
Abstract
Congenital anomalies of the kidney and urinary tract (CAKUT) account for up to 30 % of all congenital anomalies diagnosed (Schedl A. Nat Rev Genet 8(10):791–802, 2007). These anomalies are often prenatal diagnoses by screening prenatal ultrasound, but can present in childhood or adolescence with a urinary tract infection (UTI) and less commonly with pain. CAKUT diseases are responsible for around 50 % of end-stage renal disease (ESRD) in children in North America (Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Pediatr Nephrol 27(3):363–373, 2012). In this chapter we will discuss the relevant embryology, the scope of congenital anomalies of the kidney and urinary tract (CAKUT), the approach to prenatal diagnosis and imaging of fetuses with CAKUT, and the approach to postnatal diagnosis and imaging of children with CAKUT. More time will be allotted to the more common congenital anomalies.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others



Keywords
- Autosomal Dominant Polycystic Kidney Disease
- Renal Dysplasia
- Autosomal Recessive Polycystic Kidney Disease
- Renal Vein Thrombosis
- Febrile Urinary Tract Infection
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) account for up to 30 % of all congenital anomalies diagnosed [1]. These anomalies are often prenatal diagnoses by screening prenatal ultrasound, but can present in childhood or adolescence with a urinary tract infection (UTI) and less commonly with pain. CAKUT diseases are responsible for around 50 % of end-stage renal disease (ESRD) in children in North America [2]. In this chapter we will discuss the relevant embryology, the scope of congenital anomalies of the kidney and urinary tract (CAKUT), the approach to prenatal diagnosis and imaging of fetuses with CAKUT, and the approach to postnatal diagnosis and imaging of children with CAKUT. More time will be allotted to the more common congenital anomalies.
Embryology of Normal Kidney and Bladder Development
To help understand congenital anomalies of the kidney and urinary tract, an understanding of embryologic development is needed.
Kidney Development
In humans, three kidneys develop in the course of gestation with the first two regressing in utero and the third becoming the definitive kidney. All of the three kidneys develop from the intermediate mesoderm. The first kidney, the pronephros, develops near the beginning of the fourth week of gestation and is a nonfunctional system composed of 5–7 paired segments located in the region of the future head and neck. The pronephros develops cranial to caudal and degenerates by the end of the fourth week of gestation.
The mesonephros is the next rudimentary kidney that develops in the fifth week of development. It also develops cranial to caudal and is composed of 40–42 pairs of tubules. The mesonephros does produce a small amount of urine starting around 10 weeks of gestation, and the mesonephric duct contributes to development of the bladder and male genital ducts. By the end of 12 weeks of gestation, most of the mesonephros has degenerated except for the mesonephric duct, which in males develops into the vas deferens, seminal vesicles, epididymis, and ejaculatory ducts, and some cranial tubules that develop into the efferent ductules of the testis. In females, the cranial and caudal portions of the mesonephric duct form the vestigial structures epoophoron and paroophoron. Parts of the mesonephric duct form the ureter and part of the bladder as discussed in the next section.
The final stage in kidney development involves development of the metanephros which begins around 5–6 weeks of gestation. The metanephros is composed of the metanephric mesenchyme derived from the intermediate mesoderm in the pelvis and the ureteric bud derived from the distal part of the mesonephric duct. Complex interactions between the metanephric mesenchyme and the ureteric bud induce nephron development in the metanephric mesenchyme and collecting duct development from ureteric bud branching [1]. The metanephric mesenchyme develops into the glomeruli, proximal tubules, loops of Henle, and distal tubules, while the ureteric bud branching develops into the collecting ducts, calyces, renal pelvis, and ureters. The developing kidney migrates from the pelvis to its final position in the lumbar region by the eighth week and begins making urine by the ninth week. During renal ascent, the kidney rotates so the pelvis, which starts oriented anteriorly, is medially oriented when it completes ascent. Kidney maturation continues after birth in humans, but nephron formation is complete in utero.
Ureter and Bladder Development
Around the fifth week of gestation, the mesonephric duct that gives rise to the ureteric bud elongates and dilates and fuses with the endoderm-lined cloaca. After the mesonephric duct fuses with the cloaca, the urogenital septum divides the cloaca into the urogenital sinus and the anorectal canal with the mesonephric duct fused with the urogenital sinus. The urogenital sinus develops into the bladder and urethra in both sexes. Part of the mesonephric duct that fused with the urogenital sinus is incorporated in a caudal to cranio-lateral direction. The bladder trigone forms from the fusion and incorporation of the mesonephric ducts. The location of the ureteral orifice in the bladder is related to the location of the ureteric bud on the mesonephric duct; the more caudal the ureteric bud on the mesonephric duct, the more cranial and lateral the ureteral orifice is in the bladder [3].
The ureter develops from the non-branching part of the ureteric bud closest to the mesonephric duct. The ureter is a simple cuboidal epithelial-lined tube with a lumen by 4 weeks of gestation. It is thought that the lumen of the ureter closes briefly and then reopens by week 7 when urine production ensues. The ureter attains its mature transitional epithelium by 14 weeks, and muscle fibers are seen by 12 weeks.
The bladder is composed of two parts that arise from different structures embryologically, the trigone and the bladder body. The trigone is derived from the incorporation and fusion of the distal part of the mesonephric duct into the urogenital sinus. The bladder body is formed from the endoderm-lined urogenital sinus and the surrounding mesenchyme. The bladder apex is the urachus which connects to the allantois. The urachus degenerates by week 12 of gestation into the median umbilical ligament. Similar to the ureter, the bladder has primitive cuboidal epithelium around week 7 and transitional-appearing epithelium by 14 weeks. The bladder wall muscle also develops during this time and may be dependent upon fetal urine production for a change from a low-compliance system to a high-compliance system [4].
Normal Kidney and Collecting System Anatomy
Normal Kidney Anatomy
The mature kidney is located in the retroperitoneum, anterior to the psoas muscle, with the upper pole at T12 to L1 spinal column level and lower pole at L3 level. The left kidney is slightly higher than the right kidney usually. Adult kidneys are around 10–12 cm cranial to caudal, 5–7 cm wide, and 3–4 cm anterior to posterior.
The kidney is divided into the cortex and medulla. The medulla is made up of pyramid-shaped areas called medullary pyramids. The cortex contains glomeruli and nephron tubules, while the medullary pyramids contain the collecting ducts. The collecting ducts drain urine into the renal calyx in areas called renal papilla. The calyceal system generally contains 7–9 renal papilla, but can range from 4 to 18 [5]. The upper pole of the kidney generally contained 3–4 calyces, the middle pole 1–2, and the lower pole 3–4 calyces. Compound calyces occur when 2 renal papilla fuse, which tends to occur more frequently in the upper and lower poles. Each minor calyx narrows to an infundibulum and then combines with 2–3 other infundibulum to form the major calyces, of which there are generally 2–3 (Fig. 9.1). The major calyces drain into the renal pelvis which can vary in size from small and nondilated intrarenal pelvis to a large, dilated extrarenal pelvis. There is a wide range of normal configurations for the renal pelvicalyceal collecting system. The number and size of the minor and major calyces can vary, the renal pelvis can be bifid, and the collecting system may be completely bifid with two separate ureters draining into the bladder.

Left image: Intravenous pyelogram demonstrating normal configuration of a left kidney with upper-pole (white arrows), mid-pole (red arrows), and lower-pole (blue arrows) calyces. Right image: MR urogram of normal kidneys with distinguishable upper, mid, and lower poles
Renal vasculature is highly variable with up to 40 % of kidneys not having the classic renal blood supply with one renal vein and one renal artery. The renal artery branches into 4–5 segmental arteries which are end arteries supplying a distinct area of the kidney with no collateral arterial blood flow. The segmental arteries branch into lobar arteries which branch into the interlobar arteries which give rise to the arcuate arteries. The arcuate arteries run along the corticomedullary junction and branch into the interlobular arteries which give rise to the afferent arterioles that supply the glomerulus. From the glomerulus, the efferent arterioles give rise to the vasa recta. The venous drainage parallels the arterial supply with the major difference that there is collateral venous drainage. The main renal vein is generally anterior to the renal artery.
Normal Kidney Imaging
Antenatally and early in life, the kidneys show corticomedullary differentiation on renal ultrasound which can be confused with hydronephrosis of the calyces (Fig. 9.2). In addition, fetal lobulation of the kidney is seen at birth and generally disappears by 1 year of age but may persist (Fig. 9.3). Generally, the pelvicalyceal anatomy is not seen well on ultrasound unless there is significant hydronephrosis (Fig. 9.4). A normal kidney on ultrasound is slightly less echogenic than the liver with the difference in echogenicity generally increasing with age (Fig. 9.5).

Ultrasound of the left kidney in a 1-week-old infant demonstrating corticomedullary differentiation. The hypoechoic areas are the renal pyramids (one pyramid identified by white arrows)

Renal ultrasound of the right kidney in a 1-year-old demonstrating fetal lobulations (left images) with the renal contour highlighted in the lower image. CT scan with IV contrast in a 10-year-old demonstrating persistence of fetal lobulations into childhood (right)

Normal renal ultrasound of the left kidney in a 1-year-old demonstrating inability to discern pelvicalyceal anatomy (asterisk denote renal polar length)

Normal renal ultrasound of the right kidney in a 1-year-old demonstrating normal echogenicity compared to the liver (left). Normal renal ultrasound of the right kidney in a 10-year-old demonstrating that difference in echogenicity generally becomes more pronounced (right)
The normal kidney on the noncontrast phase of a CT scan should have a radiodensity measured in Hounsfield units of around 30. The renal cortex, medulla, and calyces are generally not distinguishable on a noncontrast phase. After infusion of intravenous contrast, the cortex is able to be distinguished from the medulla on an early nephrogram phase. On a later nephrogram phase, the cortex and medulla may be less distinguishable, and the contrast enhancement should be symmetric between the two kidneys. On the excretory phase, the renal calyces should be crisp and not dilated (Fig. 9.6).

Axial CT scan of the kidneys with intravenous contrast. The upper left image shows an early nephrogram phase, note how the medulla of the kidney has yet to fully enhance with contrast and is distinguishable from the cortex (arrow). The upper right image shows a later nephrogram phase of the same patient at another time where the contrast has now reached the medulla. The lower image shows the excretory or urogram phase with contrast easily visible within the renal collecting systems (arrow)
In a normal kidney imaged by T1-weighted MRI, the cortex has a higher signal intensity than the medulla and therefore appears brighter than the medulla allowing the cortex and medulla to be differentiated (Fig. 9.7). On T2-weighted MRI, the medulla has a higher signal intensity and appears brighter than the cortex. In addition, the collecting system has a higher signal intensity on T2-weighted MRI and can be somewhat visualized. Visualization of the collecting system and vascular supply can be improved with the addition of intravenous contrast such as gadolinium (Fig. 9.7).

MRI of a 4-year-old with solitary left kidney. Upper left image is a T1-weighted MRI image, note the cortex has a higher signal intensity than the medulla (arrow). On the T2-weighted MRI image in the upper right, the medulla (arrow) has a higher signal intensity than the cortex, and the collecting system is somewhat discernable. The lower left image is a three-dimensional reconstruction of an arterial phase demonstrating MR angiography; the arrow denotes the renal artery. The lower right image is a three-dimensional reconstruction of the excretory phase after addition of intravenous contrast demonstrating the collecting pelvicalyceal collecting system
The normal kidneys on radionuclide scans should uptake radiotracer symmetrically and homogeneously throughout the kidneys (Fig. 9.8). In the case of a MAG3 renal scan, the radiotracer should begin excretion into the collecting system within 10 min and should wash out promptly if furosemide is given. A time for half of the radiotracer to wash out (T ½) under 10–15 min is considered unobstructed. The differential function on a MAG3 renal scan is determined by the amount of radiotracer taken up by the kidneys within the first 2–3 min after radiotracer infusion.

Normal DMSA scan of a 4 year old female with vesicoureteral reflux. The kidneys uptake the radiotracer symmetrically and homogeneously on the posterior images (upper images). Posterior oblique images (lower images) give an asymmetrical appearance
Normal Ureter Anatomy
The renal pelvis drains to the ureter whose origin is normally posterior to the renal artery. The ureter courses caudally and anterior to the psoas muscle toward the pelvis. The ureter crosses the iliac vessels close to its bifurcation. Once in the pelvis, the ureter passes posteroinferiorly along the lateral walls of the pelvis and then courses anteromedially to come into contact with the bladder. The ureter travels 2–3 cm within the bladder wall before the ureteral orifice. There are three points of anatomical narrowing in the ureter that are common sites of ureteral nephrolithiasis: the ureteropelvic junction, the iliac vessels, and the ureterovesical junction. The normal ureter size in adults is 1.8 mm (SD 0.9 mm) with 96 % of ureters less than 3 mm [6]. The blood supply of the upper ureter comes medially from the aorta, and the blood supply of the lower ureter comes laterally from the iliac vessels.
Normal Ureter Imaging
Most ureters are not able to be visualized their entire length by ultrasonography unless there is hydroureter. Normal ureters can be visualized on CT and MRI, and their visualization is enhanced on the delayed excretory phase of a contrast-enhanced study (Fig. 9.9). A retrograde pyelogram can also be used to study the ureters but requires cystoscopy and anesthesia (Fig. 9.10).

Normal ureters on excretory phase of MRI (left image). It is normal for the entire ureter not to be visualized on the excretory phase as some parts of the ureter may be undergoing peristalsis. The right image shows a normal left kidney and a right kidney with hydronephrosis and thin parenchyma on excretory phase of MRI

Normal right ureter on retrograde pyelogram in a 10-year-old (left image). Left kidney with UPJ obstruction in a 1-year-old (right image), the ureter has evidence of fetal folds which are a normal variant in young children
Normal Bladder Anatomy
The bladder is a retroperitoneal pelvic organ. The superior surface is covered with peritoneum, and the urachus attaches the bladder to the anterior abdominal wall. When full, the bladder is an ovoid shape.
Normal Bladder Imaging
The bladder can be visualized by ultrasound, and the volume of urine can be estimated. The normal bladder wall thickness in adults is around 3 mm [7]. In school-age children, the normal bladder wall thickness is 1–2 mm [8]. Another study found that the normal bladder wall thickness for all ages of children from newborn to teenager was around 2 mm [9]. A VCUG can demonstrate the size of a bladder as well as its contour. The bladder contour should be smooth and ovoid shaped without trabeculations and diverticulum (Fig. 9.11).

Normal appearance of the bladder on VCUG with no trabeculations and a smooth, ovoid shape
Congenital Anomalies of the Kidney and Urinary Tract
CAKUT represents a wide variety of malformations and disorders that result from abnormal development of the kidney and collecting system. They can be divided in to three major categories:
-
1.
Anomalies of renal parenchyma development
-
2.
Anomalies of embryonic renal migration and fusion
-
3.
Anomalies of collecting system development
Anomalies of Renal Parenchyma Development
Anomalies of renal parenchyma development result in renal dysplasia. Renal dysplasia is characterized by abnormal nephron and collecting system development that leads to malformed kidney tissue with pathologic findings such as primitive-appearing tubules, fibrosis or scarring, and metaplastic tissue such as cartilage being present. This process can lead to several different clinical diagnoses such as renal hypoplasia, renal dysplasia or hypodysplasia, multicystic dysplastic kidney, polycystic kidney disease, or renal agenesis. The genes associated with renal dysplasia are often genes expressed during kidney development [10]. Exposure to angiotensin converting enzyme inhibitors in utero has been associated with renal dysplasia and is an example of an environmental exposure associated with renal dysplasia [11].
Renal Hypoplasia
Renal hypoplasia is a rare entity that results in congenitally small kidneys with normal architecture but reduced number of nephrons. Renal hypoplasia may be unilateral with contralateral compensatory hypertrophy. Bilateral renal hypodysplasia can be part of genetic syndromes such as branchio-oto-renal syndrome and renal-coloboma syndrome [12, 13]. When bilateral, renal hypoplasia often results in end-stage renal disease (ESRD) with pathology consistent with focal segmental glomerular sclerosis and interstitial fibrosis presumably from hyperfiltration injury of the reduced number of nephrons [14].
The diagnosis of renal hypoplasia is a clinical diagnosis when an imaging study, typically renal ultrasonography, demonstrates a kidney smaller than 2 standard deviations of the mean kidney size by age (Fig. 9.12) [15]. Absence of renal scarring on a 99mTc-dimercaptosuccinic acid (DMSA) radionuclide scan is expected (Fig. 9.13).

Renal ultrasound of a 13-year-old with right renal hypoplasia. The right kidney (left image) is 2 standard deviations smaller than the mean size for age, and the left kidney demonstrates compensatory hypertrophy

DMSA radionuclide renal scan of a 13-year-old from Fig. 9.12 with right renal hypoplasia with absence of renal scarring. The right kidney contributes 30 % of overall renal function
Renal Dysplasia
Renal dysplasia is more common than renal hypoplasia and is characterized by a kidney that is often smaller than normal depending on the size of associated cysts. Renal dysplasia occurs in about 3 of every 1,000 births and can be bilateral or unilateral with males affected 30–90 % more frequently than females [16]. Collecting system anomalies such as VUR, megaureter, ureteral atresia, and duplicated ureters occur at an increased incidence in kidneys with dysplasia [17–19]. Therefore, a voiding cystourethrogram (VCUG) is often obtained along with a renal ultrasound. As with renal hypoplasia, unilateral renal dysplasia is often associated with contralateral renal hypertrophy, and bilateral renal dysplasia is often associated with ESRD.
In children, ultrasonography is often the first imaging modality to evaluate renal dysplasia. Cystic areas, echogenic renal parenchyma, small kidneys, and hydronephrosis with thin renal parenchyma are examples of ultrasound findings with dysplasia but findings can be quite variable (Fig. 9.14). Findings on CT scan for renal dysplasia can also be quite variable (Fig. 9.15). A static renal scan such as a DMSA scan will show heterogenous, decreased uptake of radionuclide and possibly a small kidney representing poor renal function compared to a normal contralateral kidney (Fig. 9.16). As stated above, a VCUG is often obtained to evaluate for VUR which is often associated with renal dysplasia.

Renal dysplasia can have many different appearances on renal ultrasound. An 8-day-old with areas of cystic renal dysplasia (Top left). A 4-year-old with echogenic renal cortex and hydronephrosis (Top right). A 2-year-old with renal hypodysplasia with cystic areas (bottom left). A 6-month-old with thin renal parenchyma and hydronephrosis (bottom right)

Small areas of cystic renal dysplasia (left image). Bilateral renal dysplasia with hypoplastic kidneys demonstrating heterogeneous uptake of contrast and cystic areas (right image)

DMSA scans demonstrating heterogeneous uptake of radiotracer with renal dysplasia. Left image is a 4-year-old with small congenitally dysplastic left kidney associated with vesicoureteral reflux. The middle image is a 7-year-old with dysplastic and hydronephrotic right kidney associated with ectopic ureter. The right image is a 4-month-old with a duplex right kidney with the upper pole associated with an ectopic ureter
Multicystic Dysplastic Kidney
A multicystic dysplastic kidney (MCDK) is a very severe form of renal dysplasia characterized by a kidney that is non-reniform in shape, composed of noncommunicating cysts, lacking functional renal tissue, and has an absent or atretic ureter [20]. Most cases of MCDK are detected antenatally by ultrasound and are found between 0.3 and 1 in 1,000 live births [21]. As with other forms of renal dysplasia, if the contralateral kidney is normal, it often shows compensatory hypertrophy. Children with unilateral MCDK have a low risk of any significant sequelae such as hypertension, proteinuria, significant renal impairment, or contralateral abnormalities such as VUR or ureteropelvic junction obstruction (UPJO) that require surgical correction [22, 23]. Historical concerns about an increased risk of malignancy in children with unilateral MCDK are not supported by current literature [24, 25].
On ultrasound, a MCDK generally appears as a collection of noncommunicating cystic structures of variable size. MCDK can be quite large in neonates and may even be palpable, but the majority of children with unilateral MCDK show involution of the MCDK over the first few years of childhood (Fig. 9.17). Cross-sectional imaging with CT scan will demonstrate similar findings as ultrasound as well as contralateral compensatory hypertrophy (Fig. 9.18). DMSA radionuclide scan demonstrates no significant update to the MCDK (Fig. 9.19). Grossly, a MCDK does not have the typical reniform shape, contains multiple cysts, and the ureter may be atretic (Fig. 9.20).

A large multicystic dysplastic kidney in a newborn infant (left). Note the multiple noncommunicating cysts of varying size. The same child at age 2 with involuting MCDK (right)

CT scan with right MCDK (arrows) and compensatory renal hypertrophy on left

DMSA scan of a 9-year-old child with right MCDK and normal left kidney

Gross image of MCDK at time of removal. This particular example has a well-defined ureter, but the ureter may also be atretic
Autosomal Recessive Polycystic Kidney Disease
Autosomal recessive polycystic kidney disease (ARPKD), previously called infantile polycystic kidney disease, has an estimated incidence of 1 per 10,000–40,000 births [26, 27]. Mutations in the PKHD1 gene on chromosome 6p12 that encodes for the protein fibrocystin are responsible for ARPKD with over 200 mutations identified [28]. The kidneys are increased in size and are filled with small cysts (<3 mm) arising from the collecting ducts that appear to radiate from the medulla to the cortex. The liver is also affected in ARPKD with evidence of congenital fibrosis due to malformation of the biliary system and may show hepatomegaly, increased echogenicity, and dilation of bile ducts on ultrasonography [29]. The clinical spectrum of ARPKD varies widely from death in utero or in infancy due to complications of oligohydramnios, to bilateral nephrectomies at birth with dialysis, and to delayed diagnosis into early adulthood with only signs of hepatic fibrosis [30]. In general, the earlier the diagnosis is discovered, the more severe the renal impairment will be. Most severe cases of ARPKD are detected by prenatal ultrasound.
On ultrasound, the kidneys are large most commonly have a reniform shape but will be filled with multiple small cysts causing the echotexture to be hyperechoic (Fig. 9.21). Cross-sectional imaging can demonstrate the cysts that arise in the cortex and radiate from the medulla to the cortex (Fig. 9.22). A newborn with severe ARPKD will have oligohydramnios, and the kidneys can be quite large and may need to be removed for any potential for survival with dialysis (Fig. 9.23).

Ultrasound examples of a newborn with ARPKD. Multiple small cysts in a reniform-shaped kidney that are both over 11 cm in length. The kidney also has increased echogenicity

MRI image of ARPKD in a 7-year-old. The kidneys retain a reniform shape, and multiple small cysts are seen radiating from the medulla to the cortex

Newborn female with ARPKD, oligohydramnios, renal failure, and pulmonary insufficiency (ultrasound shown in Fig. 9.20). Gross images after bilateral native nephrectomy
Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease (ADPKD), previously adult polycystic kidney disease, may present in childhood. ADPKD is much more common than ARPKD with 1 case occurring every 400–1,000 live births. The mutation for ADPKD occurs in the PKD1 or PKD2 gene with PKD1 mutations accounting for 85 % of the cases [31]. The PKD1 and PKD2 genes encode the polycystin-1 and polycystin-2 proteins, respectively. Pathologically, the kidneys develop cysts of variable size in all parts of the nephron. Since the number of cysts generally increases with age, children may only have a few cysts. The natural history of ADPKD is a progressive increase in the number and size of cysts with corresponding deterioration of renal function resulting in renal replacement in middle age to late adulthood. Most children with ADPKD are asymptomatic, but occasionally complications associated with the later stages of ADPKD such as ESRD, hypertension, pain, hematuria, or cyst infection can occur [32]. Other significant organ involvement occurs including hepatic, pancreatic, or splenic cysts; cerebral aneurysms in up to 10 % of patients; and increased left ventricular size [33, 34].
Renal ultrasound in children with ADPKD can be normal, have a small number of cysts, or demonstrate kidneys that are filled with cysts (Fig. 9.24). The cysts on cross-sectional imaging often have varying signal intensity due to hemorrhage or infection history (Fig. 9.24).

ADPKD in a 9-year-old (left image) with a small number of cysts. The same patient at 20 years old (right image) showing progression and the right kidney now almost entirely filled with cysts. The lower T2-weighted MRI images of a 4-year-old demonstrate that ADPKD in children can be quite severe. The cysts have varying signal intensity likely due to hemorrhage
Isolated Renal Cysts
Isolated renal cysts are common incidental findings with the prevalence increasing with increasing age. Given how common isolated renal cysts are and the concern about risk of malignancy with complex renal cysts, the Bosniak classification system was created in which cysts are classified into one of five categories based upon morphologic and enhancement characteristics with CT scanning [35].
-
Category I: Benign simple cysts with thin wall and without septations, calcifications, or solid components. It has the density of water and does not enhance with contrast (Fig. 9.25).
Fig. 9.25 
CT scan images of a category I simple renal cyst (arrows). They cyst is smooth and round without any calcifications or solid components. The fluid within cyst is homogenous with a density similar to water (Hounsfield units less than 20). No contrast enhancement exists
-
Category II: Benign cystic lesions that may have a few thin septa, fine calcifications, or a short segment of thickened calcification. No contrast enhancement. Also includes lesions less than 3 cm in size with higher attenuation than water but without enhancement.
-
Category IIF: More complicated cystic lesions than category II but without contrast enhancement. Multiple thin septa may be present as well as thick calcifications. Also includes lesions greater than 3 cm in size with higher attenuation than water but without enhancement. Considered nonmalignant but follow-up required.
-
Category III: Cystic lesions that can be complex with measurable contrast enhancement. Approximately 50 % are malignant with low-grade renal cell carcinoma as the most common diagnosis (Fig. 9.26) [36].
Fig. 9.26 
CT scan image showing category III renal cystic lesion (red arrow) in a 13-year-old. Note the presence of septations with measurable contrast enhancement. Contrast enhancement of septations (Bosniak III) and soft tissue components (Bosniak IV) are the main distinguishing factors for renal cysts considered to have a high risk of malignancy
-
Category IV: These lesions are similar to category III lesions, and they contain contrast-enhancing soft tissue components. Greater than 75 % chance of malignancy with category IV lesions.


The Bosniak classification system has been evaluated on a limited scale in children with complex renal cysts. In one study, 5 of 39 children with complex renal cysts underwent resection; three had a benign cyst (Category IIF in 2, Category III in 1), and two had renal cell carcinoma (Category III in 1 and Category IV in 1) [37]. In another study of 22 children, all lesions classified as Category I or II were benign, and all malignant lesions were classified as Category III or IV [38].
The most common type of isolated renal cyst seen in children and adults is the simple renal cyst. Among adults, the prevalence of simple renal cysts increases from approximately 5 % in the fourth decade of life to over 30 % in the eighth decade of life [39]. The prevalence among people age 15–30 years old is less than 1 % [40]. One study evaluated over 16,000 renal ultrasounds of children from birth to age 18 found that the prevalence of simple renal cysts was 0.22 %, which was relatively stable across all age groups [41]. The average cyst size was 1 cm (range 0.3–7 cm), and of the 23 cysts followed, only 2 were slightly larger and only 1 cyst required treatment with percutaneous drainage [41].
Ultrasound is used most commonly in children to evaluate simple renal cysts, and a CT scan is not needed if a cyst meets ultrasound criteria for a simple cyst. On ultrasound a simple renal cyst is defined as being round and sharply demarcated with smooth walls, no internal echoes within the cyst, and there is a posterior wall echo with enhanced transmission beyond the cyst (Fig. 9.27) [42]. Rarely, a large simple cyst will cause pain and will require treatment such as laparoscopic cyst decortication or percutaneous drainage (Fig. 9.28).

Examples of ultrasound images of simple cysts in four different children. The cysts are round with smooth walls, there are no internal echoes, and there is a posterior wall echo with enhanced transmission beyond the cyst (arrows showing enhanced transmission)

Preoperative evaluation prior to laparoscopic decortication of a symptomatic left lower-pole renal cyst. The renal ultrasound shows a large lower-pole cystic structure with questionable thick walls (upper left). The MRI (upper right) and CT scan (lower left) demonstrate a non-enhancing simple cyst. The MAG3 renal scan demonstrated a left kidney that functions well but had an area in the lower pole with lack of contrast uptake (arrow) that corresponded to the cyst (lower right)
Renal Agenesis
The congenital absence of renal parenchymal tissue, renal agenesis, results from an early disruption of metanephric development and occurs approximately every 1 per 3,000–5,000 births [1, 43]. Renal agenesis is thought to be due to a failure of ureteral bud induction or development of the mesonephric duct [1]. Most patients are asymptomatic and are diagnosed during screening antenatal ultrasound or for imaging performed for other reasons showing an absent renal fossa and contralateral compensatory renal hypertrophy. A postnatal ultrasound and radionuclide scan confirm the diagnosis. Other congenital anomalies such as VUR, UPJO, and ureterovesical junction obstruction (UVJO) are more common in the remaining kidney [44, 45]. Unilateral renal agenesis in males can be associated with abnormalities or absence of structures derived from the mesonephric duct such as the seminal vesicles, vas deferens, and epididymis [46]. Unilateral renal agenesis in females is associated with an increased risk of Mullerian abnormalities such as uterine didelphys, duplicated vagina, and obstructed hemivagina [47].
Anomalies of Renal Migration and Fusion
Renal migration and fusion anomalies result in renal ectopia and renal fusion anomalies, respectively. The kidney normally ascends during development from the pelvis to the lumbar position by week 8 of gestation. As the kidney ascends, the kidney obtains its blood supply from nearby vessels. As a result, ectopic kidneys have an anomalous blood supply. The kidney also normally rotates medially 90° as it ascends causing the renal hilum to be oriented medially. Fusion anomalies occur when the developing kidneys fuse abnormally prior to ascent. All forms of ectopic or fused kidneys are associated with an increased risk of ipsilateral and contralateral renal abnormalities such as VUR, UPJO, hydronephrosis, kidney stones, and urinary tract infections [48].
Renal Ectopia
The most common position for an ectopic kidney is below the ipsilateral pelvic brim. These pelvic kidneys are found in 1 in 500–1,200 births [46]. Pelvic kidneys also fail to rotate properly and tend to have an anteriorly directed blood supply and renal pelvis. The blood supply is variable and can come from a one or a combination of the internal iliac, external iliac, common iliac, or aorta. Rarely, an ectopic kidney can be located in the thoracic region [49]. There is an increased incidence of other urologic abnormalities in the ectopic kidney as well as the contralateral kidney with vesicoureteral reflux to the ectopic kidney being the most common [48]. In females, Mullerian abnormalities have been associated with ectopic kidneys such as Mayer-Rokitansky-Kuster-Hauser syndrome or ipsilateral uterine agenesis [50, 51].
Simple renal ectopia is often evaluated with ultrasonography or cross-sectional imaging (Fig. 9.29). A DMSA scan can be obtained to evaluate function of the ectopic kidney (Fig. 9.30). A VCUG is commonly obtained to evaluate for VUR in the ectopic or contralateral kidney.

CT and ultrasound images of a 10-year-old with right ectopic kidney (pelvic kidney)

DMSA scan of the right pelvic kidney. Note the decreased function that is typical of ectopic kidneys with the right pelvic kidney contributing 16 % of total renal function
Crossed Renal Ectopia
Crossed renal ectopia occurs in 1 in 1,000–2,000 births and is associated with some fusion to the other kidney 90–95 % of the time [46, 52]. Most commonly, the fusion is between the upper pole of the crossed ectopic kidney and the lower pole of the kidney on the appropriate side. The ureter of the crossed ectopic kidney generally crosses back to the other side and inserts into the bladder trigone. Other rare forms of crossed renal ectopic include crossed unfused renal ectopia and bilateral crossed renal ectopia.
Crossed renal ectopia can also be detected with ultrasonography and cross-sectional imaging, and an excretory phase can be obtained to further characterize the anatomy if needed (Fig. 9.31). Radionuclide scans are also helpful in determining the anatomy and relative function of the kidneys (Fig. 9.32).

Ultrasound and CT imaging of crossed fused ectopia found incidentally after trauma (note free fluid on CT images). The ectopic kidney has anteriorly facing renal pelvis

DMSA scan of crossed fused renal ectopia. The ectopic kidney has decreased function compared to the non-ectopic kidney
Horseshoe Kidney
Horseshoe kidneys are the most common renal fusion abnormality occurring in 1 in 600 individuals [53]. The horseshoe kidney occurs when the lower poles of the developing kidneys fuse prior to renal ascent. The part of the horseshoe kidney that connects the two sides is called the isthmus and can contain functional renal tissue or just be a fibrous band depending on how early the fusion event occurred. The fused kidneys are not able to rotate properly, and ascent is limited by the isthmus coming into contact with the inferior mesenteric artery. As a result, a horseshoe kidney has its distinct horseshoe shape, is located more caudal than normal kidneys, has an anteriorly directly renal pelvis, and has a highly variable blood supply from the aorta and iliac vessels [54, 55]. The presence of a horseshoe kidney is often asymptomatic but is associated with an increased risk of VUR, hydronephrosis, UPJO, kidney stones, and UTIs [21].
Ultrasonography can usually detect the isthmus of a horseshoe kidney (Fig. 9.33), but sometimes cross-sectional imaging is needed (Fig. 9.34). In addition, studies such as a VCUG or MAG3 furosemide renal scan are obtained if there is the presence of hydronephrosis and/or urinary tract infections (Fig. 9.35).

Ultrasound images of a 7-year-old with a horseshoe kidney. Right kidney (upper left), left kidney (upper right), and isthmus indicated by arrows (bottom images)

CT images of horseshoe kidney demonstrating prominent isthmus in a 10-year-old

VCUG demonstrating bilateral vesicoureteral reflux into a horseshoe kidney. Note the medially directed calyces (red arrows) typical of a horseshoe kidney
Anomalies of Collecting System Development
Anomalies of collecting system development constitute some of the more common CAKUT diseases. Anomalies of the renal pelvis include congenital hydronephrosis, congenital UPJ obstruction, and megacalycosis. Anomalies of the ureter include primary megaureter, ectopic ureter, duplicated ureter, ureteroceles, and vesicoureteral reflux. Anomalies of the bladder and urethra include bladder exstrophy and epispadias complex, prune-belly syndrome, posterior urethral valves, bladder diverticulum, duplicated urethra, and urethral atresia.
Congenital Hydronephrosis
Antenatal hydronephrosis is found in approximately 1–2 % of all fetuses screened by prenatal ultrasound and seems to be more common in male fetuses [56, 57]. Clinical outcomes are related to severity of hydronephrosis, and two main grading systems are used. The Society for Fetal Urology (SFU) criteria grade antenatal hydronephrosis and postnatal hydronephrosis based upon the degree and location of hydronephrosis as well as presence of thinned renal parenchyma [58].
-
Grade 0: Normal examination with no dilatation of the renal pelvis
-
Grade 1: Mild dilatation of renal pelvis only (Fig. 9.36)
Fig. 9.36 
Hydronephrosis grades. Grade 1 (upper left), grade 2 (upper right), grade 3 (lower left), and grade 4 (lower right). The distinction between grade 3 and 4 is the presence of thinning of the renal parenchyma in grade 4
-
Grade 2: Moderate dilatation of the renal pelvis including a few calyces (Fig. 9.36)
-
Grade 3: Dilatation of the renal pelvis with visualization of all the calyces, which are uniformly dilated, and normal renal parenchyma (Fig. 9.36)
-
Grade 4: Similar appearance of the renal pelvis and calyces as Grade 3 plus thinning of the renal parenchyma (Fig. 9.36)

The other method of grading antenatal hydronephrosis is measurement of the anterior-posterior renal pelvis diameter (RPD). Several studies have found an association with increasing RPD and increasing risk of postnatal UTI, diagnosis of a significant CAKUT, and undergoing surgery [59–64]. A RPD of >4 mm and <10 mm is generally considered mild renal pelvis dilatation, and >15 mm is considered severe (Fig. 9.37). For mild renal pelvis dilatation, over 75 % of the cases are likely to resolve spontaneously [64]. Mild renal pelvis dilatation is the most commonly seen type of antenatal hydronephrosis, so the etiology is transient hydronephrosis accounts for around 50 % of all cases. The next most common causes of congenital hydronephrosis are physiologic hydronephrosis without obstruction (15 %), UPJO (11 %), and VUR (9 %) [65].

Prenatal ultrasound of a 32-week-old fetus with bilateral grade 3 hydronephrosis. The left kidney has an anterior-posterior diameter of 10 mm (upper left image) with no parenchymal thinning (upper right image). The right kidney has an anterior-posterior diameter of 11 mm (lower left image) with no parenchymal thinning (lower right image)
Ureteropelvic Junction Obstruction (UPJO)
As mentioned in the previous section, UPJO is the most common diagnosis given for children with antenatal hydronephrosis. Congenital UPJO is caused by an anatomical or functional intrinsic stenosis of the UPJ or less commonly by an extrinsic compression of the UPJ usually by a lower-pole renal artery. The exact mechanism of intrinsic stenosis of the UPJ is not known. The reported incidence of UPJO is 1 in 500–1,500 births [66]. UPJO can be bilateral in a minority of cases, and boys are more frequently affected [66].
Most congenital UPJO cases are detected by prenatal ultrasound. The diagnosis is supported by the presence of hydronephrosis, but that is not sufficient (Fig. 9.38). The diagnosis is confirmed by diuretic renography, such as a MAG3 diuretic renal scan, that demonstrates delayed washout of radiotracer from the renal pelvis (Fig. 9.39). The washout time of interest is the number of minutes it takes for the amount of radiotracer in the renal pelvis to decrease in half. A washout time of <15 min is considered unobstructed, and a washout time of >20 is considered consistent with obstruction. A washout time between 15 and 20 min is considered intermediate. Another important piece of information obtained from diuretic renography is the split function of the two kidneys. If the split function is equal in a patient with a unilateral kidney with a washout time consistent with obstruction, that kidney can often be observed conservatively and surgery avoided [67]. However, an initial split renal function <40 % and severe hydronephrosis are associated with increased risk of undergoing surgery [67].

Ultrasound images of a 10-month-old with left ureteropelvic junction obstruction with grade 4 hydronephrosis. No hydroureter was seen (images not shown). The presence of hydronephrosis suggests obstruction, but is not diagnostic

MAG3 diuretic renal scan demonstrating left ureteropelvic junction obstruction in a 10-month-old. The left kidney has decreased relative function at 35 % and a very prolonged T ½ washout time (arrows) even after diuretic was given at 55 min. No radiotracer is seen along the course of the left ureter
Ureteral Fibroepithelial Polyps
Fibroepithelial polyps are rare causes of UPJ obstruction in children. They are benign tumors that can occur anywhere in the urinary tract, but the most common locations are the UPJ, upper ureter, and posterior urethra [68]. While seen rarely, these lesions are often encountered unexpectedly at time of UPJ repair (Fig. 9.40).

Gross image: benign fibroepithelial polyp (arrows) in the proximal ureter (arrowheads) seen at time of UPJ repair in a child. Ultrasound image: the presence of the polyp was not known prior to surgery, but a polyp with blood flow was retrospectively appreciated on preoperative ultrasound
The diagnosis of a fibroepithelial polyp prior to surgery requires an index of suspicion. Often the ultrasound will have the appearance of a typical UPJ obstruction with hydronephrosis without hydroureter, and the diagnosis is not suspected. One study reported that subtle finding of a fibroepithelial polyp was detected by ultrasound preoperatively in 62 % of 35 children undergoing UPJ repair [68]. The authors report that blood flow was detected in all polyps that were suspected prior to surgery. Retrospective analysis of preoperative imaging of the child in Fig. 9.40 demonstrates blood flow to a polyp in the region of the UPJ (Fig. 9.40).
Primary Megaureter
A megaureter is a descriptive term given to a ureter >7 mm in diameter in children <12 years old and neonates greater than 30 weeks gestation (Fig. 9.41) [69]. There are four types of megaureter based upon the presence or absence of obstruction and reflux [70].

: Megaureter in a 10-month-old. The ureter (U) is dilated distally near the bladder (B) on the left image and proximally near the kidney on the right image
-
Refluxing, non-obstructed megaureter: this is a common type of megaureter caused by high-grade vesicoureteral reflux that leads to a dilated ureter.
-
Refluxing, obstructed megaureter: this is generally associated with an ectopic ureter that inserts into the external sphincter and is relatively rare.
-
Nonrefluxing, non-obstructed megaureter: this is a common variant of megaureter and is also called primary dilated megaureter.
-
Nonrefluxing, obstructed megaureter: this type of primary megaureter is relatively rare and often leads to a decrease in renal function, infection, or pain.
The refluxing, non-obstructed megaureter will be discussed in the section on vesicoureteral reflux, and the refluxing, obstructed megaureter will be discussed in the section on ectopic ureters. This section focuses on nonrefluxing, primary megaureter where the pathology is due to an intrinsic abnormality of the ureter and not bladder outlet obstruction or high intravesical pressures.
Nonrefluxing primary megaureter is a common reason for evaluation for potential urinary tract obstruction. Boys are affected more frequently than girls, the left side is more common, bilateral cases occur in 25 %, and the contralateral kidney can be absent or dysplastic in 10–15 % of cases [71]. The cause of primary megaureter is thought to be a defect in the muscle and collagen development in the distal ureter resulting in an aperistaltic segment near the ureterovesical junction which can lead to obstruction (Fig. 9.42) [71]. The main question with primary megaureter is whether there is significant obstruction and risk of renal deterioration. As in evaluation for UPJO, primary megaureter is evaluated with diuretic renography with the area of interest including the ureter (Fig. 9.43). If the split function is equal and UTIs are not a significant problem, primary megaureters are often observed even if the washout time is greater than 20 min. A high percentage of primary megaureters does not lead to renal deterioration and resolves spontaneously (Fig. 9.44) [72–75].

Retrograde pyelogram of left megaureter demonstrating distal aperistaltic segment (red arrows) (upper image). Gross appearance of distal aperistaltic segment (black arrows) at time of ureter reimplantation for nonrefluxing, obstructed megaureter (lower image)

MAG3 diuretic renal scan images of nonrefluxing, non-obstructed megaureter. The left and right kidney have symmetrical uptake of radiotracer with approximately equal split function (left image). The left kidney has a megaureter (arrow) with delayed drainage, but no obstruction

Natural history of megaureters by intravenous pyelogram. The right image shows an infant with bilateral non-refluxing, non-obstructed megaureters. The middle image is at 5 years old and right image is at 8 years old demonstrating marked improvement on the left side and resolution on the right
Ectopic Ureter
An ectopic ureter drains to an abnormal location. In males, the ureteral orifice of an ectopic ureter is always proximal to the external urethral sphincter and can be located distal to the trigone, in the posterior urethra, seminal vesicle, vas deferens, ejaculatory duct, and epididymis. The reason for these locations is explained embryologically (see above). Given the association of duplex kidneys and upper-pole ectopic ureters, ultrasonography often reveals a duplex kidney with upper-pole hydronephrosis (Fig. 9.45). There can be significant dysplasia associated with ectopic ureters, and the associated kidneys or upper-pole segments are often poorly functioning (Fig. 9.46). The location of the ectopic ureter insertion is often not able to be determined by ultrasonography. If the ectopic ureteral orifice is in the external urethral sphincter, a unique situation can occur where an obstructed, refluxing megaureter is found. The reflux in these ureters is usually seen only during the voiding phase when the external sphincter is relaxed (Fig. 9.47).

Upper pole (UP) of duplex kidney with ectopic ureter (upper left image) and distal ureter (U) behind the bladder (B) (upper right) in a 4-month-old. The same patient at 14 months old after laparoscopic upper-pole heminephrectomy demonstrating removal of upper pole (lower left image) and persistence of some distal hydroureter behind the bladder (lower right image)

DMSA scan demonstrating dysplasia and lack of function of the upper pole (arrow) of the right kidney that is often associated with an ectopic ureter

Example of refluxing, obstructed megaureter in a duplex kidney of a male child. The reflux is seen as the child starts to void (left image) and does not drain after completion of voiding (right image). Some prostatic ducts (arrows) are seen suggesting the ectopic ureter inserts near ejaculatory duct in the prostate
Ureterocele
A cystic dilation of the distal ureter where it inserts into the bladder or urethra is called a ureterocele (Fig. 9.48). Ureteroceles are most commonly associated with the upper pole in a duplex kidney and occur more frequently in females [76]. The reported incidence of ureteroceles is 1 in every 1,000 births [77]. The risk of UTI in children with ureteroceles may be as high as 50 % [78]. Ureteroceles are classified as intravesical if they are entirely within the bladder or ectopic if part of the ureterocele extends into the urethra. The exact embryologic explanation for ureteroceles is unknown, but one popular theory is that there is a membrane between the mesonephric duct and the ureteric bud that does not completely break down and leads to a stenotic ureteral orifice and obstruction [79].

Gross image of ureterocele in a bladder at time of surgery. The left kidney is a duplex kidney with a ureteral stent in the lower-pole ureter (yellow arrow) and upper pole associated with a ureterocele (black arrow)
Ureteroceles can be detected antenatally by screening ultrasound (Fig. 9.49) or postnatally after a urinary tract infection. Most ureteroceles are diagnosed by ultrasonography that demonstrates a cystic mass within the bladder and potentially hydroureteronephrosis (Fig. 9.50). A VCUG is generally performed as well because of the increased incidence of ipsilateral lower-pole reflux in duplex systems. Early images of the bladder with the VCUG will often demonstrate the ureterocele (Fig. 9.50). As with ectopic ureters associated with duplex kidneys, renal scans are also used clinically in determining the relative function of the different poles in a duplex kidney.

Prenatal diagnosis of bilateral ureteroceles. The right kidney is a duplex kidney (upper left) with an upper-pole segment associated with a ureterocele (bottom left). The left kidney is a single system (upper right) associated with a ureterocele (lower right). The ureteroceles within the bladder are highlighted by arrows; there are clearly two cystic structures in this case of bilateral ureteroceles

Right-sided ureterocele. On early filling images of VCUG, the ureterocele is visible as a filling defect (left image). Ultrasound demonstrates a cystic structure within the bladder (red arrow) (right image)
Duplex Kidney
The most common congenital anomaly of the urinary tract is complete or partial duplication of the collecting system with an estimated incidence of 1–5 % [80]. Partial duplication is more common than complete duplication. In collecting systems that have complete duplication, separate ureters drain the upper and lower poles of the duplex kidney. As mentioned in prior sections, the upper-pole ureteric bud arises higher on the mesonephric duct and may lead to an ectopic ureter or ureterocele, while the lower-pole ureteric bud arises lower on the mesonephric duct and can lead to VUR and a more craniolateral insertion into the bladder. In addition, the lower pole in a duplex kidney can develop a UPJ obstruction. However most completely duplicated systems do not lead to ectopic ureters, ureteroceles, UPJ obstructions, or VUR. The upper-pole ureter simply inserts medial and caudal to the lower-pole ureter. This relationship is referred to the Weigert-Meyer rule, but rare exceptions exist [81, 82].
Duplicated collecting systems are often detected by ultrasonography (Fig. 9.51) or incidentally on cross-sectional imaging (Fig. 9.51). In cases of UTI in children, a VCUG will often demonstrate lower-pole VUR (Fig. 9.52) or an upper-pole ectopic ureter or ureterocele discussed in the previous sections. Renal scans for kidneys with complete duplication can have the area of interest drawn around the upper and lower poles of a kidney to obtain the relative function of each.

Ultrasound of a 10-month-old with duplex right kidney without hydronephrosis (Top images). The duplex kidney is longer than a normal kidney, and a strip of kidney parenchyma can be seen that divides the poles. CT scan of an 11-year-old with right duplex kidney (lower left) and CT scan of a 10-year-old with left duplex kidney (lower right). The upper pole (UP) and lower pole (LP) of the duplex kidneys are highlighted in two of the images

Duplex kidney with lower-pole (LP) reflux. Note the grade 4 hydronephrosis in the lower pole on ultrasound (upper left) and the decreased function seen on MAG3 renal scan of the lower pole (upper right). The reflux is grades 4–5 and the drooping lily appearance of the lower-pole calyces is demonstrated (lower images)
Vesicoureteral Reflux
Vesicoureteral reflux (VUR) is the flow of urine retrograde from the bladder to the ureters. The two most common presentations for VUR are a neonate with a history of antenatal hydronephrosis or a young child with a febrile UTI. VUR is found in approximately 1 % of newborns and 30–45 % of children with a febrile UTI [83]. Caucasians and females are at increased risk of being diagnosed with VUR. VUR is divided into primary and secondary. Primary VUR is the most common form of VUR and is thought to occur because the ureterovesical junction (UVJ) is incompetent or the intravesical tunnel of the ureter is too short to allow closure with voiding. Secondary VUR occurs in the setting of high bladder pressures often associated with bladder outlet obstruction or neurogenic bladder.
The diagnosis of VUR is most commonly confirmed by a voiding cystourethrogram (VCUG) or a radionuclide cystogram (RNC). A VCUG is performed by placing a catheter into the bladder and filling the bladder retrograde with contrast. Images of the bladder are obtained during the filling and voiding phase. The International Reflux Study Group developed a 5-level grading method for VUR detected by VCUG [84].
-
Grade 1: Reflux into ureter only without dilation (Fig. 9.53).
Fig. 9.53 
Reflux grades on VCUG. Grade I (upper left). Grade II (upper middle). Grade III (upper right). Grade IV (lower left). Grade V (lower right)
-
Grade 2: Reflux into ureter and renal collecting system without dilation (Fig. 9.53).
-
Grade 3: Reflux fills and mildly dilates the ureter and renal collecting system with mild blunting of the calyces (Fig. 9.53).
-
Grade 4: Reflux fills and grossly dilates the ureter and renal collecting system with blunting of the calyces. The ureter is also somewhat tortuous (Fig. 9.53).
-
Grade 5: Reflux massively dilates the ureter and renal collecting system with blunted calyces. The ureter is significantly tortuous (Fig. 9.53).

The International Reflux Study Group also developed a grading method for RNC [85].
-
Mild VUR – reflux into ureter only without dilation (Fig. 9.54)
Fig. 9.54 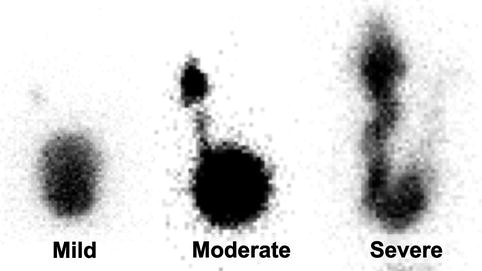
Reflux grades on RNC. Mild VUR (left). Moderate VUR (middle). Severe VUR (left)
-
Moderate VUR – reflux into ureter and renal collecting systems with or without mild dilation and blunting of calyces (Fig. 9.54)
-
Severe VUR – reflux that grossly dilates the ureter and renal colleting system with tortuosity in the ureter (Fig. 9.54)

The VCUG is the most widespread test used to diagnose VUR but has the disadvantage of more radiation exposure than the RNC. In RNC, one possible disadvantage is decreased anatomical detail. Several groups have attempted to use ultrasonography to diagnose VUR using ultrasound contrast media, but its use is not widespread [86].
Megacalycosis
Megacalycosis is a very rare entity characterized by an increased number of renal calyces [12–20], caliectasis in the absence of obstruction, and hypoplasia of the medullary pyramids [87]. The cause of megacalycosis is not known. Megacalycosis can present as prenatal hydronephrosis, and the diagnosis is confirmed by postnatal ultrasonography which shows the increased number of renal calyces with caliectasis and diuretic renography without obstruction (Fig. 9.55).

Megacalycosis in a 15-year-old. Ultrasonography of megacalycosis (upper left image) will show an increased number of calyces as well as CT (upper right image). Perhaps the best demonstration of the increased number of calyces (arrows) is a plain film obtained during excretory phase of CT scan (lower left image). In megacalycosis, diuretic renography typically demonstrates lack of obstruction with radiotracer washout (arrows) after diuretic administration (lower right image)
Bladder Exstrophy and Epispadias Complex (BEEC)
BEEC composes rare and complex congenital anomalies including epispadias, bladder exstrophy, and cloacal exstrophy. Epispadias is defined by an open urethra dorsally on the penis in males or a bifid clitoris in females. Isolated cases of epispadias can occur. Classic bladder exstrophy is characterized by an open bladder and posterior urethra in the lower abdomen, diastasis of the symphysis pubis, and epispadias. Cloacal exstrophy is the most severe and least common of the BEEC diseases and involves an open bladder and hindgut through a large abdominal wall defect, anal atresia, omphalocele, and genital anomalies.
BEEC complex diseases can be diagnosed antenatally by ultrasonography. Classic bladder exstrophy can be detected prenatally with ultrasonography with findings such as absence of bladder filling, pubic symphysis diastasis, small genitalia in males, and low-set umbilicus. However, prenatal MRI may also be useful in the diagnosis (Fig. 9.56) [88]. Around 3 % of bladder exstrophy patients will have renal anomalies such as duplex kidney, UPJO, pelvic kidney, and MCDK that are usually diagnosed by renal ultrasonography [89].

Classic bladder exstrophy. The findings on prenatal ultrasound done at 20 weeks included a small penis (arrow) with no bladder visualized (upper left image). A prenatal MRI performed at 21 weeks visualized the abdominal wall defect with the herniated bladder (circle) typical of classic bladder exstrophy (upper right image). The bottom image shows the appearance just prior to surgical reconstruction at 3 weeks of age with a small, wide penis with epispadias and abdominal wall defect with open bladder
Prune Belly
Prune-belly or Eagle-Barrett syndrome (PBS) is a rare congenital syndrome of unknown etiology characterized by a triad of findings: abdominal muscle deficiency, genitourinary tract anomalies, and bilateral undescended testis. Typical genitourinary tract anomalies seen in PBS include dilated and tortuous ureters, enlarged but non-obstructed bladder, prostate hypoplasia, and variable renal dysplasia. The incidence of ESRD is around 25–30 % in PBS and is related to the amount of renal dysplasia [90, 91].
Antenatal ultrasound may show dilated ureters and bladder and the lack of abdominal wall muscle. The appearance of prune-belly syndrome antenatally can often be confused with posterior urethral valves.
Posterior Urethral Valves
Posterior urethral valves (PUV) are rare congenital anomalies of the urethra that can severely affect the urinary tract. PUV themselves appear to be membranous folds in the posterior urethra that are the result of persistence of the urogenital membrane and deficient cannulization of the urethra [92]. The PUV leads to obstruction of the urinary tract with resulting collecting system anomalies including abnormal bladder function, hydroureteronephrosis, VUR, renal dysplasia, and ESRD.
The majority of PUV cases are detected by prenatal ultrasound that may show unilateral or bilateral hydroureteronephrosis and dilated bladder and posterior urethra (Fig. 9.57). Postnatally, PUV cases are evaluated with a VCUG, which shows the characteristic dilated posterior urethra during the voiding phase and often trabeculated bladder (Fig. 9.58). VUR is present in up to 50 % of all patients with PUV [93]. Renal dysplasia and differential renal function can be evaluated by radionuclide scans such as the DMSA scan.

Antenatal ultrasound of posterior urethral valves. This fetus had bilateral hydroureteronephrosis (upper image); however, unilateral or no hydroureteronephrosis is also possible with posterior urethral valves. The bladder (lower image) had the typical appearance of posterior urethral valves with a dilated posterior urethra (arrow) known as the keyhole sign

Postnatal VCUG of three different infants with posterior urethral valves. Vesicoureteral reflux is also a common finding on VCUG
Antenatal and Postnatal CAKUT Evaluation
Antenatal Evaluation
The majority of clinically significant CAKUT diseases are detected by fetal ultrasonography. The most common time to obtain a screening antenatal ultrasound is between 16 and 20 weeks of gestation. At this time there is good visualization of urinary tract anatomy with a high sensitivity for detecting CAKUT diseases. In addition, it is early enough in the gestation where other prenatal testing such as karyotype can be performed as well as termination of the pregnancy.
The fetal kidneys and bladder can be detected by the 15th week of gestation [94, 95]. The fetal ureters are normally not seen on prenatal ultrasound. The amount of hydronephrosis, if present, can be graded by SFU grade or the RPD measured. The bladder wall should be thin. The decrease in the volume of amniotic fluid (oligohydramnios) is a sensitive marker for abnormal fetal renal function because by 20 weeks gestation fetal urine accounts for the majority of amniotic fluid volume [96, 97]. There are several methods for assessing amniotic fluid volume by prenatal ultrasound with the single deepest pocket (SDP) measurement and amniotic fluid index (AFI) being the most common. The SDP measurement is obtained by measuring the vertical length of the largest pocked of amniotic fluid measured at a right angle to the uterus. The SDP can be interpreted accordingly: 0–2 cm, oligohydramnios; 2.1–8 cm, normal; and greater than 8 cm, polyhydramnios [98]. The AFI is a slightly more complicated measurement that is obtained by dividing the uterus into four quadrants and then obtaining the vertical length of the largest amniotic fluid pocket in each quadrant then summing the four measurements. The AFI can be interpreted accordingly: 0 to less than 4 cm, oligohydramnios; 5–25 cm, normal; and greater than 25 cm, polyhydramnios [99]. A Cochrane Collaboration meta-analysis comparing the SDP and AFI found that no method was superior in prevention of poor peripartum outcomes. However, the AFI diagnosed oligohydramnios more frequently, which led to more labor inductions and cesarean section deliveries. Therefore, the SDP was recommended [100].
Fetal MRI is increasing in use in the evaluation of CAKUT diseases [101, 102]. In one report, 35 women with 37 fetuses underwent fetal MRI with a gestational age range of 17–35 weeks. Fetal MRI was able to evaluate the kidneys and bladder in all infants and the perineum in 27 of the 35 pregnancies. In four cases of renal agenesis, the fetal MRI was able to detect the kidneys. Fetal MRI was able to distinguish between PUV and PBS by detecting the dilated posterior urethra in all cases of PUV and no posterior urethral dilation in the PBS cases. The authors suggest that fetal MRI has advantages in pregnancies with significant oligohydramnios and cases where evaluation of the perineum is important [102]. MRI can be a useful adjunct to ultrasound when the diagnosis is inconclusive on ultrasound [103].
Postnatal Evaluation
The postnatal evaluation strategy varies by the severity of antenatal findings and presumed diagnosis. A detailed pregnancy and maternal history should be performed as well as a physical examination of the newborn for all infants with a CAKUT finding prenatally. In infants with severe oligohydramnios, the infant’s pulmonary function and efforts toward pulmonary resuscitation will be most important. Some CAKUT diseases will be obvious such as prune-belly syndrome or BEEC syndromes.
Postnatal radiologic evaluation of CAKUT diseases usually begins with abdominal ultrasonography. In infants with severe prenatal findings, oligohydramnios, an abnormal solitary kidney, or bilateral disease an ultrasound within the first 24 h of life is appropriate. In infants with only unilateral involvement, it is recommended to perform ultrasonography after 48 h but within the first week of life. The reason to wait 48 h is that infants are relatively dehydrated, and severity of findings such as hydronephrosis may be underestimated earlier. A VCUG is often performed within the first week of life in infants with severe findings, oligohydramnios, an abnormal solitary kidney, or those with severe disease. However, it is controversial whether a VCUG should be performed in infants with bilateral or unilateral low-grade hydronephrosis. If there is severe hydronephrosis or megaureter, a MAG3 diuretic renal scan is often performed within the first weeks of life to assess for obstruction. A static renal scan such as a DMSA scan can also be performed within the first few weeks of life to evaluate for severity of dysplasia and differential function. In many cases of mild hydronephrosis, only serial ultrasounds are performed as the hydronephrosis is very likely to resolve spontaneously [61].
Other imaging modalities such as MRI or CT are performed occasionally to evaluate complex CAKUT malformations. However, ultrasonography, VCUG, and radionuclide renal scans remain the main imaging modalities for postnatal evaluation of CAKUT diseases.
Diagnosis of CAKUT Diseases in Childhood or Adolescence
Ureteropelvic Junction Obstruction
The most common presentations of a UPJO in childhood or adolescence are episodes of pain called Dietl’s crises, after evaluation for UTI, after trauma, or incidentally. Usually, the first test obtained is a renal ultrasound which shows dilation of the renal pelvis and calyces, but not hydroureter as mentioned in previous sections. A MAG3 diuretic renal scan is then obtained to evaluate the kidneys for differential function and obstruction. In older children, the concern for a crossing lower-pole renal vessel is higher than in infants, so occasionally CT or MR angiography is sometimes obtained to look for lower-pole crossing vessels (Fig. 9.59).

Three-dimensional reconstruction of CT angiography images showing vascular supply to kidneys in a child with a history consistent with left UPJ obstruction. A lower-pole crossing vessel was detected on the left (arrow)
Vesicoureteral Reflux
Vesicoureteral reflux (VUR) is commonly found after a VCUG is performed in a child with a febrile UTI. Approximately 30–45 % of children with a febrile UTI will be found to have VUR [83]. VUR in childhood is more common in females and Caucasians [104]. Most cases of VUR resolve spontaneously with lower-grade VUR resolves at a higher rate than high-grade VUR [105]. VUR can be associated with renal scarring or dysplasia with higher grades of VUR having a higher risk of abnormalities seen on DMSA scan (Fig. 9.60).

DMSA scan of child with bilateral VUR demonstrating renal scarring in upper and lower poles of right kidney and scarring in the lower pole of the left kidney
Congenital Renal Vein Thrombosis
Congenital (neonatal) renal vein thrombosis (RVT) is a rare entity occurring in 2–3 infants per 100,000 live births [106]. The exact etiology is often unclear, but perinatal dehydration and/or asphyxia are thought to be risk factors as well as hereditary prothrombotic states such as lupus anticoagulant, prothrombin gene mutations, factor V Leiden mutation, and others. Hereditary prothrombotic states are found in around 50 % of cases of RVT [107]. The classic presentation includes the triad of gross hematuria, palpable abdominal mass, and thrombocytopenia although a minority of patients present with the classic triad of findings [108]. There is no consensus on the optimal management of neonates with RVT [107].
Ultrasound is a common method for diagnosing congenital RVT [107]. The kidneys affected by RVT are enlarged and with increased echogenicity and loss of corticomedullary differentiation. In addition, a renal vein thrombus may be able to be seen on ultrasound. Blood flow in the main renal vein may still be present in kidneys with RVT, but an increased resistance to flow may be seen in the renal arteries. Given the propensity for renal scarring with RVT, radionuclide scans are often used to assess the kidneys in follow-up.
Key Points to Remember
-
Congenital anomalies of the kidney and urinary tract are the most common causes of end-stage renal disease in childhood.
-
Understanding the embryology of kidney development helps understand the pathology seen in CAKUT diseases.
-
Anomalies in renal parenchymal development can result in renal dysplasia, renal hypoplasia, and cystic renal diseases including multicystic dysplastic kidney.
-
Anomalies in renal fusion or ascent during development can result in ectopic kidneys, horseshoe kidneys, and crossed fused ectopic kidneys.
-
Anomalies in collecting system development can result in ectopic ureter, vesicoureteral reflux, ureteropelvic junction obstruction, ureterocele, megaureter, and other collecting system anomalies.
-
Anomalies in collecting system development are commonly associated with renal dysplasia.
-
Anomalies in renal fusion or ascent are commonly associated with renal dysplasia and anomalies in collecting system development.
-
Many CAKUT diseases are diagnosed prenatally by ultrasonography.
-
Renal ultrasonography is the first imaging modality for most CAKUT diseases with radionuclide scanning and voiding cystourethrograms utilized frequently. MRI is increasing in use for CAKUT diseases.
References
Schedl A. Renal abnormalities and their developmental origin. Nat Rev Genet. 2007;8(10):791–802.
Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Epidemiology of chronic kidney disease in children. Pediatr Nephrol. 2012;27(3):363–73.
Pope JC, Brock 3rd JW, Adams MC, Stephens FD, Ichikawa I. How they begin and how they end: classic and new theories for the development and deterioration of congenital anomalies of the kidney and urinary tract, CAKUT. J Am Soc Nephrol. 1999;10(9):2018–28.
Baskin L, Meaney D, Landsman A, Zderic SA, Macarak E. Bovine bladder compliance increases with normal fetal development. J Urol. 1994;152(2 Pt 2):692–5; discussion 696–7.
Anderson JK, Kabalin JN, Cadeddu JA. Chapter 1: Surgical Anatomy of the Retroperitoneum, Adrenals, Kidneys, and Ureters. Campbell-Walsh Urology. 9th ed. Philadelphia: Saunders Elsevier; 2007.
Zelenko N, Coll D, Rosenfeld AT, Smith RC. Normal ureter size on unenhanced helical CT. AJR Am J Roentgenol. 2004;182(4):1039–41.
Hakenberg OW, Linne C, Manseck A, Wirth MP. Bladder wall thickness in normal adults and men with mild lower urinary tract symptoms and benign prostatic enlargement. Neurourol Urodyn. 2000;19(5):585–93.
Uluocak N, Erdemir F, Parlaktas BS, Caglar MK, Hasiloglu Z, Etikan I. Bladder wall thickness in healthy school-aged children. Urology. 2007;69(4):763–6; discussion 766.
Jequier S, Rousseau O. Sonographic measurements of the normal bladder wall in children. AJR Am J Roentgenol. 1987;149(3):563–6.
Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17(10):2864–70.
Cooper WO, Hernandez-Diaz S, Arbogast PG, Dudley JA, Dyer S, Gideon PS, et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354(23):2443–51.
Weaver RG, Cashwell LF, Lorentz W, Whiteman D, Geisinger KR, Ball M. Optic nerve coloboma associated with renal disease. Am J Med Genet. 1988;29(3):597–605.
Fraser FC, Ling D, Clogg D, Nogrady B. Genetic aspects of the BOR syndrome–branchial fistulas, ear pits, hearing loss, and renal anomalies. Am J Med Genet. 1978;2(3):241–52.
Broyer M, Soto B, Gagnadoux MF, Adi M, Rica C, Gubler MC. Oligomeganephronic renal hypoplasia. Adv Nephrol Necker Hosp. 1997;26:47–63.
Sanna-Cherchi S, Caridi G, Weng PL, Scolari F, Perfumo F, Gharavi AG, et al. Genetic approaches to human renal agenesis/hypoplasia and dysplasia. Pediatr Nephrol. 2007;22(10):1675–84.
Harris J, Robert E, Kallen B. Epidemiologic characteristics of kidney malformations. Eur J Epidemiol. 2000;16(11):985–92.
Murawski IJ, Gupta IR. Vesicoureteric reflux and renal malformations: a developmental problem. Clin Genet. 2006;69(2):105–17.
Mackie GG, Stephens FD. Duplex kidneys: a correlation of renal dysplasia with position of the ureteral orifice. J Urol. 1975;114(2):274–80.
Risdon RA. Renal dysplasia. I. A clinico-pathological study of 76 cases. J Clin Pathol. 1971;24(1):57–71.
Ismaili K, Avni FE, Alexander M, Schulman C, Collier F, Hall M. Routine voiding cystourethrography is of no value in neonates with unilateral multicystic dysplastic kidney. J Pediatr. 2005;146(6):759–63.
Eckoldt F, Woderich R, Smith RD, Heling KS. Antenatal diagnostic aspects of unilateral multicystic kidney dysplasia–sensitivity, specificity, predictive values, differential diagnoses, associated malformations and consequences. Fetal Diagn Ther. 2004;19(2):163–9.
Aslam M, Watson AR. Unilateral multicystic dysplastic kidney: long term outcomes. Arch Dis Child. 2006;91(10):820–3.
Kuwertz-Broeking E, Brinkmann OA, Von Lengerke HJ, Sciuk J, Fruend S, Bulla M, et al. Unilateral multicystic dysplastic kidney: experience in children. BJU Int. 2004;93(3):388–92.
Narchi H. Risk of Wilms’ tumour with multicystic kidney disease: a systematic review. Arch Dis Child. 2005;90(2):147–9.
Beckwith JB. Should asymptomatic unilateral multicystic dysplastic kidneys be removed because of the future risk of neoplasia? Pediatr Nephrol. 1992;6(6):511.
Shaikewitz ST, Chapman A. Autosomal recessive polycystic kidney disease: issues regarding the variability of clinical presentation. J Am Soc Nephrol. 1993;3(12):1858–62.
Zerres K, Hansmann M, Mallmann R, Gembruch U. Autosomal recessive polycystic kidney disease. Problems of prenatal diagnosis. Prenat Diagn. 1988;8(3):215–29.
Bergmann C, Kupper F, Dornia C, Schneider F, Senderek J, Zerres K. Algorithm for efficient PKHD1 mutation screening in autosomal recessive polycystic kidney disease (ARPKD). Hum Mutat. 2005;25(3):225–31.
Wen J. Congenital hepatic fibrosis in autosomal recessive polycystic kidney disease. Clin Transl Sci. 2001;4(6):460–5.
Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006;85(1):1–21.
Peters DJ, Spruit L, Saris JJ, Ravine D, Sandkuijl LA, Fossdal R, et al. Chromosome 4 localization of a second gene for autosomal dominant polycystic kidney disease. Nat Genet. 1993;5(4):359–62.
Mekahli D, Woolf AS, Bockenhauer D. Similar renal outcomes in children with ADPKD diagnosed by screening or presenting with symptoms. Pediatr Nephrol. 2010;25(11):2275–82.
Belz MM, Hughes RL, Kaehny WD, Johnson AM, Fick-Brosnahan GM, Earnest MP, et al. Familial clustering of ruptured intracranial aneurysms in autosomal dominant polycystic kidney disease. Am J Kidney Dis. 2001;38(4):770–6.
Cadnapaphornchai MA, McFann K, Strain JD, Masoumi A, Schrier RW. Increased left ventricular mass in children with autosomal dominant polycystic kidney disease and borderline hypertension. Kidney Int. 2008;74(9):1192–6.
Israel GM, Bosniak MA. An update of the Bosniak renal cyst classification system. Urology. 2005;66(3): 484–8.
Harisinghani MG, Maher MM, Gervais DA, McGovern F, Hahn P, Jhaveri K, et al. Incidence of malignancy in complex cystic renal masses (Bosniak category III): should imaging-guided biopsy precede surgery? AJR Am J Roentgenol. 2003;180(3):755–8.
Wallis MC, Lorenzo AJ, Farhat WA, Bagli DJ, Khoury AE, Pippi Salle JL. Risk assessment of incidentally detected complex renal cysts in children: potential role for a modification of the Bosniak classification. J Urol. 2008;180(1):317–21.
Peng Y, Jia L, Sun N, Li J, Fu L, Zeng J, et al. Assessment of cystic renal masses in children: comparison of multislice computed tomography and ultrasound imaging using the Bosniak classification system. Eur J Radiol. 2010;75(3):287–92.
Terada N, Ichioka K, Matsuta Y, Okubo K, Yoshimura K, Arai Y. The natural history of simple renal cysts. J Urol. 2002;167(1):21–3.
Ravine D, Gibson RN, Donlan J, Sheffield LJ. An ultrasound renal cyst prevalence survey: specificity data for inherited renal cystic diseases. Am J Kidney Dis. 1993;22(6):803–7.
McHugh K, Stringer DA, Hebert D, Babiak CA. Simple renal cysts in children: diagnosis and follow-up with US. Radiology. 1991;178(2):383–5.
Curry NS. Small renal masses (lesions smaller than 3 cm): imaging evaluation and management. AJR Am J Roentgenol. 1995;164(2):355–62.
Parikh CR, McCall D, Engelman C, Schrier RW. Congenital renal agenesis: case-control analysis of birth characteristics. Am J Kidney Dis. 2002;39(4): 689–94.
Song JT, Ritchey ML, Zerin JM, Bloom DA. Incidence of vesicoureteral reflux in children with unilateral renal agenesis. J Urol. 1995;153(4):1249–51.
Kaneyama K, Yamataka A, Satake S, Yanai T, Lane GJ, Kaneko K, et al. Associated urologic anomalies in children with solitary kidney. J Pediatr Surg. 2004;39(1):85–7.
Singer A, Simmons MZ, Maldjian PD. Spectrum of congenital renal anomalies presenting in adulthood. Clin Imaging. 2008;32(3):183–91.
Smith NA, Laufer MR. Obstructed hemivagina and ipsilateral renal anomaly (OHVIRA) syndrome: management and follow-up. Fertil Steril. 2007;87(4):918–22.
Guarino N, Tadini B, Camardi P, Silvestro L, Lace R, Bianchi M. The incidence of associated urological abnormalities in children with renal ectopia. J Urol. 2004;172(4 Pt 2):1757–9; discussion 1759.
Murphy JJ, Altit G, Zerhouni S. The intrathoracic kidney: should we fix it? J Pediatr Surg. 2012;47(5):970–3.
D’Alberton A, Reschini E, Ferrari N, Candiani P. Prevalence of urinary tract abnormalities in a large series of patients with uterovaginal atresia. J Urol. 1981;126(5):623–4.
Fedele L, Bianchi S, Agnoli B, Tozzi L, Vignali M. Urinary tract anomalies associated with unicornuate uterus. J Urol. 1996;155(3):847–8.
McDonald JH, McClellan DS. Crossed renal ectopia. Am J Surg. 1957;93(6):995–1002.
Weizer AZ, Silverstein AD, Auge BK, Delvecchio FC, Raj G, Albala DM, et al. Determining the incidence of horseshoe kidney from radiographic data at a single institution. J Urol. 2003;170(5):1722–6.
O’Brien J, Buckley O, Doody O, Ward E, Persaud T, Torreggiani W. Imaging of horseshoe kidneys and their complications. J Med Imaging Radiat Oncol. 2008;52(3):216–26.
Boatman DL, Cornell SH, Kolln CP. The arterial supply of horseshoe kidneys. Am J Roentgenol Radium Ther Nucl Med. 1971;113(3):447–51.
Ek S, Lidefeldt KJ, Varricio L. Fetal hydronephrosis; prevalence, natural history and postnatal consequences in an unselected population. Acta Obstet Gynecol Scand. 2007;86(12):1463–6.
Sairam S, Al-Habib A, Sasson S, Thilaganathan B. Natural history of fetal hydronephrosis diagnosed on mid-trimester ultrasound. Ultrasound Obstet Gynecol. 2001;17(3):191–6.
Fernbach SK, Maizels M, Conway JJ. Ultrasound grading of hydronephrosis: introduction to the system used by the Society for Fetal Urology. Pediatr Radiol. 1993;23(6):478–80.
Tombesi MM, Alconcher LF. Short-term outcome of mild isolated antenatal hydronephrosis conservatively managed. J Pediatr Urol. 2011;8(2):129–33.
de Kort EH, Bambang Oetomo S, Zegers SH. The long-term outcome of antenatal hydronephrosis up to 15 millimetres justifies a noninvasive postnatal follow-up. Acta Paediatr. 2008;97(6):708–13.
Coelho GM, Bouzada MC, Pereira AK, Figueiredo BF, Leite MR, Oliveira DS, et al. Outcome of isolated antenatal hydronephrosis: a prospective cohort study. Pediatr Nephrol. 2007;22(10):1727–34.
Coelho GM, Bouzada MC, Lemos GS, Pereira AK, Lima BP, Oliveira EA. Risk factors for urinary tract infection in children with prenatal renal pelvic dilatation. J Urol. 2008;179(1):284–9.
Wollenberg A, Neuhaus TJ, Willi UV, Wisser J. Outcome of fetal renal pelvic dilatation diagnosed during the third trimester. Ultrasound Obstet Gynecol. 2005;25(5):483–8.
Lee RS, Cendron M, Kinnamon DD, Nguyen HT. Antenatal hydronephrosis as a predictor of postnatal outcome: a meta-analysis. Pediatrics. 2006;118(2):586–93.
Woodward M, Frank D. Postnatal management of antenatal hydronephrosis. BJU Int. 2002;89(2):149–56.
Liang CC, Cheng PJ, Lin CJ, Chen HW, Chao AS, Chang SD. Outcome of prenatally diagnosed fetal hydronephrosis. J Reprod Med. 2002;47(1):27–32.
Chertin B, Pollack A, Koulikov D, Rabinowitz R, Hain D, Hadas-Halpren I, et al. Conservative treatment of ureteropelvic junction obstruction in children with antenatal diagnosis of hydronephrosis: lessons learned after 16 years of follow-up. Eur Urol. 2006;49(4):734–8.
Wang XM, Jia LQ, Wang Y, Wang N. Utilizing ultrasonography in the diagnosis of pediatric fibroepithelial polyps causing ureteropelvic junction obstruction. Pediatr Radiol. 2012;42(9):1107–11.
Cussen LJ. Dimensions of the normal ureter in infancy and childhood. Invest Urol. 1967;5(2): 164–78.
Report of working party to establish an international nomenclature for the large ureter. Birth Defects Orig Artic Ser. 1977;13(5):3–8.
Hodges SJ, Werle D, McLorie G, Atala A. Megaureter. ScientificWorldJournal. 2010;10:603–12.
Arena F, Baldari S, Proietto F, Centorrino A, Scalfari G, Romeo G. Conservative treatment in primary neonatal megaureter. Eur J Pediatr Surg. 1998;8(6):347–51.
Liu HY, Dhillon HK, Yeung CK, Diamond DA, Duffy PG, Ransley PG. Clinical outcome and management of prenatally diagnosed primary megaureters. J Urol. 1994;152(2 Pt 2):614–17.
Baskin LS, Zderic SA, Snyder HM, Duckett JW. Primary dilated megaureter: long-term followup. J Urol. 1994;152(2 Pt 2):618–21.
Shukla AR, Cooper J, Patel RP, Carr MC, Canning DA, Zderic SA, et al. Prenatally detected primary megaureter: a role for extended followup. J Urol. 2005;173(4):1353–6.
Shokeir AA, Nijman RJ. Ureterocele: an ongoing challenge in infancy and childhood. BJU Int. 2002;90(8):777–83.
Jelen Z. The value of ultrasonography as a screening procedure of the neonatal urinary tract: a survey of 1021 infants. Int Urol Nephrol. 1993;25(1):3–10.
Besson R, Ngoc BT, Laboure S, Debeugny P. Incidence of urinary tract infection in neonates with antenatally diagnosed ureteroceles. Eur J Pediatr Surg. 2000;10(2):111–13.
Coplen DE, Duckett JW. The modern approach to ureteroceles. J Urol. 1995;153(1):166–71.
Decter RM. Renal duplication and fusion anomalies. Pediatr Clin North Am. 1997;44(5):1323–41.
Zamani R, Martinez R, Reddy MP. Duplicated collecting system: not following Weigert-Meyer rule as a result of renal stone obstructing the lower ureteropelvic junction. Clin Nucl Med. 2004;29(6):386–7.
Jain P, Parelkar S, Shah H, Sanghavi B, Mishra P. Uncrossed complete ureteral duplication with dysplastic lower moiety: a violation of the Weigert-Meyer law. J Pediatr Urol. 2008;4(5):404–6.
Hoberman A, Charron M, Hickey RW, Baskin M, Kearney DH, Wald ER. Imaging studies after a first febrile urinary tract infection in young children. N Engl J Med. 2003;348(3):195–202.
Medical versus surgical treatment of primary vesicoureteral reflux: report of the International Reflux Study Committee. Pediatrics. 1981;67(3):392–400.
Willi U, Treves S. Radionuclide voiding cystography. Urol Radiol. 1983;5(3):161–73, 175.
Darge K. Diagnosis of vesicoureteral reflux with ultrasonography. Pediatr Nephrol. 2002;17(1): 52–60.
Pieretti-Vanmarcke R, Pieretti A, Pieretti RV. Megacalycosis: a rare condition. Pediatr Nephrol. 2009;24(5):1077–9.
Gearhart JP, Ben-Chaim J, Jeffs RD, Sanders RC. Criteria for the prenatal diagnosis of classic bladder exstrophy. Obstet Gynecol. 1995;85(6):961–4.
Stec AA, Baradaran N, Gearhart JP. Congenital renal anomalies in patients with classic bladder exstrophy. Urology. 2012;79(1):207–9.
Noh PH, Cooper CS, Winkler AC, Zderic SA, Snyder 3rd HM, Canning DA. Prognostic factors for long-term renal function in boys with the prune-belly syndrome. J Urol. 1999;162(4):1399–401.
Reinberg Y, Manivel JC, Pettinato G, Gonzalez R. Development of renal failure in children with the prune belly syndrome. J Urol. 1991;145(5):1017–19.
Krishnan A, de Souza A, Konijeti R, Baskin LS. The anatomy and embryology of posterior urethral valves. J Urol. 2006;175(4):1214–20.
DeFoor W, Clark C, Jackson E, Reddy P, Minevich E, Sheldon C. Risk factors for end stage renal disease in children with posterior urethral valves. J Urol. 2008;180(4 Suppl):1705–8; discussion 1708.
Dugoff L. Ultrasound diagnosis of structural abnormalities in the first trimester. Prenat Diagn. 2002;22(4):316–20.
Cohen HL, Kravets F, Zucconi W, Ratani R, Shah S, Dougherty D. Congenital abnormalities of the genitourinary system. Semin Roentgenol. 2004;39(2):282–303.
Vanderheyden T, Kumar S, Fisk NM. Fetal renal impairment. Semin Neonatol. 2003;8(4):279–89.
Leibovitch L, Kuint J, Rosenfeld E, Schushan-Eisen I, Weissmann-Brenner A, Maayan-Metzger A. Short-term outcome among term singleton infants with intrapartum oligohydramnios. Acta Paediatr. 2012;101(7):727–30.
Chamberlain PF, Manning FA, Morrison I, Harman CR, Lange IR. Ultrasound evaluation of amniotic fluid volume. I. The relationship of marginal and decreased amniotic fluid volumes to perinatal outcome. Am J Obstet Gynecol. 1984;150(3):245–9.
Rutherford SE, Phelan JP, Smith CV, Jacobs N. The four-quadrant assessment of amniotic fluid volume: an adjunct to antepartum fetal heart rate testing. Obstet Gynecol. 1987;70(3 Pt 1):353–6.
Nabhan AF, Abdelmoula YA. Amniotic fluid index versus single deepest vertical pocket as a screening test for preventing adverse pregnancy outcome. Cochrane Database Syst Rev. 2008;3:CD006593.
Chauvin NA, Epelman M, Victoria T, Johnson AM. Complex genitourinary abnormalities on fetal MRI: imaging findings and approach to diagnosis. AJR Am J Roentgenol. 2012;199(2):W222–31.
Caire JT, Ramus RM, Magee KP, Fullington BK, Ewalt DH, Twickler DM. MRI of fetal genitourinary anomalies. AJR Am J Roentgenol. 2003;181(5):1381–5.
Alamo L, Laswad T, Schnyder P, Meuli R, Vial Y, Osterheld MC, et al. Fetal MRI as complement to US in the diagnosis and characterization of anomalies of the genito-urinary tract. Eur J Radiol. 2010;76(2):258–64.
Vates TS, Shull MJ, Underberg-Davis SJ, Fleisher MH. Complications of voiding cystourethrography in the evaluation of infants with prenatally detected hydronephrosis. J Urol. 1999;162(3 Pt 2):1221–3.
Estrada Jr CR, Passerotti CC, Graham DA, Peters CA, Bauer SB, Diamond DA, et al. Nomograms for predicting annual resolution rate of primary vesicoureteral reflux: results from 2,462 children. J Urol. 2009;182(4):1535–41.
Bokenkamp A, von Kries R, Nowak-Gottl U, Gobel U, Hoyer PF. Neonatal renal venous thrombosis in Germany between 1992 and 1994: epidemiology, treatment and outcome. Eur J Pediatr. 2000;159(1–2):44–8.
Lau KK, Stoffman JM, Williams S, McCusker P, Brandao L, Patel S, et al. Neonatal renal vein thrombosis: review of the English-language literature between 1992 and 2006. Pediatrics. 2007;120(5): e1278–84.
Winyard PJ, Bharucha T, De Bruyn R, Dillon MJ, Van’t Hoff W, Trompeter RS, et al. Perinatal renal venous thrombosis: presenting renal length predicts outcome. Arch Dis Child Fetal Neonatal Ed. 2006;91(4):F273–8.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media New York
About this chapter
Cite this chapter
Schlomer, B.J., Cohen, R.A., Baskin, L.S. (2014). Renal Imaging: Congenital Anomalies of the Kidney and Urinary Tract. In: Palmer, L., Palmer, J. (eds) Pediatric and Adolescent Urologic Imaging. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-8654-1_9
Download citation
DOI: https://doi.org/10.1007/978-1-4614-8654-1_9
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-8653-4
Online ISBN: 978-1-4614-8654-1
eBook Packages: MedicineMedicine (R0)