
Abstract
Vascular disease as evidenced by aberrant endothelial and vascular smooth muscle cell physiology represents one of the major complications of diabetes. Although the metabolic disturbances such as oxidative stress, inflammation, and hyperlipidemia have been well described as main players in the process of vascular dysfunction, epigenetic modifications of gene expression also occur under the hyperglycemic state and modulate cardiovascular homeostasis. The main epigenetic mechanisms that can modify chromatin structure and gene expression include chromatin remodeling via histone modifications or DNA methylation, and gene silencing by small noncoding RNA molecules termed microRNAs. Recent studies have suggested that these epigenetic events either alone or in concert are capable of modulating the expression of multiple target genes involved in redox homeostasis, vascular cell proliferation, and migration, as well as in proinflammatory pathways associated with vascular dysfunction. This review highlights some epigenetic changes induced by hyperglycemic and oxidative states in the vascular system and discusses their potential role in the pathogenesis of diabetes-associated vascular complications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
- Hyperglycemia
- Oxidative stress
- Endothelial dysfunction
- Gene silencing
- Inflammation
- MicroRNAs
- Vessel remodeling
- DNA methylation
- Atherosclerosis
- Histone deacetylases
- Histone demethyltransferases
1 Introduction
The cluster of cardiovascular complications that occur during diabetes mellitus includes atherosclerosis, peripheral vascular disease, cardiomyopathy, and stroke [1]. Although they are distinct with regard to their pathophysiological features (endothelial dysfunction, cell proliferation and migration, myocyte hypertrophy, vessel narrowing, and apoptosis), these pathologies share the same underlying mechanisms that mainly involve major intracellular processes taking place under hyperglycemic conditions and for which oxidative stress plays a central role [2]. First, the increase in glucose accumulation leads to an increased activation of the polyol pathway that consists of the reduction of glucose or other carbonyl compounds into sorbitol and other corresponding sugar alcohols. This reaction requires the catalytic activity of the enzyme aldose reductase that utilizes the cofactor nicotinamide adenine dinucleotide phosphate (NAD(P)H) [3]. Second, hyperglycemia also induces protein glycosylation that contributes to the increase in the quantity of advanced glycation end product (AGE) and reactive oxygen species (ROS) [4]. AGE binding to their receptors (RAGE) on cell surfaces can activate NAD(P)H oxidase and thereby increase ROS production. Hyperglycemia also activates protein kinase C (PKC) isoforms partly by AGE receptor activation as well as from ROS-dependent increase in intracellular diacylglycerol, the upstream activator of PKC [5]. In diabetes, PKC activation induces massive ROS generation mainly via the stimulation of NAD(P)H, resulting in increased redox signaling [6–9]. Thus, oxidative stress via hyperactivation of mitogenic, inflammatory, and proliferative signaling cascades plays a critical role in vascular complications associated with diabetes [5, 10, 11]. In addition to a role of hyperglycemia and oxidative stress-mediated signaling pathways in inducing diabetic vascular complications, implication of epigenetic mechanisms in the pathophysiology of cardiovascular dysfunction has also been suggested recently [12, 13]. The purpose of this chapter is to highlight some key epigenetic signals that are activated by hyperglycemia and oxidative stress in the diabetic state and to discuss their potential role in vascular dysfunction in diabetes.
2 Basic Epigenetic Mechanisms
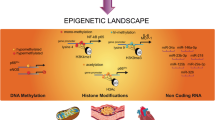
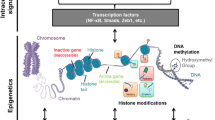
Epigenetic changes modify the patterns of gene expression without affecting the DNA structure. These changes occur in response to a variety of stimuli in the physical or physiological environment. Epigenetic mechanisms include reversible posttranslational modifications of histone structure within the nucleosomes in the chromatin, methylation of specific sites of the DNA strand, and gene silencing by small sequences of noncoding RNA (microRNAs) [14].
2.1 Histone Modifications
The chromatin is constructed of multiple groups consisting of four pairs of histones (H3, H4, H2A, H2B) wrapped by a 147-base-pair fragment of DNA and linked together with a fifth type of histone, H1 [14]. Each monomer of histone at the center of a nucleosome extends as a histone tail with a C-terminal domain and an N-terminal tail. With regard to gene expression, the standard requirement for an active transcription is a loosened chromatin opened for the binding of transcription factors; such conformation is called euchromatin. In contrast, a compact chromatin, called heterochromatin, disables transcriptional events. The transition from heterochromatin to euchromatin is achieved through reversible posttranscriptional modifications within the N-terminal fragment of histone tails. Such modifications mainly include acetylation and methylation, catalyzed by histone acetyltransferases (HATs) and histone methyltransferases (HMTs), respectively; these enzymes transfer acetyl or methyl groups mostly to lysine, serine, or arginine residues within the N-terminal tail of histone. These reactions are reversible because of the presence of other enzymes with opposite effects, histone deacetylases (HDACs) and histone demethyltransferases (HDMTs). Altogether, chromatin changes at the histone level form a histone code that can be read by other proteins and translated into a functional signal depending on the underlying context [14]. Implication of such a code in vascular biology has been suggested and is being further explored in the context of diabetic cardiovascular dysfunction [15].
2.2 DNA Methylation
Methylation of DNA is another form of epigenetic modification that occurs at the 5′-position of a cytosine residue separated by a phosphate group from a guanosine residue in the longitudinal DNA sequence (CpG island) [14]. Hypomethylation or absence of methylation favors gene transcription whereas hypermethylation or methylation of previously unmethylated CpG causes gene silencing. CpG methylation is catalyzed by DNA methyltransferases (DNMTs). It is the best studied epigenetic mechanism, and to date, three types of DNMTs have been identified: DNMT1 is responsible for the heritable DNA methylation state as it helps to preserve the parental methylation patterns after DNA replication whereas DNMTs 3a and 3b are de novo methyltransferases that methylate previously unmethylated fragments of DNA [14]. The loss of DNMT1 has been shown to be lethal in mice [16]. Attempts have been made to understand the correlation between DNA methylation state and diabetic nephropathy in patients with type 1 diabetes [17], as well as to identify and to replicate the alterations of DNA methylation signatures of diabetic nephropathy in renal mesangial and epithelial cells, suggesting the importance of this process in diabetes and cardiovascular homeostasis [18].
2.3 Gene Silencing by MicroRNAs
The central dogma in molecular biology requires that DNA is transcribed into RNA and that RNA is translated into proteins. However, it has been found that there are tiny RNA fragments that are expressed in the nucleus but remain untranslated. These fragments are termed microRNAs (miRNAs, named as miR-specific number) that are frequently referred to as “micromanagers of gene expression” [19]. They are broadly expressed in eukaryotic cells, and it is estimated that approximately 1,000 miRNAs are encoded by the human genome [20]. Mature miRNAs are made up of approximately 22 nucleotides. Primary miRNA genes located in the introns of either coding or noncoding genes [21] are transcribed by RNA polymerase II or III in the nucleus to form large pre-miRNA transcripts. These transcripts remain in the nucleus and are processed by an RNase III enzyme, Drosha, and by a double-stranded RNA-binding protein, Pasha, into approximately 70 nucleotide pre-miRNAs. Two transporters, RanGTP and exportin 5, export the pre-miRNA into the cytoplasm where it is digested by another RNase III, Dicer, to form a transient 18- to 24-nucleotide duplex. The duplex is incorporated as a single strand into a multiprotein RNA-induced silencing complex and forms the mature miRNA. miRNAs function through binding with complementary sites in the mRNA transcript to induce either translational repression or gene silencing by RNA degradation [22]. miRNA function has been associated with chromatin modulation as well as with genome stability [23], resulting in the regulation of multiple genes involved in the control of cardiovascular functions [24].
3 HDACs- and HATs-Mediated Responses in Diabetic Cardiovascular Complications
Depending on their domain organization or sequence identity, HDACs are divided into four classes: I, IIA, IIB, and III. They are expressed in multiple atherosclerosis-relevant cell types in the vasculature including monocytes, endothelial cells, and smooth muscle cells [25]. Inflammation, monocyte adhesion, and aberrant smooth muscle cell migration and proliferation are among the critical events that contribute to the atherogenic process. Several groups have investigated the roles of HDAC- and HAT-mediated deacetylation or acetylation of histone and non-histone proteins in the events associated with diabetic complications. For example, de Kreutzenberg et al. reported that high glucose downregulated the expression of Sirt-1, a protein that belongs to the class III HDACs, in human peripheral blood mononuclear cells [26]. In these studies, the downregulation of Sirt-1 gene and protein expression was associated with increased acetylation of p53, a cell-cycle regulator, and enhanced phosphorylation of the stress signaling molecule c-Jun N-terminal kinase (JNK) [26]. Because of a negative association of Sirt-1 expression and the carotid intima-media thickness, a potential role of Sirt-1 in regulating early atherosclerosis was suggested from these studies [26]. Downregulation of Sirt-1 was also observed in endothelial cells in response to high glucose [27]. In fact, hyperglycemia was shown to induce atherosclerotic endothelial cell aging through reduction of Sirt-1 expression [27]. Sirt-1 has also the ability to modify oxidative stress by its effects on the transcription of enzymes implicated in regulating ROS generation. Sirt-1, via the deacetylation of the transcription factor FOXO [28], positively regulates the transcription of the manganese superoxide dismutase (MnSOD), an antioxidant enzyme that attenuates the oxidative stress induced by hyperglycemia [29]. Hyperglycemia-dependent Sirt-1 downregulation, through a regulatory feedback interaction between Sirt-1 and HAT p300, stimulates the acetylation of FOXO and decreases MnSOD levels, resulting in increased oxidative stress [30]. Thus, in diabetic endothelial senescence leading to vascular dysfunction, a persistent redox state is maintained by high glucose-induced change in the acetylation state of the non-histone protein FOXO, and by modulation of the epigenetic markers Sirt-1 and p300 that exhibit HDAC and HAT properties, respectively [30]. In accordance with this, decreased glucose concentrations have been associated with the increase in Sirt-1 in cancer cells [31].
The thioredoxin interacting protein (TXNIP), which is an endogenous antagonist of the ROS scavenging protein thioredoxin, is known to be upregulated under hyperglycemic conditions [32] and contributes to hyperglycemia-induced oxidative stress as well as vascular complications [33, 34]. An elegant study reported that glucose-induced upregulation of the TXNIP gene expression is dependent on the recruitment of the HAT p300 at the TXNIP promoter and its histone H4 acetylation [35]. These studies demonstrated that H4 acetylation plays an important role in the control of TXNIP transcription and showed that a pharmacological HDAC inhibitor, trichostatin A (TSA), increased TXNIP expression in pancreatic beta cells highlighting the role of histone acetylation in TXNIP gene transcription [35].
Moreover, hyperglycemia has also been shown to induce the expression of TXNIP as well as several pro-inflammatory genes such as cyclooxygenase 2 (COX-2), vascular endothelial growth factor (VEGF), and the intercellular adhesion molecule type 1 (ICAM-1) in retinal capillary endothelial cells [36]. In these studies, hyperglycemia-induced TXNIP gene expression also contributed to the histone H3 lysine 9 acetylation of the COX-2 gene promoter via a p38-dependent signaling pathway [36]. Such hyperglycemia-induced histone acetylation has not only been described in the context of inflammation but also in other atherosclerotic events such as the expression of vasoactive factors and extracellular matrix proteins [37, 38]. For example, high glucose-induced HAT activity via p300 upregulation has been shown to be an essential upstream component for glucose-induced expression of endothelin-1, fibronectin, and VEGF in vascular smooth muscle cells (VSMCs) and endothelial cells [37]. Increased H3 acetylation was associated with these glucose-induced responses and, interestingly, overexpression of the HAT p300 potently induced glucose-like effects in these cells [37]. Moreover, because the HAT p300 is one of the primary targets of the nuclear factor kappa B (NF-κB) [39], a marker of glucose-induced inflammation [40, 41], it has been proposed to participate in the expression of the pro-inflammatory cytokine interleukin 6 (IL-6). Accordingly, p300 was shown to mediate the lysine acetylation at IL-6 as well as the tumor necrosis factor-α (TNF-α) gene promoters and enhance their transcription in monocytes [42]. Similarly, hyperglycemia-induced recruitment of HAT and lysine acetylation at key inflammatory gene promoters were also observed in monocytes from patients with either type 1 or type 2 diabetes, suggesting a contribution of chromatin remodeling in diabetes-induced inflammation [13, 43].
Oxidative stress, as observed in diabetes, is an inhibitor of HDAC2, a member of the class 1 HDACs, and this inhibition was shown to stimulate the expression of another pro-inflammatory cytokine, interleukin 8 (IL-8) [44]. Experimental production of peroxynitrite as well as treatment of human airway epithelial cells with TSA was reported to attenuate HDAC2 activity and expression, resulting in elevated production of IL-8 in response to IL-1β [44]. More importantly, a similar attenuation of HDAC activity by TSA in the context of dyslipidemia has been shown to exacerbate the atherogenic process; this was observed in hyperlipidemic mice in which treatment with TSA was shown to enhance the levels of atherogenic markers such as the scavenger receptor A, the lymphocyte CD36, TNF-α, and vascular cell adhesion molecule 1 (VCAM-1) [45], suggesting a critical role of HDAC activity for the maintenance of vascular homeostasis in pathophysiological circumstances. The inhibition of HDAC2 by hyperglycemia-induced redox signaling could also be deleterious per se because HDAC2 itself is an antagonist of the atherosclerotic plaque formation. HDAC2 acts in this sense by deacetylating the class II transactivator (CIITA), whose role is to repress the collagen promoter and to activate the major histocompatibility complex promoter in smooth muscle cells and macrophages [46]. Thus, it appears that, in these cell types, HDAC2 is a good antagonist of transcriptional activity, as well as a positive regulator of collagen formation, both mechanisms contributing to a delay in the atherogenic events [46, 47].
In view of the importance of the histone acetylation state in vascular abnormalities, epigenetic therapy by reversal or mimicking of HAT or HDAC activities, respectively, has generated much interest for the treatment of diabetic cardiovascular complications. In this regard, dietary polyphenols have attracted some attention because of their ability to modulate HDAC or HAT activity and, subsequently, atherosclerotic gene expression [48, 49]. Among these polyphenolic compounds, curcumin, a derivative of the curry spice turmeric, has been shown to exhibit cardiovascular protective, as well as antiinflammatory, antithrombotic and antioxidant properties in the context of type 2 diabetes [50–53]. Interestingly, curcumin has been reported to inhibit p300 HAT activity [54]. It has been shown to inhibit high glucose-induced relA/p65 activation, the critical regulatory subunit of NF-κB, in monocytes through suppression of HAT activity and upregulation of the HDAC activity [42]. RelA/p65 is regulated on one hand through its own acetylation and deacetylation but on the other hand through histone acetylation at the NF-κB promoter [42, 55]. Furthermore, in the context of VSMC physiology, curcumin has been demonstrated to inhibit growth of rat and human VSMCs through the induction of the antiproliferative enzyme heme oxygenase via the translocation of the nuclear transcription factor E2-related factor-2 (Nrf-2) and the subsequent activation of the antioxidant response element (ARE) within the heme oxygenase promoter [56]. This effect of curcumin on Nrf-2 translocation could also arise from its HAT inhibitory property because it has recently been reported that TSA-mediated inhibition of HDAC is a potent inducer of Nrf-2 expression, translocation, and binding to ARE on the heme oxygenase [57].
Another interesting compound with epigenetic properties is resveratrol, found in red grape skin as a polyphenolic phytoalexin, that exhibits the ability to activate Sirt-1 [58, 59]. Because of its antioxidant and antiinflammatory properties, resveratrol has been broadly demonstrated to be beneficial for cardiovascular health and disease [60–62] and also to protect against diabetic nephropathy [63] as well as diabetic atherogenic events in the vasculature [64]. In vessels isolated from alloxan-induced diabetic rabbits, resveratrol attenuated the production of ROS, improved vascular reactivity to acetylcholine, and helped in maintaining the integrity of the endothelium under diabetic conditions [65]. It has been reported that high glucose-induced mitochondrial ROS production was decreased in a resveratrol-Sirt-1-dependent fashion in human coronary arterial endothelial cells (CAECs) [66]. In these studies, siRNA- or electroporation-mediated silencing of Sirt-1 in hyperglycemic CAECs abolished the resveratrol-induced increase in the pro-oxidant molecules, MnSOD and glutathione, as well as the resveratrol-induced reduction in intracellular H2O2 [66]. Such positive effect on endothelial cell physiology was also recently reported from studies using bovine aortic endothelial cells (BAECs) under hyperglycemic conditions [67]. Resveratrol was also shown to attenuate glucose-induced early atherosclerosis events such as endothelial hyperpermeabilty and overexpression of caveolin as well as the glucose-induced expression of VEGF and its receptor [67]. It should be noted that VEGF induction has been previously reported to be Sirt-1 dependent in HT1080 cell line [68]. Furthermore, a positive effect of resveratrol in reversing the p53 acetylation induced by high glucose and high palmitate in human monocytes was reported and demonstrated to be a Sirt-1-dependent process, suggesting the antiapoptotic effect of resveratrol [26]. Thus, given the potential of natural HDAC/HAT modulators in regulating cardiovascular homeostasis and cardiovascular protection, further investigations are required to appropriately assign them a beneficial role in diabetic vascular dysfunction.
4 MicroRNA-Mediated Responses in Diabetic Atherosclerosis
Multiple studies have assessed the implication of microRNAs in diabetes pathobiology [12, 69, 70], suggesting a possible involvement of these molecules in diabetic cardiovascular diseases. The process of diabetic atherosclerosis is a result of endothelial dysfunction, chronic inflammation, and vessel remodeling [71].
4.1 Endothelial Dysfunction
Depending on their specificity, microRNAs can be considered as either positive or negative modulators of diabetic endothelial dysfunction. Caporali et al. have assessed the role of miR-503 on endothelial cell function in diabetic conditions and demonstrated that hyperglycemia resulted in increased levels of miR-503 in cultured endothelial cells [72]. They also showed that lentiviral overexpression of miR-503 reduced endothelial cell proliferation and migration; these functional capacities were upregulated by inhibition of miR-503 suggesting the negative involvement of this miRNA in diabetic-induced endothelial dysfunction. Their observations were further strengthened by the finding that heightened levels of miR-503 are expressed in ischemic muscular tissues from diabetic patients [72]. Similarly, high glucose was previously shown to induce an increase of miR-221 in human umbilical vein endothelial cells [73]. Hyperglycemia is known to alter the transmigratory capacity of endothelial cells that is essential for angiogenesis following vascular damage, and a lack of this capacity exacerbates endothelial dysfunction as seen in numerous cardiovascular disorders. In the latter study [73], the authors found that c-kit, a specific stem-cell receptor that plays a role in endothelial cell transmigration, was downregulated under hyperglycemic conditions. Antisense oligonucleotide-mediated inhibition of miR-221 was able to prevent this downregulation and to restore the endothelial transmigratory capacities altered by high glucose, suggesting a role of miR-221 in facilitating endothelial dysfunction. These two studies proposed that miR-221 and miR-503 inhibition in endothelial cells can reinforce their functional capacities under diabetic conditions and help in preventing atherosclerotic disease. However, some opposite actions of miRNAs have also been reported. For example, a study from Meng et al. showed that diabetes was associated with reduced levels of miR-126, miR-21, miR-27a, miR-27b, and miR-130a in endothelial progenitor cells (EPC) [74]; among these, miR-126 was the most affected. Overexpression of miR-126 resulted in enhanced EPC function and reduced EPC apoptosis through the downregulation of Spred-1 gene, a known angiogenic inhibitor in endothelial cells. These observations were in accordance with another study where miR-126 was demonstrated to be a positive regulator of vascular integrity [75]. Endothelial cell-specific deletion of miR-126 in mice resulted in the impairment of vascular function as illustrated by defective cell migration, proliferation, and angiogenesis [75]. Thus, to clarify the role of various miRNAs in vascular dysfunction, more in-depth studies on the contribution of miRNAs in the endothelial system pathobiology under diabetic conditions are needed.
4.2 Inflammation
MicroRNAs also play a role in chronic inflammation that occurs in the vessels of diabetic patients. Molecular hallmarks of inflammation include the heightened production of adhesion molecules and proinflammatory enzymes such as COX-2.
COX-2 mRNA is a target of miR-16 in endothelial cells. Past studies have demonstrated that under diabetic conditions, inflammation and atherosclerosis are in part caused by the binding of RAGE with their ligand, S100b, leading to their subsequent activation [76]. Recent work from Shanmugam et al. has proposed that this is achieved through the S100b-dependent inhibition of the binding of miR-16 with COX-2 mRNA [77]. S100b was in fact shown to displace the nuclear ribonucleoprotein K into the cytoplasm, where it interacts with COX-2 mRNA and disables miR-16 attachment [77]. These observations support an antiinflammatory role of miR-16 in diabetes. Moreover, several other microRNAs have been implicated in endothelial atherosclerotic inflammation through pro-inflammatory gene repression. Overexpression of miR-155 and miR-121/122 effectively lowered angiotensin-II induced vascular cell adhesion molecule 1 and monocyte chemoattractant protein 1 (MCP-1) in a study [78]. It was concluded that these microRNAs participate in endothelial cell inflammation and migration by targeting angiotensin II receptor type 1 or the transcription factor Ets [78]. However, in contradiction to this, miR-200b has been demonstrated to exhibit pro-inflammatory properties in diabetic VSMCs [79]. The overexpression of this miRNA enhanced COX-2 promoter activity and induced the expression of MCP-1. In the same study, monocyte binding in diabetic VSMCs was also stimulated by miR-200b, suggesting a positive regulation of inflammatory processes by this miRNA [79]. Another group has also addressed the role of miR-125b in the epigenetic regulation of inflammatory genes in VSMCs [80]. They observed that miR-125b was upregulated in diabetic mice concomitantly to a downregulation of histone methyltransferase Suv39H1. Overexpression of miR-125b reduced Suv39H1 transcription and resulted in pro-inflammatory diabetic phenotype, as illustrated by the enhanced monocyte binding [80]. This last set of studies demonstrates that, on the basis of their pro-inflammatory properties, some miRNAs contribute to the development of diabetic atherosclerosis.
4.3 Vessel Remodeling
Vessel remodeling is a common feature of cardiovascular diseases and vascular dysfunction that involves the aberrant growth and proliferation of vascular cells and rearrangement of the extracellular matrix. miRNAs are involved in VSMC proliferation and hypertrophy, leading to vessel remodeling [81, 82]. In a recent study [83], miR-208 was shown to mediate insulin-induced VSMC proliferation through the downregulation of the cell-cycle regulator protein p21, suggesting a role of this miRNA in diabetic neointima formation. In addition, a role of miR-143 and miR-145 in the maintenance of the beneficial contractile VSMC phenotype, and in turn in vascular homeostasis, has recently been suggested [82, 84–86]. It has been demonstrated that miR-143 and miR-145 control VSMC proliferation, migration, and differentiation, as well as overall vessel reactivity [87]. In fact, overexpression of miR-143/145 in VSMCs resulted in significant attenuation of DNA synthesis [87]. In addition, VSMCs isolated from miR-143/145 knockout mice exhibited a heightened migratory response to the platelet-derived growth factor (PDGF) and presented decreased levels of the VSMC differentiation markers smooth muscle myosin heavy chain and smooth muscle α-actin [87]. It has also been postulated that miR-145 could regulate the atherogenic processes in relationship to plaque formation and stability. This hypothesis was recently confirmed in an atherosclerotic animal model, the apolipoprotein E knockout mouse, where a VSMC-targeted overexpression of miR-145 decreased the risk of plaque rupture by reducing its size and by promoting the VSMC differentiation toward the contractile phenotype [88]. Similar conclusions were made from studies that focused on the role of miR-133 in vascular physiology [89]. Adenoviral transfection of miR-133 in rat carotid aorta led to a reduction of neointima formation, VSMC proliferation, and the phenotypic switch from the contractile to the synthetic state after balloon injury, all of these via the repression of the transcription factor Sp-1, a mediator of smooth muscle gene expression [89]. Therefore, given the altered expression of multiple miRNAs in diabetes, it can be postulated that miRNAs represents potential targets for the modulation of diabetic atherosclerosis as they can interfere with endothelial dysfunction, inflammatory processes, and vessel remodeling.
5 DNA Methylation in Diabetic Cardiovascular Complications
The DNA methylation process has not been very well explored in the context of diabetic cardiovascular complications; however, in addition to genome-wide studies that have attempted to characterize the DNA methylation signatures in diabetes and to establish a correlation with the risk of diabetic nephropathy in patients with type 1 diabetes [17], few studies have also associated DNA hypomethylation with atherosclerosis in human atherosclerotic smooth muscle cells as well as in obese animal models such as high-fat-fed rabbits and ApoE null mice [90–92]. Normal chow-fed mice ApoE null mice were also shown to exhibit altered DNA methylation patterns in leukocytes before the onset of atherosclerosis [93]. Furthermore, global DNA methylation in peripheral blood leukocytes from patients with chronic kidney disease was associated with inflammation and increased mortality, suggesting the involvement of DNA methylation state in the exacerbation of kidney disease and related vascular disorders [94]. An alteration in the DNA methylation of several vascular homeostasis-relevant genes such as c-fos, p53, matrix metalloproteinase, endothelial nitric oxide synthase, hypoxia-induced factor 1, and growth factors has been reported in VSMCs and endothelial cells isolated from animal models, suggesting a role of these modifications in the development of vascular dysfunction [25, 95]. Although a direct role of DNA methylation in diabetes and its complications remains unclear, studies using animal models have implicated increased methylation at key gene promoters in islet dysfunction and diabetes [96, 97]. However, no differences in DNA methylation have been noticed at the promoters of high glucose-induced genes in streptozotocin-induced type 1 diabetic rats [18, 98], suggesting further studies are required to better enlighten the role of this epigenetic process in diabetes-associated disorders.
6 Conclusions
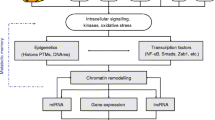
Epigenetic modulation of gene expression focused on the effect of DNA methylation and histone modifications has been studied for a long time in the context of cancer. However, it is only recently that there has been a surge of interest in investigating the involvement of epigenetic pathways in the pathogenesis of vascular dysfunction in diabetes. As a result, evidence has accumulated to show that histone acetylation, methylation, and miRNA-induced processes modulate the expression and function of the genes that are linked to ROS generation, inflammation, cell-cycle regulation, migration, proliferation, and other proatherogenic events (Fig. 1). Nevertheless, more in-depth studies on the epigenetic modifications in the vascular system of various experimental models of diabetes and in humans are needed to precisely establish a role of these modifications in the pathogenesis of vascular dysfunction associated with diabetes.

Schematic model depicting the interplay between hyperglycemia, reactive oxygen species, and epigenetic events in diabetes-associated vascular complications. Diabetes/hyperglycemia promotes redox signaling through an increase in the generation of reactive oxygen species (ROS). ROS activity can either be modulated by epigenetic events that result from hyperglycemia or exacerbate them, leading to alterations in the expression and function of the genes that are linked to ROS generation, inflammation, cell-cycle regulation, migration, proliferation, and other proatherogenic events. The consequences of these events are activation of the proinflammatory cascades in the vessel wall as well as the impairment of endothelial and vascular smooth muscle physiology, both contributing to aberrant vascular functions
References
Grundy SM, Benjamin IJ, Burke GL et al (1999) Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation 100:1134–1146
Niedowicz DM, Daleke DL (2005) The role of oxidative stress in diabetic complications. Cell Biochem Biophys 43:289–330
Chung SS, Ho EC, Lam KS, Chung SK (2003) Contribution of polyol pathway to diabetes-induced oxidative stress. J Am Soc Nephrol 14:S233–S236
Cantero AV, Portero-Otin M, Ayala V et al (2007) Methylglyoxal induces advanced glycation end product (AGEs) formation and dysfunction of PDGF receptor-beta: implications for diabetic atherosclerosis. FASEB J 21:3096–3106
Giacco F, Brownlee M (2010) Oxidative stress and diabetic complications. Circ Res 107:1058–1070
Inoguchi T, Li P, Umeda F et al (2000) High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49:1939–1945
Inoguchi T, Sonta T, Tsubouchi H et al (2003) Protein kinase C-dependent increase in reactive oxygen species (ROS) production in vascular tissues of diabetes: role of vascular NAD(P)H oxidase. J Am Soc Nephrol 14:S227–S232
Singh U, Jialal I (2006) Oxidative stress and atherosclerosis. Pathophysiology 13:129–142
Srivastava A, Anand-Srivastava M (2008) Role of hyperglycemia and redox-induced signaling in vascular complications of diabetes. In: Srivastava A, Anand-Srivastava M (eds) Signal transduction in the cardiovascular system in health and disease, 3rd edn. Springer, New York, pp 177–192
Forbes JM, Cooper ME (2013) Mechanisms of diabetic complications. Physiol Rev 93: 137–188
Srivastava AK (2002) High glucose-induced activation of protein kinase signaling pathways in vascular smooth muscle cells: a potential role in the pathogenesis of vascular dysfunction in diabetes. Int J Mol Med 9:85–89
Muhonen P, Holthofer H (2009) Epigenetic and microRNA-mediated regulation in diabetes. Nephrol Dial Transplant 24:1088–1096
Villeneuve LM, Reddy MA, Natarajan R (2011) Epigenetics: deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol 38:451–459
Korkmaz A, Manchester LC, Topal T, Ma S, Tan DX, Reiter RJ (2011) Epigenetic mechanisms in human physiology and diseases. J Exp Integr Med 1:139–147
Yla-Herttuala S, Glass CK (2011) Review focus on epigenetics and the histone code in vascular biology. Cardiovasc Res 90:402–403
Li E, Bestor TH, Jaenisch R (1992) Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69:915–926
Bell CG, Teschendorff AE, Rakyan VK, Maxwell AP, Beck S, Savage DA (2010) Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med Genomics 3:33
Brennan EP, Ehrich M, O’Donovan H et al (2010) DNA methylation profiling in cell models of diabetic nephropathy. Epigenetics 5:396–401
Bartel DP, Chen CZ (2004) Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet 5:396–400
Berezikov E, Guryev V, van de Belt J, Wienholds E, Plasterk RH, Cuppen E (2005) Phylogenetic shadowing and computational identification of human microRNA genes. Cell 120:21–24
Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A (2004) Identification of mammalian microRNA host genes and transcription units. Genome Res 14:1902–1910
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
van Wolfswinkel JC, Ketting RF (2010) The role of small non-coding RNAs in genome stability and chromatin organization. J Cell Sci 123:1825–1839
Natarajan R, Putta S, Kato M (2012) MicroRNAs and diabetic complications. J Cardiovasc Transl Res 5:413–422
Matouk CC, Marsden PA (2008) Epigenetic regulation of vascular endothelial gene expression. Circ Res 102:873–887
de Kreutzenberg SV, Ceolotto G, Papparella I et al (2010) Downregulation of the longevity-associated protein sirtuin 1 in insulin resistance and metabolic syndrome: potential biochemical mechanisms. Diabetes 59:1006–1015
Mortuza R, Chen S, Feng B, Sen S, Chakrabarti S (2013) High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS One 8:e54514
Daitoku H, Sakamaki J, Fukamizu A (2011) Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochim Biophys Acta 1813:1954–1960
Malik AI, Storey KB (2011) Transcriptional regulation of antioxidant enzymes by FoxO1 under dehydration stress. Gene 485:114–119
Han L, Zhou R, Niu J, McNutt MA, Wang P, Tong T (2010) SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res 38:7458–7471
Mousa SA, Gallati C, Simone T et al (2009) Dual targeting of the antagonistic pathways mediated by Sirt1 and TXNIP as a putative approach to enhance the efficacy of anti-aging interventions. Aging (Albany NY) 1:412–424
Shalev A, Pise-Masison CA, Radonovich M et al (2002) Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGF-beta signaling pathway. Endocrinology 143:3695–3698
Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT (2004) Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem 279:30369–30374
Turturro F, Friday E, Welbourne T (2007) Hyperglycemia regulates thioredoxin-ROS activity through induction of thioredoxin-interacting protein (TXNIP) in metastatic breast cancer-derived cells MDA-MB-231. BMC Cancer 7:96
Cha-Molstad H, Saxena G, Chen J, Shalev A (2009) Glucose-stimulated expression of Txnip is mediated by carbohydrate response element-binding protein, p300, and histone H4 acetylation in pancreatic beta cells. J Biol Chem 284:16898–16905
Perrone L, Devi TS, Hosoya K, Terasaki T, Singh LP (2009) Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol 221:262–272
Chen S, Feng B, George B, Chakrabarti R, Chen M, Chakrabarti S (2010) Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. Am J Physiol Endocrinol Metab 298:E127–E137
Kaur H, Chen S, Xin X, Chiu J, Khan ZA, Chakrabarti S (2006) Diabetes-induced extracellular matrix protein expression is mediated by transcription coactivator p300. Diabetes 55:3104–3111
Vanden Berghe W, De BK, Boone E, Plaisance S, Haegeman G (1999) The nuclear factor-kappaB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J Biol Chem 274:32091–32098
Shanmugam N, Reddy MA, Guha M, Natarajan R (2003) High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes 52:1256–1264
Guha M, Bai W, Nadler JL, Natarajan R (2000) Molecular mechanisms of tumor necrosis factor alpha gene expression in monocytic cells via hyperglycemia-induced oxidant stress-dependent and -independent pathways. J Biol Chem 275:17728–17739
Yun JM, Jialal I, Devaraj S (2011) Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem 22:450–458
Miao F, Gonzalo IG, Lanting L, Natarajan R (2004) In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem 279:18091–18097
Ito K, Hanazawa T, Tomita K, Barnes PJ, Adcock IM (2004) Oxidative stress reduces histone deacetylase 2 activity and enhances IL-8 gene expression: role of tyrosine nitration. Biochem Biophys Res Commun 315:240–245
Choi JH, Nam KH, Kim J et al (2005) Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol 25:2404–2409
Kong X, Fang M, Li P, Fang F, Xu Y (2009) HDAC2 deacetylates class II transactivator and suppresses its activity in macrophages and smooth muscle cells. J Mol Cell Cardiol 46:292–299
Fernandez AZ, Siebel AL, El-Osta A (2010) Atherogenic factors and their epigenetic relationships. Int J Vasc Med 2010:437809
Rahman I, Biswas SK, Kirkham PA (2006) Regulation of inflammation and redox signaling by dietary polyphenols. Biochem Pharmacol 72:1439–1452
Wolfram S (2007) Effects of green tea and EGCG on cardiovascular and metabolic health. J Am Coll Nutr 26:373S–388S
Khajehdehi P, Pakfetrat M, Javidnia K et al (2011) Oral supplementation of turmeric attenuates proteinuria, transforming growth factor-beta and interleukin-8 levels in patients with overt type 2 diabetic nephropathy: a randomized, double-blind and placebo-controlled study. Scand J Urol Nephrol 45:365–370
Usharani P, Mateen AA, Naidu MU, Raju YS, Chandra N (2008) Effect of NCB-02, atorvastatin and placebo on endothelial function, oxidative stress and inflammatory markers in patients with type 2 diabetes mellitus: a randomized, parallel-group, placebo-controlled, 8-week study. Drugs R D 9:243–250
Wongcharoen W, Phrommintikul A (2009) The protective role of curcumin in cardiovascular diseases. Int J Cardiol 133:145–151
Kapakos G, Youreva V, Srivastava AK (2012) Cardiovascular protection by curcumin: molecular aspects. Indian J Biochem Biophys 49:306–315
Marcu MG, Jung YJ, Lee S et al (2006) Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem 2:169–174
Kiernan R, Bres V, Ng RW et al (2003) Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J Biol Chem 278:2758–2766
Pae HO, Jeong GS, Jeong SO et al (2007) Roles of heme oxygenase-1 in curcumin-induced growth inhibition in rat smooth muscle cells. Exp Mol Med 39:267–277
Wang B, Zhu X, Kim Y et al (2012) Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med 52:928–936
Brooks CL, Gu W (2009) How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer 9:123–128
Borra MT, Smith BC, Denu JM (2005) Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 280:17187–17195
Petrovski G, Gurusamy N, Das DK (2011) Resveratrol in cardiovascular health and disease. Ann N Y Acad Sci 1215:22–33
Das M, Das DK (2010) Resveratrol and cardiovascular health. Mol Aspects Med 31:503–512
Markus MA, Morris BJ (2008) Resveratrol in prevention and treatment of common clinical conditions of aging. Clin Interv Aging 3:331–339
Sharma S, Anjaneyulu M, Kulkarni SK, Chopra K (2006) Resveratrol, a polyphenolic phytoalexin, attenuates diabetic nephropathy in rat. Pharmacology 76:69–75
Zang M, Xu S, Maitland-Toolan KA et al (2006) Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 55:2180–2191
Akar F, Pektas MB, Tufan C et al (2011) Resveratrol shows vasoprotective effect reducing oxidative stress without affecting metabolic disturbances in insulin-dependent diabetes of rabbits. Cardiovasc Drugs Ther 25:119–131
Ungvari Z, Labinskyy N, Mukhopadhyay P et al (2009) Resveratrol attenuates mitochondrial oxidative stress in coronary arterial endothelial cells. Am J Physiol Heart Circ Physiol 297:H1876–H1881
Tian C, Zhang R, Ye X et al (2013) Resveratrol ameliorates high-glucose-induced hyperpermeability mediated by caveolae via VEGF/KDR pathway. Genes Nutr 8:231–239
Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38:864–878
Gallagher IJ, Scheele C, Keller P et al (2010) Integration of microRNA changes in vivo identifies novel molecular features of muscle insulin resistance in type 2 diabetes. Genome Med 2:9
Lovis P, Roggli E, Laybutt DR et al (2008) Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 57:2728–2736
Gonzalez-Chavez A, Elizondo-Argueta S, Gutierrez-Reyes G, Leon-Pedroza JI (2011) Pathophysiological implications between chronic inflammation and the development of diabetes and obesity. Cir Cir 79:209–216
Caporali A, Meloni M, Vollenkle C et al (2011) Deregulation of microRNA-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation 123:282–291
Li Y, Song YH, Li F, Yang T, Lu YW, Geng YJ (2009) MicroRNA-221 regulates high glucose-induced endothelial dysfunction. Biochem Biophys Res Commun 381:81–83
Meng S, Cao JT, Zhang B, Zhou Q, Shen CX, Wang CQ (2012) Downregulation of microRNA-126 in endothelial progenitor cells from diabetes patients, impairs their functional properties, via target gene Spred-1. J Mol Cell Cardiol 53:64–72
Wang S, Aurora AB, Johnson BA et al (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15:261–271
Bucciarelli LG, Wendt T, Qu W et al (2002) RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 106:2827–2835
Shanmugam N, Reddy MA, Natarajan R (2008) Distinct roles of heterogeneous nuclear ribonuclear protein K and microRNA-16 in cyclooxygenase-2 RNA stability induced by S100b, a ligand of the receptor for advanced glycation end products. J Biol Chem 283:36221–36233
Zhu N, Zhang D, Chen S et al (2011) Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 215:286–293
Reddy MA, Jin W, Villeneuve L et al (2012) Pro-inflammatory role of microrna-200 in vascular smooth muscle cells from diabetic mice. Arterioscler Thromb Vasc Biol 32:721–729
Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L, Natarajan R (2010) Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes 59:2904–2915
Quintavalle M, Condorelli G, Elia L (2011) Arterial remodeling and atherosclerosis: miRNAs involvement. Vascul Pharmacol 55:106–110
Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L (2011) miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet 4:197–205
Zhang Y, Wang Y, Wang X et al (2011) Insulin promotes vascular smooth muscle cell proliferation via microRNA-208-mediated downregulation of p21. J Hypertens 29:1560–1568
Norata GD, Pinna C, Zappella F et al (2012) MicroRNA 143–145 deficiency impairs vascular function. Int J Immunopathol Pharmacol 25:467–474
Boettger T, Beetz N, Kostin S et al (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119:2634–2647
Cordes KR, Sheehy NT, White MP et al (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460:705–710
Elia L, Quintavalle M, Zhang J et al (2009) The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ 16:1590–1598
Lovren F, Pan Y, Quan A et al (2012) MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation 126:S81–S90
Torella D, Iaconetti C, Catalucci D et al (2011) MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res 109:880–893
Laukkanen MO, Mannermaa S, Hiltunen MO et al (1999) Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol 19:2171–2178
Hiltunen MO, Yla-Herttuala S (2003) DNA methylation, smooth muscle cells, and atherogenesis. Arterioscler Thromb Vasc Biol 23:1750–1753
Hiltunen MO, Turunen MP, Hakkinen TP et al (2002) DNA hypomethylation and methyltransferase expression in atherosclerotic lesions. Vasc Med 7:5–11
Lund G, Andersson L, Lauria M et al (2004) DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem 279:29147–29154
Stenvinkel P, Karimi M, Johansson S et al (2007) Impact of inflammation on epigenetic DNA methylation—a novel risk factor for cardiovascular disease? J Intern Med 261:488–499
Turunen MP, Aavik E, Yla-Herttuala S (2009) Epigenetics and atherosclerosis. Biochim Biophys Acta 1790:886–891
Park JH, Stoffers DA, Nicholls RD, Simmons RA (2008) Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest 118:2316–2324
Ling C, Del GS, Lupi R et al (2008) Epigenetic regulation of PPARGC1A in human type 2 diabetic islets and effect on insulin secretion. Diabetologia 51:615–622
Williams KT, Garrow TA, Schalinske KL (2008) Type I diabetes leads to tissue-specific DNA hypomethylation in male rats. J Nutr 138:2064–2069
Acknowledgments
This work was supported by funding from the Canadian Institutes of Health Research (CIHR) operating grant number 67037 to A.K.S. E.R.S.C. is a recipient of a studentship from the Faculty of Graduate and Postdoctoral Studies of the University of Montreal.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Cheyou, E.R.S., Srivastava, A.K. (2014). Hyperglycemia, Oxidative Stress, and Vascular Complications: Role of Epigenetic Mechanisms. In: Turan, B., Dhalla, N. (eds) Diabetic Cardiomyopathy. Advances in Biochemistry in Health and Disease, vol 9. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-9317-4_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-9317-4_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-9316-7
Online ISBN: 978-1-4614-9317-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)