
Abstract
Dissolution test is an extremely valuable and powerful tool in understanding the parameters that control dissolution of amorphous formulations as well as in revealing supersaturation effect and the solid-state transformations that might occur after oral administration. Supersaturated/non-sink and biorelevant dissolution conditions could guide in designing the solid dispersion in order to select optimal polymer(s), drug loading, and downstream processing for the desired in vivo performance as well as shelf life stability. The focus of this chapter is to review and describe the different models being used to characterize dissolution of solid oral dosage forms containing amorphous drugs in aqueous and biorelevant media. Dissolution case studies of weak base, weak acid, and neutral compounds are presented, illustrating a rational approach for the development and implementation of the desired dissolution characteristics for the amorphous drug product performance. Finally, case studies of successful in vitro–in vivo correlations of amorphous formulations are presented. This chapter can serve as guidance for the development and understanding of dissolution of amorphous solid dispersions.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Biorelevant
- Dissolution
- Formulation design
- In vivo
- precipitation
- Solution-mediated transformation
- Supersaturation
- Two-stage dissolution
1 Introduction
The dissolution test has long served as the scientific, industrial, and regulatory standard for measuring the rate of drug solubilization or drug release from solid oral drug products and other dosage forms. The monitoring of the rate of drug solubilization or release from the dosage form is particularly important for drug products in which drug release is the rate-determining step for absorption and pharmacodynamic effect. Therefore, variations in dissolution rate may result in potential variability in bioavailability and safety or efficacy concerns. The dissolution test is a holistic measure of critical material attributes of the composition of the drug product (drug substance and excipients), manufacturing process, and physicochemical changes in the drug product upon aging or exposure to stress (e.g., tablet hardening, loss of disintegrant functionality, physical form change, etc.). For drug products containing amorphous solid dispersions, the properties of the carrier and, consequently, its dissolution behavior will be important to the in vitro dissolution performance of the active drug/drug product. Indeed, the dissolution test , when properly designed, is a powerful tool to assist in the selection of the polymer carrier by virtue of its ability to discriminate between polymers with respect to its ability to facilitate dissolution, to maintain a supersaturated solution, and to influence solid-state stability of the amorphous state of the drug substance. It is also noted that the dissolution test is model dependent. That is, the results are influenced by test parameters such as test media pH, hydrodynamics, and test apparatus. As such, it is not surprising to note standardized test descriptions and apparatus described in the USP (USP 2009a), Ph. Eur. (PhEur 2011), and JP (JP 2006), and various regulatory guidances published by the major health authorities, (FDA 1997) and (EMA 2010), covering dissolution testing. USP<1092 > provides detailed requirements for the development and validation of dissolution methods (USP 2009b). As noted in the guidance, recommendations are made for sink condition to be established for the test whereby sink condition is defined as the volume of medium at least three times that required in order to form a saturated solution of drug substance. When sink conditions are present, it is more likely that dissolution results will reflect the properties of the dosage form. USP<1092 > allows for non-sink conditions if the method is shown to be more discriminating or otherwise appropriately justified. The guidances indicate that a small amount of surfactant is justified as needed to establish sink condition for the test.
2 Regulatory Quality Control Dissolution Method
As amorphous characteristic is a critical quality attribute of the amorphous solid dispersion-based drug product, it must be monitored and controlled to ensure quality of the product for the desired in vivo product performance/bioavailability throughout its shelf life. The regulatory quality control (QC) dissolution method should be capable of discriminating substantial changes in the amorphous characteristic of a drug product, corroborating results obtained by spectroscopic methods such as X-ray powder diffraction (XRPD), near-infrared chemical imaging (NIR-CI), and infrared (IR). The QC dissolution method must be able to discriminate presence of the crystalline drug in the amorphous solid dispersion-based drug product. This discriminatory power of the dissolution method allows monitoring of batch-to-batch consistency as well as physical form stability of an amorphous solid dispersion-based drug product. The QC dissolution method should be validated as per the regulatory standards with the additional emphasis on the amorphous–crystalline discriminatory power.
Where dissolution or drug release is the rate-limiting step to drug absorption, it may be possible to establish an in vitro–in vivo correlation (IVIVC) whereby validated predictions of in vivo performance may be made from in vitro dissolution data.
For the dissolution testing of an amorphous drug product, any of the test apparatus typically used for testing of solid oral dosage forms may be selected based on its demonstrated capability to provide meaningful drug-release data and to discriminate for critical factors. Typically, the basket and paddle apparatus are the most commonly used. However, the reciprocating cylinder apparatus and the flow-through apparatus also may be used.
When the drug product contains an amorphous solid dispersion, it is important not only to characterize the dissolution rate but also to determine the ability of the formulation to maintain a supersaturated state upon dissolution of the drug. This prolongation of the supersaturated state is an important aspect of the dissolution test when applied towards the selection of polymers for the solid dispersion during the formulation development phase. Once the formulation has been established, a dissolution test which incorporates sink condition in the test for monitoring lot-to-lot differences in release rate is appropriate.
3 Amorphous State and Solid Dispersion
As described in previous chapters, solid forms are classified into crystalline and amorphous states based on the order of molecular packing (Raumer et al. 2006; Brittain et al. 1991; Ossi 2003). Molecules aggregate together with long-range order in the crystalline state, but this is not the case for the amorphous state (Shalaev and Zografi 2002; Raumer et al. 2006).
The dissolution and bioavailability of a poorly water-soluble drug can be enhanced when formulated as an amorphous form (Goldberg et al. 1966; Chiou and Riegelman 1970; Hancock and Parks 2000; Six et al. 2004; Leuner and Dressman 2000).
Solid dispersion is defined as one type of method to produce an amorphous compound by incorporating a hydrophobic drug into a hydrophilic carrier (Chiou and Riegelman 1971). It is one of the most studied methods to solubilize and to enhance dissolution rate of biopharmaceutical classification system (BCS) class 2 compounds. For instance, a solid dispersion of ritonavir (Law et al. 2001), ER-3421 (a dual 5-lipoxygenase/cyclooxygenase inhibitor; Kushida et al. 2002), was found to have a much higher dissolution rate than the crystalline counterpart and resulted in higher area under curve (AUC) and \({C}_{\rm max}\) in the in vivo study.
4 Theoretical Aspects of Dissolution Testing of Amorphous Drugs
4.1 Solubility and Dissolution of Amorphous Compounds and Solid Dispersions
The amorphous form has attracted increasing interest within the pharmaceutical field because of its higher kinetic solubility in aqueous solvents and faster dissolution rate, which may result in faster absorption rate and increased bioavailability of poorly water-soluble compounds (Leuner and Dressman 2000). The solubility increment of amorphous forms over crystalline states depends on the potential energy difference between these physical states (Hancock and Zografi 1997; Gupta et al. 2004a). It was estimated that 10–1600 folds of solubility increment can be achieved by applying the amorphous form (El-Zein et al. 1998; Fawaz et al. 1996; Ali and Gorashi 1984; Kohri et al. 1999; Hancock and Parks 2000). However, the measured solubility of amorphous compounds was much less, due to rapid crystallization of amorphous materials upon contact with water. Thus, it is important to store amorphous solids protected from moisture.
It is well established that amorphous solids will generally exhibit faster rates of dissolution and higher solubility than the stable crystalline form of the drug. Furthermore, the “enhanced” solubility observed with amorphous solids is transient. Over time, the concentration of dissolved drug will decrease to a concentration consistent with the thermodynamic solubility of the stable crystalline form in the test medium, which is attributed to the precipitation of the dissolved drug from the supersaturated solution as crystalline precipitate. Additionally, solid-state transformation from the amorphous state to the crystalline state may occur due to the effect of solvent on the glass transition temperature (\({T}_{\rm g}\)) of the amorphous solid. These characteristics are also typical of amorphous solid dispersions, although the dissolution rate, extent of dissolution, ability to maintain a supersaturated solution, and the \({T}_{\rm g}\) of the solid dispersion are largely dependent upon the choice of the polymer in the solid dispersion.
It is generally believed that a solid dispersion can enhance the dissolution rate of a poorly soluble drug through one or a combination of factors (Chiou and Riegelman 1971; Ford 1986; Bloch and Speiser 1987; Craig 1990; Torrado et al. 1996; Fawaz et al. 1996; Serajuddin 1999; Kohri et al. 1999; Leuner and Dressman 2000; Kushida et al. 2002; Gohel and Patel 2003; Miller et al. 2008b). In the case of amorphous solid dispersions, a homogeneous molecular dispersion of the drug in the hydrophilic carrier is formed. Since the drug is already present in the molecular state as a “solid solution,” the step of drug dissolution is bypassed.
The dissolution rate of a solid is usually described by the Noyes and Whitney (1897) model (Noyes and Whitney 1897) and the Nernst (1904) model (Nernst 1904). However, the dissolution rate of a two-component system or binary mixture of a drug within a carrier is more complex which led Higuchi et al. (Higuchi et al. 1965) to investigate a uniform, intimate, non-disintegrating mixture of two dissolving compounds both in crystalline state. But the Higuchi model was claimed to not adequately explain the dissolution process in the amorphous solid dispersion model (Van Drooge et al. 2004).
A model proposed by Van Drooge et al. (2004) claimed to describe more accurately the dissolution of amorphous solid dispersions compared to Higuchi’s model as it considers phase transition during dissolution from amorphous to crystalline phase (Van Drooge et al. 2004). Moreover, this model is able to deal with altered dissolution behavior due to crystallization before dissolution that was also reported by other researchers (Torrado et al. 1996; Moneghini et al. 1998; Forster et al. 2001). The model was based on slow release profile of diazepam from solid dispersion tablets with amorphous disaccharide as carrier. During the first phase, drug release is slow, but is gradually accelerated and both carrier and drug dissolve according to a nonlinear profile. In the second phase, a linear release at higher rate than during the first phase is observed that stops when the entire amount of carrier is dissolved. In this phase, dissolution of the solid dispersion results in transportation of drug to the bulk. During the third phase, the crystalline drug dissolves slowly. The geometry of the undissolved crystalline drug skeleton determines the release profile. Apparently, this is not enough to form a robust skeleton tablet: drug particles will be dispersed in the dissolution medium and dissolve according to a first-order release profile. On the other hand, when the carrier is dissolved, over 80 or 90 % of the drug is still undissolved: a skeleton of crystalline drug is formed, yielding a zero-order release.
Similar behavior was reported previously by Allen and coworkers for tablets prepared from dispersions of small saccharides like sucrose, mannitol, and sorbitol (Allen et al. 1978). The faster release phase was attributed to a molecular incorporated fraction and the slow release to a crystalline fraction of the lipophilic drug, which was present due to incomplete dissolution of the drug in the molten sugar during preparation (Allen et al. 1977). The results imply that crystallization occurred during dissolution of disaccharide solid dispersions.
Another model was proposed to describe the dissolution behavior of solid dispersions and the process of crystallization (Craig 2002). It was suggested that the release behavior of drug molecules from a biphasic solid dispersion as being either carrier mediated or drug mediated (Fig. 15.1)

Release modes of amorphous drug inclusion complexes during dissolution of a solid dispersion. (Reproduced with permission from Moore and Wildfong 2009)
Initially, a polymer-rich diffusion layer is formed between the solid dispersion and the dissolution medium. In a carrier-mediated dissolution (Fig. 15.1a), after diffusion of drug into the polymer-rich phase, drug is further released into the dissolution medium either as solvated molecules or as amorphous particles at a rate dictated by the carrier. In a drug-mediated dissolution (Fig. 15.1b), the high solubility of the carrier in the dissolution medium does not allow the formation of a polymer-rich phase and drug is dissolved through diffusion of the amorphous complex from the dispersion to the dissolution medium at a rate proportional to the aqueous solubility of the amorphous drug. Agglomeration can occur, resulting in instability in the dissolution (Fig. 15.1c). When the amorphous molecules diffuse through the polymer-rich diffusion layer too rapidly and their dissolution rate in the aqueous medium is relatively slow, crystallization may occur, which creates a high-energy interfacial boundary that slows down the dissolution rate. The reduction in specific surface area due to agglomeration would slow the dissolution rate at a greater extent, permitting a longer time frame for crystallization (Craig 2002). The model also implied that the relative aqueous solubility of the carrier component may induce physical instability and the effect of manufacturing methods will affect release behavior (critical manufacturing variables) (Moore and Wildfong 2009). Intermolecular interactions between drug and carrier on drug release were not taken into account in this model.
In biorelevant media, according to Friesen et al. (2008), solid drug dispersions (SDDs) rapidly dissolve and/or disperse and produce a wide variety of potential species—categorized based on their size and composition: (1) free or solvated drug, (2) drug in bile salt micelles, (3) free or solvated polymer, (4) polymer colloids, (5) amorphous drug/polymer nanostructures, (6) small aggregates of amorphous drug/polymer nanostructures (termed “nanoaggregates”), and (7) large amorphous particles/precipitate (Fig. 15.2; Friesen et al. 2008).

Species formed when SDDs are added to aqueous solutions, simulating duodenal and intestinal contents. (Reproduced with permission from Friesen et al. 2008)

Concentration in the solution and the solid compositions as a function of time during the solution-mediated phase transformation. (Reproduced with permission from Zhang et al. 2009)
The rapid formation of drug/polymer nanostructures and nanoaggregates upon introduction of solid dispersion to an aqueous solution which are stable in aqueous suspension can justify the enhanced oral drug absorption. These nanostructures and nanoaggregates produce a free drug concentration higher than the solubility of crystalline drug which is sustained by replacing free drug as it is absorbed (Friesen et al. 2008).
4.2 Solution-Mediated Transformation During Dissolution
One of the issues relating to the stability of the amorphous state, particularly in vivo, is its solution-mediated transformation characteristic. Solution-mediated transformation of amorphous to crystalline state is the conversion of metastable solids such as amorphous solids to the crystalline state when the solids are exposed to a solvent, in this case water. The transformation to the thermodynamically stable crystalline state occurs at a higher rate in the presence of solvents than in the dry state because of higher molecular mobility in the presence of solvents.
Characterization of solution-mediated transformations in the amorphous state can give an insight into amorphous crystallization (Zhang et al. 2009). The importance of the phase transition kinetics, molecular interpretations, and process implications has been emphasized in numerous studies (Cardew and Davey 1985; Davey et al. 1986, 1997a, 1997b; Rodriguez-Hornedo et al. 1992; Blagden et al. 1998).
The presence of the solvent does not change the thermodynamics and stability relationship, unless a solvate/hydrate is formed with the solvent. However, owing to the much higher mobility in the solution state than in the solid, transformation to the stable phase is much faster. This process is analogous to the effect of catalysts for chemical reactions (Zhang et al. 2009). The schematic representation of concentrations in the solution, as well as the solid compositions as a function of time, is shown in Fig. 15.3 for a typical solution-mediated transformation process (Zhang et al. 2009).
As depicted in Fig. 15.3, three consecutive steps are involved in a solution-mediated transformation (Cardew and Davey 1985; Zhang et al. 2002; Rodriguez-Hornedo et al. 1992): (1) initial dissolution of the metastable phase into the solution to reach and exceed the solubility of the stable phase, (2) nucleation of the stable phase, and (3) crystal growth of the stable phase coupled with the continuous dissolution of the metastable phase. Step (2) or (3) is usually the slowest of the steps. For the overall solution-mediated transformation process, step (2) is usually the slowest. Therefore, this is the rate-determining step as nucleation is involved. When step (2) is the rate determinant, factors such as the solubility and the solubility difference between the phases, processing temperature, contact surfaces, agitation, and soluble excipients/impurities that affect nucleation will influence the overall transformation. When step (3) is the rate-controlling step, the kinetics of the conversion is determined by solubility difference, solid/solvent ratio, agitation, processing temperature, particle size of the original phase, and soluble excipients/impurities.
4.3 Crystallization During Dissolution
While it is acknowledged that orally administered amorphous formulations may supersaturate and crystallize in vivo (when exposed to the gastrointestinal (GI) fluids in the human body), the ability to study this phenomenon in the in vivo setting has been hampered by the dearth of analytical tools, the complexity of GI fluids, and the inaccessibility of the GI tract where the measurements need to be made. In contrast, there are many reported studies on proposed mechanisms and kinetics devoted to crystallization in vitro (Cardew and Davey 1985; Davey et al. 1986, 1997a, 1997b; Weissbuch et al. 1987; Rodriguez-Hornedo et al. 1992; Weissbuch et al. 1994; Zhu and Grant 1996; Zhu et al. 1996; Blagden et al. 1998; Davey et al. 2002).
The crystallization of amorphous drugs during dissolution could be shown by the nearly 20 % difference between the predicted and experimentally measured solubility ratios of amorphous drug. For instance, in the case of indomethacin (IMC) and griseofulvin particles, the predicted solubility ratio in water ranged from 25 to 104 and 38 to 441, respectively, whereas the experimentally measured value was only 4.5 (IMC) and 1.4 (griseofulvin; Hancock and Parks 2000; Matteucci et al. 2008; Elamin et al. 1994).
The concentration of the solution at equilibrium will decrease after a period of time to the level equivalent to the solubility of the most stable crystalline form. The duration of the period of increased (metastable) solubility is generally thought to be controlled by the rate of nucleation and, thus, the rate of growth of the more stable phase (Clarkson et al. 1992).
The suggested mechanism behind transformation from disordered structure to ordered thermodynamically stable structure is mainly through surface solid-state transition (Mosharraf et al. 1999). This would result in a very slow reduction in the apparent solubility plateau level down to the thermodynamically stable value. The investigation of the relationship between equilibrium solubility, the amount of solute added to the solvent, and the proportion of disordered or amorphous structures on the surface of the particles can provide valuable information which can be used to predict and control the solubility and dissolution behavior of sparingly soluble hydrophobic drugs (Mosharraf et al. 1999).
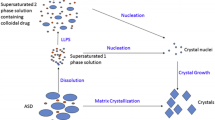
Dissolution, precipitation, and crystallization that can occur during dissolution of an amorphous system are summarized in Fig. 15.4 (Alonzo et al. 2010). Modified Noyes and Whitney equation is used to describe the dissolution pathway, where dc/dt represents the dissolution rate which is directly proportional to the surface area (A) and the difference between the solution concentration (C) and the equilibrium concentration (\({C}_{\rm eq}\); Alonzo et al. 2010). In the nucleation path, J represents the nucleation rate, which is proportional to the degree of supersaturation (S). For the growth path, the rate of crystal growth is also proportional to the difference between the actual solution concentration and the equilibrium concentration (Alonzo et al. 2010).

Schematic illustration of the competition between dissolution and crystallization via the solid or solution state for amorphous systems. (Reproduced with permission from Alonzo et al. 2010)
4.4 Supersaturation of Amorphous Systems During Dissolution
In recent pharmaceutical literature, the terms “equilibrium solubility” and “kinetic or apparent solubility” are often used for the systems with stable and metastable equilibria, respectively (Lipinski et al. 2001; Huang and Tong 2004).
A solid phase is crystallized from solution if the chemical potential of the solid phase is less than that of the dissolved component. A solution in which the chemical potential of the solute is the same as that of the corresponding solid phase and is in equilibrium with the solid phase under the given conditions (temperature, pH, and concentration) is referred to as a saturated solution (Gamsjager et al. 2008). In order for crystallization from solution to occur, this equilibrium concentration or solubility must be exceeded. This excess concentration or chemical potential , called the supersaturation, is the driving force for nucleation and crystal growth (Strickley et al. 2007).
For poorly water-soluble drugs, the maximum achievable intraluminal drug concentration may limit absorption. However, the intraluminal concentration of a drug is not necessarily limited by its solubility in GI fluids (Brouwers et al. 2009). Drugs may be in solution at a concentration above their saturation solubility, that is, in a state of supersaturation. The degree of supersaturation can be expressed by the supersaturation ratio, S (Eq. 15.1; Brouwers et al. 2009):
with C and \({C}_{\rm eq}\) representing the solubility and equilibrium solubility (saturation), respectively.
A solution is defined as unsaturated, saturated, or supersaturated based on the following relationships: S<1, S=1, or S>1, respectively (Brouwers et al. 2009).
Amorphous drugs are high-energy solid systems that are capable of reaching higher kinetic solubility values (supersaturation) than would be expected from the equilibrium solubility of a crystalline material (Wei-Qin 2009). A supersaturated drug solution is thermodynamically unstable compared with the equilibrium condition (saturation). Thus, it has the tendency to return to the equilibrium state by drug precipitation. The higher the supersaturation, the more precipitation will take place as the former is the driving force for the latter (Six et al. 2004). The accelerated dissolution and the higher apparent solubility provided by amorphous systems could induce the generation of supersaturated solutions in the GI lumen that can result in increased absorption (Brouwers et al. 2009).
The dissolution characteristics of solid dispersions depend to a large extent on the physical state (ideally: amorphous), drug dispersivity (ideally: molecular dispersion), and particle size (Brouwers et al. 2009). Supersaturation can be achieved through:
-
1.
The in vivo GI conditions: effect of pH and content changes from stomach to intestine.
Due to the pH gradient in the GI lumen (pH 1.5–2 in the stomach compared with pH 5–8 in the intestine), the solubility of weak bases in gastric fluid (ionized form) typically exceeds their solubility in the intestinal fluid (unionized form). Therefore, higher dissolution of poorly water-soluble weak bases in the stomach before transfer to the intestine may result in supersaturation (Brouwers et al. 2009; Bevernage et al. 2010).
-
2.
High-energy and rapidly dissolving solid forms.
As mentioned earlier, amorphous state requires less energy to dissolve, resulting in higher apparent solubility and increased dissolution rates (Hancock and Parks 2000). Methods such as particle size reduction through milling and co-grinding that can form an amorphous state may increase dissolution rates by enhancing the surface area available for dissolution (Sarode et al. 2013; Brouwers et al. 2009).
This higher initial solubility may be sufficient to ensure increased and more rapid absorption for a drug with good permeability such as BCS class 1 and 2 drugs. But a more thermodynamically stable form may crystallize at any time inside the GI tract, and the crystallization would have a major impact on the product performance in vivo (Strickley et al. 2007; Sarode et al. 2013). The higher dissolution rate and apparent solubility of an amorphous drug usually causes supersaturation during in vivo dissolution. It should be noted though that rapid dissolution and supersaturation could prove counterproductive in some cases, for example, for drugs that precipitate during transit from the stomach to upper small intestine where an increase in the pH is observed. This precipitation in the GI tract may compromise oral bioavailability (Brouwers et al. 2009; Overhoff et al. 2008).
If a crystallization-inhibitory polymer is incorporated into the amorphous solid dispersion, the in vivo precipitation may be delayed or completely eliminated, resulting in much improved oral absorption. It is ideal if the polymeric carrier can function as a precipitation (crystallization) inhibitor during in vivo dissolution (Zhang et al. 2009).
The commercially available Sporanox\(^{\textregistered}\) capsule formulation is a solid dispersion relying on the principle of supersaturation to enhance the intestinal absorption of the antifungal itraconazole (ITR), a weak base (pKa=4) with an extremely low and pH-dependent aqueous solubility (ca. 1 ng/mL in water, 6 mg/mL in 0.1 M HCl). This formulation comprises a molecular dispersion of ITR in a hydroxypropylmethylcellulose (HPMC) matrix, which is coated onto inert sugar spheres. Dissolution of HPMC in media simulating the gastric environment results in supersaturated concentrations which are maintained for at least 4 h. HPMC is believed to prevent ITR from precipitation in the stomach and in the intestine, resulting in significant absorption (maximum fraction absorbed ca. 85 %) and oral bioavailability (ca. 55 %; Brewster et al. 2008).
Yamashita et al. (2003) investigated the dissolution in acidic medium of solid dispersions containing the macrolide lactone tacrolimus in an amorphous state comparing three different polymers (HPMC, PVP, and PEG 6000) as the carrier. Rapid dissolution and supersaturated concentrations of tacrolimus up to 25-fold higher than the equilibrium solubility (2 mg/mL) were observed. Even though the polymer choice did not affect the maximum degree of supersaturation, it was only HPMC that could fully inhibit precipitation for up to 24 h.
The changes of pH in stomach and intestine and in fasted and fed state will affect the solubility and dissolution of weak bases , for example, ITR. The GI pH may also change the performance of precipitation inhibitors, especially their solubility and hydrogen bonding between the H donor and acceptor. Surface tension, viscosity of GI fluids, and presence of bile salts and phospholipids may influence the solubility and dissolution of the drug (Dressman et al. 2007) and subsequently the intraluminal supersaturation and drug-precipitation kinetics (Brouwers et al. 2009). Supersaturation may be affected by the hydrodynamics and the composition of the GI fluids (Brouwers et al. 2009). Thus, for the in vitro dissolution, testing the extent of supersaturation following acidic-to-neutral pH transition must be considered in order to correlate the in vitro dissolution with the in vivo absorption (Miller et al. 2008a). Furthermore, ITR absorption mostly occurs in the proximal small intestine (Miller et al. 2007; Six et al. 2005); hence, immediate release (IR) formulations are provided with a small window for absorption because supersaturated levels of ITR in the gastric environment rapidly precipitate upon exit from the stomach (Miller et al. 2007; Six et al. 2005). A study was performed using various types of ITR amorphous formulations (including Sporanox\(^{\textregistered}\)) and reported finding revealed inconsistency between the in vitro dissolution performance in simulated gastric fluid (SGF) and the in vivo absorption. It was suggested that faster release and increased supersaturation in the acidic medium along with differences in crystallization rate upon transfer to the small intestine correlated with the lower predicted bioavailability (Six et al. 2005). Precipitation can decrease the driving force for transport across the biological membrane and limit the time available for absorption which complicates development of IVIVC with amorphous compound (Overhoff et al. 2008). The importance of simulating the Gl pH shift during supersaturation dissolution testing of amorphous compound to evaluate whether supersaturation is maintained in the small intestine was described (Six et al. 2005).
5 Factors That Influence Dissolution of Amorphous Drug Products
5.1 Formulation Factors
As with conventional formulations of IR solid oral drug products containing crystalline drug, dissolution behavior of amorphous drug products are greatly influenced by the composition of the formulation as well as the manufacturing process. Of note is the polymer used as the carrier for the solid dispersion. The dissolution rate of the dosage form is determined by the characteristic of the carrier (Leuner and Dressman 2000). Such carrier systems include cellulose-based polymers such as HPMC and its acidic derivative hydroxypropylmethylcellulose acetate succinate (HPMC-AS); polyethylene glycol (PEG), polyvinylpyrrolidone (PVP) and its copolymers; acrylate polymers (Eudragit), sugars and their derivatives; emulsifiers, organic acids and its derivatives. There are only a handful of oral pharmaceutical products containing amorphous active pharmaceutical ingredient (API) that have been successfully marketed despite several decades of effort in research and development (Chap. 3). However, recently launched products such as Incivek\(^{\textregistered}\), Kalydeco\(^{\textregistered}\), and Zelboraf\(^{\textregistered}\) demonstrate a great versatility of the amorphous solid dispersions in increasing the rate of solubilization and bioavailability .
Whereas, for conventional formulations, where the dissolution of the drug may occur from the disintegrated granule (i.e., drug substance particle), dissolution of amorphous drug products occur from the solid dispersion consisting of the amorphous drug and the polymer. Therefore, the dissolution behavior of the polymer plays a key role in the dissolution of the amorphous drug product. Ionizable polymers, such as those of the acrylates or hypromellose acetate succinate will dissolve through salt formation at pH above the pKa. Additionally, the dissolution rates of different grades of ionizable polymers will differ depending on the degree of substitution. Unlike ionizable polymers, the dissolution rate of non-ionizable polymers (e.g., copovidone) is pH independent. The viscosity of the polymer will also influence the dissolution rate of the amorphous solid dispersion and the final drug product.
Drug load, expressed as the ratio of the drug to polymer in the amorphous solid dispersion, will also play a significant role in the dissolution and physical stability of the supersaturated solution. Lower drug load (i.e., higher polymer content) will generally result in enhanced dissolution and stability of the supersaturated solution.
5.2 Manufacturing Factors Which Influence Solid-State Properties
The process for the manufacture of the drug product, particularly the processing step for the amorphous solid dispersion and those that impact the solid-state properties of the solid dispersion, will have a major influence on the dissolution of the drug product. Amorphous processing technologies are described in a different chapter of this book and, therefore, will not be covered in detail here. It should be mentioned, however, that these technologies generally will result in material with different solid-state properties, most notably particle size, shape, porosity and density. In addition to the particulate properties, there may be potential differences in the type and extent of interactions of the drug and polymer as a function of the processing technology used to prepare the solid dispersion. Systematic research in this area is lacking; however, it is anticipated that solvent-based processes may be conducive to certain interactions that may not be feasible with non-solvent-based technologies such as hot-melt extrusion (HME).
Material produced from spray-drying processes are generally spherical and hollow due to process in which the solution of dissolved drug and polymer are sprayed as fine droplets and then rapidly dried in an inert stream of warm air. Spray-dried dispersions are oftentimes porous and fragile due to the escape of solvent through the solid matrix. Processing factors that influence the particle properties of the spray-dried dispersion (drying temperature, spray rate, droplet size, air flow, etc.) will influence the dissolution rate.
In contrast, there are no solvents used in HME processes for manufacturing amorphous solid dispersions. Instead, the mixture of crystalline drug and polymer are heated to a temperature at which the components melt or form a eutectic, and then flash cooled, resulting in a dense amorphous glassy solid. The solid is then milled to achieve uniform particle size distribution, which is then processed into the final formulation. Therefore, the milling step will determine particle size and surface area, which in turn is related to the dissolution rate of the solid.
A newer, innovative processing technology for the manufacture of amorphous solid dispersions is the microprecipitation method in which an organic solution of the drug and polymer is introduced into a miscible anti-solvent in which the drug and polymer are insoluble or less soluble (e.g., water), causing the precipitation of the drug and polymer as a microprecipitated bulk powder (MBP) . As there is water and solvent involved in the process, a subsequent drying step and milling step are required. Materials made by the MBP process tend to be porous. Manufacturing processes that influence the porosity and particle size of the MBP (rate of precipitation, drying temperature, MBP milling) will significantly impact the dissolution of the MBP and the final drug product.
It is noted that discussion has centered on the processing technologies and parameters that will influence the MBP, and no mention is made of the drug substance. This should be inherently clear that the solid-state properties of the starting drug substance is not that important to the dissolution of the drug product, as the crystalline drug substance is converted to an amorphous solid dispersion, and it is the properties of the amorphous solid dispersion that is responsible for the dissolution behavior of the drug product. It should also be mentioned that the above factors that influence dissolution are discussed independently, whereas it is the combination of these factors, that is, composition and process factors which influence the chemical and physical properties of the amorphous solid dispersion, which will determine the dissolution rate of the solid dispersion.
6 Dissolution Case Studies to Guide Formulation Development
In addition to the physical form stability of the amorphous drug, a sound understanding of the chemical form of the drug in the amorphous solid dispersion and its behavior during and after dissolution are important elements of the quality-by-design approach to development of an amorphous solid dispersion drug product. The chemical form of a drug (weak acid, weak base, or neutral) and its pH-dependent solubility is known to impact in vivo performance of the amorphous solid dispersion product due to its interplay with the pH gradient of the GI tract. Weak acids may rapidly precipitate, crystallize, or gel in the stomach at low pH, while weak bases may rapidly dissolve at the lower pH of the stomach but precipitate at the higher pH in the lower GI upon transiting into the intestine. It is therefore very important to consider the impact of the drug’s chemical form and pKa while designing the amorphous solid dispersion with respect to selection of stabilizing polymer (nonionic vs. ionic, and pH-dependent release), drug loading , manufacturing technology, as well as downstream processing (IR vs. eroding vs. enteric coating) to maximize the duration of in vivo supersaturation . In order to guide the design of amorphous solid dispersions of new chemical entities (NCEs) with respect to these elements, researchers have recently successfully utilized in vitro dissolution testing in non-sink and biorelevant media as well as two-stage media as a predictive tool for in vivo precipitation , kinetic solubility, and supersaturation. Dissolution medium representing 100 % saturation is preferred for formulation screening; however, several successful examples of use of less than 100 % saturation media during early design of amorphous solid dispersion have been published. Formulations that are able to sustain supersaturation for at least 2 h (physiologically relevant) would represent viable formulations that can be investigated in vivo. Formulation that can maintain supersaturation for less than 60 min would need careful evaluation as these may be more prone to higher pharmacokinetic (PK) variability. The following examples illustrate various in vitro dissolution methodologies utilized to guide amorphous formulation development with high predictive power for the desired in vivo performance.
6.1 Between Amorphous and Crystalline Phase
Solid-state changes that may occur during dissolution of amorphous carbamazepine (CBZ) were studied in phosphate buffer pH 7.2 at room temperature using in situ Raman spectroscopy (off-line; Savolainen et al. 2009). The findings of this study confirmed that the surface of the CBZ samples crystallize immediately upon contact with the dissolution medium. The transition from the amorphous form to crystalline anhydrate (form I) of CBZ and then a solution-mediated transformation from form I to dihydrate, as previously demonstrated by Murphy et al. (Murphy et al. 2002), was proposed. The transition from the amorphous form to the crystalline anhydrate is likely a solid-state transition as amorphous CBZ has been shown to crystallize to an anhydrate form in dry conditions (25 °C and<10 % RH; Patterson et al. 2005). To validate the instant crystallization of CBZ, the dissolution test in phosphate buffer was performed and no significant improvement on the dissolution rates was noted (Savolainen et al. 2009). Dissolution from most of the amorphous samples was even slightly slower than from the dihydrate compacts. Analysis of the remaining sample after the dissolution experiment confirmed that the sample surface had converted to the dihydrate (crystalline form) during dissolution (Savolainen et al. 2009).
In another case, dissolution of crystalline and amorphous ciclesonide was studied (Feth et al. 2008). Crystalline and amorphous ciclesonide exhibit the same saturation solubility (Feth et al. 2008). For the crystalline ciclesonide, within the first 60 min of dissolution, the concentration measured (50 mg/L) was significantly lower than its saturation solubility (90.1 ± 2.2 mg/L). On the contrary, for the amorphous form, concentration increased almost instantaneously to values up to four times higher than the saturation solubility determined for amorphous ciclesonide (91.6 ± 5.2 mg/L). After 24 h, for both forms, concentrations were approaching the saturation concentration. It was suggested that the amorphous ciclesonide was able to form a stable supersaturated solution in water for at least 60 min due to the absence of inoculation crystals in the amorphous phase. On the other hand, in crystalline ciclesonide, the crystal itself acts as crystallization seed in the slurry which speeds up the process.
6.2 Between Different Temperatures and Polymers
Both temperature and polymer (which act as a crystallization inhibitor) can greatly affect the solubility of amorphous drugs (Alonzo et al. 2009). The concentration–time (dissolution) profiles attained with solid dispersions may be higher than those achieved with the pure amorphous API (Gupta et al. 2004b), suggesting that certain polymers are able to further enhance solution concentrations relative to pure amorphous drug. The increased solution concentrations observed following dissolution of amorphous solid dispersions have been attributed to the inhibition of API crystallization from the supersaturated solution by the polymer (Gupta et al. 2004b; Tanno et al. 2004) and increased equilibrium solubility of the API due to solution complexation with the polymer (Usui et al. 1997; Acartürk et al. 1992; Loftsson et al. 1996).
In order to guide design of solid dispersions, a simple supersaturation test/dissolution study for rank ordering of polymers and drug loading was reported without actually making solid dispersions (Konno et al. 2008). Dissolution of amorphous IMC at different temperatures (25 °C, 37 °C) in simulated intestinal fluid without pancreatin showed that IMC underwent solution-mediated transformation with higher concentration and lower temperature. The inclusion of polymer inhibited crystallization (Konno and Taylor 2008; Alonzo et al. 2009). For the amorphous felodipine, extensive crystallization was observed in 10–15 min at 25 °C, whereas instant crystallization was observed at 37 °C. A supersaturated solution was not formed when polymer was not included (Konno et al. 2008; Alonzo et al. 2009).
6.3 Dissolution of Salts
In the stomach and intestine, drug solubility can be enhanced by food and bile components such as bile salts, lecithin, and fatty acids. Supersaturation in the intestinal fluid is an important property that can play a significant role in drug absorption. For compounds with poor intrinsic solubility in the intestinal fluid, solubility is often a limiting factor for absorption. For many of these compounds, it may not be possible to enhance the saturation solubility to the extent required such that the whole dose is dissolved in the GI fluid. In this case, creating or maintaining supersaturation in the intestinal fluid can be an effective way to enhance absorption of these compounds (Wei-Qin 2009).
6.3.1 Weak Bases
In most cases , supersaturation is induced from solubilized formulations or formulations that contain a high-energy state of the drug. However, for weakly basic drugs, even intake of the crystalline powder may result in supersaturation in the small intestine. Due to the pH gradient in the GI lumen in fasted state conditions (pH 1.5–2 in the stomach vs. pH 5–8 in the intestine), the gastric solubility of weak bases (ionized form) typically exceeds their intestinal solubility (unionized form). Hence, after dissolution of poorly water-soluble weak bases in the stomach, transfer to the intestine may result in supersaturated concentrations and an increased flux across the intestinal mucosa (Wei-Qin 2009). By simulating the GI pH shift during dissolution experiments, this behavior can be monitored. For instance, Kostewicz et al. (Kostewicz et al. 2004) evaluated the behavior of three weakly basic drugs (dipyridamole, BIBU 104 XX, and BIMT 17 BS) in an in vitro system simulating both the pH gradient between stomach and intestine and the presence of bile salts and phospholipids in the intestine. Upon transfer of a solution of the drug in an acidic medium (pH 2, simulating fasted state gastric conditions), supersaturated concentrations of the weak bases were observed in both fasted- and fed-state simulated intestinal fluids (FaSSIF and FeSSIF). Presumably, this mechanism plays an important role in the intestinal absorption of various poorly water-soluble weak bases .
Depending on the properties of the salt and its corresponding base or acid, the fate of the salt in the GI tract may vary significantly. When the salt of the basic drug gets in the GI tract, it may dissolve in the stomach and remain either in solution or precipitate out as the free base when it is emptied into the intestine. It may also convert to the hydrochloride salt if the hydrochloride salt is less soluble, especially with the influence of the common-ion effect. In this case, the dissolution in the intestine is in reality the dissolution of the precipitated hydrochloride salt. When salt conversion happens in vivo, it can precipitate out as either the crystalline or the amorphous form with different particle size that will affect solubility and dissolution (Wei-Qin 2009; Li et al. 2005).
Since transport across the biological membrane of weak bases will be more pronounced in the small intestine (uptake of the unionized form), sufficient precipitation inhibition (polymer) is required upon transfer of the supersaturated solution to the intestine. Therefore, one cannot rely on dissolution studies at constant acidic pH to predict the performance of formulations of weak bases in vivo (Miller et al. 2007). For instance, Six et al. (2005) observed a discrepancy between the results of in vitro dissolution tests in acidic medium and in vivo absorption for four solid dispersions of ITR: faster release and increased supersaturation in acidic medium correlated with lower bioavailability. Presumably, this effect can be explained by differences in crystallization rate upon transfer to the small intestine (increased driving force for precipitation in case of higher supersaturation). Thus, it is crucial to simulate the GI pH shift during supersaturation dissolution testing of weak bases to evaluate whether supersaturation is maintained in the small intestine.
Use of a GI pH shift model was also employed in the development of a propriety weak base (compound A) which exhibited high solubility at gastric pH, but very low solubility at intestinal pH, and high PK variability in humans. A two-stage dissolution test in which the drug product is tested in pH 2 media (HCl) or pH 4.5 media (phosphate buffer) for 30 min followed by testing in a dissolution medium containing sodium taurocholate and lecitihin at pH 6.5 (fasted state simulated intestinal fluid, FaSSIF) was used to evaluate the performance of an IR tablet containing the thermodynamically stable crystalline-free base. The results showed that although very high solubility is observed at low pH (2), at elevated gastric pH (4.5) the drug exhibits very low solubility. Moreover, upon transitioning from pH 2 to pH 6.5 (FaSSIF), the solubility drops quickly to the measured solubility for the amorphous drug. These results suggested that significant PK variability observed in patients may be attributable to differences in gastric pH associated with the disease state and with concomitant gastric pH-elevating medications, and that an amorphous formulation may improve the PK variability while also potentially enhancing bioavailability. Two tablet formulations containing amorphous solid dispersion with 35 % drug load and 60 % drug load in HPMC-AS polymer made by spray drying were developed and evaluated using this two-stage dissolution test. The results shown (Figs. 15.5 and 15.6) support the hypothesis of improved variability and potential enhancement of bioavailability.

Two-stage dissolution of tablet formulations made with crystalline drug substance and amorphous solid dispersion in media simulating normal gastric pH

Two-stage dissolution of tablet formulations made with crystalline drug substance and amorphous solid dispersion in media simulating elevated gastric pH
Two tablet formulations containing crystalline drug substance and 35 % amorphous solid dispersion were evaluated in humans. The results show substantial enhancement (1.7 fold) in the bioavailability of the drug and improved PK variability (Coefficient of Variation (CV) 80 % → 18 %) with the tablet made with amorphous solid dispersion (Roche in-house data). The results from this study illustrate the utility of dissolution test conditions that are carefully chosen to simulate physiological conditions (i.e., biorelevant) when applied to testing of a poorly water-soluble weak base and the potential for improved absorption through amorphous formulation technologies.
6.3.2 Weak Acids
Dissolution of the salt of an acidic compound has its own complications. The salt is likely to convert to the free acid. When this happens, the liberated free acid may coat the surface of the remaining drug particles or nucleate on other particle surfaces, leading to a slowdown of dissolution (Wei-Qin 2009). As described in the earlier sections, weak acids may rapidly precipitate/crystallize or gel in stomach before transit to the lower GI. It is therefore important to select amorphous solid dispersion and downstream technology, yielding a drug product with optimal supersaturation at physiologically relevant pH for absorption.
6.4 Biorelevant Dissolution Testing of Amorphous Solid Dispersions
To better predict the in vivo behavior after oral administration and estimate the impact of solubility, degree of supersaturation, and dissolution on absorption, the in vitro dissolution method should be physiologically relevant (biorelevant), taking into account the contents and the transit through the GI tract.
For example, in the case of in vitro dissolution of amorphous ITR, the initial burst in FaSSGF, a biorelevant medium containing sodium taurocholate and lecithin Vertzoni et al. 2005 was far greater than in SGF and led to higher dissolution (Ghazal et al. 2009). Furthermore, the presence of lecithin and sodium taurocholate in FaSSIF and FeSSIF enhanced ITR’s dissolution rate, with a greater increase in the lower pH and more bile salts and lecithin of FeSSIF, in accordance to the in vivo food effect of the drug.
The precipitation behavior of the amorphous JNJ-25894934 from three different liquid formulations in phosphate buffer, FaSSIF, and FeSSIF was investigated (Dai et al. 2007). The solubility of the drug was similar in FaSSIF and FeSSIF, but was eightfold lower in the phosphate buffer. Differences in precipitation were observed between the different media, especially for the fast-precipitating formulation. While precipitation from this formulation was immediate and complete in phosphate buffer, a metastable zone containing about 20 and 80 % dissolved drug, was maintained during 8 h in FaSSIF and FeSSIF, respectively, and precipitation continued after 8 h. As there was no difference in drug solubility in FaSSIF versus FeSSIF, enhanced concentrations in the metastable zone in FeSSIF clearly suggest specific precipitation-inhibiting interactions that are more effective in FeSSIF, presumably due to the increased bile salt/phospholipid concentrations. The in vitro precipitation profiles in FeSSIF correlated better with in vivo absorption than those in phosphate buffer and FaSSIF (Dai et al. 2007).
Another example of the usefulness of biorelevant media, in this case FaSSIF, could be seen in the development of Zelboraf\(^{\textregistered}\) (vemurafinib), an IR tablet drug product made with amorphous solid dispersion (Shah et al. 2013). The initial clinical formulation was an IR capsule containing a fast dissolving metastable crystalline form of the vemurafinib. In order to achieve higher exposures in the clinical studies, IR capsule prototypes made with amorphous solid dispersion were prepared. Whereas dissolution results for the three formulations suggested comparable exposures; the actual exposures obtained in humans revealed the amorphous formulations to be far superior to the formulation made with the metastable crystalline drug substance (Fig. 15.7). However, when the formulations were tested using biorelevant media (FaSSIF), a clear distinction could be made between the formulations, which corresponded to the observed differences in vivo (Fig. 15.8). Further investigation into the underlying reason for the substantial differences found evidence of solid-state transformation of the metastable crystalline form of the drug substance (form 1) to its thermodynamically stable, insoluble crystalline form (form 2).

Plasma concentration time profiles of vemurafinib, given as a single dose of capsule formulations made with metastable crystalline drug substance (form 1) or amorphous MBP (MBP-1 and MBP-2) to healthy volunteers. (Reproduced with permission from Shah et al. 2013)

Dissolution profiles of capsule formulations made with metastable crystalline drug substance (form 1) and amorphous solid dispersions (MBP-1 and MBP-2) obtained using paddle apparatus and fasted state simulated intestinal fluid (FaSSIF). (Reproduced with permission from Shah et al. 2013)
The same test conditions were used to demonstrate the effect of stressing on the amorphous formulation (Fig. 15.9), which showed lower rate and extent of dissolution of the amorphous formulation when stressed by heat and humidity. The diminished dissolution observed in the stressed sample was determined to be due to stress-induced crystallization of the amorphous product (Shah et al. 2013).
Although it is not possible to measure intraluminal supersaturation in the GI tract as mentioned previously, more reliable prediction of intraluminal supersaturation of amorphous solid dispersions can be gained by performing supersaturation/dissolution assays in biorelevant media. It is important to evaluate supersaturation not only in gastric but also in intestinal conditions, as discussed previously, and simulation of the GI pH gradient would be essential (Brouwers et al. 2009). A pH shift can be simulated by multi-compartmental dissolution techniques (Kostewicz et al. 2004;

Dissolution profiles of MBP and crystalline vemurafenib in USP apparatus 2: a unstressed vemurafenib MBP, b stressed vemurafenib MBP, c metastable crystalline vemurafenib, and d stable crystalline vemurafenib. (Reproduced with permission from Shah et al. 2013)
Gu et al. 2005) or simple manual transfer of gastric medium into intestinal medium (Mellaerts et al. 2008). Understanding of the intraluminal factors that affect supersaturation in both fasted and fed state would allow the development/modification of more appropriate biorelevant media for dissolution testing of amorphous solid dispersions. As endogenous and exogenous components such as bile salts, phospholipids, and food digestion products alter the dissolution and solubility of drugs (Dressman et al. 2007), more studies are needed in order to understand their influence on the rate and extent of intraluminal supersaturation as currently little is known regarding the impact of these factors (Brouwers et al. 2009). The discrepancy in supersaturation of amorphous compounds between in vitro studies and in vivo results (DiNunzio et al. 2008) presents another biopharmaceutical consideration in the development of IVIVCs.
7 IVIVCs of Amorphous Formulations
A few successful examples of IVIVC for amorphous formulation have been reported in the literature. A level A correlation was developed for disintegration-controlled amorphous nilvadipine matrix tablet (DCMT; Tanaka et al. 2006) (Fig. 15.10). Two DCMT amorphous nilvadipine formulations, DCMT-1 and DCMT-2, with different compositions of disintegrant were prepared and tested in vitro (USP apparatus 2 (100 rpm) in Japanese Pharmacopoeia (JP) first medium (pH 1.2, 37 ± 0.5 °C)) and in vivo (fasted beagle dogs; in vivo absorption calculated with numerical deconvolution).

Level A IVIVCs of DCMT-1 (a) and DCMT-2 (b) amorphous nilvadipine formulation. The reported linear regressions were y=0.995x (a) and y=0.9512x (b). (Reproduced with permission from Tanaka et al. 2006)
A successful multiple level C correlation between \({C}_{\rm max}\) and AUC with % released at 5 min (Q\(_{5 \rm min}\)) and at 60 min (Q\(_{60 \rm min}\)) was developed for poly(ethylene glycol) (PEG)–ritonavir amorphous solid dispersion formulations (Law et al. 2004). In this study, in vitro dissolution was conducted with a USP apparatus 1 (50 rpm, 37 ± 0.5 °C) in 900 mL of 0.1 N hydrochloric acid. The in vivo data was obtained from beagle dogs.
8 Conclusion
The dissolution test is still the only in vitro test that can potentially serve as a surrogate for in vivo performance. A well-designed test can serve as a valuable tool for the development of amorphous solid dispersions and the drug products containing amorphous solid dispersions. Appropriate test conditions may be developed to discriminate for composition, drug load, manufacturing process, and solid-state properties of the solid dispersion. Use of biorelevant and two-stage dissolution methods as well as supersaturation and sink versus non-sink conditions should be carefully considered during design of amorphous solid dispersion. The test may also offer insight into supersaturation and crystallization behavior as the drug is exposed to water during dissolution. The QC dissolution method must be able to discriminate between the presence and absence of the crystalline drug in the amorphous solid dispersion-based drug product. As a final note, consideration should be given to new developments in the area of integrated modeling and simulation , which offer the opportunity to integrate solubility, dissolution, precipitation, and absorption with simulations of dissolution behavior and impact on bioavailability of amorphous formulations.
References
Acartürk F, Kislal Ö, Çelebi N (1992) The effect of some natural polymers on the solubility and dissolution characteristics of nifedipine. Int J Pharm 85:1–6
Ali AA, Gorashi AS, (1984) Absorption and dissolution of nitrofurantoin from different experimental formulations. Int J Pharm 19:297–306
Allen LV Jr, Yanchick VA, Maness DD (1977) Dissolution rates of corticosteroids utilizing sugar glass dispersions. J Pharm Sci 66:494–497
Allen LV, Levinson RS, Martono DD (1978) Dissolution rates of hydrocortisone and prednisone utilizing sugar solid dispersion systems in tablet form. J Pharm Sci 67:979–981
Alonzo DE, Gao Y, Taylor LS (2009) Crystallization behavior of amorphous pharmaceuticals during dissolution. In: AAPS annual meeting and exposition. AAPS Journal, Los Angeles
Alonzo DE, Zhang GGZ, Zhou DL, Gao Y, Taylor LS (2010) Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm Res 27:608–618
Bevernage J, Brouwers J, Clarysse S, Vertzoni M, Tack J, Annaert P, Augustijns P (2010) Drug supersaturation in simulated and human intestinal fluids representing different nutritional states. J Pharm Sci 99(11):4525–4534
Blagden N, Davey RJ, Rowe R, Roberts R (1998) Disappearing polymorphs and the role of reaction by-products: the case of sulphathiazole. Int J Pharm 172:169–177
Bloch DW, Speiser PP (1987) Solid dispersions—fundamentals and examples. Pharm Acta Helv 62:23–27
Brewster ME, Vandecruys R, Peeters J, Neeskens P, Verreck G, Loftsson T (2008) Comparative interaction of 2-hydroxypropyl-beta-cyclodextrin and sulfobutylether-beta-cyclodextrin with itraconazole: phase-solubility behavior and stabilization of supersaturated drug solutions. Eur J Pharm Sci 34:94–103
Brittain HG, Bogdanowich SJ, Bugay DE, Devincentis J, Lewen G, Newman AW (1991) Physical characterization of pharmaceutical solids. Pharm Res 8:963–973
Brouwers J, Brewster ME, Augustijns P (2009) Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci 98:2549–2572
Cardew PT, Davey RJ (1985) The kinetics of solvent-mediated phase transformations. Proc Royal Soc Lond A 398:415–428
Chiou WL, Riegelman S (1970) Oral absorption of griseofulvin in dogs: increased absorption via solid dispersion in polyethylene glycol 6000. J Pharm Sci 59:937–942
Chiou WL, Riegelman S (1971) Pharmaceutical applications of solid dispersion systems. J Pharm Sci 60:1281–1302
Clarkson JR, Price TJ, Adams CJ (1992) Role of metastable phases in the spontaneous precipitation of calcium-carbonate. J Chem Soc Faraday Trans 88:243–249
Craig DQM (1990) Polyethyelene glycols and drug release. Drug Dev Ind Pharm 16:2501–2526
Craig DQM (2002) The mechanisms of drug release from solid dispersions in water-soluble polymers. Int J Pharm 231:131–144
Dai WG, Dong LC, Shi X, Nguyen J, Evans J, Xu Y, Creasey AA (2007) Evaluation of drug precipitation of solubility-enhancing liquid formulations using milligram quantities of a new molecular entity (NME). J Pharm Sci 96:2957–2969
Davey RJ, Cardew PT, Mcewan D, Sadler DE (1986) Rate controlling processes in solvent-mediated phase-transformations. J Cryst Growth 79:648–653
Davey RJ, Black SN, Goodwin AD, Mackerron D, Maginn SJ, Miller EJ (1997a) Crystallisation in polymer films: control of morphology and kinetics of an organic dye in a polysilicone matrix. J Mater Chem 7:237–241
Davey RJ, Blagden N, Potts GD, Docherty R (1997b) Polymorphism in molecular crystals: stabilization of a metastable form by conformational mimicry. J Am Chem Soc 119:1767–1772
Davey RJ, Allen K, Blagden N, Cross WI, Lieberman HF, Quayle MJ, Righini S, Seton L, Tiddy GJT (2002) Crystal engineering-nucleation, the key step. Cryst Eng Comm 4:257–264
Dinunzio JC, Miller DA, Yang W, Mcginity JW, Williams RO (2008) Amorphous compositions using concentration enhancing polymers for improved bioavailability of itraconazole. Mol Pharm 5:968–980
Dressman JB, Vertzoni M, Goumas K, Reppas C (2007) Estimating drug solubility in the gastrointestinal tract. Adv Drug Deliv Rev 59:591–602
El-Zein H, Riad L, El-Bary AA (1998) Enhancement of carbamazepine dissolution: in vitro and in vivo evaluation. Int J Pharm 168:209–220
Elamin AA, Ahlneck C, Alderborn G, Nyström C (1994) Increased metastable solubility of milled griseofulvin, depending on the formation of a disordered surface structure. Int J Pharm 111:159–170
EMA (2010) Guideline on the investigation of bioequivalence, appendix I: dissolution testing and similarity of dissolution EMA 2010 profiles CPMP/EWP/QWP/1401/98 Rev. 1/ Corr. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2010/01/WC500070039.pdf
Fawaz F, Bonini F, Guyot M, Bildet J, Maury M, Lagueny AM (1996) Bioavailability of norfloxacin from peg 6000 solid dispersion and cyclodextrin inclusion complexes in rabbits. Int J Pharm 132:271–275
FDA (1997) Guidance for industry: dissolution testing of immediate release solid oral dosage forms. Rockville, md: US department of health and human services, food and drug administration, center for drug evaluation and research (CDER)
Feth MP, Volz J, Hess U, Sturm E, Hummel RP (2008) Physicochemical, crystallographic, thermal, and spectroscopic behavior of crystalline and X-ray amorphous ciclesonide. J Pharm Sci 97:3765–3780
Ford JL (1986) The current status of solid dispersions. Pharm Acta Helv 61:69–88
Forster A, Hempenstall J, Tucker I, Rades T (2001) The potential of small-scale fusion experiments and the gordon-taylor equation to predict the suitability of drug/polymer blends for melt extrusion. Drug Dev Ind Pharm 27:549–560
Friesen DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JAS (2008) Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Mol Pharm 5:1003–1019
Gamsjager H, Lorimer JW, Scharlin P, Shaw DG (2008) Glossary of terms related to solubility. Pure Appl Chem 80:233–276
Ghazal HS, Dyas AM, Ford JL, Hutcheon GA (2009) In vitro evaluation of the dissolution behaviour of itraconazole in bio-relevant media. Int J Pharm 366:117–123
Gohel MC, Patel LD (2003) Processing of nimesulide-peg 400-pg-pvp solid dispersions: preparation, characterization, and in vitro dissolution. Drug Dev Ind Pharm 29:299–310
Goldberg AH, Gibaldi M, Kanig JL, Mayersohn M (1966) Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures IV. Chloramphenicol–urea system. J Pharm Sci 55:581–583
Gu CH, Rao D, Gandhi RB, Hilden J, Raghavan K (2005) Using a novel multicompartment dissolution system to predict the effect of gastric ph on the oral absorption of weak bases with poor intrinsic solubility. J Pharm Sci 94:199–208
Gupta P, Chawla G, Bansal AK (2004a) Physical stability and solubility advantage from amorphous celecoxib: the role of thermodynamic quantities and molecular mobility. Mol Pharm 1:406–413
Gupta P, Kakumanu VK, Bansal AK (2004b) Stability and solubility of celecoxib-pvp amorphous dispersions: a molecular perspective. Pharm Res 21:1762–1769
Hancock BC, Parks M (2000) What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res 17:397–404
Hancock BC, Zografi G (1997) Characteristics and significance of the amorphous state in pharmaceutical systems. J Pharm Sci 86:1–12
Higuchi WI, Mir NA, Desai SJ (1965) Dissolution rates of polyphase mixtures. J Pharm Sci 54:1405–1410
Huang LF, Tong WQ (2004) Impact of solid state properties on developability assessment of drug candidates. Adv Drug Deliv Rev 56:321–334
JP XV (2006) Japanese pharmacopoeia, Chapter 6.10 dissolution test.
Kohri N, Yamayoshi Y, Xin H, Iseki K, Sato N, Todo S, Miyazaki K (1999) Improving the oral bioavailability of albendazole in rabbits by the solid dispersion technique. J Pharm Pharmacol 51:159–164
Konno H, Taylor LS (2008) Ability of different polymers to inhibit the crystallization of amorphous felodipine in the presence of moisture. Pharm Res 25:969–978
Konno H, Handa T, Alonzo DE, Taylor LS (2008) Effect of polymer type on the dissolution profile of amorphous solid dispersions containing felodipine. Eur J Pharm Biopharm 70:493–499
Kostewicz ES, Wunderlich M, Brauns U, Becker R, Bock T, Dressman JB (2004) Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J Pharm Pharmacol 56:43–51
Kushida I, Ichikawa M, Asakawa N (2002) Improvement of dissolution and oral absorption of er-34122, a poorly water-soluble dual 5-lipoxygenase/cyclooxygenase inhibitor with anti-inflammatory activity by preparing solid dispersion. J Pharm Sci 91:258–266
Law D, Krill SL, Schmitt EA, Fort JJ, Qiu YH, Wang WL, Porter WR (2001) Physicochemical considerations in the preparation of amorphous ritonavir-poly(ethylene glycol) 8000 solid dispersions. J Pharm Sci 90:1015–1025
Law D, Schmitt EA, Marsh KC, Everitt EA, Wang WL, Fort JJ, Krill SL, Qiu YH (2004) Ritonavir-peg 8000 amorphous solid dispersions: in vitro and in vivo evaluations. J Pharm Sci 93:563–570
Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50:47–60
Li S, Wong S, Sethia S, Almoazen H, Joshi YM, Serajuddin ATM (2005) Investigation of solubility and dissolution of a free base and two different salt forms as a function of ph. Pharm Res 22:628–635
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26
Loftsson T, Fririksdóttir H, Gumundsdóttir TK (1996) The effect of water-soluble polymers on aqueous solubility of drugs. Int J Pharm 127:293–296
Matteucci ME, Miller MA, Williams RO, Johnston KP (2008) Highly supersaturated solutions of amorphous drugs approaching predictions from configurational thermodynamic properties. J Phys Chem B 112:16675–16681.
Mellaerts R, Mols R, Kayaert P, Annaert P, Van Humbeeck J, Van Den Mooter G, Martens JA, Augustijns P (2008) Ordered mesoporous silica induces ph-independent supersaturation of the basic low solubility compound itraconazole resulting in enhanced transepithelial transport. Int J Pharm 357:169–179
Miller DA, Mcconville JT, Yang W, Williams RO, Mcginity JW (2007) Hot-melt extrusion for enhanced delivery of drug particles. J Pharm Sci 96:361–376
Miller DA, Dinunzio JC, Yang W, Mcginity JW, Williams RO (2008a) Enhanced in vivo absorption of itraconazole via stabilization of supersaturation following acidic-to-neutral ph transition. Drug Dev Ind Pharm 34:890–902
Miller DA, Mcginity JW, Williams III RO (2008b) Solid dispersion technologies. In: Williams III RO, Taft DR (eds) Advanced drug formulation design to optimize therapeutic outcomes. Informa Healthcare, New York
Moneghini M, Carcano A, Zingone G, Perissutti B (1998) Studies in dissolution enhancement of atenolol. Part I. Int J Pharm 175:177–183
Moore MD, Wildfong PLD (2009) Aqueous solubility enhancement through engineering of binary solid composites: pharmaceutical applications. J Pharm Innov 4:36–49
Mosharraf M, Sebhatu T, Nystrom C (1999) The effects of disordered structure on the solubility and dissolution rates of some hydrophilic, sparingly soluble drugs. Int J Pharm 177:29–51
Murphy D, Rodriguez-Cintron F, Langevin B, Kelly RC, Rodriguez-Hornedo N (2002) Solution-mediated phase transformation of anhydrous to dihydrate carbamazepine and the effect of lattice disorder. Int J Pharm 246:121–134
Nernst W (1904) Theorie der reaktionsgeschwindigkeit in heterogenen systemen. Z Phys Chem 47:52–102
Noyes AA, Whitney WR (1897) The rate of solution of solid substances in their own solutions. J Am Chem Soc 19:930–934
Ossi PM (2003) Structural changes induced by swift heavy ions in non-metallic compounds. Beam interactions with materials and atoms. Nucl Instrum Methods Phys Res 209:55–61
Overhoff KA, Mcconville JT, Yang W, Johnston KP, Peters JI, Williams RO (2008) Effect of stabilizer on the maximum degree and extent of supersaturation and oral absorption of tacrolimus made by ultra-rapid freezing. Pharm Res 25:167–175
Patterson JE, James MB, Forster AH, Lancaster RW, Butler JM, Rades T (2005) The influence of thermal and mechanical preparative techniques on the amorphous state of four poorly soluble compounds. J Pharm Sci 94:1998–2012
PhEUR (2011) European pharmacopoeia, Chapter 2.9.3 Dissolution test for solid dosage forms, strasbourg, france, council of europe, european directorate for the quality of medicines and healthcare
Raumer MV, Hilfiker R, Blatter F (2006) Relevance of solid-state properties for pharmaceutical products. In: Hilfiker R (ed) Polymorphism in the pharmaceutical industry. Wiley VCH, Weinheim
Rodriguez-Hornedo N, Lechuga-Ballesteros D, Wu HJ (1992) Phase transition and heterogeneous/epitaxial nucleation of hydrated and anhydrous theophylline crystals. Int J Pharm 85:149–162
Sarode AL, Sandhu H, Shah N, Malick W, Zia H (2013) Hot melt extrusion for amorphous solid dispersions: temperature and moisture activated drug–polymer interactions for enhanced stability. Mol Pharm 10:3665–3675
Savolainen M, Kogermann K, Heinz A, Aaltonen J, Peltonen L, Strachan C, Yliruusi J (2009) Better understanding of dissolution behaviour of amorphous drugs by in situ solid-state analysis using raman spectroscopy. Eur J Pharm Biopharm 71:71–79
Serajuddin ATM (1999) Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci 88:1058–1066
Shah N, Iyer RM, Mair H-J, Choi DS, Tian H, Diodone R, Fähnrich K, Pabst-Ravot A, Tang K, Scheubel E, Grippo JF, Moreira SA, Go Z, Mouskountakis J, Louie T, Ibrahim PN, Sandhu H, Rubia L, Chokshi H, Singhal D, Malick W (2013) Improved human bioavailability of vemurafenib, a practically insoluble drug, using an amorphous polymer-stabilized solid dispersion prepared by a solvent-controlled coprecipitation process. J Pharm Sci 102:967–981
Shalaev E, Zografi G (2002) The concept of structure in amorphous solids from the perspective of the pharmaceutical sciences. Amorphous food and pharmaceutical systems. The Royal Society Of Chemistry, Cambridge
Six K, Verreck G, Peeters J, Brewster M, Van Den Mooter G (2004) Increased physical stability and improved dissolution properties of itraconazole, a class II drug, by solid dispersions that combine fast- and slow-dissolving polymers. J Pharm Sci 93:124–131
Six K, Daems T, De Hoon J, Van Hecken A, Depre M, Bouche MP, Prinsen P, Verreck G, Peeters J, Brewster ME, Van Den Mooter G (2005) Clinical study of solid dispersions of itraconazole prepared by hot-stage extrusion. Eur J Pharm Sci 24:179–186
Strickley RG, Oliyai R (2007) Solubilizing vehicles for oral formulation development. In: Augustijins P, Brewster ME (eds) Solvent systems and their selection in pharmaceutics and biopharmaceutics. Biotechnology: pharmaceutical aspects, vol VI. Springer, New York, pp 257–308
Tanaka N, Imai K, Okimoto K, Ueda S, Tokunaga Y, Ibuki R, Higaki K, Kimura T (2006) Development of novel sustained-release system, disintegration-controlled matrix tablet (DCMT) with solid dispersion granules of nilvadipine (II): in vivo evaluation. J Control Release 112:51–56
Tanno F, Nishiyama Y, Kokubo H, Obara S (2004) Evaluation of hypromellose acetate succinate (HPMCAS) as carrier in solid dispersions. Drug Dev Ind Pharm 30:9–17
Torrado S, Torrado S, Torrado JJ, Cadórniga R (1996) Preparation, dissolution and characterization of albendazole solid dispersions. Int J Pharm 140:247–250
USP (2009a) The United States pharmacopeia and the national formulary, <711> Dissolution. The Official Compendia of Standards USP 32–NF 27
USP (2009b) The United States pharmacopeia and the national formulary, <1092> The dissolution procedure: development and validation. The Official Compendia of Standards USP 32–NF 27
Usui F, Maeda K, Kusai A, Nishimura K, Keiji Y (1997) Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int J Pharm 154:59–66
Van Drooge DJ, Hinrichs WL, Frijlink HW (2004) Anomalous dissolution behaviour of tablets prepared from sugar glass-based solid dispersions. J Control Release 97:441–452
Vertzoni M, Dressman J, Butler J, Hempenstall J, Reppas C (2005) Simulation of fasting gastric conditions and its importance for the in vivo dissolution of lipophilic compounds. Eur J Pharm Biopharm 60:413–417
Wei-Qin T (2009) Salt screening and selection: new challenges and considerations in the modern pharmaceutical research and development paradigm. Developing solid oral dosage forms. Academic Press, San Diego
Weissbuch I Leisorowitz L, Lahav M (1994) Tailor-made and charge-transfer auxiliaries for the control of the crystal polymorphism of glycine. Adv Mater 6:952–956
Weissbuch I, Zbaida D, Addadi L, Leiserowitz L, Lahav M (1987) Design of polymeric inhibitors for the control of crystal polymorphism—induced enantiomeric resolution of racemic histidine by crystallization at 25-Degrees-C. J Am Chem Soc 109:1869–1871
Yamashita K, Nakate T, Okimoto K, Ohike A, Tokunaga Y, Ibuki R, Higaki K, Kimura T (2003) Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int J Pharm 267:79–91
Zhang GGZ, Gu C, Zell MT, Burkhardt RT, Munson EJ, Grant DJW (2002) Crystallization and transitions of sulfamerazine polymorphs. J Pharm Sci 91:1089–1100
Zhang GGZ, Zhou D, Yihong Q, Yisheng C, Geoff GZZ, Lirong L, William RP (2009) Crystalline and amorphous solids. Developing solid oral dosage forms. Academic Press, San Diego
Zhu HJ, Grant DJW (1996) Influence of water activity in organic solvent plus water mixtures on the nature of the crystallizing drug phase 2. Ampicillin. Int J Pharm 139:33–43
Zhu HJ, Yuen CM, Grant DJW (1996) Influence of water activity in organic solvent plus water mixtures on the nature of the crystallizing drug phase 1. Theophylline. Int J Pharm 135:151–160
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Controlled Release Society
About this chapter
Cite this chapter
Fotaki, N., Long, C., Tang, K., Chokshi, H. (2014). Dissolution of Amorphous Solid Dispersions: Theory and Practice. In: Shah, N., Sandhu, H., Choi, D., Chokshi, H., Malick, A. (eds) Amorphous Solid Dispersions. Advances in Delivery Science and Technology. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-1598-9_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1598-9_15
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-1597-2
Online ISBN: 978-1-4939-1598-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)