
Abstract
Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of all birth defects As a group, they are the cause of most cases of end-stage renal disease (ESRD) in children. Additionally, they are the most frequent malformations detected by prenatal ultrasound. CAKUT occur in association with nonrenal malformations in about 30 % of cases. In the majority of patients, CAKUT are sporadic; however, mutations in several renal development genes have been identified as etiologic factors. The widespread use and increased sensitivity of fetal ultrasound in identifying CAKUT has led to the frequent diagnosis of these anomalies in utero. It is important to diagnose and initiate therapy in affected patients to minimize renal damage and prevent or delay the onset ESRD. In this chapter, I will present an overview of issues related to the etiology, pathobiology, diagnosis, and clinical management of CAKUT, which will serve as a foundation for more detailed presentation in subsequent chapters.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
Introduction
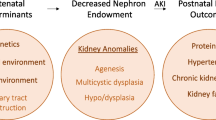
Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of all birth defects, constituting 23 % of all such defects [1]. As a group, CAKUT are the cause of 30–50 % of all cases of end-stage renal disease (ESRD) in children [2]. Further, they are the most frequent malformations detected by ultrasound in utero [3]. Lower urinary tract abnormalities can be identified in approximately 50 % of affected patients and include vesicoureteral reflux (VUR) (25 %), ureteropelvic junction obstruction (11 %), and ureterovesical junction obstruction (11 %) [4]. Renal malformations, other than mild antenatal pelviectasis, occur in association with nonrenal malformations in about 30 % of cases [3]. This chapter is an overview of issues related to the etiology, pathobiology, diagnosis, and clinical management of CAKUT and serves as a foundation for more detailed presentation in subsequent chapters.
Clinical Classification
Renal–urinary tract malformations are classified under the rubric congenital anomalies of the kidney and urinary tract (CAKUT). An overarching classification for these malformations was proposed due to recognition that (1) multiple structures within one or both kidney–urinary tract units may be affected within any given affected individual, (2) mutation in a particular gene is associated with different urinary tract anomalies in different affected individuals, and (3) mutations in different genes give rise to similar renal and lower urinary tract phenotypes. Within the CAKUT rubric, a spectrum of phenotypes exist ranging from aplasia (agenesis), defined as congenital absence of kidney tissue; to simple hypoplasia, defined as renal length <2 s.d. below the mean for age and normal renal architecture; dysplasia ± cysts, defined as malformation of tissue elements; and isolated dilatation of the renal pelvis ± ureters (collecting system). Any malformation phenotype can be observed for a kidney in an orthotopic (normal) position or an ectopic kidney.
Pathogenesis of CAKUT
Genetic Mechanisms
The genetics of CAKUT are complex (Refer to Chap. 15). The incidence of gene mutations in patients with CAKUT is unknown since population-based genome-wide sequencing studies are only now being performed. In the majority of affected patients, congenital renal malformations occur as sporadic events . In approximately 30 % of affected individuals, CAKUT occurs as part of a multiorgan genetic syndrome. Over 200 distinct genetic syndromes feature some type of kidney and urinary tract malformation. More than 30 genes have been identified as mutant in multiorgan syndromes with CAKUT (Table 1.1). Incomplete penetrance with variable expressivity is frequent in affected families. Studies of patients with CAKUT but without evidence of a multiorgan syndrome indicate that a minority of such patients will manifest mutations in genes which have been associated with genetic syndromes. For example, a study in which a small number of genes were examined in 100 patients with renal hypodysplasia and renal insufficiency demonstrated a gene mutation in genes including TCF2 and PAX2 in 16 % of affected individuals [5]. Some of the mutations were de novo mutations, explaining the sporadic appearance of CAKUT. Careful clinical analysis of patients with TCF2 and PAX2 mutations revealed the presence of extrarenal symptoms in only 50 %, supporting previous reports that TCF2 and PAX2 mutations can be responsible for isolated renal tract anomalies or at least CAKUT malformations with minimal extrarenal features [6, 7]. It is not uncommon for first-degree relatives of individuals with bilateral renal agenesis or bilateral renal dysgenesis and without evidence of a genetic syndrome or a family history to have ultrasound evidence of a renal–urinary tract malformation of some type. Studies have suggested an incidence ranging from 9 to 23 % [8, 9].
Embryologic Mechanisms
CAKUT arises from disrupted renal development. Formation of renal–urinary tract structures is initiated at 5-week gestation and concludes by about 34-week gestation. Here, the morphologic and genetic events that control kidney development are summarized. At 5-week gestation in humans, the ureteric duct is induced to undergo lateral outgrowth from the Wolffian duct and to invade the adjacent metanephric mesenchyme. After invading the metanephric mesenchyme, the ureteric bud then undergoes repetitive branching events, so termed because each event consists of expansion of the advancing ureteric bud branch at its leading tip and division of the ampulla, resulting in formation of new branches and elongation of the newly formed branches. This process results in formation of approximately 65,000 collecting ducts. During the latter stages of kidney development, tubular segments formed from the first five generations of ureteric bud branching undergo remodeling to form the kidney pelvis and calyces [10].
Identification of genes mutated in humans with CAKUT coupled with analyses of genes expressed in the developing kidney and urinary tract has provided critical insights into the mechanisms that govern mammalian renal–urinary tract morphogenesis in health and disease. Here, examples of how the study of genes mutated in human CAKUT has informed our understanding of renal development are discussed as a framework for a more detailed discussion of such studies elsewhere in this book.
Outgrowth of a single ureteric bud in the correct position is a critical initial stage of renal development. Without this process, induction of the metanephric mesenchyme does not occur. The budding process is dependent on a signaling axis comprised of Ret, a proto-oncogene and tyrosine kinase receptor, and its ligand, Gdnf. RET is expressed on the surface of ureteric cells [11], while GDNF is expressed by metanephric mesenchyme cells [12]. Homozygous deletion of either Ret or Gdnf in mice causes failure of ureteric outgrowth and renal agenesis. Patients with CAKUT have mutations in the RET/GDNF signaling pathway [13–16]. A study of 122 patients with CAKUT identified heterozygous deleterious sequence variants in GDNF or RET in 6/122 patients, 5 %, while another group screened 749 families from all over the world and identified three families with heterozygous mutations in RET [13]. Similar findings have been reported in studies of fetuses with bilateral or unilateral renal agenesis [14, 16].
The site of ureteric bud outgrowth from the Wolffian duct is normally invariant and the number of outgrowths is limited to one. Outgrowth of more than one ureteric bud can result in renal malformations including a double collecting system and duplication of the ureter. The position at which the ureteric bud arises from the Wolffian duct relative to the metanephric mesenchyme influences the interactions between the ureteric bud and the metanephric mesenchyme; ectopic positioning of the ureteric bud is associated with renal dysplasia and is also thought to contribute to the integrity of the ureterovesical junction. Mackie and Stephens postulated [17] that an abnormal position of the ureteral orifice in the bladder is associated with vesicoureteral reflux in humans. This hypothesis is supported by the discovery that mutations in ROBO2, a cell surface receptor expressed in the metanephric mesenchyme, are associated with vesicoureteral reflux in humans [18, 19]. Mice deficient in Robo2 exhibit ectopic ureteric bud formation, multiple ureters, and hydroureter [20].
Branching of the ureteric bud is initiated immediately following invasion of the metanephric mesenchyme by the ureteric bud. The number of ureteric bud branches elaborated is considered to be a major determinant of final nephron number since each ureteric bud branch tip induces a discrete subset of metanephric mesenchyme cells to undergo nephrogenesis. Regulation of ureteric branch number has been informed by complementary studies in humans and mice. Mutations in PAX2 cause renal coloboma syndrome (also named papillo-renal syndrome), an autosomal dominant disorder characterized by the association of renal hypoplasia, vesicoureteric reflux, and optic nerve coloboma [21]. During renal development, Pax2 is expressed in the Wolffian duct, the ureteric bud, and the metanephric mesenchyme. Studies in the 1Neu mouse strain, which is characterized by a Pax2 mutation, demonstrated decreased ureteric branching in association with decreased nephron number. Decreased ureteric branch number and nephron number are rescued by inhibition of apoptosis in the ureteric lineage [22, 23]. Studies in normal term newborns suggest that loss of PAX2 function may also contribute to generating a lower number of nephrons within the range of nephron number (approximately 250,000–1,600,000) observed in humans [24]. Goodyer hypothesized that gene polymorphisms that generate loss of PAX2 function could contribute to mild reductions in nephron number and discovered that a PAX2 haplotype (PAX2 AAA) is associated with an approximately 10 % decrease in kidney volume in a cohort of newborn infants [25].
As discussed above, GDNF expression by metanephric mesenchyme cells is critical to ureteric branching. In the metanephric mesenchyme, Sall1, Eya1, and Six1 positively control Gdnf expression. Sall1, a member of the Spalt family of transcriptional factors [26], is expressed in the metanephric mesenchyme prior to and during ureteric bud invasion. Mutational inactivation of Sall1 in mice causes renal agenesis or severe dysgenesis and a marked decrease in GDNF expression [27]. Mutations in SALL1 are associated with Townes–Brock syndrome, an autosomal dominant malformation syndrome characterized by imperforate anus, preaxial polydactyly and/or triphalangeal thumbs, external ear defects, sensorineural hearing loss, and, less frequently, kidney, urogenital, and heart malformations [28, 29]. EYA1, a DNA-binding transcription factor, is expressed in metanephric mesenchyme cells in the same spatial and temporal pattern as GDNF. EYA1 functions in a molecular complex with SIX1 [30] to control expression of Gdnf [31]. Both EYA1 and SIX1 are also expressed in developing otic and branchial tissues [32, 33]. Mice with EYA1 deficiency demonstrate renal agenesis and failure of GDNF expression [32]. Mutations in EYA1 and SIX1 occur in humans with branchio-oto-renal (BOR) syndrome [30, 34], which consists, in its classic form, of conductive and/or sensorineural hearing loss, branchial defects, ear pits, and renal anomalies [35, 36]. Renal malformations include unilateral or bilateral renal agenesis, hypodysplasia, as well as malformation of the lower urinary tract including vesicoureteral reflux, pyeloureteral obstruction, and ureteral duplication.
While the genome was originally conceived as consisting of two copies of each gene, the situation is more complex. Within the genome, there exist stretches of DNA that exist in less than or more than two copies. These genomic regions are termed copy number variants (CNV) and are defined as stretches of DNA that are larger than 1kb in length. Rare CNVs, that is, CNVs that are detected with a very low frequency in a human population, have recently been implicated in syndromes with CAKUT [37, 38]. For example, Sanna-Cherchi et al. examined the frequency of rare CNVs in individuals with CAKUT and identified such variants in 10 % of affected individuals compared to 0.2 % of population controls [38]. Deletions at the HNF1 locus (chromosome 17q12 ) and the locus for DiGeorge syndrome (chromosome 22q11 ) were most frequently identified, suggesting these are “hotspots” for copy number variation. Interestingly, 90 % of the CNVs associated with congenital renal malformations were previously reported to predispose to developmental delay or neuropsychiatric disease, suggesting that there are shared pathways implicated in renal and central nervous system development. Similarly, Handrigan et al. demonstrated that copy number variants at chromosome 16q24.2 are associated with autism spectrum disorder, intellectual disability, and congenital renal malformations [37].
Mechanisms Related to the Environment and Exposures in Utero
A substantial body of evidence, derived from human epidemiological studies and animal models, demonstrates an important role for the intrauterine environment in the pathogenesis of renal hypoplasia and predisposition to later kidney disease (reviewed in [39]). Renal hypoplasia with low nephron number is associated with low birth weight or intrauterine growth retardation (IGUR) and maternal undernutrition in animals [40, 41]. While the underlying mechanisms are not well defined, there is some evidence suggesting that the maternal diet programs the expression of critical genes required for embryonic kidney development, cell survival, and renal function [42–44].
Maternal diabetes is associated with renal hypoplasia in the absence of reduced birth weight. In animal models, offspring of hyperglycemic or diabetic mothers demonstrate a significant nephron deficit [45]. In utero exposure to drugs and alcohol has also been associated with renal hypoplasia . Maternal intake of angiotensin-converting enzyme inhibitors during the first trimester in humans is associated with an increased risk of renal dysplasia as well as cardiovascular and central nervous system malformations [46]. Human infants exposed to cocaine in utero have an increased risk of renal tract anomalies [47]. Similarly, infants with fetal alcohol syndrome have a higher incidence of CAKUT [48].
Diagnosis of CAKUT in Utero
The human kidney does not exhibit a capacity to accelerate the rate of nephron formation in children born prematurely or to extend the period of nephrogenesis beyond the equivalent of 34-week gestation [49]. Thus, the integrity of nephron formation in utero is absolutely critical to postnatal life. The number of functional nephrons formed by 32–34-week gestation has been implicated in short- and long-term renal function. Infants with a moderate to severe degree of hypodysplasia exhibit renal insufficiency. A more subtle deficiency in nephron number has been associated with adult-onset hypertension [50], consistent with the “Barker hypothesis,” which is based on epidemiologic evidence showing a correlation between birth weight and the incidence of cardiovascular diseases and proposes that adult-onset diseases such as hypertension have a fetal origin [51, 52]. Growth of renal tubules and expansion of glomerular cross-sectional area in utero and after birth is critical to renal functional capacity. The observation in animal models that tubule number, cross-sectional area, and cellular maturation are abnormal in renal dysgenesis is consistent with clinical observations that infants with moderate to severe renal hypoplasia or dysplasia demonstrate a limitation of GFR and tubular function.
The widespread use and 80 % sensitivity of fetal ultrasound in identifying renal–urinary tract anomalies has led to the frequent diagnosis of these anomalies in utero [53]. The fetal kidney can be visualized at 12–15 weeks of human gestation. Corticomedullary differentiation is distinct by 25 weeks of gestation and sometimes earlier. The fetal ureters are not normally detected by ultrasound. Visualization of ureters may be indicative of ureteric or bladder obstruction, or VUR. A urine-filled bladder is normally identified at 13–15-week gestation [54]. Development of the kidney in utero is commonly assessed using fetal renal length standardized for gestational age as a surrogate marker [55]. The volume of amniotic fluid is a surrogate measure of renal function. Fetal urine production begins at 9 weeks of gestation. By 20-week gestation and thereafter, fetal urine is the primary source of amniotic fluid volume [56]. A decrease in amniotic fluid volume, termed oligohydramnios, at or beyond the 20th week of gestation is an excellent indicator of a critical defect in both kidneys, for example, bilateral renal dysplasia (or a critical defect in one kidney where a solitary kidney exists), bilateral ureteral obstruction, or obstruction of the bladder outlet. Severe oligohydramnios in the second trimester can result in lung hypoplasia since an adequate amniotic fluid volume is critical for lung development [57].
Fetal urine is also used as a marker of kidney function in utero and after birth. Levels of sodium and beta-2-microglobulin in fetal urine decrease with increasing gestational age, while urine osmolality increases [58, 59]. Impaired resorption occurs in fetuses with bilateral renal dysplasia or severe bilateral obstructive uropathy, resulting in abnormal high urine levels of sodium and beta-2-microglobulin and high urine osmolality [60]. In general, sodium and chloride concentration greater than 90 meq/l (90 mmol/l), urinary osmolality greater than 210 mosmol/kg H2O (210 mmol/kg H2O), and urinary beta-2-microglobulin levels >6 mg/l raise concern as to postnatal renal prognosis [61, 62]. However, the predictive value of these indices is by no means 100 %, providing motivation for the development of other biomarkers to predict renal function. A recent study of fetuses with posterior urethral values demonstrates the promise of such approaches. Analysis of the fetal urine proteome in affected fetuses vs. controls generated a peptide profile that correctly predicted postnatal renal function with 88 % sensitivity and 95 % specificity in affected fetuses and was superior to fetal urine biochemistry and fetal ultrasound in this group of patients [63].
Clinical Sequelae and Management of CAKUT
Because CAKUT play a causative role in 30–50 % of cases of CKD in children [64], it is important to diagnose and initiate therapy to minimize renal damage, prevent or delay the onset of ESRD, and provide supportive care to avoid complications of ESRD. Counseling of families during pregnancy is a key element in the management of CAKUT. Coordinated consultation among professionals in the disciplines of obstetrics, pediatric nephrology, pediatric urology, and neonatology is critical. Consistent and clear clinical information regarding diagnosis and prognosis should be provided during pregnancy and after birth. The level of certainty regarding the severity of the diagnosis and prognosis has a major impact on decision-making during pregnancy and in the immediate postnatal period . To date, little evidence exists that relief of urinary tract obstruction in utero prevents the development of associated renal dysplasia or renal scarring . In contrast, insertion of a bladder–amniotic cavity shunt in the fetus with obstruction below the bladder neck can rescue oligohydramnios and pulmonary hypoplasia [65, 66]. Diagnostic and therapeutic management after birth should be anticipated via the coordinated actions of obstetricians, neonatologists, pediatric nephrologists, and pediatric urologists and should include an immediate assessment in the postnatal period of the need for specialized imaging, assessment of renal function, and management of nutrition and electrolytes.
After delivery, a detailed history and careful physical examination should be performed in all infants with an antenatally detected renal malformation . The examination should include the respiratory system to assess the presence of pulmonary insufficiency; the abdomen to detect the presence of a mass that could represent an enlarged kidney due to obstructive uropathy or multicystic dysplastic kidney or a palpable enlarged bladder, which could suggest posterior urethral valves; the ears, since outer ear abnormalities are associated with an increased risk of CAKUT; and the umbilicus, since a single umbilical artery is also associated with an increased risk of CAKUT.
In newborns with bilateral renal malformation, a solitary malformed kidney, or a history of oligohydramnios, an abdominal ultrasound is recommended within the first 24 h of life since an intervention such as decompression of the bladder with a transurethral catheter may be required. Newborn infants with unilateral involvement do not need immediate attention. In these infants, a renal ultrasound is generally performed after 72 h of age and within the first week of life. Ultrasound examination before 72 h of age may not detect collecting system dilatation since a newborn is relatively volume contracted during this period of time [67]. The serum creatinine estimates the extent of renal impairment and should be utilized when there is bilateral renal disease or an affected solitary kidney. The serum creatinine concentration at birth is similar to that in the mother (usually ≤1.0 mg/dl [88 μmol/l]). Thus, serum creatinine should be measured after the first 24 h of life. It declines to normal values (serum creatinine 0.3–0.5 mg/dl [27–44 μmol/l]) within approximately 1 week in term infants and 2–3 weeks in preterm infants.
Management of CAKUT is further guided by the characteristics of specific phenotypes.
Renal anomalies are frequently associated with collecting system abnormalities including VUR. Because of the frequent association of upper urinary tract anomalies including dysplasia and ectopy with a collecting system anomaly in the affected and in an apparently normal contralateral renal unit, a VCUG should be considered in such patients. A DMSA radionuclide scan can provide further information on the differential function of each kidney, which may be useful in management decisions regarding surgical interventions. Also refer to Chap. 14.
Clinical Outcomes of CAKUT
Clinical outcomes in CAKUT vary widely from no symptoms whatsoever to CKD, resulting in a need for renal replacement during a period ranging from the newborn period to the 4th and 5th decades of life. Risk factors for mortality during infancy and early childhood include coexistence of renal and nonrenal disease, prematurity, low birth weight, oligohydramnios, and severe forms of CAKUT (agenesis, hypodysplasia) [68]. In a case series of 822 children with prenatally detected CAKUT that were followed for a median time of 43 months, Quirino et al. reported a mortality of 1.5 % and morbidities including urinary tract infection, hypertension, and CKD in 29, 2.7, and 6 % of surviving children, respectively [69]. A faster rate of decline of renal function in patients with CAKUT and CKD has been associated with a urine albumin to creatinine ratio greater than 200 mg/mmol compared to less than 50 mg/mmol (eGFR: −6.5 ml/min/1.73 m2/year vs. −1.5 ml/min/1.73 m2/year), and with more than two (vs. <2) febrile urinary tract infections (eGFR −3.5 ml/min/1.73m2 vs. −2 ml/min/1.73 m2 year). A greater decline in eGFR occurs during puberty (eGFR: −4 ml/min/1.73 m2/year vs. −1.9 ml/min/1.73m2/year) [70]. A study examining the risk for dialysis in patients with CAKUT demonstrated a significantly higher risk for patients with a solitary kidney compared to non-disease controls [71]. These results raise the possibility that the prognosis for a solitary apparently normal kidney may not be as “normal” as previously thought. Finally, a study of CAKUT patients receiving some form of replacement therapy and registered within the European Dialysis and Transplant Association Registry showed that some of these patients only require renal replacement in the 3rd, 4th, or 5th decade of life. The finding that the mean age at which patients with CAKUT require dialysis and/or transplantation is 31 years indicates that children with CAKUT are at risk of developing a requirement for dialysis and/or transplantation as adults [72].
Conclusions
A majority of CAKUT can be identified in utero. However, the ability to predict the natural history of particular phenotypes is limited, and therapies, beyond surgical correction that treats the primary cause of these disorders , are nonexistent. New developments in human genetics and rapid evolution of DNA sequencing technology provide a basis to identify genetic variants in affected individuals using tools such as next-generation genomic sequencing. The knowledge gained will inform new genotype–phenotype correlation and natural history studies and the development of nongenetic disease biomarkers. This, in turn, can provide a basis for biological signatures that inform the pathobiology of specific disorders, predict their natural history, and guide personalized therapy. Further, with such knowledge, it may be possible to design new interventions to extend the current repertoire of interventions which can be used to relieve or correct urinary tract obstruction but are otherwise limited in their ability to address tissue malformation at more fundamental level using regenerative medicine strategies.
Abbreviations
- CAKUT:
-
Congenital anomalies of the kidney and urinary tract
- CKD:
-
Chronic kidney disease
- ESRD:
-
End-stage renal disease
- VCUG:
-
Voiding cystourethrogram
- VUR:
-
Vesicoureteral reflux
References
Loane M, Dolk H, Kelly A, Teljeur C, Greenlees R, Densem J, et al. Paper 4: EUROCAT statistical monitoring: identification and investigation of ten year trends of congenital anomalies in Europe. Birth Defects Res A Clin Mol Teratol. 2011;91 Suppl 1:S31–43.
Ardissino G, Dacco V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E, et al. Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics. 2003;111(4 Pt 1):e382–7. Epub 2003/04/03.eng.
Wiesel A, Queisser-Luft A, Clementi M, Bianca S, Stoll C. Prenatal detection of congenital renal malformations by fetal ultrasonographic examination: an analysis of 709,030 births in 12 European countries. Eur J Med Genet. 2005;48:131–44.
Piscione TD, Rosenblum ND. The malformed kidney: disruption of glomerular and tubular development. Clin Genet. 1999;56:343–58.
Weber S, Moriniere V, Knuppel T, Charbit M, Dusek J, Ghiggeri GM, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol. 2006;17:2864–70.
Salomon R, Tellier AL, Attie-Bitach T, Amiel J, Vekemans M, Lyonnet S, et al. PAX2 mutations in oligomeganephronia. Kidney Int. 2001;59:457–62.
Ulinski T, Lescure S, Beaufils S, Guigonis V, Decramer S, Morin D, et al. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J Am Soc Nephrol. 2006;17:497–503.
Roodhooft AM, Jason MD, Birnholz JC, Holmes LB. Familial nature of congenital absence and severe dysgenesis of both kidneys. N Eng J Med. 1984;310:1341–4.
Bulum B, Ozcakar ZB, Ustuner E, Dusunceli E, Kavaz A, Duman D, et al. High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr Nephrol. 2013;28:2143–7.
Rosenblum ND. Developmental biology of the human kidney. Semin Fetal Neonatal Med. 2008;13:125–32. Epub 2007/12/22.eng.
Pachnis V, Mankoo B, Costantini F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development. 1993;119:1005–17.
Hellmich HL, Kos L, Cho ES, Mahon KA, Zimmer A. Embryonic expression of glial cell-line derived neurotrophic factor (GDNF) suggests multiple developmental roles in neural differentiation and epithelial-mesenchymal interactions. Mech Dev. 1996;54:95–105.
Chatterjee R, Ramos E, Hoffman M, VanWinkle J, Martin DR, Davis TK, et al. Traditional and targeted exome sequencing reveals common, rare and novel functional deleterious variants in RET-signaling complex in a cohort of living US patients with urinary tract malformations. Hum Genet. 2012;131:1725–38. Pubmed Central PMCID: 3551468, Epub 2012/06/26. eng.
Skinner MA, Safford SD, Reeves JG, Jackson ME, Freemerman AJ. Renal aplasia in humans is associated with RET mutations. Am J Hum Genet. 2008;82:344–51.
Yang D, Zhang J, Chen C, Xie M, Sperling S, Fang F, et al. BMPR IA downstream genes related to VSD. Pediatr Res. 2008;63:602–6.
Jeanpierre C, Mace G, Parisot M, Moriniere V, Pawtowsky A, Benabou M, et al. RET and GDNF mutations are rare in fetuses with renal agenesis or other severe kidney development defects. J Med Genet. 2011;48:497–504. Epub 2011/04/15.eng.
Mackie GG, Stephens FD. Duplex kidneys: a correlation of renal dysplasia with position of the ureteral orifice. J Urol. 1975;114:274–80.
Bertoli-Avella AM, Conte ML, Punzo F, de Graaf BM, Lama G, La Manna A, et al. ROBO2 gene variants are associated with familial vesicoureteral reflux. J Am Soc Nephrol. 2008;19:825–31.
Piper M, Georgas K, Yamada T, Little M. Expression of the vertebrate Slit gene family and their putative receptors, the Robo genes, in the developing murine kidney. Mech Dev. 2000;94:213–7.
Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Dev Cell. 2004;6:709–17.
Weaver RG, Cashwell LF, Lorentz W, Whiteman D, Geisinger KR, Ball M. Optic nerve coloboma associated with renal disease. Am J Med Genet. 1988;29:597–605.
Porteous S, Torban E, Cho N-P, Cunliffe H, Chua L, McNoe L, et al. Primary renal hypoplasia in humans and mice with PAX2 mutations: evidence of increased apoptosis in fetal kidneys of Pax21Neu +/− mutant mice. Hum Mol Genet. 2000;9:1–11.
Dziarmaga A, Eccles M, Goodyer P. Suppression of ureteric bud apoptosis rescues nephron endowment and adult renal function in Pax2 mutant mice. J Am Soc Nephrol. 2006;17:1568–75.
Nyengaard JR, Bendtsen TF. Glomerular number and size in relation to age, kidney weight, and body surface in normal man. Anat Rec. 1992;232:194–201.
Quinlan J, Lemire M, Hudson T, Qu H, Benjamin A, Roy A, et al. A common variant of the PAX2 gene is associated with reduced newborn kidney size. J Am Soc Nephrol. 2007;18:1915–21.
Kohlhase J, Wischermann A, Reichenbach H, Froster U, Engel W. Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat Genet. 1998;18:81–3.
Nishinakamura R, Matsumoto Y, Nakao K, Nakamura K, Sato A, Copeland NG, et al. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128:3105–15.
Townes PL, Brocks ER. Hereditary syndrome of imperforate anus with hand, foot, and ear anomalies. J Pediatr. 1972;81:321–6.
O'Callaghan M, Young ID. The Townes-Brocks syndrome. J Med Genet. 1990;27:457–61.
Ruf RG, Xu PX, Silvius D, Otto EA, Beekmann F, Muerb UT, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A. 2004;101:8090–5.
Sajithlal G, Zou D, Silvius D, Xu PX. Eya1 acts as a critical regulator for specifying the metanephric mesenchyme. Dev Biol. 2005;284:323–36.
Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat Genet. 1999;23:113–7. Epub 1999/09/02.eng.
Ozaki H, Watanabe Y, Ikeda K, Kawakami K. Impaired interactions between mouse Eyal harboring mutations found in patients with branchio-oto-renal syndrome and Six, Dach, and G proteins. J Hum Genet. 2002;47:107–16. Epub 2002/04/13.eng.
Abdelhak S, Kalatzis V, Heilig R, Compain S, Samson D, Vincent C, et al. A human homologue of the Drosophila eyes absent gene underlies Branchio-Oto-Renal (BOR) syndrome and identifies a novel gene family. Nat Gen. 1997;15:157–64.
Chen A, Francis M, Ni L, Cremers CW, Kimberling WJ, Sato Y, et al. Phenotypic manifestations of branchio-oto-renal syndrome. Am J Med Genet. 1995;58:365–70.
Chang EH, Menezes M, Meyer NC, Cucci RA, Vervoort VS, Schwartz CE, et al. Branchio-oto-renal syndrome: the mutation spectrum in EYA1 and its phenotypic consequences. Hum Mutat. 2004;23:582–9.
Handrigan GR, Chitayat D, Lionel AC, Pinsk M, Vaags AK, Marshall CR, et al. Deletions in 16q24.2 are associated with autism spectrum disorder, intellectual disability and congenital renal malformation. J Med Genet. 2013;50:163–73. Epub 2013/01/22.eng.
Sanna-Cherchi S, Kiryluk K, Burgess KE, Bodria M, Sampson MG, Hadley D, et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet. 2012;91:987–97. Pubmed Central PMCID: 3516596, Epub 2012/11/20. eng.
Denton KM. Can adult cardiovascular disease be programmed in utero? J Hypertens. 2006;24:1245–7. Epub 2006/06/24.eng.
Wlodek ME, Mibus A, Tan A, Siebel AL, Owens JA, Moritz KM. Normal lactational environment restores nephron endowment and prevents hypertension after placental restriction in the rat. J Am Soc Nephrol. 2007;18:1688–96.
Zohdi V, Moritz KM, Bubb KJ, Cock ML, Wreford N, Harding R, et al. Nephrogenesis and the renal renin-angiotensin system in fetal sheep: effects of intrauterine growth restriction during late gestation. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1267–73. Epub 2007/06/22.eng.
Abdel-Hakeem AK, Henry TQ, Magee TR, Desai M, Ross MG, Mansano RZ, et al. Mechanisms of impaired nephrogenesis with fetal growth restriction: altered renal transcription and growth factor expression. Am J Obstet Gynecol. 2008;199:252 e1–7. Epub 2008/07/22.eng.
Gilbert JS, Lang AL, Grant AR, Nijland MJ. Maternal nutrient restriction in sheep: hypertension and decreased nephron number in offspring at 9 months of age. J Physiol. 2005;565:137–47. Epub 2005/03/26. eng.
Welham SJ, Riley PR, Wade A, Hubank M, Woolf AS. Maternal diet programs embryonic kidney gene expression. Physiol Genomics. 2005;22:48–56. Epub 2005/04/14. eng.
Amri K, Freund N, Vilar J, Merlet-Benichou C, Lelievre-Pegorier M. Adverse effects of hyperglycemia on kidney development in rats: in vivo and in vitro studies. Diabetes. 1999;48:2240–5. Epub 1999/10/27.eng.
Cooper WO, Hernandez-Diaz S, Arbogast PG, Dudley JA, Dyer S, Gideon PS, et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med. 2006;354:2443–51.
Battin M, Albersheim S, Newman D. Congenital genitourinary tract abnormalities following cocaine exposure in utero. Am J Perinatol. 1995;12:425–8. Epub 1995/11/01.eng.
Taylor CL, Jones KL, Jones MC, Kaplan GW. Incidence of renal anomalies in children prenatally exposed to ethanol. Pediatrics. 1994;94:209–12. Epub 1994/08/01.eng.
Potter EL. Normal and abnormal development of the kidney. Chicago: Year Book Medical Publishers Inc; 1972. 305 p.
Keller G, Zimmer G, Mall G, Ritz E, Amann K. Nephron number in patients with primary hypertension. New Engl J Med. 2003;348:101–8.
Barker DJ, Osmond C, Golding J, Kuh D, Wadsworth ME. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ. 1989;298:564–7. Epub 1989/03/04. eng.
Barker DJ, Bagby SP, Hanson MA. Mechanisms of disease: in utero programming in the pathogenesis of hypertension. Nat Clin Pract Nephrol. 2006;2:700–7.
Vanderheyden T, Kumar S, Fisk NM. Fetal renal impairment. Semin Neonatol. 2003;8:279–89. Epub 2004/03/06.eng.
Cohen HL, Kravets F, Zucconi W, Ratani R, Shah S, Dougherty D. Congenital abnormalities of the genitourinary system. Semin Roentgenol. 2004;39:282–303. Epub 2004/05/18.eng.
Cohen HL, Cooper J, Eisenberg P, Mandel FS, Gross BR, Goldman MA, et al. Normal length of fetal kidneys: sonographic study in 397 obstetric patients. Am J Roentgenol. 1991;157:545–8. Epub 1991/09/01.eng.
Gilbert WM, Brace RA. Amniotic fluid volume and normal flows to and from the amniotic cavity. Semin Perinatol. 1993;17:150–7. Epub 1993/06/01.eng.
Potter EL. Bilateral renal agenesis. J Pediatr. 1946;29:68–76. Epub 1946/07/01.eng.
Nicolini U, Fisk NM, Rodeck CH, Beacham J. Fetal urine biochemistry: an index of renal maturation and dysfunction. Br J Obstet Gynecol. 1992;99:46–50.
Muller F, Dommergues M, Bussieres L, Lortat-Jacob S, Loirat C, Oury JF, et al. Development of human renal function: reference intervals for 10 biochemical markers in fetal urine. Clin Chem. 1996;42:1855–60. Epub 1996/11/01.eng.
Muller F, Dommergues M, Mandelbrot L, Aubry MC, Nihoul-Fekete C, Dumez Y. Fetal urinary biochemistry predicts postnatal renal function in children with bilateral obstructive uropathies. Obstet Gynecol. 1993;82:813–20.
Glick PL, Harrison MR, Golbus MS, Adzick NS, Filly RA, Callen PW, et al. Management of the fetus with congenital hydronephrosis II: prognostic criteria and selection for treatment. J Pediatr Surg. 1985;20:376–87. Epub 1985/08/01.eng.
Morris RK, Quinlan-Jones E, Kilby MD, Khan KS. Systematic review of accuracy of fetal urine analysis to predict poor postnatal renal function in cases of congenital urinary tract obstruction. Prenat Diagn. 2007;27:900–11. Epub 2007/07/05.eng.
Klein J, Lacroix C, Caubet C, Siwy J, Zurbig P, Dakna M, et al. Fetal urinary peptides to predict postnatal outcome of renal disease in fetuses with posterior urethral valves (PUV). Sci Transl Med. 2013;5:198ra06.
Seikaly MG, Ho PL, Emmett L, Fine RN, Tejani A. Chronic renal insufficiency in children: the 2001 Annual Report of the NAPRTCS. Pediatr Nephrol. 2003;18:796–804.
Elder JS, Duckett Jr JW, Snyder HM. Intervention for fetal obstructive uropathy: has it been effective? Lancet. 1987;2(8566):1007–10. Epub 1987/10/31.eng.
Freedman AL, Johnson MP, Smith CA, Gonzalez R, Evans MI. Long-term outcome in children after antenatal intervention for obstructive uropathies. Lancet. 1999;354(9176):374–7.
Bueva A, Guignard JP. Renal function in preterm neonates. Pediatr Res. 1994;36:572–7.
Melo BF, Aguiar MB, Bouzada MC, Aguiar RL, Pereira AK, Paixao GM, et al. Early risk factors for neonatal mortality in CAKUT: analysis of 524 affected newborns. Pediatr Nephrol. 2012;27:965–72. Epub 2012/03/10.eng.
Quirino IG, Diniz JS, Bouzada MC, Pereira AK, Lopes TJ, Paixao GM, et al. Clinical course of 822 children with prenatally detected nephrouropathies. Clin J Am Soc Nephrol. 2012;7:444–51. Pubmed Central PMCID: 3302677, Epub 2012/01/24.eng.
Gonzalez Celedon C, Bitsori M, Tullus K. Progression of chronic renal failure in children with dysplastic kidneys. Pediatr Nephrol. 2007;22:1014–20.
Sanna-Cherchi S, Ravani P, Corbani V, Parodi S, Haupt R, Piaggio G, et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009;76:528–33. Epub 2009/06/19.eng.
Wuhl E, van Stralen KJ, Verrina E, Bjerre A, Wanner C, Heaf JG, et al. Timing and outcome of renal replacement therapy in patients with congenital malformations of the kidney and urinary tract. Clin J Am Soc Nephrol. 2013;8:67–74. Pubmed Central PMCID: 3531653, Epub 2012/10/23.eng.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Rosenblum, N.D. (2016). Congenital Anomalies of the Kidney and Urinary Tract: An Overview. In: Barakat, A., Rushton, H. (eds) Congenital Anomalies of the Kidney and Urinary Tract. Springer, Cham. https://doi.org/10.1007/978-3-319-29219-9_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-29219-9_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-29217-5
Online ISBN: 978-3-319-29219-9
eBook Packages: MedicineMedicine (R0)