
Abstract
The genome is under constant assault from both endogenous and exogenous sources such as reactive oxygen species and ionizing radiation capable of inducing a wide array of mutagenic changes [1]. To maintain genomic integrity cells have evolved elegant mechanisms to recognize DNA damage, arrest the cell cycle, and activate specific repair pathways. One of the most cytotoxic lesions that a cell must contend with is a double-strand break (DSB) because even a single unrepaired DSB is capable of inducing cell death [2]. To repair a DSB, cells have at least four mechanisms at their disposal: homologous recombination (HR), single-strand annealing (SSA), nonhomologous end-joining (NHEJ), and microhomology-mediated end joining (MMEJ) (Fig. 1) [3]. HR relies on the sister chromatid as a template to fill in damaged or missing DNA, restoring the chromosome to its original condition. In cells with competent DNA repair mechanisms, HR is the preferred pathway of repair during the S and G2 phase of the cell cycle when the sister chromatid is available [4]. SSA, a variant of HR that is thought to play a minor role in the repair DSBs, utilizes homologous repeats surrounding a DSB to anneal the broken ends resulting in the deletion of the intervening sequence. In contrast, NHEJ and MMEJ both operate throughout the cell cycle and directly ligate two ends of a DSB; however, MMEJ always introduces small deletions at broken ends to produce a region of microhomology to facilitate ligation [5]. The important point to note is that HR is considered an error-free pathway whereas SSA, NHEJ, and MMEJ are error-prone because they can create gross chromosomal aberrations if ligation occurs incorrectly—potentially leading to neoplastic transformation [1].
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
The genome is under constant assault from both endogenous and exogenous sources such as reactive oxygen species and ionizing radiation capable of inducing a wide array of mutagenic changes [1]. To maintain genomic integrity cells have evolved elegant mechanisms to recognize DNA damage, arrest the cell cycle, and activate specific repair pathways. One of the most cytotoxic lesions that a cell must contend with is a double-strand break (DSB) because even a single unrepaired DSB is capable of inducing cell death [2]. To repair a DSB, cells have at least four mechanisms at their disposal: homologous recombination (HR), single-strand annealing (SSA), nonhomologous end-joining (NHEJ), and microhomology-mediated end joining (MMEJ) (Fig. 1) [3]. HR relies on the sister chromatid as a template to fill in damaged or missing DNA, restoring the chromosome to its original condition. In cells with competent DNA repair mechanisms, HR is the preferred pathway of repair during the S and G2 phase of the cell cycle when the sister chromatid is available [4]. SSA, a variant of HR that is thought to play a minor role in the repair DSBs, utilizes homologous repeats surrounding a DSB to anneal the broken ends resulting in the deletion of the intervening sequence. In contrast, NHEJ and MMEJ both operate throughout the cell cycle and directly ligate two ends of a DSB; however, MMEJ always introduces small deletions at broken ends to produce a region of microhomology to facilitate ligation [5]. The important point to note is that HR is considered an error-free pathway whereas SSA, NHEJ, and MMEJ are error-prone because they can create gross chromosomal aberrations if ligation occurs incorrectly—potentially leading to neoplastic transformation [1].

Functional domains and interacting partners of human BRCA1 and BRCA2 proteins. Only domains (listed above) and protein partners (drawn below) that were discussed in this review are described. Proteins are color-coded with its corresponding interacting domain
BRCA1 and BRCA2 are tumor suppressors essential for the faithful repair of DSBs by HR [6]. However, BRCA1 also participates in other cellular functions important in maintaining genomic integrity including the assembly of the mitotic spindle [7], centrosome duplication [8], cell-cycle control [9–14], chromatin remodeling at sites of DSBs [15, 16], and DNA decantenation [17]. In contrast, the role of BRCA2 is primarily to regulate RAD51 filament formation, which is a critical step in catalyzing strand invasion and homologous recombination (Fig. 1).

Schema describing DNA repair pathways following a double-strand break (DSB). Homologous recombination is the preferred pathway during S and G2 phases of the cell cycle and is considered an error-free pathway. NHEJ, MMEJ, and SSA, on the other hand, are thought to be error-prone pathways because they introduce deletions at broken-ends and may promiscuously ligate nonadjacent ends creating gross chromosomal aberrations. XRCC1/4 X-ray repair complementing defective repair in Chinese hamster cells 1/4. DNA-PK DNA dependent protein kinase catalytic subunit. MRN Mre11-Rad50-Nbs1
Cells lacking BRCA1 or BRCA2 are unable to repair DSBs by HR and must resort to more error-prone pathways such as MMEJ and SSA. These cells display gross chromosomal rearrangements such as large deletions, translocations, and fusions during successive rounds of cell division [18]. While the vast majority of these lesions result in cell death, the genetic instability caused by loss of competent HR leads to a dramatically increased number of genetic alterations, which provide a rich background for Darwinian forces to act at the level of the tumor microenvironment, promoting the emergence of multiple clones, some of which have the capability to divide autonomously and metastasize [19]. The importance of BRCA genes in maintaining genomic integrity is underscored by patients who harbor germ-line mutations in BRCA1 or BRCA2 and have a markedly increased predisposition to develop, among others, breast and ovarian cancers [20].
Since the discovery of BRCA1 and BRCA2 more than 15 years ago [21, 22], understanding their function has been of primary importance and much progress has been made. In this review, we summarize the role BRCA1 and BRCA2 play in homologous recombination and how this knowledge can be utilized to target tumors deficient in this cellular pathway in hereditary as well as sporadic cancers.
2 Structure and Function of BRCA1
BRCA1 is composed of 1,863 amino acids and contains three functionally important domains (Fig. 2). At its amino terminal is a RING-finger domain with E3 ubiquitin ligase activity (Box 1). It is normally found in association with its heterodimeric protein partner BARD1 (which is itself a RING E3 ubiquitin ligase). This interaction stabilizes the complex, preventing its degradation [23] and enhances its E3 ligase function [24]. In addition, the ubiquitin ligase activity of BRCA1 is activated upon two post-modificational processes: auto-ubiquitination [25] and SUMOylation [26, 27]. It is not yet clear how the ubiquitin ligase activity of BRCA1 is increased; however, two possible scenarios can be envisaged. One is that ubiquitination or SUMOylation directly alters the conformation of the RING-finger domain increasing enzymatic activity. A second possibility could be that posttranslational modifications increase affinity for the E2 conjugating enzyme UbcH5a, accelerating ubiquitin transfer. BRCA1 has been shown to ubiquitinate various proteins including histones (H2A, H2AX, and H2B) [25, 28], CtIP [29], γ-tubulin [8], nucleophosmin [30], RNA polymerase II [31, 32], and ERα [33]. How ubiquitination of these target proteins modifies their function is unclear; however, germ-line mutations derived from patients with breast cancer that abolish RING finger ligase activity are observed to result in checkpoint deregulation and sensitivity to ionizing radiation [34, 35]. Strikingly these effects are independent of homologous recombination [36]. To reconcile this apparent paradox, Zhu et al. propose that BRCA1 acts in vivo to regulate expression of satellite DNA that is normally silenced by ubiquitination of H2A and that overexpression of satellite transcripts is linked to genomic instability [28]. However, the function of satellite transcripts and how its aberrant expression leads to tumor development are currently unknown.
At the carboxyl end of BRCA1 are tandem BRCT domains which contain a phosphate binding core providing an interface for phosphorylated proteins [37, 38]. Phosphorylation, mediated primarily by the kinases ATM and CHEK2, is an important spatiotemporal regulator of proteins involved in check-point control and DNA repair. The tandem BRCT domain of BRCA1 helps localize it to nuclear foci by binding to different phosphorylated intermediates including Abraxas [10, 14, 39], CtIP [40], and BRIP1 [13]. These protein complexes form three distinct entities during HR and each has important functions at sites of DNA breakage. For instance BRCA1 in association with Abraxas and RAP80 has been shown to regulate the G2-M checkpoint. When BRCA1 is bound to CtIP coupled with MRN, however, it regulates DNA end-resection diverting the pathway away from MMEJ towards HR (Fig. 2) [41].
BRCA1 also plays a more central role in HR. BRCA1 contains a coiled-coil domain present near the carboxyl terminal which binds PALB2 (Partner and Localizer of BRCA2) [42, 43]. PABL2 physically bridges BRCA1 to BRCA2. This complex in turn mediates the final enzymatic step of RAD51 assembly, and strand exchange between homologous chromosomes (described below).
4 Structure and Function of BRCA2
Although bearing similar names, BRCA2 is structurally and functionally distinct from BRCA1. It is a much larger (3,418 amino acids) protein containing eight BRC motifs, which enable binding to RAD51 [44, 45] and another distinct RAD51-binding domain at its terminal end [46, 47] (Fig. 1). Full-length human BRCA2 had never been purified to sufficient quantities due to its large molecular size. As such, its functions could only be derived from studying BRCA2 orthologues and smaller protein fragments. Recently, however, three teams using different approaches have managed to obtain purified full-length human BRCA2, providing an unprecedented in vitro analysis of its molecular functions [48–50]. All three papers were able to demonstrate that BRCA2 mediates loading of RAD51 onto ssDNA while displacing RPA (a protein that binds ssDNA preventing secondary DNA structures from forming). In addition, Jensen et al. and Thorslund et al. show that BRCA2 prevents RAD51 association to dsDNA, which would inhibit HR, and favors RAD51 association to ssDNA or dsDNA with ssDNA tails. Jensen et al. and Liu et al. demonstrate that BRCA2 inhibits RAD51 hydrolysis of ATP, which stabilizes the nucleoprotein filament. Much more is still to be learned about BRCA2. For example, there is direct evidence to demonstrate that, despite being evolutionary conserved domains, not all BRC motifs are required for competent HR to be elicited in the presence of DNA DSBs, suggesting that they may have alternate or modulatory roles in HR [51]. Understanding how its interacting partners—such as PALB2 and BRCA1—affect BRCA2 function is also unclear. With full-length BRCA2 at hand characterization of its complex molecular functions will be more readily answered.
5 Fanconi Anemia and Homologous Repair
An interstrand cross-link (ICL) is another highly cytotoxic lesion that prevents separation of complementary strands of DNA during replication. A specialized pathway is necessary to recognize and remove a cross-link but in so doing, a DSB is generated, requiring the HR machinery to complete the repair (Fig. 3) [52]. Patients with defects in ICL repair develop a rare genetic condition known as Fanconi anemia (FA), characterized by aplastic anemia, multiple congenital defects, susceptibility to both hematologic and solid malignancies, and sensitivity to ICL agents such as platinum drugs and mitomycin C. It is a heterogeneous disease caused by defects (either by recessive or X-linked mutations) in 1 of 13 genes, three of which—BRIP1, PALB2, and BRCA2—are also proteins involved in HR, providing further evidence that these two pathways are closely interrelated. See chapter “Repair of DNA Interstrand Cross-links Produced by Cancer Chemotherapeutic Drugs” for a detailed review of ICL repair.

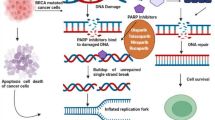
Schema describing DNA repair pathways following a single-strand break and interstrand cross-link (ICL). A single-strand break is normally repaired by base-excision repair (BER). If PARP is inhibited, however, BER is defective and a double-strand break is induced during the S phase of the cell cycle requiring homologous recombination (HR) to mediate repair and regenerate the replication fork. Tumor cells unable to properly repair DNA damage by both BER and HR, resort to more error prone mechanisms such as NHEJ, MMEJ and SSA, which induces genomic instability and ultimately cell death. Interstrand cross-links require both intact Fanconi anemia (FA) and HR pathways to mediate its repair. A defect in any one of these pathways leads to chromosome breakage and cell death
6 Targeting BRCA1 and BRCA2-Mutated Tumors
Nearly all ovarian carcinomas and most breast cancers derived from patients with germ-line BRCA1 and BRCA2 mutations have lost their remaining wild-type allele and thus the ability to repair DSBs by HR [53, 54]. These cancers instead rely on complementary pathways such as NHEJ, to maintain some degree of genomic stability. By contrast, “healthy” cells with only one functional BRCA gene still have an intact HR pathway, a biological difference that can be exploited. In cells with a defective HR pathway, agents that introduce ICLs, such as mitomycin C and platinum drugs, are not effectively repaired and induce cell death, while those still capable of repairing DSBs by HR are relatively spared. Early data from platinum-based regimens on carriers of BRCA1 mutations have suggested some efficacy in treating breast cancer in the neoadjuvant setting [55]; however, stronger data will be needed before its clinical use in treating BRCA-associated cancers can be routinely proposed [56]. Anthracyclines, which intercalate DNA causing DSBs, may also be effective [57, 58]; however, reports from cell and clinical data are conflicting [55, 59]. Based on in vitro data, taxanes (traditionally used in the treatment of sporadic breast and ovarian cancer) may be of lesser benefit in hereditary cancers, at least for those lacking BRCA1 [60, 61]. Nevertheless, available clinical data do not support this view.
A novel class of drugs called PARP (Poly ADP-ribose polymerase) inhibitors was developed to target cells deficient in HR pathways [62, 63]. By inhibiting PARP, base-excision repair (BER) is impaired leading to the accumulation of unrepaired single-strand breaks, which during S phase lead to stalling and/or collapse of replication forks, and eventually degenerate into DSBs (Fig. 3). While normal cells have the capacity to compensate for PARP inhibitor-mediated loss of BER via HR, cells without the means to repair the damage by HR have to resort to error prone mechanisms (i.e., SSA, NHEJ, and MMEJ) to repair the DNA DSBs. These observations have led to the development of synthetic lethal approaches to target BRCA1 and BRCA2 deficient cancers [62, 63]. An extended phase I study [64] and two phase 2 clinical trials in BRCA1 and BRCA2 carriers have shown promise in the treatment of both metastatic breast [65] and ovarian cancers [66]. Further clinical trials are underway to evaluate whether PARP inhibitors act synergistically in combination with other chemotherapeutic agents such as cisplatin.
A caveat to cisplatin and PARP therapy is the cancer’s inevitable progression towards drug resistance. Studies have described the mechanism of drug resistance in BRCA1- and BRCA2-mutant tumors as intragenic deletions and secondary mutations induced by error-prone repair pathways such as NHEJ and SSA that restore an open-reading frame resulting in the expression of a functional BRCA1 or BRCA2 protein [51, 67–69]. It would seem that while loss of BRCA1 or BRCA2 is advantageous early in the progression of tumor development, the presence of BRCA1 or BRCA2 in its later stages may have little if any effect on tumor viability. In addition, it is thought that mutations are stochastic events and therefore the larger the tumor population the greater the likelihood that a revertant mutation will arise. Taken together, this would suggest that treatment with cisplatin or PARP inhibitors in the very early stages of cancer would have the greatest chance of eliminating disease, while treatment beyond a certain stage of development will likely end in relapse.
7 Targeting Sporadic Cancers Lacking Homologous Recombination
Do sporadic cancers harbor defects in HR and FA pathways, and if they do, would targeting them with cisplatin and PARP inhibitors be effective? A logical first step in answering this question would be to determine whether BRCA1 and BRCA2 are mutated in sporadic cases. About 20% of high grade serous ovarian carcinomas [70] and a similar percentage in triple-negative breast cancers (TNBC) [71] have germ line or somatic mutations in BRCA1/2; however, BRCA1/2 are also found to be down-regulated by other means such as epigenetic silencing [72–76] and transcriptional repression [77, 78]. In the latter example, the hypothesized role of EMSY amplification and BRCA2 suppression has been called into question as it appears that EMSY amplification in cancer cell lines is not associated with impaired HR function or increased sensitivity to cisplatin or PARP inhibition [79]. It has been previously suggested that the consequences of early BRCA1 deficiency dictate tumor lineage and phenotype [80] and that cell phenotype or “BRCAness” may be used as a surrogate marker for an underlying BRCA1 mutation [81]. Cells with a BRCA2 deficiency, however, seem not to follow a particular lineage, which is reflected by a lack of an association for BRCA2-associated tumors to a histopathologic phenotype that distinguishes them from sporadic cancers.
BRCA1 deficient breast cancers are characteristically “triple-negative” meaning they lack estrogen and progesterone receptors and do not over-express HER2 [82]; a tendency that could be explained by a haploinsufficiency of BRCA1 leading to a failure of luminal-progenitor cells to differentiate [83] thus creating a comparatively larger pool of basal-like stem cells that have the potential to give rise to a triple-negative phenotype [84, 85]. The unique biology of BRCA1 may underlie the phenotype seen in sporadic TNBCs providing the rational for clinical trials targeting TNBC with cisplatin [86] and PARP inhibitors [87]. However, promising results in a phase 2 trial for the novel therapeutic drug iniparib [87], a previously ascribed PARP inhibitor, failed to meet clinical outcomes in a subsequent phase 3 trial (http://en.sanofi-aventis.com/research_innovation/rd_key_figures/rd_key_figures.asp). Although the mechanism by which iniparib achieves its antitumor effects is unclear, its failure in the phase 3 trial may be due to the plasticity by which BRCA1 is down-regulated allowing tumor cells to more readily reactivate BRCA1 function leading to earlier resistance. Another possibility could be because TNBC is a convergent phenotype of a heterogeneous disease with only a small subgroup having an underlying BRCA1 defect. More reliable methods at predicting HR and FA function are being sought such as gene expression profiling [88] and radiation-induced RAD51 foci formation [89]; however, it is expected that next-generation sequencing technologies may ultimately prove to be the “gold standard” in the prediction of the ability to repair DNA. A genomic landscape not only characterizes all the mutations found within HR and FA related genes, but also describes the genetic signature of HR dysfunction. A comprehensive understanding of tumor biology however will rely on more than just genomic data. As a testament to the rapid advances made in sequencing technology and bioinformatics, a recent paper demonstrated the monumental task of analyzing 466 tumors across different platforms, integrating copy number variation, exomic, epigenomic, transcriptomic and proteomic data, providing a comprehensive understanding cancer drivers and drugable targets for the major breast cancer subtypes [90].
8 Conclusion
Studying the molecular pathways underlying hereditary breast and ovarian cancers has elucidated the processes that drive tumor progression, processes that are also common to sporadic cancers. Novel therapies are available to target cells defective in HR and FA pathways; however, determining which tumors have an underlying HR and FA defect is complex with no single method capable of providing a complete picture. As we begin to enter the genomic age, next-generation sequencing should allow full molecular characterization of cancer architecture and function including the tumor’s ability to respond to DNA damage—setting the stage for personalized medicine.
References
Jackson SP, Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461:1071–1078
Bennett CB, Lewis AL, Baldwin KK, Resnick MA (1993) Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci USA 90:5613–5617
Helleday T, Lo J, van Gent DC, Engelward BP (2007) DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst) 6:923–935
Rothkamm K, Krüger I, Thompson LH, Löbrich M (2003) Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 23:5706–5715
McVey M, Lee SE (2008) MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet 24:529–538
Narod SA, Foulkes WD (2004) BRCA1 and BRCA2: 1994 and beyond. Nat Rev Cancer 4:665–676
Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, Walter JC, Livingston DM (2006) The BRCA1/BARD1 heterodimer modulates Ran-dependent mitotic spindle assembly. Cell 127:539–552
Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, Gygi SP, Parvin JD (2004) BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol 24:8457–8466
Kumaraswamy E, Shiekhattar R (2007) Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol 27:6733–6741
Liu Z, Wu J, Yu X (2007) CCDC98 targets BRCA1 to DNA damage sites. Nat Struct Mol Biol 14:716–720
Kim H, Chen J, Yu X (2007) Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 316:1202–1205
Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA (2007) RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316:1198–1202
Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA et al (2001) BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105:149–160
Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ (2007) Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316:1194–1198
Yarden RI, Brody LC (1999) BRCA1 interacts with components of the histone deacetylase complex. Proc Natl Acad Sci USA 96:4983–4988
Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, Wang W, Kashanchi F, Shiekhattar R (2000) BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell 102:257–265
Lou Z, Minter-Dykhouse K, Chen J (2005) BRCA1 participates in DNA decatenation. Nat Struct Mol Biol 12:589–593
Venkitaraman AR (2002) Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108:171–182
Ashworth A, Lord CJ, Reis-Filho JS (2011) Genetic Interactions in Cancer Progression and Treatment. Cell 145:30–38
Foulkes WD (2008) Inherited susceptibility to common cancers. N Engl J Med 359:2143–2153
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W (1994) A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 266:66–71
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G (1995) Identification of the breast cancer susceptibility gene BRCA2. Nature 378:789–792
Joukov V, Chen J, Fox EA, Green JB, Livingston DM (2001) Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc Natl Acad Sci USA 98:12078–12083
Xia Y, Pao GM, Chen H-W, Verma IM, Hunter T (2003) Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J Biol Chem 278:5255–5263
Mallery DL, Vandenberg CJ, Hiom K (2002) Activation of the E3 ligase function of the BRCA1/BARD1 complex by polyubiquitin chains. EMBO J 21:6755–6762
Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T et al (2009) The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462:886–890
Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462:935–939
Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, Gage FH, Verma IM (2011) BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 477:179–184
Yu X (2006) BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev 20:1721–1726
Sato K, Hayami R, Wu W, Nishikawa T, Nishikawa H, Okuda Y, Ogata H, Fukuda M, Ohta T (2004) Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. J Biol Chem 279:30919–30922
Wu W, Nishikawa H, Hayami R, Sato K, Honda A, Aratani S, Nakajima T, Fukuda M, Ohta T (2007) BRCA1 ubiquitinates RPB8 in response to DNA damage. Cancer Res 67:951–958
Starita LM, Horwitz AA, Keogh M-C, Ishioka C, Parvin JD, Chiba N (2005) BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J Biol Chem 280:24498–24505
Eakin CM, Maccoss MJ, Finney GL, Klevit RE (2007) Estrogen receptor alpha is a putative substrate for the BRCA1 ubiquitin ligase. Proc Natl Acad Sci USA 104:5794–5799
Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T (2001) The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem 276:14537–14540
Ruffner H, Joazeiro CA, Hemmati D, Hunter T, Verma IM (2001) Cancer-predisposing mutations within the RING domain of BRCA1: loss of ubiquitin protein ligase activity and protection from radiation hypersensitivity. Proc Natl Acad Sci USA 98:5134–5139
Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng J-T, Jasin M, Baer R, Ludwig T (2008) E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci USA 105:20876–20881
Yu X (2003) The BRCT domain is a phospho-protein binding domain. Science 302:639–642
Manke IA, Lowery DM, Nguyen A, Yaffe MB (2003) BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science 302:636–639
Kim H, Huang J, Chen J (2007) CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat Struct Mol Biol 14:710–715
Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R (1998) The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem 273:25388–25392
Yun MH, Hiom K (2009) CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459:460–463
Sy SM-H, Huen MSY, Chen J (2009) PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci USA 106:7155–7160
Zhang F, Fan Q, Ren K, Andreassen PR (2009) PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res 7:1110–1118
Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (1997) RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem 272:31941–31944
Galkin VE, Esashi F, Yu X, Yang S, West SC, Egelman EH (2005) BRCA2 BRC motifs bind RAD51-DNA filaments. Proc Natl Acad Sci USA 102:8537–8542
Davies OR, Pellegrini L (2007) Interaction with the BRCA2 C terminus protects RAD51-DNA filaments from disassembly by BRC repeats. Nat Struct Mol Biol 14:475–483
Esashi F, Galkin VE, Yu X, Egelman EH, West SC (2007) Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol 14:468–474
Jensen RB, Carreira A, Kowalczykowski SC (2010) Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467:678–683
Thorslund T, McIlwraith MJ, Compton SA, Lekomtsev S, Petronczki M, Griffith JD, West SC (2010) The breast cancer tumor suppressor BRCA2 promotes the specific targeting of RAD51 to single-stranded DNA. Nat Struct Mol Biol 17:1263–1265
Liu J, Doty T, Gibson B, Heyer W-D (2010) Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol 17:1260–1262
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A (2008) Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451:1111–1115
D’Andrea AD (2010) Susceptibility pathways in Fanconi’s anemia and breast cancer. N Engl J Med 362:1909–1919
Berchuck A, Heron KA, Carney ME, Lancaster JM, Fraser EG, Vinson VL, Deffenbaugh AM, Miron A, Marks JR, Futreal PA et al (1998) Frequency of germline and somatic BRCA1 mutations in ovarian cancer. Clin Cancer Res 4:2433–2437
Osorio A, de la Hoya M, Rodríguez-López R, Martínez-Ramírez A, Cazorla A, Granizo JJ, Esteller M, Rivas C, Caldés T, Benítez J (2002) Loss of heterozygosity analysis at the BRCA loci in tumor samples from patients with familial breast cancer. Int J Cancer 99:305–309
Byrski T, Gronwald J, Huzarski T, Grzybowska E, Budryk M, Stawicka M, Mierzwa T, Szwiec M, Wiśniowski R, Siolek M et al (2010) Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol 28:375–379
Carey LA (2010) Targeted chemotherapy? Platinum in BRCA1-dysfunctional breast cancer. J Clin Oncol 28:361–363
Chappuis PO, Goffin J, Wong N, Perret C, Ghadirian P, Tonin PN, Foulkes WD (2002) A significant response to neoadjuvant chemotherapy in BRCA1/2 related breast cancer. J Med Genet 39:608–610
Brodie SG, Xu X, Qiao W, Li WM, Cao L, Deng CX (2001) Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene 20:7514–7523
Tassone P, Tagliaferri P, Perricelli A, Blotta S, Quaresima B, Martelli ML, Goel A, Barbieri V, Costanzo F, Boland CR et al (2003) BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br J Cancer 88:1285–1291
Lafarge S, Sylvain V, Ferrara M, Bignon YJ (2001) Inhibition of BRCA1 leads to increased chemoresistance to microtubule-interfering agents, an effect that involves the JNK pathway. Oncogene 20:6597–6606
Quinn JE, Kennedy RD, Mullan PB, Gilmore PM, Carty M, Johnston PG, Harkin DP (2003) BRCA1 functions as a differential modulator of chemotherapy-induced apoptosis. Cancer Res 63:6221–6228
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434:917–921
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434:913–917
Fong P, Boss D, Yap T, Tutt A, Wu P (2009) Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 361(2):123–134
Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK et al (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376:235–244
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N et al (2010) Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376:245–251
Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T (2008) Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 68:2581–2586
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, Karlan BY, Taniguchi T, Swisher EM (2011) Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol 29:3008–3015
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ et al (2008) Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 451:1116–1120
Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474:609–615
Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, Lanchbury JS, Stemke-Hale K, Hennessy BT, Arun BK et al (2011) Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res 17:1082–1089
Hilton JL, Geisler JP, Rathe JA, Hattermann-Zogg MA, DeYoung B, Buller RE (2002) Inactivation of BRCA1 and BRCA2 in ovarian cancer. J Natl Cancer Inst 94:1396–1406
Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA et al (2000) Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 92:564–569
Catteau A, Harris WH, Xu CF, Solomon E (1999) Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene 18:1957–1965
Baldwin RL, Nemeth E, Tran H, Shvartsman H, Cass I, Narod S, Karlan BY (2000) BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res 60:5329–5333
Geisler JP, Hatterman-Zogg MA, Rathe JA, Buller RE (2002) Frequency of BRCA1 dysfunction in ovarian cancer. J Natl Cancer Inst 94:61–67
Beger C, Pierce LN, Kruger M, Marcusson EG, Robbins JM, Welcsh P, Welch PJ, Welte K, King MC, Barber JR et al (2001) Identification of Id4 as a regulator of BRCA1 expression by using a ribozyme-library-based inverse genomics approach. Proc Natl Acad Sci USA 98:130–135
Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin S-F, Milner J, Brown LA, Hsu F, Gilks B et al (2003) EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 115:523–535
Wilkerson PM, Dedes KJ, Wetterskog D, MacKay A, Lambros MB, Mansour M, Frankum J, Lord CJ, Natrajan R, Ashworth A et al (2011) Functional characterization of EMSY gene amplification in human cancers. J Pathol 225:29–42
Foulkes WD (2004) BRCA1 functions as a breast stem cell regulator. J Med Genet 41:1–5
Turner N, Tutt A, Ashworth A (2004) Hallmarks of “BRCAness” in sporadic cancers. Nat Rev Cancer 4:814–819
Foulkes WD, Smith IE, Reis-Filho JS (2010) Triple-negative breast cancer. N Engl J Med 363:1938–1948
Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, Gilmore H, Tung N, Naber SP, Schnitt S et al (2011) Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell 8:149–163
Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, Asselin-Labat M-L, Gyorki DE, Ward T, Partanen A et al (2009) Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 15:907–913
Molyneux G, Geyer FC, Magnay F-A, McCarthy A, Kendrick H, Natrajan R, MacKay A, Grigoriadis A, Tutt A, Ashworth A et al (2010) BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7:403–417
Silver DP, Richardson AL, Eklund AC, Wang ZC, Szallasi Z, Li Q, Juul N, Leong CO, Calogrias D, Buraimoh A et al (2010) Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J Clin Oncol 28:1145–1153
O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, Koo IC, Sherman BM, Bradley C (2011) Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med 364:205–214
Konstantinopoulos PA, Spentzos D, Karlan BY, Taniguchi T, Fountzilas E, Francoeur N, Levine DA, Cannistra SA (2010) Gene expression profile of BRCAness that correlates with responsiveness to chemotherapy and with outcome in patients with epithelial ovarian cancer. J Clin Oncol 28:3555–3561
Graeser M, McCarthy A, Lord CJ, Savage K, Hills M, Salter J, Orr N, Parton M, Smith IE, Reis-Filho JS et al (2010) A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res 16:6159–6168
Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490(7418):61–70
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Shuen, A.Y., Reis-Filho, J.S., Foulkes, W.D. (2013). The Role of BRCA1 and BRCA2 in Anticancer Drug Therapy. In: Panasci, L., Aloyz, R., Alaoui-Jamali, M. (eds) Advances in DNA Repair in Cancer Therapy. Cancer Drug Discovery and Development, vol 72. Springer, New York, NY. https://doi.org/10.1007/978-1-4614-4741-2_6
Download citation
DOI: https://doi.org/10.1007/978-1-4614-4741-2_6
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4614-4740-5
Online ISBN: 978-1-4614-4741-2
eBook Packages: MedicineMedicine (R0)