
Abstract
Solid dispersion technology has been used over the last three decades to improve the dissolution and oral absorption of poorly soluble compounds. While the characterization of dissolution performance of crystalline pharmaceutical systems has long been established, the dynamic nature of the amorphous dissolution processes requires the use of unique methodologies. The in vitro differentiation of the drug and drug-containing species of these systems is crucial to accomplishing the measurement of the critical-to-performance free drug concentrations as a function of time. This chapter describes the theoretical aspects of amorphous dissolution and recent examples applying free drug dissolution testing to the oral bioavailability assessment of solid dispersion formulations.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Free Drug
- Dissolution Profile
- Dissolution Testing
- Biopharmaceutics Classification System
- Free Drug Concentration
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Amorphous solid dispersions (ASDs) Footnote 1 have long been noted for their potential advantages to the pharmaceutical industry (Chiou and Reigelman 1971 ; Goldberg et al. 1965 ). More than ever before, however, ASDs are being embraced in the formulation development and manufacturing of oral drug candidates that have solubility-limited absorption. Such insoluble Biopharmaceutics Classification System (BCS) II and IV compounds (Amidon et al. 1995 ) are increasingly filling drug development pathwaysFootnote 2 as newer and better biological targets demand hydrophobic interactions for selectivity and therapeutically balanced potency. It is well known that ASDs can enhance dissolution , provide large supersaturation , and maintain that supersaturation in aqueous media for these poorly soluble, but otherwise promising, active pharmaceutical ingredients (APIs; reviewed in Janssens and Van der Mooter 2009 ; Yu 2001 ; Leuner and Dressman 2000).
Commercial products recently launched with ASD solubilization technology are shown in Table 8.1,Footnote 3 and many more are reported to have entered human clinical trials (Friesem et al. 2008 ). The most used commercial processing methods to manufacture ASDs are hot-melt extrusion (HME; Repka et al. 2007 ; Crowley et al. 2007; DiNunzio et al. 2008; Breitenbach 2002) and spray drying (SD; Patel and Suthar 2009 ; Ronald 1997; Oakley 1994; Shoyele and Cawthorne 2006; Vehring 2008) because these processes are mature (Breitenbach 2002 ; Patel and Suthar 2009; Ronald 1997 ), scalable (Breitenbach 2002 ; Oakley 1994 ), and controllable (Repka et al. 2007 ; Crowley et al. 2007 ; Shoyele and Cawthorne ; Vehring 2008). Additional process methodologies to produce ASDs exist, but until recently, only SD and HME were employed on a commercial scale. The current exception is the use of antisolvent controlled precipitation processes to make Roche’s Zelboraf® (vemurafenib) ,Footnote 4 the most recently launched ASD product (see Table 8.1). It is anticipated that ASDs will continue to gain momentum in the industry as enabling formulation strategies owing to the recent commercial launches of ASDs on the market, the well-known oral bioavailability enhancements to be gained, and the increasing amount of promising, but poorly soluble, candidates entering drug development pipelines.
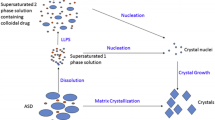
From formulation design and development, to cGMP manufacturing and commercialization, amorphous molecular dispersions (AMDs) undergo similar treatment and testing to that of traditional drug formulations. These ASDs, however, require special considerations that extend to the material’s critical-to-performance attributes, such as dissolution , physicochemical properties, and stability. For example, Fig. 8.1 depicts the intestinal dissolution events for a low soluble drug formulated as a crystalline drug (Fig. 8.1a), and formulated as an ASD (Fig. 8.1b). As a consequence of the formulation-derived drug species (Fig. 8.1b, box), the dissolution performance of a low soluble drug in an ASD (and any formulations thereof) follow more complex dissolution pathways than crystalline forms.

Simple illustration of drug speciation absorption for a low soluble, orally administered drug in a crystalline form (unformulated) and b amorphous solid dispersion (ASD) formulation. Total formulation-derived drug species upon disintegration are shown in box for drug in ASD
The dissolution rate to produce freely solvated drug (i.e., “free drug”) is often the rate-limiting step to overall oral drug absorption for low soluble drugs (e.g., BCS II compounds). This rate can therefore greatly impact the oral bioavailability . Fundamental factors affecting this rate for crystalline material are described by the well-known modified Noyes–Whitney expression:
where dm/dt is the rate expressed at the change in the amount of freely dissolved drug (m) per unit time (t), D D is the drug’s diffusivity in a uniform boundary layer, S is the surface area, V is the volume of dissolution media, h is the thickness of the uniform boundary layer, and C s − C t describes the difference in the concentration of drug and the drug’s saturation solubility in the medium. Expressed as the integral, free drug amounts can be written as:
Limitations of this for describing ASDs are well known (Martinez and Amidon 2002 ; Balakrishnan et al. 2004 ). Static surface areas and boundary layers, for example, may not be fixed for ASD-formulated drugs as the primary particles disintegrate into colloidal drug-containing species (see Fig. 8.1b) and these transit down the gastrointestinal (GI) tract. While a crystalline drug deposited in the GI lumen is limited in solubility by the crystalline equilibrium (see Fig. 8.1a), drug presented in the higher energy amorphous state can achieve supersaturation that can be described thermodynamically as:
where S a /S c is the ratio of the amorphous solubility to crystalline solubility, ∆ H f is the heat of fusion, and T m is the melting point of the crystalline material.
Successful ASD-formulated drugs are designed to achieve this supersaturation , but this places the free drug in a thermodynamic regime that will promote rapid recrystallization or amorphous precipitation unless interference occurs. It is therefore critical to understand and manipulate the precipitation rate of amorphous drug at supersaturations achieved by ASDs since the sustainment of any solubility enhancements can greatly impact drug absorption for BCS II compounds. The “spring and parachute” dissolution profile (Guzmán et al. 2007 ) from ASD formulations (and other insoluble drug formulations) is currently used to describe the events of drug dissolution from a supersaturating formulation. This includes the rapid dissolution (“spring”) of the amorphous form followed by the inhibition (“parachute”) of the compound’s propensity to rapidly return to its most thermodynamically stable form. Proper in vitro dissolution testing of these systems remains the important empirical approach to accurately determine free drug enhancement and differentiation among different ASDs. This chapter will focus on the in vitro dissolution performance testing of ASDs to measure the absorbable free drug species in solution separately from the other drug-containing species in solution.
2 Sink In Vitro Dissolution
For oral, immediate-release final drug products , the importance of dissolution testing is exemplified by the regulated use of United States Pharmacopeia (USP) method < 711> .Footnote 5 Guidelines take into consideration the apparatus, sample preparation, sampling procedure, and other aspects of the methodology. In the context of quality control testing of final drug product per USP methodology and guidelines (US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) 1997, 2000), the goal is twofold. First, testing the dissolution-rate consistency of the product provides some insight on the potential in vivo consistency. Second, dissolution testing also provides a performance metric for the manufacturing processes used to produce the ASD. Currently, USP methodology is the gold standard for in vitro dissolution testing of final drug product. Consequently, final drug products containing ASD solubilization technology are tested for complete dissolution over the course of a practical testing duration.
In order to establish this complete release metric, USP dissolution testing for final drug product is developed under sink conditions. The in vitro volumes are based on the accepted values for human GI volumes. For example, in the fasted state, the volumes are considered to be around 250 mL. A soluble compound as determined by the BCS categorization (i.e., BCS I, III; Amidon et al. 1995) is therefore considered one that is soluble in less than 250 mL of test media at its highest dose. Insoluble compounds, on the other hand, are those that are not soluble in these media volumes at their expected doses. For these compounds, sink dissolution conditions cannot be achieved in conventional aqueous media like 0.1 or 0.01 N HCl (gastric mimic pH = 1 or 2, respectively), or phosphate-buffered saline (PBS, intestinal mimic pH = 6.5).
Sink conditions are established during analytical method development to ensure that the drug product completely releases the API as free drug over a brief period of time (e.g., < 3 h). For insoluble compounds, the appropriate use of ionic and nonionic surfactants in the media to achieve sink conditions has been discussed (Gowthamarajan and Singh 2010 ). Likewise, the merits of the four established apparatuses (i.e., apparatus 1, basket; apparatus 2, paddle; apparatus 3, reciprocating cylinder; and apparatus 4, flow-through cell) to test products containing low soluble compounds under sink conditions have also been reported (Uddin et al. 2011 ; Brown et al. 2004). Understanding these parameters for the release testing of clinical trial materials (CTMs) that contain AMDs and their insoluble molecules continues to be discussed. Currently, however, this is addressed similarly to release testing of soluble drug products .
3 Physiologically Relevant Dissolution
In vitro dissolution testing is not relegated to testing final drug products , though. This valuable performance assessment is utilized extensively at all stages of oral drug development . Table 8.2 lists the rationale for dissolution testing at various stages of drug/formulation development . Even discovery groups are utilizing dissolution testing of formulations for lead compound selection (Padden et al. 2011 ). By measuring the drug candidate’s ability to disintegrate, dissolve, and release freely solvated drug release free drug into physiologically relevant media, performance can be established and tracked against a number of design, development, and manufacturing variables.
In the best-case scenario, the in vitro dissolution testing predicts the in vivo performance. Toward this end, there has been considerable attention paid to in vitro–in vivo correlation (IVIVC; Cardot et al. 2007 ). In general, IVIVC efforts continue to focus on dissolution apparatus and methodology (Fotaki and Vertzoni 2010 ), media (Taupitz et al. 2013; Otsuka et al. 2013; Arndt et al. 2013; Reppas and Vertzoni 2012; Bevernage et al. 2010), and physiologically based pharmacokinetic (PBPK) modeling and statistical analyses (Zhao et al. 2011 ). In some cases, remarkable correlations have been reported; however, achieving universal methods for accurate IVIVC remains elusive.
Nevertheless, it is reasonable to still strive toward the development of physiologically relevant dissolution procedures. This is especially important during the design and development of performance-enhancing solubilized formulations like ASDs. By incorporating in vitro performance assays during formulation development , comparisons between formulations, and between formulated and unformulated API can be assessed for rank ordering, process-to-performance metrics, and stability sample performance. Rank ordering formulations based on in vitro performance can greatly reduce the number of in vivo studies necessary to screen different formulations. During downstream formulation development and manufacturing scale-up, in vitro dissolution of ASDs can be used to assess performance improvements or detriments of altering the formulation composition or unit operation processes.
4 Nonsink In Vitro Dissolution
In order to accomplish in vitro dissolution performance for insoluble drug candidates, nonsink conditions should be utilized since this more closely represents the physiological situation. For example, if a moderate-to-low potency compound is to be examined in a fasted-state simulated intestinal fluid (FaSSIF), the projected highest dose may be ≥ 250 mg, especially for in vivo (nonhuman) PK and toxicology studies. This would require the solubility of the API to be ≥ 1.0 mg/mL in the dissolution media to be considered sink conditions. Compounds requiring solubilization formulation strategies such as those incorporated into ASDs, are typically much lower in solubility and it is often not possible to adjust the media to reach these levels without the concomitant loss of relevance to in vivo physiology. Thus, nonsink dissolution assays are most often applied to test poorly soluble API during the formulation development stages.
Using nonsink dissolution procedures to examine the performance of ASDs provides the dissolution rate, amorphous API solubility supersaturation enhancement, and the potential sustainment inherent to successful ASDs. Importantly, the nonsink dissolution affords insight into the biorelevant API precipitation rates. For example, Fig. 8.2 shows the dissolution profiles for the amorphous insoluble compound fenofibrate presented in a 1:3 API:HPMCAS-M ASD (blue line) made via SD and comparatively as the unformulated API (red line). The profile exhibited by the ASD has been described as a “spring and parachute” dissolution profile (Brouwers et al. 2009 ). The hallmarks of this profile are that the higher energy amorphous API dissolves rapidly, reaches a supersaturation state that is sustained for some period of time, and eventually precipitates as a lower energy crystalline form, or as an amorphous solid. In contrast, the unformulated API only reaches a very low solubility of about 0.35 µg/mL after 60 min in the dissolution media.

In vitro nonsink dissolution of 1:3 fenofibrate:HPMCAS-M (blue line) and unformulated fenofibrate (red line)
5 Drug Speciation of AMDs and Free Drug Analyses
An important aspect to consider when testing the in vitro dissolution performance of ASDs is the different drug-containing species that can occur in the media. Figure 8.1 differentiates this drug speciation as it occurs for crystalline API during dissolution (Fig. 8.1a), and as it transpires for AMD dissolution (Fig. 8.1b). In the case of the crystalline API, two or three physiologically relevant drug species can be present during testing. These are crystalline drug, freely dissolved drug (or “free drug”), and drug in micelles, if micelles are present in the media. It is critical to separate the crystalline drug from the mixture and to measure the free drug as a function of time in the media since it is only the free drug that is absorbed in vivo. This is relatively straightforward to do and is typically accomplished by filtering and/or centrifugation steps in the sample processing.
In contrast to the dissolution of crystalline API, Fig. 8.1b shows the various drug species that are formed during the dissolution of drug from an ASD. As shown, the ASD produces similar drug species as that of unformulated API (i.e., free drug, crystalline drug, drug in micelles); however, there also exists drug–excipient interactions that can lead to physiologically relevant API–excipient complexes and colloid species during dissolution. These colloidal materials form during the wetting and disintegration of the primary particles of the ASD and vary in size and density according to the physicochemical and solution properties of the particular drug–excipient interaction (Bikiaris 2011 ). For example, a high degree of API–polymer interaction (e.g., hydrophobic or hydrogen bonding) in one polymeric ASD can lead to a longer duration of API chain disentanglement and therefore a potentially greater amount of colloidal material, as compared to an ASD made from a different polymer (Balata et al. 2010 ).
Removing the colloidal species during the sample preparation, and separately measuring free drug as a function of time, can be challenging. Traditional in vitro dissolution sample preparation methodologies may not sufficiently remove the resulting submicron and/or nanometer-sized colloidal material that can form. Instrument-intensive methodologies such as nuclear magnetic resonance (NMR) have been employed to examine complex dissolution profiles for API–excipient systems (Kojima et al. 2012 ; Abhishek and Chandrakumar 2011; Dahlberg 2010; Zhang et al. 2011), but it is desirable to be able to measure free drug using more practical dissolution setups. More recently, several authors have adapted diffusion and partitioning methodologies to the assessment of free drug in solution. In a study by Alonzo et al. (2011 ), researchers used dialysis bags to conduct free drug dissolution testing by measuring the diffusion across the membrane. While effective at determining the free drug concentration, dialysis methods similar to this can be time and labor intensive thus limiting the throughput of the technology. Other, more resource-efficient methods have also been reported, including the use of immiscible liquids to determine the free drug concentration via partitioning into the water immiscible phase (Shi et al. 2010) . Although researchers have shown acceptable correlations, many concerns, immiscible phase, including the incorporation of colloidal material into the water immiscible phase remain. Despite the difficulties in assaying for free drug during ASD dissolution, proprietary methods have been developed that can greatly impact the formulation design of polymeric ASDs.
6 Case Study 1
It is often the case that the formulation scientist will utilize an in vitro dissolution assessment to screen different ASDs during formulation development studies. Screening efforts can help rank order a larger set of formulations and reduce the scope of in vivo studies by selecting a smaller set of materials to test. It is therefore important to measure the free drug, in addition to measuring the total drug species, during in vitro performance assessments of ASDs (see also Fig. 8.2). Fenofibrate is currently marketed as an adjunctive therapeutic to treat hypertriglyceridemia or mixed dyslipidemia in adult patients. Fenofibrate is considered a BCS II molecule because of its dose and physicochemical properties (e.g., maximum dose of 120 mg/day; aqueous solubility < < 480 µg/mL; log P ≈ 5.3). Five amorphous molecular dispersions of fenofibrate and polymer were produced at a 25 % API loading by weight using laboratory-scale SD techniques (Smithey et al. 2010 ) and these compositions are shown in Table 8.3.
The 1:4 fenofibrate:polymer ASDs were assessed for in vitro performance in PBS (pH = 6.5) media, and dosed as dry powders at 1 mg active equivalent (4 mg ASD) per milliliter of media (i.e., nonsink conditions) in scintillation vials stirred at approximately 150 rpm. Aliquots of media were removed at set time points (i.e., T = 10, 20, 40, 90, 120, 180 min), and both the total drug and free drug concentrations (see Fig. 8.1b) were processed and measured separately. Total drug was assessed using a procedure similar to microcentrifuge methodologies previously described (Friesem et al. 2008). Free drug was measured by proprietary means.
Figure 8.3 depicts the Cmax results of the total drug and free drug analyses for these five ASDs. Although there is some differentiation of the different solubilized formulations when assessing total drug species only, the Cmax for these dispersions are very similar (see Fig. 8.3a). If a rank order were to be produced from the total drug analysis, it would be HPMCAS-M > CAP > HPMCP-H55 > Soluplus > PVP-VA > > > crystalline API. In contrast, when free drug is assessed (see Fig. 8.3b), there is a much greater distinction among the ASDs. Moreover, the rank ordering results are completely different than that which arise from the total drug analysis. Using the free drug concentrations at Cmax, the rank ordering is Soluplus > HPMCP-H55 > CAP > HPMCAS-M > PVP-VA > crystalline API.

Cmax values for five amorphous molecular dispersions (AMDs) of 1:3 fenofibrate:polymer as determined for in vitro nonsink dissolution measurements of a total drug species and b free drug species
By obtaining the concentrations of the true free drug portion of the drug speciation that occurs during nonsink dissolution of ASDs, more accurate computational calculations can also be made than if total drug values are used. Using the fenofibrate free drug concentrations of the five compositions, the calculated fraction absorbed can be estimated as a function of free drug concentrations and dose. Figure 8.4 displays these calculations using the physiological parameters for beagle dogs. The fraction absorbed for unformulated, crystalline fenofibrate is also calculated, and an overlay of the range of in vivo performance is estimated from previous reports (Chen et al. 2009 ).

Calculated fraction absorbed (%) of 1:3 fenofibrate:polymer amorphous molecular dispersions (AMDs) and crystalline API as a function of the average in vitro free drug concentrations (µg/mL) and dose (mg/kg) for beagle dog physiology. The shaded box is the range of in vivo performance as estimated from literature values
7 Case Study 2
Clearly the in vitro nonsink dissolution free drug analyses can be important during the selection of excipients in the formulation design stages. These measurements can also be used to determine the extent of performance advantages and disadvantages of different API loadings in ASDs. Figure 8.5 shows the in vitro and in vivo dissolution profiles for two ASDs, each produced with the same polymeric excipient, but at two different API loadings (i.e., 25 and 40 % w/w). As in the previous fenofibrate example, the in vitro performance was measured in PBS (pH = 6.5), at a dry powder dose of 1 mg active equivalent per milliliter of media (i.e., nonsink conditions). The in vivo PK analysis was carried out on Sprague Dawley rats (n = 3) at a 30 mg/kg dose of SDI suspension.

In vitro performance of total drug species and free drug species concentrations and their in vivo relationship for two amorphous molecular dispersions (AMDs) of the same active pharmaceutical ingredient (API) and polymeric excipient, but at two different drug loadings
In Fig. 8.5, the total drug analyses would suggest that little or no performance differences would be expected between the 25 and 40 % API loadings. The free drug analysis, however, forecasts greater performance for the ASD with the lower API loading. This result is not surprising because lower drug loading often leads to a dose-equivalent enhanced performance for ASDs made with the same excipient. This is due, in part, because there is more polymer present in solution to help sustain the API supersaturation state. It is therefore also no surprise that the in vivo study results show a better relationship with the in vitro free drug analysis as compared to the total drug analysis.
The aforementioned case studies demonstrate the importance of proper in vitro dissolution studies for ASDs. When accomplished in nonsink conditions, and analyzed for free drug concentrations as a function of time in the dissolution media, the results can be used to make informative decisions regarding excipient choice and drug loading. This can also be extended to monitoring any process changes during formulation development and scale-up.
8 Conclusion
The number of poorly soluble compounds in the pharmaceutical development pipelines is increasing. Concomitantly, so too are the drug delivery systems suited for oral delivery of promising, but low soluble, drug candidates. ASDs , however, remain well known for their potential formulation advantages for BCS II and IV compounds. More than ever before, ASD formulations are found in the marketplace as an answer for formulating drugs with solubility-limited absorption. Critical-to-performance attributes of ASDs, such as in vitro dissolution , require specialized knowledge and testing procedures to accurately determine their potential for bioavailability enhancements during formulation design and development, manufacturing, and commercial production.
An important aspect to consider when testing the in vitro dissolution performance of ASDs is the different drug-containing species that can occur in the media. Removing the colloidal species during the sample preparation, and separately measuring free drug as a function of time, can be challenging; however, accurately determining the amount of free drug that is available for absorption remains an important empirical measurement. Case studies discussed in this chapter are examples of how the data obtained from such measurements can lend insight into the potential physiological impacts of ASD formulations. Such test results allow for the proper formulation design and selection, the efficient adjustments during formulation development , and the monitoring of quality attributes during manufacturing.
Notes
- 1.
Amorphous solid dispersion (ASD) is used to describe a homogenous dispersion of noncrystalline API and excipient(s) at molecular compositions. Similar systems are often described as solid dispersions, amorphous molecular dispersions, solid solutions, solid liquids, and others.
- 2.
- 3.
- 4.
Center for Drug Evaluation and Research Application No. 202429, Clinical Pharmacology and Biopharmaceutics Review(s), www.accessdata.fda.gov.
- 5.
References
Abhishek B, Chandrakumar N (2011) Real-time in vitro drug dissolution studies of tablets using volume-localized NMR (MRS). Appl Magn Reson 40:251–259
Alonzo DE, Gao Y, Zhou D, Mo H, Zhang GGZ, Taylor LS (2011) Dissolution and precipitation behavior of amorphous solid dispersions. J Pharm Sci 100:3316–3331
Amidon GL, Lennernäs H, Shah VP, Crison JR (1995) A theoretical basis for a biopharmaceutical drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 12:413–420
Arndt M, Chokshi H, Tang K, Parrott NJ, Reppas C, Dressman JB (2013, Aug) Dissolution media simulating the proximal canine gastrointestinal tract in the fasted state. Eur J Pharm Biopharm 84(3):633–641
Balakrishnan A, Rege BD, Amidon GL, Polli JE (2004) Surfactant-mediated dissolution: contributions of solubility enhancement and relatively low micelle diffusivity. J Pharm Sci 93:2064–2075
Balata G, Mahdi M, Bakera RA (2010) Improvement of solubility and dissolution properties of ketoconazole by solid dispersion and inclusion complexes. Asian J Pharm Sci 5:1–12
Benet LZ, Wu C-Y (2006) Using a biopharmaceutics drug disposition classification system to predict bioavailability and elimination characteristics of new molecular entities. New Jersey Drug Metabolism Discussion Group (NJDMDG), Somerset, NJ. October 5:2006
Bevernage J, Brouwers J, Clarysse S, Vertzon M, Tack J, Annaert P, Augustijns P (2010) Drug supersaturation in simulated and human intestinal fluids representing different nutritional states. J Pharm Sci 99:4525–4534
Bikiaris DN (2011) Solid dispersions, part I: recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin Drug Deliv 8:1501–1519
Bottorf KJ, Katstra JP, Gasper F (2007) US Patent 2007/0218138. Sept. 20th, 2007
Breitenbach J (2002) Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm 54:107–117
Brouwers J, Brewster ME, Augustijns P (2009) Supersaturating drug delivery systems: the answer to solubility-limited oral bioavailability? J Pharm Sci 98:2549–2572
Brown CK, Chokshi HP, Nickerson B, Reed RA, Rohrs BR, Shah PA (2004) Acceptable analytical practices for dissolution testing of poorly soluble compounds. Pharm Technol 28:56–65
Cardot J-M, Beyssac E, Alric M (2007) In vitro–in vivo correlation: importance of dissolution in IVIVC. Dissolution Technol 14:15–19
CDER (2011) NDA submission application #202429. March 21, 2011
Chen Y, Lu Y, Chen J, Lai J, Sun J, Hu F, Wu W (2009) Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing bile salt. Int J Pharm 376:153–160
Chiou WL, Reigelman S (1971) Pharmaceutical applications of solid dispersion systems. J Pharm Sci 60:1281–1302
Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, McGinity JW, Martin C (2007) Pharmaceutical applications of hot melt extrusion: part I. Drug Dev Ind Pharm 33:909–926
Dahlberg C (2010) Doctoral thesis, Royal Institute of Technology, Stockholm, Sweden
DiNunzio JC, Miller DA, Yang W, McGinity JW, Williams RO (2008) Amorphous compositions using concentrating enhancing polymers for improved bioavailability of itraconazole. Mol Pharm 5:968–980
Dong W (2005) Multiparticulate drug delivery system for lipophilic drugs and macromolecules. PhD dissertation in Chemistry, Freie Universität, Berlin
Fotaki N, Vertzoni M (2010) Biorelevant dissolution methods and their application in in vitro–in vivo correlations for oral formulations. Open Drug Deliv J 4:2–13
Friesem DT, Shanker R, Crew M, Smithey DT, Curatolo WJ, Nightingale JS (2008) Hydroxypropyl methylcellulose acetate succinate-based spray-dried dispersions: an overview. Mol Pharm 5:1003–1019
Goldberg AH, Gibaldi M, Kanig JL (1965) Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures I. Theoretical consideration and discussion of the literature. J Pharm Sci 54:1145–1148
Gowthamarajan K, Singh SK (2010) Dissolution testing for poorly soluble drugs: a continuing perspective. Dissolution Technol 17:24–32
Guzmán HR, Tawa M, Zhang Z, Ratanabanangkoon P, Shaw P, Gardner CR, Chen H, Moreau J-P, Almarsson Ö, Remenar JF (2007) Combined use of crystalline salt forms and precipitation inhibitors to improve oral absorption of celecoxib from solid oral formulations. J Pharm Sci 96:2686–2702
Janssens S, Van der Mooter G (2009) Review: physical chemistry of solid dispersions. J Pharm Pharmacol 61:1571–1586
Kojima T, Higashi K, Suzuki T, Tomono K, Moribe K, Yamamoto K (2012) Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with EUDRAGIT® EPO. Pharm Res 29:2777–2791
Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm 50:47–60
Martinez MN, Amidon GL (2002) A mechanistic approach to understanding the factors affecting drug absorption: a review of the fundamentals. J Clin Pharmacol 42:620–643
Oakley DE (1994) Scale-up of spray dryers with the aid of computational fluid dynamics. Dry Technol 12:217–233
Otsuka K, Shono Y, Dressman JB (2013) Coupling biorelevant dissolution, methods with physiologically based pharmacokinetic modelling to forecast in-vivo performance of solid oral dosage forms. J Pharm Pharmacol 65:937–952
Padden BE, Miller JM, Robbins T, Zocharski PD, Prasad L, Spence JK, LaFountaine J (2011) Amorphous solid dispersions as enabling formulations for discovery and early development. Am Pharm Rev 14:66–73
Patel RP, Suthar AM (2009) Spray drying technology: an overview. Indian J Sci Technol 2:44–47
Pomerantz RJ (2007) Combining biomedical research within academia and industry in the 21st century (keynote address), AAPS Annual Meeting, 2007, San Diego, CA
Repka MA, Battu SK, Upadhye SB, Thumma S, Crowley MM, Zhang F, Martin C, McGinity JW (2007) Pharmaceutical applications of hot melt extrusion: part II. Drug Dev Ind Pharm 33:1043–1057
Reppas C, Vertzoni M (2012) Biorelevant in-vitro performance testing of orally administered dosage forms. J Pharm Pharmacol 64:919–930
Ronald CD (1997) Spray drying: innovative use of an old process. Des Elem 7:97–113
Rosenberg J, Reinhold U, Liepold B, Breitenbach J, Alani LL, Ghosh S (2008) US Patent 2008/0299203 A1. Dec. 4th, 2008
Shi Y, Gao P, Gong Y, Ping H (2010) Application of a biphasic test for characterization of in vitro drug release of immediate release formulations of Celecoxib and its relevance to in vivo absorption. Mol Pharm 7:1458–1465
Shoyele SA, Cawthorne S (2006) Particle engineering techniques for inhaled biopharmaceuticals. Adv Drug Deliv Rev 58:1009–1029
Smithey D, Fennewald J, Gautschi J, Crew M, Ali S, Lan Y, Langley N (2010) Evaluation of the polymer Soluplus® for spray-dried dispersions of poorly soluble compounds, AAPS 2010, Poster R6081
Taupitz T, Dressman JB, Klein S (2013) In vitro tools for evaluating novel dosage forms of poorly soluble, weakly basic drugs: case example of ketoconazole. J Pharm Sci (Epub ahead of print)
Uddin R, Saffoon N, Bishwajit SK (2011) Dissolution and dissolution apparatus: a review. Int J Cur Biomed Phar Res 1:201–207
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) (1997) Guidance for industry, dissolution testing of immediate release solid oral dosage forms, August 1997
US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER) (2000) Guidance for industry, waiver of in vivo bioavailability and bioequivalence studies for immediate-release solid oral dosage forms based on a biopharmaceutics classification system, August 2000
Waterbeemd H van de, Gifford E (2003) ADMET in silico modelling: towards prediction paradise? Nat Rev Drug Disc 2:192–204
Vehring R (2008) Pharmaceutical particle engineering via spray drying. Pharm Res 25:999–1022
Yu L (2001) Amorphous pharmaceutical solids: preparation, characterization, and stabilization. Adv Drug Deliv Rev 48:27–42
Zhang Q, Gladden L, Avalle P, Mantle M (2011) In vitro quantitative 1H and 19F nuclear magnetic resonance spectroscopy and imaging studies of fluvastatin™ in Lescol® XL tablets in a USP-IV dissolution cell. J Control Release 156:345–354
Zhao P, Zhang L, Crillo JA, Liu Q, Bullock JM, Moon YJ, Song P, Crar SS, Madabushi R, Wu TC, Booth BP, Rahman NA, Reynolds KS, Gil Berglund E, Lesko LJ, Huang S-M (2011) Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther 89:259–267
Acknowledgments
The author would like to thank the following individuals and organizations: Marshall Crew, Dan Smithey, and James Fennewald at Agere Pharmaceuticals, Inc. for contributions to the in vitro drug speciation dissolution studies, and Shaukat Ali and Nigel Langley at BASF for the generous samples of Soluplus® polymeric excipient.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 American Association of Pharmaceutical Scientists
About this chapter
Cite this chapter
Gautschi, J. (2013). Nonsink In Vitro Dissolution Testing of Amorphous Solid Dispersions. In: Repka, M., Langley, N., DiNunzio, J. (eds) Melt Extrusion. AAPS Advances in the Pharmaceutical Sciences Series, vol 9. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8432-5_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8432-5_8
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8431-8
Online ISBN: 978-1-4614-8432-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)