose therapy and autologous haematopoietic stem-cell transplantation for HIV-1-associated lymphoma. Lancet. 2000;355:1071–2.
Campbell P, Iland H, Gibson J, Joshua D. Syngeneic stem cell transplantation for HIV-related lymphoma. Br J Haematol. 1999;105:795–8.
Hoffmann C, Stellbrink HJ, Dielschneider T, Degen O, Stoehr A, et al. Adoptive transfer of syngeneic T cells in HIV-1 discordant twins indicates rapid regulation of T-cell homeostasis. Br J Haematol. 2007;136:641–8.
Allers K, Hutter G, Hofmann J, Loddenkemper C, Rieger K, et al. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood. 2011;117:2791–9.
Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–8.
Hutter G, Thiel E. Allogeneic transplantation of CCR5-deficient progenitor cells in a patient with HIV infection: an update after 3 years and the search for patient no. 2. AIDS. 2011;25:273–4.
Biti R, Ffrench R, Young J, Bennetts B, Stewart G, et al. HIV-1 infection in an individual homozygous for the CCR5 deletion allele. Nat Med. 1997;3:252–3.
Cohen OJ, Paolucci S, Bende SM, Daucher M, Moriuchi H, et al. CXCR4 and CCR5 genetic polymorphisms in long-term nonprogressive human immunodeficiency virus infection: lack of association with mutations other than CCR5-Delta32. J Virol. 1998;72:6215–7.
Michael NL, Chang G, Louie LG, Mascola JR, Dondero D, et al. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nat Med. 1997;3:338–40.
Zimmerman PA, Buckler-White A, Alkhatib G, Spalding T, Kubofcik J, et al. Inherited resistance to HIV-1 conferred by an inactivating mutation in CC chemokine receptor 5: studies in populations with contrasting clinical phenotypes, defined racial background, and quantified risk. Mol Med. 1997;3:23–36.
Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91:6064–8.
Smih F, Rouet P, Romanienko PJ, Jasin M. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995;23:5012–9.
Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188:773–82.
Handel EM, Cathomen T. Zinc-finger nuclease based genome surgery: it's all about specificity. Curr Gene Ther. 2011;11:28–37.
Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D. Efficient gene targeting in Drosophila with zinc-finger nucleases. Genetics. 2006;172:2391–403.
Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–8.
Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol. 2008;26:695–701.
Li H, Haurigot V, Doyon Y, Li T, Wong SY, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–21.
Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, et al. Precise genome modification in the crop species Zea mays using zinc-finger nucleases. Nature. 2009;459:437–41.
Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, et al. High-frequency modification of plant genes using engineered zinc-finger nucleases. Nature. 2009;459:442–5.
Geurts AM, Cost GJ, Freyvert Y, Zeitler B, Miller JC, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433.
Merlin C, Beaver LE, Taylor OR, Wolfe SA, Reppert SM. Efficient targeted mutagenesis in the monarch butterfly using zinc-finger nucleases. Genome Res. 2013;23:159–68.
Young JJ, Cherone JM, Doyon Y, Ankoudinova I, Faraji FM, et al. Efficient targeted gene disruption in the soma and germ line of the frog Xenopus tropicalis using engineered zinc-finger nucleases. Proc Natl Acad Sci U S A. 2011;108:7052–7.
Holt N, Wang J, Kim K, Friedman G, Wang X, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–47.
Lombardo A, Genovese P, Beausejour CM, Colleoni S, Lee YL, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–306.
Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16.
Gaj T, Guo J, Kato Y, Sirk SJ, Barbas 3rd CF. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods. 2012;9:805–7.
Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–23.
Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8:765–70.
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–12.
Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501.
Mussolino C, Cathomen T. TALE nucleases: tailored genome engineering made easy. Curr Opin Biotechnol. 2012;23:644–50.
Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, et al. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39:9283–93.
Mussolino C, Alzubi J, Fine EJ, Morbitzer R, Cradick TJ, et al. TALENs facilitate targeted genome editing in human cells with high specificity and low cytotoxicity. Nucleic Acids Res. 2014;42(10):6762–73.
Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, et al. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther. 2013;21:1718–26.
Osborn MJ, Starker CG, McElroy AN, Webber BR, Riddle MJ, et al. TALEN-based gene correction for epidermolysis bullosa. Mol Ther. 2013;21:1151–9.
Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41:e63.
Makarova KS, Haft DH, Barrangou R, Brouns SJ, Charpentier E, et al. Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol. 2011;9:467–77.
Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482:331–8.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10:957–63.
Jinek M, East A, Cheng A, Lin S, Ma E, et al. RNA-programmed genome editing in human cells. Elife. 2013;2:e00471.
Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23.
Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31(3):230–2.
Ebina H, Misawa N, Kanemura Y, Koyanagi Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 2013;3:2510.
Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31:822–6.
Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–32.
Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42(11):7473–85.
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–9.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–84.
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32(6):569–76.
Symons J, Vandekerckhove L, Hutter G, Wensing AM, van Ham PM, et al. Dependence on the CCR5 co-receptor for viral replication explains the lack of rebound of CXCR4-predicted HIV-variants in the Berlin patient. Clin Infect Dis. 2014;59(4):596–600.
Didigu CA, Wilen CB, Wang J, Duong J, Secreto AJ, et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors ccr5 and cxcr4 protects CD4+ T cells from HIV-1 infection. Blood. 2014;123:61–9.
Peterson CW, Younan P, Jerome KR, Kiem HP. Combinatorial anti-HIV gene therapy: using a multipronged approach to reach beyond HAART. Gene Ther. 2013;20:695–702.
Morgan RA, Walker R, Carter CS, Natarajan V, Tavel JA, et al. Preferential survival of CD4+ T lymphocytes engineered with anti-human immunodeficiency virus (HIV) genes in HIV-infected individuals. Hum Gene Ther. 2005;16:1065–74.
Mitsuyasu RT, Merigan TC, Carr A, Zack JA, Winters MA, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med. 2009;15:285–92.
Amado RG, Mitsuyasu RT, Rosenblatt JD, Ngok FK, Bakker A, et al. Anti-human immunodeficiency virus hematopoietic progenitor cell-delivered ribozyme in a phase I study: myeloid and lymphoid reconstitution in human immunodeficiency virus type-1-infected patients. Hum Gene Ther. 2004;15:251–62.
Truong L, Wood T, Henley J, Ya-Li L, Kim K, et al. Autologous hematopoietic stem/progenitor cell (HSPC) therapy for monogenic blood disorders: scalable, cGMP-compliant process for generating highly efficient genome edited HSPC. Blood. 2013;122:4213.
Tebas P, Stein D, Tang WW, Frank I, Wang SQ, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370:901–10.
Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–51.
Yuan J, Wang J, Crain K, Fearns C, Kim KA, et al. Zinc-finger nuclease editing of human cxcr4 promotes HIV-1 CD4(+) T cell resistance and enrichment. Mol Ther. 2012;20:849–59.
Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996;382:635–8.
Qu X, Wang P, Ding D, Li L, Wang H, et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013;41:7771–82.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 American Society of Gene and Cell Therapy
About this chapter
Cite this chapter
Cornu, T.I., Mussolino, C., Bloom, K., Cathomen, T. (2015). Editing CCR5: A Novel Approach to HIV Gene Therapy. In: Berkhout, B., Ertl, H., Weinberg, M. (eds) Gene Therapy for HIV and Chronic Infections. Advances in Experimental Medicine and Biology(), vol 848. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2432-5_6
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2432-5_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2431-8
Online ISBN: 978-1-4939-2432-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)