
Abstract
Studies of the complex interactions in intermediary metabolism between neurons and glia in cell cultures and animal models are important in order to understand normal physiology, and the origin and development of psychiatric and neurological diseases in humans. This chapter will give examples of how cell culture studies using glutamatergic neurons from cerebellum, GABAergic neurons from cerebral cortex and astrocytes from both areas can be used to probe the importance of the tricarboxylic acid cycle, transamination reactions and glutamine synthesis. The middle cerebral artery occlusion model of ischemic stroke, the pentylene tetrazole model of epileptic seizures, the pilocarpine and kainic acid models of mesial temporal lobe epilepsy in rats and Genetic Absence Epilepsy Rats from Strasbourg (GAERS), will be used to describe what 13C nuclear magnetic resonance spectroscopy analysis of rat brain extracts can tell us about metabolism in the diseased brain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Nuclear magnetic resonance (NMR) spectroscopy is widely applied to study glial-neuronal interaction (Fig. 8.1). The necessity of this interaction arises from the fact that essential enzymes for glutamate and GABA synthesis are located in different cell types. Pyruvate carboxylase, the major anaplerotic enzyme in the brain is present exclusively in astrocytes, the same holds for glutamine synthetase whereas glutamate decarboxylase is mostly in GABAergic neurons (for details see: McKenna et al. 2011).
The nuclei most commonly used in NMR spectroscopy are: 1H, 31P, and 13C. Since 1H and 31P have a high natural abundance, the most common methods of study involve examining differences in the natural abundance of 1H or 31P spectra of samples (subjects) with various metabolic and genetic alterations. As indicated in Fig. 8.1, 13C has a natural abundance of 1.1%. This disadvantage normally makes detection difficult and 13C NMR spectroscopy is thus of limited use for studies on endogenous metabolites unless they occur in large amounts. However, the low natural abundance can be an advantage in that 13C-enriched precursors can be used for pathway mapping with little or no background interference from endogenous metabolites. For detailed description of homonuclear splitting patterns in biological molecules arising from 13C labeled precursors see Cerdan et al. (1990). Quantification of 13C spectra is thoroughly described by Badar-Goffer et al. (1990) and Sonnewald et al. (2011), and a detailed allocation of resonances in biological systems has been done by Barany et al. (1985), Fan (1996) and Fan and Lane (2008).

Information from Nuclear Magnetic Resonance Spectroscopy
Bachelard (1998) pioneered the application of 13C NMR spectroscopy to the analysis of metabolic pathways in guinea pig brain slices using [1-13C]glucose both in extracts and during superfusion, whereas Shank et al. (1993) were the first to apply 13C NMR spectroscopy to study glial-neuronal interaction in rat brain extracts. [1-13C]Glucose has also been used for in vivo monitoring of cerebral metabolism and how this metabolism can be modeled is described in an excellent paper by Henry et al. (2006). Compartmentation of glutamate metabolism has been studied in brain slices using 1H and 13C NMR spectroscopy (Kauppinen et al. 1994). Using the same system, Rae et al. (2006) showed that the metabolic response to ionotropic glutamate receptor perturbations in vitro is a sensitive discriminator of metabolic function. In primary cultures of cortical astrocytes, neurons and co-cultures thereof, glial-neuronal interactions have been studied using 13C labeled substrates since 1991 (Sonnewald et al. 1991). These pioneer studies have unequivocally demonstrated that 13C NMR spectroscopy is a unique and powerful tool for the analysis of brain metabolism and metabolic trafficking between different cerebral compartments and many excellent studies have been performed since.
Studies of the complex interactions in intermediary metabolism between neurons and glia in cell cultures, animal and human brains are important in order to understand normal physiology. Furthermore, progression of psychiatric and neurological diseases is accompanied by derangement in these processes. For example, it has been shown that glutamine synthetase is decreased in hippocampi of patients with mesial temporal lobe epilepsy, and 2-oxoglutarate dehydrogenase in Alzheimer’s disease patients (Eid et al. 2004; Gibson et al. 1998). NMR spectroscopy is an excellent technique to analyze disturbances in metabolic processes, and enzyme inhibitors have been used to study the importance of a number of pathways in cell culture experiments and in vivo. In rats trifluoroacetic acid which blocks the astrocytic TCA cycle, has been used to show that glutamate synthesis is predominantly neuronal (Hassel et al. 1997). The glutamine synthetase inhibitor methionine sulfoximine (MSO) blocks glutamine synthesis and reduces glutamate labeling by 50% demonstrating the importance of the glutamate – glutamine cycle (Garcia-Espinosa et al. 2004).
2 13C NMR Spectroscopy and Cell Cultures
Cells taken directly from the organism and subsequently grown for at least 24 h in vitro are considered to be primary cultures. They are obtained after an initial mechanical and/or enzymatic dissociation of the nervous tissue of neonatal or newborn rat, mouse, or chick. Due to the relative abundance of cells, NMR spectroscopy studies are often performed on cultures consisting of excitatory cerebellar granule cells or inhibitory GABAergic cerebral cortex neurons (Schousboe et al. 1989; Hertz et al. 1989a). The choice of these particular preparations of cultured neurons should be viewed in light of the fact that 90% of the synapses in the brain utilize either glutamate or GABA as the neurotransmitter. As counterparts for the neurons, astrocytes from the same brain region may be maintained in culture (Hertz et al. 1989b). Such cultures of neuronal or glial origin may be kept not only in plastic tissue culture flasks or Petri dishes, but, alternatively, microcarriers or basement membrane gel threads may be used as matrix (Westergaard et al. 1991b; Sonnewald et al. 1992; Alves et al. 1996). This technique allows transfer of the cultures directly to the NMR spectrometer. Additionally, Westergaard et al. (1991a, 1992) have developed a technique to culture neurons on top of preformed layers of astrocytes, so called co-cultures, that have proven useful in studying neuronal-glial interactions. Using cell cultures, it is possible to distinguish intra- and extracellular metabolites, and in vivo spectra can be understood on the basis of the characteristics of the separate cell culture types contained in the tissue.
2.1 [1-13C]Glucose and [2-13C]Acetate
Both astrocytes and neurons, the main players in metabolism, can be kept in mono- and co-culture as outlined above. Using cell cultures it is possible to monitor intra- and extracellular metabolites separately. By incubating astrocytes and GABAergic neuronal cultures either alone or as co-cultures with [1-13C]glucose (for labeling pattern see Fig. 8.2 and Sect. 8.2.1) it could be shown that cortical neurons label GABA efficiently and that astrocytes not only export glutamine, but also citrate to the medium (Sonnewald et al. 1991, 1993b). [2-13C]Acetate (for labeling pattern see Fig. 8.3) did not label GABA in mono-cultures, but did so in co-cultures (Sonnewald et al. 1993b). This labeling could be decreased by MSO thereby demonstrating that glutamine is a precursor for GABA (Sonnewald et al. 1993b). Incubation of cortical neurons with [1-13C]glucose has also been used to show that GABA is synthesized in cerebellar neurons which are thought to be largely glutamatergic (Sonnewald et al. 2004).

Labeling patterns of amino acids originating from metabolism of [1-13C]glucose. Black circles indicate 13C labeled atoms

Labeling patterns of amino acids originating from metabolism of [2-13C]acetate. Black circles indicate 13C labeled atoms
2.2 [U-13C]Glucose and [U-13C]Lactate
2.2.1 Metabolism in Neurons
The advantage of uniformly labeled precursors is that detection of incorporation of label into metabolites is unambiguous due to 13C-13C spin-spin coupling patterns (Cerdan et al. 1990). The probability of having two 13C atoms in the same molecule from the 1.1% naturally abundant 13C is 0.01%, well below the detection limit for most naturally occurring substances. Uniformly labeled glucose ([U-13C]glucose) and lactate ([U-13C]lactate) have been used to probe neurotransmitter synthesis in both cerebellar and cerebral cortical neurons (Fig. 8.4) (Waagepetersen et al. 2004, 2005a; Bak et al. 2006). It is well known that glutamate exists in a vesicular as well as a cytoplasmic pool and is metabolically closely related to the TCA cycle. Glutamate released during neuronal activity is to a large extent accumulated by astrocytes surrounding the synapse. A compensatory flux from astrocytes to neurons of suitable precursors is obligatory as neurons are incapable of performing a net synthesis of glutamate from glucose (for references see McKenna et al. 2011). Glutamine appears to play a major role in this context. Employing cultured cerebellar granule cells, as a model system for glutamatergic neurons, details of the biosynthetic machinery have been investigated during control (Olstad et al. 2007) and depolarizing conditions inducing vesicular release (Waagepetersen et al. 2005a). [U-13C]Glucose, [U-13C]Glutamine or [U-13C]Glutamate were used as labeled precursors for monitoring metabolic pathways by NMR spectroscopy and/or mass spectrometry (MS), a method complementary to NMR. Surprisingly, the intracellular pool of glutamate was dependent on reuptake of released glutamate in cerebellar granule neurons, disputing the importance of astrocytic glutamate uptake for controlling glutamate levels in the synapse in this brain region.

NMR spectra of cell extracts from cultured cerebellar (top) and neocortical neurons (bottom) incubated in medium containing 12 mM [U-13C]glucose for 4 h after change of medium. Abbreviations: 1, alanine C-3; 2, lactate C-3; 3, GABA C-3; 4, glutamate C-3; 5, glutamate C-4; 6, GABA C-2; 7, aspartate C-3; 8, GABA C-4 (from Waagepetersen et al. 2005b)
2.2.2 Metabolism in Astrocytes
Pyruvate carboxylation in brain has been linked to the glial specific enzyme, pyruvate carboxylase (PC). However, also malic enzyme is in principle capable of pyruvate carboxylation and this enzyme is present both in neurons and astrocytes. In order to probe in which cell type carboxylation is taking place cerebellar neurons and astrocytes in mono-culture were incubated with [U-13C]glucose plus [U-13C]lactate (Waagepetersen et al. 2001). These labeled precursors can be metabolized into [U-13C]pyruvate and [1,2-13C]acetyl CoA (Fig. 8.5). In the first turn of the TCA cycle, an unlabeled molecule of oxaloacetate condenses with [1,2-13C]acetyl CoA leading to formation of [4,5-13C]glutamate. In astrocytes [4,5-13C]glutamate is converted to [4,5-13C]glutamine, and in GABAergic neurons to [1,2-13C]GABA. [1,2-13C]aspartate and [3,4-13C]aspartate are formed in equal amounts in the first turn due to the symmetrical succinate molecule. Label can also enter the TCA cycle via carboxylation of [U-13C]pyruvate (Fig. 8.5, only labeled acetyl CoA is shown) and thus lead to formation of [1,2,3-13C]oxaloacetate and, when acetyl CoA is labeled, [2,3,4,5-13C]glutamate, [U-13C]GABA and, in astrocytes, [2,3,4,5-13C]glutamine as well. It should be mentioned that the isotopomers generated from pyruvate carboxylation can also be formed in the 3rd turn of the TCA cycle. Therefore, Waagepetersen et al. (2001) used the succinate dehydrogenase inhibitor 3-nitrpropionic acid to block cycling of metabolites. Using this inhibitor, it could be shown that only astrocytes were capable of carboxylation (Waagepetersen et al. 2001). A overview of NMR experiments exploring pyruvate carboxylation is given in Sonnewald and Rae (2010). Also co-cultures of cerebellar neurons and astrocytes have been incubated with uniformly labeled glucose and lactate, showing that metabolism of pyruvate, the precursor for lactate, alanine and acetyl-CoA is highly compartmentalized (Bak et al. 2007).

Labeling patterns originating from metabolism of [U-13C]glucose and [U-13C]lactate. Black circles indicate 13C labeled atoms. Top: Metabolism of [U-13C]pyruvate involving the pyruvate dehydrogenase (PDH) complex. Bottom: [U-13C]pyruvate metabolism via the pyruvate carboxylation pathway
2.3 [U-13C]Glutamate and [3-13C]Glutamate
Glutamate is the most abundant neurotransmitter and derangement in glutamate homeostasis/neurotransmission is linked to a number of neurological disorders like epilepsy, stroke, amylotrophic lateral sclerosis, and Alzheimer’s disease. It is therefore important to study glutamate metabolism under normal and pathological conditions. Cell cultures can be incubated with [U-13C]glutamate (Fig. 8.6) and the distribution of label from [U-13C]glutamate can be seen in Fig. 8.7. This approach makes it possible to analyze to which extent [U-13C]glutamate is converted directly to glutamine or GABA (depending on cell type), or passed through the TCA cycle. Direct conversion will lead to [U-13C]-2-oxoglutarate, [U-13C]GABA (in GABAergic neurons) and [U-13C]glutamine (in astrocytes). [U-13C]-2-oxoglutarat can enter the TCA cycle and lead to the formation of uniformly labeled ([U-13C]) malate and oxaloacetate which in turn can give rise to [U-13C]aspartate and lactate (Fig. 8.7). After one turn of the TCA cycle, the C-4 and C-5 positions in 2-oxoglutarate will be occupied by 12C atoms from unlabeled acetyl-CoA. Glutamate and glutamine derived from this precursor will have 12C atoms in the C-4 and C-5 positions, and GABA will have 12C atoms in the C-1 and C-2 positions (Figs. 8.7 and 8.8).

NMR spectrum of cell extract from cultured cerebellar neurons incubated in medium containing 0.25 mM [U-13C]glutamate. Abbreviations: 1, glutamate C-3; 2, glutamate C-4; 3, aspartate C-3; 4, aspartate C-2; 5, glutamate C-2

Labeling patterns of amino acids originating from metabolism of [U-13C]glutamate. Black circles indicate 13C labeled atoms. Abbreviations: Asp, aspartate; Gln, glutamine; Glu, glutamate; Lac, lactate; Mal, malate; OAA, oxaloacetate

Labeling patterns of amino acids originating from metabolism of [1,2-13C]acetate. Black circles indicate 13C labeled atoms
Cell culture studies have demonstrated that [U-13C]glutamate is metabolized extensively in astrocytes and neurons (Qu et al. 2000b, 2001a, b; Sonnewald et al. 1993a). Astrocytes metabolized [U-13C]glutamate to uniformly labeled aspartate, glutamine, lactate and alanine which were exported to the medium (Sonnewald et al. 1993a). In cerebral cortical neurons [U-13C]glutamate did not label lactate, but in cerebellar granule neurons some labeling of lactate was detected (Sonnewald et al. 1996). However, it cannot be totally excluded that this lactate was formed by the small number of astrocytes present in this type of preparation. Glutamate derived from the first turn of the TCA cycle, i.e. [1,2,3-13C]glutamate (Fig. 8.7) is present in cerebellar and cortical astrocytes and neurons (Sonnewald et al. 1996). [1,2-13C]Glutamate which is derived from the second turn of the cycle was only detected in granule neurons (Fig. 8.6) showing that cerebellar neurons have a higher TCA cycle turnover than the other cell types investigated (Sonnewald et al. 1996). McKenna et al. (1996) showed that metabolism of [U-13C]glutamate in astrocytes is concentration dependent. At [U-13C]glutamate levels above 0.2 mM more glutamate entered the TCA cycle after conversion to 2-oxoglutarate than was converted directly to glutamine. As mentioned above, using enzyme inhibitors the significance of certain enzymes for metabolic processes can be probed. In astrocytes it could be shown that the transaminase inhibitor aminooxyacetic acid not only abolished the appearance of aspartate, but also that of the glutamate-isotopomer, [1,2,3-13C]glutamate, derived from the TCA cycle (Westergaard et al. 1996). In this way it could be demonstrated that transamination is necessary for the conversion of 2-oxoglutarate to glutamate, but not for entry of glutamate into the TCA cycle via 2-oxoglutarate since lactate labeling was only slightly decreased. The succinate dehydrogenase inhibitor 3-nitropropionic acid was used to abolish the labeling of aspartate and [1,2,3-13C]glutamate from [U-13C]glutamate by inhibiting TCA cycling in cerebellar granule neurons and cortical astrocytes (Sonnewald et al. 1996; Bakken et al. 1997). As a result lactate was no longer labeled, indicating that malate or oxaloacetate derived from the TCA cycle directly are the precursors, rather than citrate, which could give rise to lactate via the citrate lyase pathway. Furthermore, it was shown that cerebellar neurons were affected by 3-nitropropionic acid at lower concentrations than astrocytes. These findings have been replicated in vivo in the rat brain (Hassel and Sonnewald 1995a).
Pyruvate recycling is a pathway for complete oxidation of glutamate. The cellular location and the physiological significance of such recycling have been debated during the last decade (Cerdan et al. 1990; Hassel and Sonnewald 1995b). Using [3-13C]glutamate, pyruvate recycling was probed in neurons, astrocytes and co-cultures of astrocytes and neurons. With the sensitivity at that time, recycling was only detected in astrocytes in mono-culture (Waagepetersen et al. 2001). In a recent study, using [U-13C]glutamate analysis of the labeling pattern of the C-4 position of glutamate and the number of 13C atoms per glutamate molecule showed that pyruvate recycling is clearly present both in astrocytes and neurons (Olstad et al. 2007).
2.4 [3-13C]Malate
Malate, specifically labeled with 13C on C-3, was synthesized by chemical means and used to study malate metabolism in cortical astrocytes (Alves et al. 2000). The consumption of malate was only 0.26 μmol/mg of protein, approximately 25-fold lower than the consumption of glucose. [3-13C]Malate in combination with glucose as well as [3-13C]malate alone caused labeling of lactate, glutamine and fumarate which were the three major metabolites released to the medium. Very low and similar levels of isotopic enrichment were detected on C-2 and C-3 of lactate. Glutamine was labeled on C-2 and C-3 to a similar extent as well and labeling on C-4 of glutamine was only detected when glucose was not added. These labeling patterns clearly show that cytosolic malic enzyme is not active in primary astrocytes and demonstrate the occurrence of pyruvate recycling (Alves et al. 2000).
3 13C NMR Spectroscopy and Animal Models
3.1 Administration of Labeled Substrates and Factors Relevant to Label Incorporation
In animal studies there are several ways of obtaining 13C enrichment in the compounds of interest. The appropriate approach depends on the hypothesis under investigation.
For in vivo 13C NMR spectroscopy studies of metabolic fluxes, a continuous infusion until steady state is required (Sibson et al. 1997, 2001; Cerdan et al. 1990; Henry et al. 2003, for review see Henry et al. 2006). In these experiments plasma glucose concentration and enrichment need to be monitored in parallel with spectra acquisition in order to obtain an input function for the metabolic model used. In some instance this may pose a problem. For instance in small rats and mice obtaining serial blood samples have to be performed in a separate set of animals outside the magnet because the blood volume is very small. Furthermore, lengthy steady state studies may be problematic in many animal models of disease as the condition may progress with time, for instance in acute stroke. Also animals with certain conditions such as chronic epilepsy, may not be fit for lengthy in vivo scan protocols. The increased glucose concentration, which accompanies a steady state infusion scheme, may be deleterious in for example stroke and epilepsy models. The effect of hyperglycemia on the underlying condition rather than the primary condition per se is then studied. However, dynamic studies can also be performed in awake animals (Oz et al. 2004). This method requires more animals to be sacrificed, but sensitivity is excellent as 13C NMR spectroscopy analysis of all time points will be performed in tissue extracts. Furthermore, small specific brain regions can be investigated.
There are many instances in which a short intra venous infusion (Serres et al. 2003) or intra venous bolus injection (Haberg et al. 2001) are feasible. Short intervals of label administration lead to short period of increased plasma concentration of the substrate given. This may reduce deleterious and unwanted effects of the labeled substrate. At the same time this “snap shot” approach enables one to differentiate between metabolic processes in different brain regions and due to different pathological processes even though steady state has not been obtained (Haberg et al. 2001, 2007). Indeed, short infusion schemes have been able to elucidate aspects of brain metabolism, not detectable with steady state infusion. Using direct intracarotid bolus injection biphasic labeling in [4-13C]glutamate was found combined with rapid decline in plasma glucose levels. These results suggested that the initial glutamate labeling came from labeled plasma glucose, whereas later glutamate labeling arose from glycogen synthesized in astrocytes from the labeled glucose (Griffin et al. 1999). It is also possible to administer the labeled substrate subcutaneously (Hassel et al. 1995) or intraperitoneally (Shank et al. 1993; Kondziella et al. 2003). These methods may be less reliable than intravenous approaches because variations in local blood flow in the area of injection will influence uptake of the substrate into the circulation. The pro with subcutanouse and intraperitoneal administration is that no anesthesia is required, and that they can be performed by most researchers. Actually results from intraperitoneal injection share some characteristics with that of an intravenous bolus injection, as there is a trend for biphasic labeling of glutamate (Shank et al. 1993) similar to that reported by Griffin et al. (1999).
For studies aimed at investigating synthesis and metabolism of compounds with a very slow conversion rate it is even possible to introduce 13C label through 13C enriched chow (Karelson et al. 2003) or water (Choi and Gruetter 2003). Animal chow can be enriched with 13C labeled glucose, or with 13C enriched biomass (Karelson et al. 2003).
There are also other factors that will influence the results when giving 13C labeled precursors to animals. One important factor is systemic metabolism of labeled substrates, i.e. metabolism of the labeled compound outside the brain. This will give rise to newly labeled compounds that may be substrates for brain metabolism. In the liver label originating from glucose and/or acetate can be incorporated into glucose via gluconeogenesis and thus leads to labeled lactate and glucose. These newly formed compounds can have 13C label in positions other than the original position. Furthermore, the Cori and alanine cycles as well as pyruvate recycling and futile cycling may lead to non-symmetrical scrambling of label in glucose. In plasma, 13C from labeled glucose, acetate and lactate is detected in new positions in glucose 15 min after bolus injection in fasted rats (Haberg et al. 2001, 1998b, a; Qu et al. 2000a; Serres et al. 2003). Label was not detectable in plasma amino acids. Brain amino acids, on the other hand, were labeled. This is due to the fact that conversion of glucose or acetate into amino acid in the brain proceeds at a rate several times higher than in all other organs (Gaitonde et al. 1965; Vrba 1962). Factors like type (continuous versus bolus) and duration of infusion, the metabolic state of the animal (fasted versus non-fasted) and drugs administered will influence the extra-cerebral metabolism of the labeled substrates. Plasma samples should be obtained and analyzed in order to evaluate whether there has been synthesis of significant amounts of newly labeled substrates, which could interfere with the analysis of labeling patterns in the brain. The importance of this phenomenon is illustrated by the results obtained from plasma extract analyses from rats receiving [U-13C]lactate. Fifteen minutes after an intravenous bolus injection of [U-13C]lactate there were approximately equal amounts of [1,2,3-13C]- and [1,2-13C]glucose indicating gluconeogenesis from [U-13C]lactate. It was thus possible that the labeling seen in the cerebral amino acids originated from labeled glucose, not [U-13C]lactate. However, the presence of significantly more label in [U-13C]alanine than in [2,3-13C]alanine demonstrated that [U-13C]lactate did indeed cross the blood-brain barrier, and was metabolized further in the brain (Qu et al. 2000a).
Also anesthesia has important effects on 13C labeling of metabolic intermediates in the brain which need to be taken into account. Surgical procedures demand that the animal is under proper anesthesia. Furthermore, most in vivo NMR spectroscopy experiments using animals will require anesthesia. However, anesthetic agents reduce cerebral glucose metabolism (Cremer and Lucas 1971) and thus the incorporation of 13C label from glucose into amino acids (Shank et al. 1993; Hyder et al. 2003). Labeling from 13C acetate is not affected, but astrocytic acetate oxidation is reduced. Moreover, anesthesia affects cortical and subcortical brain regions to different degrees (Hansen et al. 1988; Hoffman et al. 1991; Haberg et al. 2001). It should also be noted that anesthetic compounds influence neurotransmitter release (Haberg et al. 2001), and that some anesthetic agents interact with neurotransmitter receptors (Freo and Ori 2004; Whitehead et al. 2004). In addition, different anesthetic agents depress the cerebral metabolism to varying degrees (Hyder et al. 2003). This implies that the metabolic changes induced by anesthesia will influence the 13C labeling patterns, and must be considered when interpreting 13C NMR spectroscopy results.
3.2 13C Labeled Glucose and Acetate
13C labeled glucose has been used widely in 13C MR spectroscopy studies since it is the main substrate in the adult brain. [1-13C]glucose is the most frequently administrated principally because it is the least expensive. However use of [1,6-13C]glucose will increase the enrichment in the metabolic intermediates with a factor of two, and thus increase the sensitivity. [2-13C]glucose has also been used to explore the importance of astrocytic pyruvate carboxylation and TCA cycle activity (Sibson et al. 2001; Mason et al. 2007). [U-13C]glucose is another alternative. It has been shown that glucose enters neurons and astrocytes equally (Nehlig et al. 2004). Still glucose is thought to be metabolized more in the neuronal TCA cycle (Fig. 8.9) (Minchin and Beart 1975b, a; Sonnewald et al. 1991). Using 13C NMR spectroscopy it has been calculated that 65% of acetyl CoA derived from glucose is metabolized in the neuronal TCA cycle in vivo (Hassel et al. 1995; Qu et al. 2000a). In the astrocyte glucose may be converted to lactate without entering the TCA cycle and then shuttled to neurons (Serres et al. 2004) (Fig. 8.10).

NMR spectrum of rat brain extract. Rats were injected with [1-13C]glucose (3 mM/kg) and [1,2-13C]acetate (3 mM/kg) and decapitated 15 min later. Abbreviations: ala, Alanine; Asp, aspartate; Gln, glutamine; Glu, glutamate; Lac, lactate; NAA, N-acetylaspartate

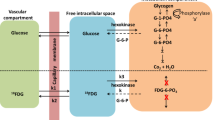
Metabolic pathways for acetate. The neuron depicted is a non-existing neuron, which releases both neurotransmitters glutamate and GABA. Acetate is only taken up by astrocytes due to the presence of a specific acetate transporter on these cells. In the astrocytes acetate is converted to acetyl CoA by acetyl CoA synthetase and can subsequently enter the tricarboxylic acid (TCA) cycle. α-Ketoglutarate can leave the TCA cycle and can be transaminated by aspartate, alanine or branched chain aminotransferases to glutamate. In the astrocytes glutamate is rapidly converted to glutamine by the action of glutamine synthetase (GS). GS is found only in astrocytes and oligodendrocytes. Glutamine is released from astrocytes and then taken up by high affinity transporters present on neurons. Inside the neuron glutamine is converted to glutamate by phosphate activated glutaminase (PAG). Astrocytic glutamine is used specifically to replenish the glutamate neurotransmitter pool in glutamatergic neurons. Glutamate released as neurotransmitter is predominantly taken up by astrocytes. In the astrocyte the neurotransmitter glutamate can either be converted directly to glutamine by GS, or enter the TCA cycle via transamination or oxidative deamination by glutamate dehydrogenase. As schematically illustrated in the neuron, glutamate derived from astrocytic glutamine can be converted to GABA by the action of glutamate decarboxylase (GAD). This enzymatic reaction is considered to take place mostly in GABAergic neurons. GABA released as neurotransmitter is mainly taken up by the GABAergic neuron, but astrocytes possess the capacity to take up and metabolize GABA
Acetate labeled in the C-2 or C-1,2 positions are also frequently used. Acetate metabolism is considered to be located to astrocytes. Acetate is selectively taken up by astrocytes by a specialized transport system, which is absent or less active in neurons (Waniewski and Martin 1998).
By simultaneous injection of [1-13C]glucose and [1,2-13C]acetate followed by 13C NMR spectroscopic analysis of brain extracts, information about both neuronal and astrocytic metabolism can be obtained in the same animal (Taylor et al. 1996) (for simplified overview of the metabolic pathways for glucose and acetate see Figs. 8.10 and 8.12). Injection of 13C labeled glucose and acetate leads to efficient labeling of many metabolites as can be seen in the brain extract spectrum in Fig. 8.9. Label from [1-13C]glucose can be quantified by analyzing the singlet peaks in the different compounds. The doublets seen in the spectrum are mostly derived from [1,2-13C]acetate and thus astrocytic metabolism. Simplified schemes of the metabolic pathways of these two substrates are shown in Figs. 8.2 and 8.8. Via glycolysis [1-13C]glucose is converted to [3-13C]pyruvate which can be converted to [3-13C]alanine or [3-13C]lactate. Alternatively, pyruvate can enter the TCA cycle via [2-13C]acetyl CoA. This will lead to the formation of [4-13C]glutamate and glutamine or [2-13C]GABA. Furthermore, pyruvate can be carboxylated by pyruvate carboxylase (PC) to oxaloacetate which will lead to the synthesis of [2-13C]glutamate and glutamine or [4-13C]GABA. PC activity is illustrated in Fig. 8.5 for [U-13C]glucose (which is analogous to [1-13C]glucose). [1,2-13C]acetate can also be converted to acetyl CoA, however, the product, [1,2-13C]acetyl CoA, has two 13C atoms (Fig. 8.8), resulting in doublet formation (Fig. 8.9). Thus [4,5-13C]glutamate and glutamine or [1,2-13C]GABA are formed. Since both acetyl CoA and oxaloacetate can be either labeled or unlabeled, the number of possible isotopomers of the metabolites derived from the TCA cycle is large. Only compounds derived from the first turn are represented in Figs. 8.2 and 8.8. By comparing the doublets with singlets, it can be seen in Fig. 8.9 that glutamine is labeled more from [1,2-13C]acetate (doublet) than from [1-13C]glucose (singlet), the opposite is the case for glutamate and GABA. Lactate, alanine and N-acetylaspartate (NAA) are mainly labeled from glucose.

Metabolic pathways for lactate. The neuron depicted is a non-existing neuron, which releases both neurotransmitters glutamate and GABA. Lactate can be taken up by both neurons and astrocytes, but there is an uneven distribution of lactate dehydrogenase (LDH) isoenzymes between the two cell types. Neurons have only the LDH1 isoform, which favors the production of pyruvate, while astrocytes have the LDH5 isoform that drives the reaction towards lactate formation and the LDH1 isoform. Consequently there is limited synthesis of pyruvate from lactate in astrocytes. In neurons, however, lactate is readily converted to pyruvate and subsequently acetyl CoA for entry into the tricarboxylic acid (TCA) cycle. α-Ketoglutarate leaves the neuronal TCA cycle and can be transaminated by aspartate, alanine or branched chain aminotransferases to glutamate. It should be noted that glutamate and GABA de novo synthesis takes place only in astrocytes because the anaplerotic enzyme in brain pyruvate carboxylse (PC) is located in astrocytes (see below). In GABAergic neurons glutamate is converted to GABA by glutamate decarboxylase activity (GAD). Neurotransmitter glutamate or GABA can subsequently be released into the synaptic cleft. Glutamate is predominantly taken up by astrocytes, and GABA by GABAergic neurons. In the astrocyte the neurotransmitter glutamate can either be converted directly to glutamine by glutamine synthetase (GS), or enter the TCA cycle via transamination or oxidative deamination by glutamate dehydrogenase. GABA is mainly taken up by the GABAergic neuron, but astrocytes possess the capacity to take up and metabolize GABA. GABA is metabolized by GABA aminotransferase and succinic-semialdehyde dehydrogenase, which allow four of the five carbon atoms from α-ketoglutarate to re-enter the TCA cycle as succinate. In astrocytes pyruvate from lactate can enter the TCA cycle via either PDH or PC activity. The entry of 13C labeled glucose into the TCA cycle via PC activity gives rise to distinct labeling patterns in glutamate, glutamine and GABA, which can be differentiated from the labeling patterns found when 13C labeled glucose enters the TCA cycle via PDH activity. Hence, in astrocytes either acetyl CoA or oxaloacetate from pyruvate is incorporated into the astrocytic TCA cycle. After additional enzymatic steps α-ketoglutarate is formed and can leave the TCA cycle and be transaminated to glutamate. In the astrocytes glutamate is rapidly converted to glutamine by the action of GS. GS is found only in astrocytes and oligodendrocytes. Glutamine is released from astrocytes and then taken up by high affinity transporters present on neurons. Inside the neuron glutamine is converted to glutamate by phosphate activated glutaminase (PAG). In GABAergic neurons GAD converts glutamate originating from glutamine to GABA

Metabolic pathways for glucose. Glucose is taken up equally by astrocytes and neurons, but at the level of acetyl CoA more glucose enters the neuronal than the astrocytic TCA cycle. In neurons pyruvate from glucose can only be incorporated into the TCA cycle via pyruvate dehydrogenase (PDH) activity. PDH converts pyruvate to Acetyl CoA which condenses with oxaloacetate and produces citrate. α-Ketoglutarate is formed after two additional enzymatic steps in the TCA cycle. α-Ketoglutarate can leave the neuronal TCA cycle for subsequent transamination with aspartate, alanine or branched chain aminotransferases to form glutamate. It should be noted that glutamate and GABA de novo synthesis takes place only in astrocytes sinc the anaplerotic enzyme in brain, pyruvate carboxylase (PC) is located in astrocytes (see below). In GABAergic neurons glutamate is converted to GABA by glutamate decarboxylase activity (GAD). Neurotransmitter glutamate or GABA can subsequently be released into the synaptic cleft. Glutamate is predominantly taken up by astrocytes, and GABA by GABAergic neurons. In the astrocyte the neurotransmitter glutamate can either be converted directly to glutamine by glutamine synthetase (GS), or enter the TCA cycle via transamination or oxidative deamination by glutamate dehydrogenase. GABA is mainly taken up by the GABAergic neuron, but astrocytes possess the capacity to take up and metabolize GABA. GABA is metabolized by GABA aminotransferase and succinic-semialdehyde dehydrogenase, which allows four of the five carbon atoms from α-ketoglutarate to re-enter the TCA cycle as succinate.In the astrocyte pyruvate from glucose can enter the TCA cycle via either PDH or PC activity. The entry of 13C labeled glucose into the TCA cycle via PC activity gives rise to distinct labeling patterns in glutamate, glutamine and GABA, which can be differentiated from the labeling patterns found when 13C labeled glucose enters the TCA cycle via PDH activity. Hence, in astrocytes either acetyl CoA or oxaloacetate from pyruvate is incorporated into the astrocytic TCA cycle. After additional enzymatic steps α-ketoglutarate is formed and can leave the TCA cycle and be transaminated to glutamate. In the astrocytes glutamate is rapidly converted to glutamine by the action of GS. GS is found only in astrocytes and oligodendrocytes. Glutamine is released from astrocytes and then taken up by high affinity transporters present on neurons. Inside the neuron glutamine is converted to glutamate by phosphate activated glutaminase (PAG). Astrocytic glutamine is used specifically to replenish the glutamate neurotransmitter pool in glutamatergic neurons. Glutamine is also taken up by GABAergic neurons, converted to glutamate via PAG and then to GABA by GAD
3.3 [U-13C]Lactate
Under normal resting conditions lactate is present in the blood at a concentration of ∼1 mM, and it was thought that the brain does not extract appreciable amounts of this potential substrate (Harada et al. 1992). However, it is now widely accepted that lactate is a fuel for neuronal metabolism and that astrocytes play a critical role in the regulation of this meatbolism (Waagepetersen et al. 2009). The astrocyte-neuron lactate shuttle hypothesis (ANLSH) describes the role of astrocytes in shuttling lactate from astrocytes to neurons in response to brain activation (Magistretti et al. 1999; Pellerin et al. 2007). The exact nature of this shuttle has yet to be fully elucidated (Waagepetersen et al. 2009), but an extension of the ANLSH was published by Cerdán et al. (2006) proposing that subcellular compartmentation of pyruvate allows neurons and astrocytes to select between glucose and lactate as alternative substrates, depending on their relative extracellular concentration and the operation of a redox switch. In neuronal cell cultures it has been shown that oxidative metabolism of lactate was more intense than that of glucose, both under resting and depolarizing conditions (Peng et al. 1994). Lactate was also shown to be an important energy fuel in cultured GABAergic neurons, and it extensively labeled amino acids synthesized from TCA cycle intermediates (Waagepetersen et al. 1998).
Several studies have appeared showing that 13C labeled lactate is a useful tool for mapping neuronal metabolism (Fig. 8.11T) (Serres et al. 2003; Qu et al. 2000a; Tyson et al. 2003). In a study by Qu et al. (2000a) fully awake rats were injected intravenously with [U-13C]lactate or [U-13C]glucose followed 15 min later by decapitation. For labeling patterns see Fig. 8.5. Incorporation of label from [U-13C]lactate was seen mainly in glutamate, GABA, glutamine, aspartate, alanine, and lactate. When [U-13C]glucose was the precursor, label incorporation was similar to that observed from [U-13C]lactate, but much larger. However, contribution from pyruvate carboxylase (astrocytic enzyme) was much more pronounced after the glucose injection. This indicated that relatively more pyruvate from lactate than glucose was metabolized in neurons. Surprisingly, the same amount of lactate was synthesized via the TCA cycle in both groups, indicating transfer of neurotransmitters from the neuronal to the astrocytic compartment as Serres et al. (2003) and Hassel et al. (1995) have shown that lactate from the TCA cycle is synthesized primarily in astrocytes. Serres et al. (2003) could come to this conclusion by analyzing 13C enrichment in lactate and Hassel et al. (1995) showed formation of lactate via the astrocytic TCA cycle by documenting lactate formation from [2-13C]acetate.
4 13C NMR Spectroscopic Analyses of Neuronal-Glial Interactions in Focal Cerebral Ischemia
4.1 Focal Ischemia
Focal cerebral ischemia or thrombo-embolic occlusion of an intracerebral artery is the cause of approximately 75% of all strokes. In focal cerebral ischemia excessive stimulation of glutamate receptors, glutamate excitotoxicity, is recognized as an important mediator of neuronal death (Rothman and Olney 1986; Choi 1994), whereas increased GABA receptor stimulation in the mature brain is considered neuroprotective (Schwartz-Bloom and Sah 2001). Astrocytes are inextricably involved in the synthesis of both neurotransmitter glutamate and GABA, and any disturbance in the metabolic interaction between astrocytes and neurons may have important implications for the final outcome after focal cerebral ischemia. Indeed, increased astrocytic oxidation of amino acids, glutamate, glutamine and GABA, combined with elevated pyruvate recycling was demonstrated in a model of combined focal and global ischemia in the hemisphere ipsilateral to the focal insult (Pascual et al. 1998). Also glutamate can be released from astrocytes in ischemic conditions, contributing to the excitotoxic damage (for review see Rossi et al. 2007).
In order to study the dynamic changes in the metabolic interactions between astrocytes and neurons during focal cerebral ischemia, we combined in vivo injection of labeled [1-13C]glucose and [1,2-13C]acetate in rats subjected to intraluminal occlusion of the middle cerebral artery (MCAO) (Longa et al. 1989). In this model of focal ischemia the cerebral blood flow is most severely reduced in the lateral caudoputamen and lower part of the frontoparietal cortex (Longa et al. 1989; Muller et al. 1995), referred to as the ischemic core. In the upper frontoparietal cortex, cerebral blood flow is moderately reduced (Memezawa et al. 1992; Muller et al. 1995), and this area is considered to represent the penumbra. Reperfusion and/or neuroprotective drug treatment is aimed mainly at salvaging the penumbra. However, it should be noted that focal cerebral ischemia is a dynamic process where the ischemic tissue gradually progresses from reversibly to irreversibly damaged. Moreover, the time-frame for this deterioration is controversial and depends on factors like temperature, reperfusion and individual variations in collateral blood supply. At different time intervals after onset of ischemia rats with permanent or transient MCAO were decapitated and their brains snap frozen. Areas considered representing the ischemic core and penumbra, were dissected and extracted for subsequent ex vivo 13C NMR spectroscopy and high performance liquid chromatography (HPLC) analysis. Cerebellum and contralateral hemisphere also undergo changes (Haberg et al. 2009), but these changes are not discussed in this chapter.
4.2 Ischemic Core
MCAO disrupted the glutamate-glutamine cycle in the ischemic core (Haberg et al. 1998b, 2001). Incorporation of label from [1-13C]glucose into [4-13C]glutamate, reflecting mainly oxidative metabolism in glutamatergic neurons, declined rapidly after MCAO. The acetate versus glucose utilization ratio which is calculated from the amount of label originating from [1,2-13C]acetate compared to that from [1-13C]glucose (Taylor et al. 1996), i.e. [4,5-13C]glutamate/[4-13C]glutamate or [1,2-13C]GABA/[2-13C]GABA, was significantly increased at all times after MCAO showing that the relative importance of astrocytic glutamine in glutamate production was increased in ischemia (Haberg et al. 1998b, 2001). Thus suggesting that astrocytic glutamine contributes significantly to excitotoxicity in the ischemic core.
In pathological states not only labeling patterns, but also tissue concentrations of the amino acids under investigation may change. The changes in label incorporation and amino acid content may not always correspond, leading to large variations in the percent enrichment of 13C in the amino acids during ongoing pathological processes in the brain. During MCAO there was a steady decline in glutamate content, most likely multifactorial in origin. Glutamate synthesis was significantly reduced, and glutamate consumption elevated since GABA and alanine levels increased with time and astrocytes continued to take up and convert glutamate to glutamine (Haberg et al. 1998b, 2001). However, the reduction in glutamate content far exceeded the aforementioned factors, implying that MCAO caused massive glutamate efflux beyond the capacity of the astrocytic glutamate transporters, and thus glutamate was lost into the systemic circulation and cerebrospinal fluid. This has been observed in patients (Castillo et al. 1996). Glutamate efflux over the blood brain barrier can only occur in instances when the blood brain barrier is damaged, as in ischemia. Under normal conditions only neutral amino acids, like glutamine, can transverse the blood brain barrier. During MCAO GABA content gradually increased, but incorporation of 13C was significantly reduced at all times after MCAO. At 240 min of MCAO no labeling above natural abundance (1.1%) was detectable in GABA. This finding implies that GABA synthesis from glutamate proceeded without functioning mitochondria, and thus death of GABAergic neurons at this time point. GABA synthesis after 240 min of MCAO was therefore direct conversion of glutamate either stored in the GABAergic neurons or originating from other neurons. The amount of glutamate stored in GABAergic neurons is, however, minimal (Ottersen and Storm-Mathisen 1984), which suggests that GABA synthesis took place in the extracellular space. Glutamate decarboxylase, GAD, which is the enzyme converting glutamate to GABA is a small enzyme and exists in to isoforms with molecular masses of approximately 65 and 67 kDa. The GABA accumulation seen during MCAO also implies impaired GABA breakdown. Ischemic conditions have been shown to stimulate GAD activity (Sze 1979; Erecinska et al. 1996), but inhibit GABA transaminase activity (Schousboe et al. 1973; Baxter and Roberts 1961).
Ischemia also immediately affected astrocytic metabolism. The first sign of reduced astrocyte metabolism was the presence of un-metabolized acetate, which was not seen in sham operated rats (Haberg et al. 2001). The enzyme acetyl CoA synthetase, necessary for acetate metabolism, is an ATP dependent enzyme. The glial specific enzymes glutamine synthetase (GS) and pyruvate carboxylase (PC), which are also ATP dependent, had significantly reduced activity at later time points. After 240 min of MCAO PC activity was only detectable in glutamate in significantly reduced amounts, and the acetate/glucose utilization ratio was markedly increased in glutamate, but not in glutamine. These findings suggest that glutamate produced from acetate or glucose in the astrocytic mitochondria was not transported over the mitochondrial membrane into the cytosol, where GS is located. This interpretation is supported by an immunocytochemical observation of mitochondrial glutamate accumulation in astrocytes in global ischemia (Torp et al. 1993).
Reperfusion of the ischemic core 120 min after MCAO did not improve either neuronal or astrocytic TCA cycle activity. Instead a steady decline in mitochondrial metabolism accompanied by lactate accumulation was observed (Haberg et al. 2006). Likewise, the lipid peroxidation inhibitor, U-101033E, considered to penetrate into mitochondrial lipid bilayers and hence prevent free radical damage to mitochondrial membranes and proteins and thereby ensure maintained TCA cycle and respiratory chain activity, did not improve mitochondrial metabolism in the ischemic core. On the contrary, the NMR spectra revealed that the drug caused a further decrease in mitochondrial pyruvate metabolism and more pronounced cytosolic pyruvate metabolism (Haberg et al. 2007).
4.3 Penumbra
Neuronal glutamate synthesis from [1-13C]glucose was significantly reduced from onset of ischemia and declined during the experimental period also in the penumbra. The amount of label in [4,5-13C]glutamate, originating from [4,5-13C]glutamine, was halved from 30 min of MCAO and remained at this level for 240 min of MCAO. This finding indicates a non-progressing functional inhibition of glutamine transporters and/or inhibition of the neuronal enzyme phosphate activated glutaminase (PAG) activity. Ammonia accumulation and/or acidosis which are known to occur in ischemia, have been shown to reduced PAG activity (Benjamin 1981; Hogstad et al. 1988; Kvamme et al. 2000). Still, with increasing duration of ischemia glutamine became a more important precursor for glutamate as neuronal glutamate synthesis from glucose continued to decline. Actually the acetate/glucose utilization ratio was doubled between 60 and 240 min of MCAO (Haberg et al. 1998b, 2001). It has been demonstrated that glutamine specifically replenishes the neurotransmitter pool of glutamate (Laake et al. 1995), and it is from this pool glutamate excitotoxicity is considered to originate in the penumbra (Takagi et al. 1993; Obrenovitch 1996). Based on the results from 13C NMR spectroscopy analysis of the ischemic penumbra (Haberg et al. 1998a, b, 2001) the glutamate neurotransmitter pool appear to be at least partly replenished from astrocytic precursors, at the same time as neuronal TCA cycle metabolism was markedly reduced. This constellation may exacerbate glutamate excitotoxicity. Indeed, GS activity inhibition has been shown to reduce infarct size in cortex in rats subjected to MCAO (Swanson et al. 1990).
In the penumbra astrocytic metabolism appeared relatively unaffected by MCAO until 90–120 min of MCAO, when un-metabolized [1,2-13C]acetate appeared in the fully awake rats (Haberg et al. 1998b, 2001). This is considerably later than in the ischemic core, and utilization of [1,2-13C]acetate for glutamine synthesis was at the normal level during the entire 240 min of MCAO, indicating that amino acid synthesis via the TCA cycle was more resistant than oxidative metabolism in astrocytes. The activity of GS appeared unaffected by ischemia, and the glutamine content increased steadily. As mentioned above, GS is an ATP dependent enzyme located in the cytosol. Continued glutamine synthesis implies that ATP was present in sufficient amounts to convert glutamate to glutamine. It is conceivable that ATP was provided partly by glycolysis, perhaps fueled by breakdown of glycogen which takes place in the astrocytic cytosol, and partly by the limited amounts of oxygen and glucose supplied to this area by a reduced, but not abolished blood flow.
Re-establishing blood flow has been shown to salvage tissue in the penumbra region. Using 13C NMR spectroscopy four metabolic events that distinguish successfully reperfused tissue have been identified (Haberg et al. 2007). (1) Improved astrocytic acetate metabolism and oxidation, (2) improved neuronal mitochondrial glucose metabolism (3) improved neuronal utilization of astrocytic PC derived metabolites, and (4) no increase in lactate synthesis. The results from reperfusion and permanent MCAO supplement each other and corroborate the notion that preservation of astrocytic metabolism is essential for neuronal survival and a predictor for recovery. Also supporting the importance of preservation of astrocytic metabolism for neuronal survival are results obtained using the lipid peroxidation inhibitor, U-101033E. In the penumbra this drug decreased both neuronal and astrocytic pyruvate metabolism via the TCA cycle, and at the same time increased the anaerobic glucose metabolism (Haberg et al. 2007). Indeed, a similar compound, tirilazad mesylate (U-74006F; Freedox), has been shown to significantly increase in the number of deaths and the degree of disability in randomized controlled trials in acute stroke in humans (Tirilazad International Steering Committee 2000). The results obtained with 13C NMR spectroscopy stress the importance of other outcome measures than ischemic tissue volume for evaluation of drug efficacy in animal models, which in turn could increase the likelihood of success in clinical trials.
5 13C NMR Spectroscopic Analyses of Neuronal-Glial Interactions in Epilepsy
Epilepsy is one of the most common neurological disorders worldwide with a prevalence of 1%. The main feature of epilepsy is the predisposition to recurrent seizures characterized by increased and synchronous neuronal activity. Seizures are associated with increased release of the excitatory neurotransmitter glutamate (Bradford, 1995), and both glutamate receptor agonists and GABA receptor antagonists have been shown to promote epileptic seizures (Hosford 1995). An imbalance of excitatory and inhibitory functions may be due to disturbed neurotransmitter metabolism (Bradford 1995) and 13C NMR spectroscopy is a useful tool to study neurotransmitter synthesis in addition to glial-neuronal interactions. The following section is a description of how 13C NMR spectroscopy in combination with different animal models can elucidate specific changes underlying epileptogenesis and specific events related to seizures.
5.1 The Pentylene Tetrazole Model
Pentylene tetrazole (PTZ) is a chemical convulsant and a GABAA receptor blocker. Given acutely in high concentration it causes generalized tonic-clonic seizures. To study glial-neuronal interactions in the early postictal stage a single PTZ injection (intra peritoneal) in rats was combined with injections (intra peritoneal) of [1-13C]glucose plus [1,2-13C]acetate 30 min later (Eloqayli et al. 2003). The animals were decapitated 45 min after PTZ injection.
13C NMR spectroscopy analyses of extracts from cortex, subcortex and cerebellum showed that astrocyte metabolism in the PTZ treated rats was unchanged. GABAergic neurons in the cerebellum showed a pronounced decrease of GABA synthesis, which might have led to decreased release of GABA. The GABAergic neurons in cortex and subcortex were unaffected. Glutamate labeling was decreased in all brain regions as a response to PTZ treatment, while the labeling of glutamine was unchanged demonstrating a reduction of the metabolic pool of glutamate and not the transmitter pool. Thus, in this model, which is used extensively in testing potential antiepileptic drugs, mainly neuronal metabolism is affected in the early phase.
Epilepsy is also a common geriatric problem (Stephen and Brodie 2000). Since the majority of epilepsy experiments with animals use young adult rodents, a new experimental model to study effects of seizures in the elderly was designed (Kondziella et al. 2002). This model uses PTZ-kindling in senescence accelerated mice (SAMP8), a genetic model of aging (Fujibayashi et al. 1994; Sato et al. 1994). Kindling is characterized by increased sensitivity to seizures after repeated administration of an initially subconvulsive electrical or chemical stimulus. Continuous application of sub-threshold doses of PTZ induces progressive development of seizures resulting in secondary generalized seizures. To investigate whether seizures induce metabolic changes and whether the effect of the anti-convulsant drug phenobarbital is age dependent, 2-month and 8-month old SAMP8 mice received PTZ, PTZ and phenobarbital or saline (20 intra peritoneal injections) every 48 h over a period of 40 days (Kondziella et al. 2003). The mice received intraperitoneal injections of [1-13C]glucose and [1,2-13C]acetate 15 min before decapitation. Extracts from the whole brain (minus cerebellum) were analyzed.
Several differences between the young and old mice, compared to their respective controls, were observed both in response to PTZ alone and PTZ combined with phenobarbital. As in the above mentioned model, the 8-month old mice treated with PTZ alone showed reduced glutamate labeling from [1-13C]glucose, indicating decreased mitochondrial activity in the glutamatergic neurons. Impaired astrocytic metabolism was apparent in the 2-month old mice since the synthesis of glutamine from [1,2-13C]acetate was decreased. PTZ activates excitatory mechanisms in brain cells, but PTZ-kindling did not increase the labeling of glutamate. The reduced seizure threshold induced by PTZ kindling could be explained by enhanced density of glutamate-binding sites on excitatory synapses (Schroeder et al. 1999) rather than increased synthesis of glutamate.
In the 2-month-old mice receiving PTZ and phenobarbital the labeling of most metabolites from both [1-13C]glucose and [1,2-13C]acetate was decreased. Since the total amount of amino acids was unchanged this indicated that turnover of metabolites in both neurons and astrocytes was decreased. Unchanged labeling of GABA showed that GABAergic neurons were less sensitive to the effect of phenobarbital. In the 8-month-old mice there was an increase in GABA labeling both from [1-13C]glucose and [1,2-13C]acetate, while the labeling in most of the other metabolites was unchanged. In summary, phenobarbital depressed cerebral metabolism in the young mice while it increased the metabolic activity of the GABAergic neurons in the older mice.
5.2 Animal Models of Mesial Temporal Lobe Epilepsy
Mesial temporal lobe epilepsy (MTLE) is one of the most common forms of focal epilepsy. The hallmark of MTLE is hippocampal sclerosis, which is characterized by neuronal loss and gliosis in CA1, CA3, and hilus, and reorganization of synaptic connections (Sommer 1880; de Lanerolle and Lee 2005). Animal models, like the kainic acid and lithium pilocarpine (Li pilo) models, are extensively used to study cellular and molecular mechanisms underlying MTLE.
The lithium-pilocarpine model was used to study neurotransmitter metabolism and neuronal-glial interactions in rats that had developed spontaneous recurrent seizures. Approximately 1 month after treatment, the rats were injected with [1-13C]glucose and [1,2-13C]acetate and extracts from cortex, cerebellum, and hippocampal formation (including hippocampus, amygdala, entorhinal and piriform cortices) were analyzed (Melø et al. 2005).
Decreased amounts of glutamate and glutathione were measured in the hippocampal formation of Li-pilo rats compared with control rats, corresponding well with the neuronal cell loss seen in this region. The amounts of [4-13C]glutamate, [3-13C]-/[2-13C]aspartate and [2-13C]GABA, derived from [1-13C]glucose, were decreased in all brain regions investigated in Li-pilo rats compared with controls. These results suggest that the activity in neuronal mitochondria is lower in the epileptic brain than in controls. This is further supported by the decreased level of NAA seen in the cortex and hippocampal formation of Li-pilo rats in comparison with controls. No changes were detected in glial-neuronal interactions in the hippocampal formation while in cortex and cerebellum the flow of glutamate to astrocytes was decreased, indicating a disturbed glutamate-glutamine cycle. In hippocampus astrocytic metabolism was unchanged despite the pronounced gliosis. The results could indicate that astrocytes in sclerotic tissue have lower mitochondrial activity per cell volume. In cortex and cerebellum, labeled glutamine from [1-13C]glucose was decreased whereas the amount of glutamine was unchanged.
In the kainic acid model, neurotransmitter metabolism and neuronal-glial interactions were investigated before (24 h and 14 days) and after (13 weeks) development of spontaneous seizures. Kainic acid was injected (intra peritoneal) into rats, and after 24 h (Qu et al. 2003) 14 days (Muller et al. 2000; Alvestad et al. 2011) the animals received an injection (intra peritoneal) of [1-13C]glucose plus [1,2-13C]acetate. After 15 min the animals were decapitated and extracts from the whole brain (minus cerebellum) were analyzed.
One day after the kainate injection there was increased labeling of glutamine and glutamate from [1,2-13C]acetate, while labeling from [1-13C]glucose was unchanged. At day 14, however, no changes were observed in the metabolites derived from [1,2-13C]acetate, but labeling of glutamate, GABA, glutamine, aspartate, and succinate from [1-13C]glucose was increased. The tissue concentration of metabolites was unchanged both after 24 h and 14 days. Thus, kainate treatment increased the amino acid turnover in astrocytes after 1 day and increased the turnover in glutamatergic and GABAergic neurons after 14 days. This indicates that early and temporary enhanced astrocytic activity may lead to altered metabolism in neurons with an increased turnover of important amino acids such as GABA and glutamate.
Thirteen weeks after injection of kainic acid, rats with spontaneous seizures were injected intra peritoneal with [1-13C]glucose (Alvestad et al. 2008). Brain extracts from hippocampal formation, entorhinal cortex, and neocortex were analyzed. The amounts of glutmate and NAA and labeling of glutamate were reduced in the hippocampal formation and entorhinal cortex of epileptic rats, indicating neuronal loss. Additionally, mitochondrial dysfunction was detected in surviving glutamatergic neurons in the hippocampal formation. In entorhinal cortex glutamine labeling and concentration were unchanged despite the reduced glutamate content and label, possibly due to decreased oxidative metabolism and conserved flux of glutamate through glutamine synthetase in astrocytes. This mechanism was not operative in the hippocampal formation, where glutamine labeling was decreased. In the neocortex, labelling and concentration of GABA were increased in the epileptic rats, possibly representing a compensatory mechanism. The changes in the hippocampus might be of pathophysiological importance and further studies aiming at resolving metabolic causes and consequences of MTLE are required.
6 Genetic Abscencee Epilpsy Rat Model
In the brain of immature Genetic Absence Epilepsy Rats from Strasbourg (GAERS), interactions between glutamatergic neurons and astrocytes appeared normal whereas increased astrocytic metabolism took place in adult GAERS, suggesting that astrocytic alterations could possibly be the cause of seizures (Melø et al. 2007).
7 13C NMR Spectroscopy in Human Pathologies
Human in vivo studies using 13C NMR spectroscopy as a tool to investigate basic brain neurochemistry have been perform in healthy volunteers (Beckmann et al. 1991; Gruetter et al. 1992; Rothman et al. 1992; Mason et al. 1995; Oz et al. 2007) and selected patient groups (Rothman et al. 1991; Blüml et al. 2001) since the early 1990s. Most human studies are dynamic studies with 13C labeled glucose, but labeled acetate (Lebon et al. 2002; Blüml et al. 2002) and hydroxybutyrate (Pan et al. 2002) have also been administrated. Studies pertaining to basic neuochemical questions in healthy volunteers are performed with protocols quite similar to those used in steady state studies of animal.
Protocols used in clinical research, on the other hand, often differ from the basic design applied in healthy volunteers and animals. Intravenous infusion times may be shorter, only one or a few spectra may be obtained, labeled substrate may be given orally (Rothman et al. 1991; Moreno et al. 2001a, b; Shic and Ross 2003; Blüml et al. 2001), and resected tissue can be extracted for in vitro 13C NMR spectroscopy in order to obtain more detailed neurochemical fingerprinting (Petroff et al. 2002). These adaptations are necessary and/or may be the only possible solution in the clinical setting. Dynamic steady-state 13C requires very cooperative subjects, able to lie still for extended periods before and after infusion of labeled substrate. Furthermore, glucose infusion may in some instances be harmful, for instance in acute stroke and epilepsy. In addition 13C labeled substrates are very expensive. The number of patients investigated with 13C MR spectroscopy thus remains low. The feasibility of clinical 13C MR spectroscopy in metabolic fingerprinting of CNS diseases has been explored in single cases and with few patients. The pathologies cover a wide spectrum; Alzheimer’s disease, epilepsy, hypoxia/ischemia, hepatic encephalopathy, leucodystrophy and a variety of metabolic diseases such as Caravan’s disease (Rothman et al. 1991; Moreno et al. 2001a, b; Blüml et al. 2001, 2002; Shic and Ross 2003). Two cases of 13C MR spectroscopy in subacute stroke/hypoxia have been reported in the literature (Rothman et al. 1991; Blüml et al. 2001). Rothman et al. (1991) demonstrated protracted elevation of lactate production from glucose in the ischemic tissue while Blüml et al. (2001) showed reduced levels of [4-13C]glutamate synthesis. Both describe reduced levels of NAA. These findings are in line with the above mentioned studies of extracts obtained from ischemic rat brain, but the level of detail is of course not comparable. The neurochemical changes in epilepsy have also been studied in vivo (Blüml et al. 2002; Shic and Ross 2003) and in extracts of hippocampi after temporal lobectomy (Petroff et al. 2002). The general trend is disturbed glutamate-glutamine cycling, but the results are conflicting as to the direction of the disturbance, i.e. both up and down regulations were reported.
Compared to other MR methods, for instance functional (fMRI) and diffusion MR imaging (DWI/DTI) that were implemented in humans at the beginning of the 1990s also, in vivo 13C MR spectroscopy has yet to become a routine investigation. In vivo 13C MR spectroscopy in humans is still limited to relatively few sites with high-field MR scanners and expert interdisplinary research groups. Clinical 13C MR spectroscopy may improve accuracy of diagnosis by offering detailed metabolic profiles of pathological tissues. Furthermore, 13C MR spectroscopy may also be a tool for monitoring treatment effects and also explore new avenues for dietary and/or pharmaceutical intervention. However, the workflow from acquisition to spectrum analysis needs to be simplified in order for this to be a clinically relevant method. It will also be necessary to prove that clinical in vivo 13C MR spectroscopy can with high specificity and sensitivity, give the correct diagnosis with additional information relevant for the clinical management of the patient not available with other methods, in order for this costly method to become a part of the standard clinical repertoire.
8 Conclusion
From the above it is evident that 13C NMR spectroscopy is a tool not only suitable to study metabolism in the intact brain, but it can also provide valuable information on changes in neuronal, astrocytic and neuronal-astrocytic interactions in the diseased state, and may thus contribute to our understanding of nervous system disease, and provide new avenues to explore in the search for new therapies. The importance of astrocytes for neuronal function and survival is clearly illustrated by numerous reports in the literature and the studies presented here. In KA induced epilepsy glia were affected earlier than neurons and in MCAO the degree of astrocytic metabolic disturbances may be predictive of neuronal survival. Also, 13C NMR spectroscopy offers an opportunity to explore the metabolic effects of drugs, and can thereby increase the likelihood of bringing the best suited drugs to clinical trials. Furthermore, it should be noted that neuronal-astrocytic interactions change with age and appropriate animal models for human disease should be used. It appears likely that 13C NMR spectroscopy will have clinical impact in the future since the dynamic picture of metabolic changes complements the rather static information obtained using 1H NMR spectroscopy and conventional MRI.
References
Alves PM, Flogel U, Brand A, Leibfritz D, Carrondo MJ, Santos H, Sonnewald U (1996) Immobilization of primary astrocytes and neurons for online monitoring of biochemical processes by NMR. Dev Neurosci 18:478–483
Alves PM, Nunes R, Zhang C, Maycock CD, Sonnewald U, Carrondo MJ, Santos H (2000) Metabolism of 3-13C-malate in primary cultures of mouse astrocytes. Dev Neurosci 22:456–462
Alvestad S, Hammer J, Eyjolfsson E, Qu H, Ottersen OP, Sonnewald U (2008) Limbic structures show altered glial-neuronal metabolism in the chronic phase of kainate induced epilepsy. Neurochem Res 33:257–266
Alvestad S, Hammer J, Qu H, Håberg A, Ottersen OP, Sonnewald U (2011) Reduced astrocytic contribution to the turnover of glutamate, glutamine, and GABA characterizes the latent phase in the kainate model of temporal lobe epilepsy. JCBFM 31(8):1675–1686
Bachelard H (1998) Landmarks in the application of 13C-magnetic resonance spectroscopy to studies of neuronal/glial relationships. Dev Neurosci 20:277–288
Badar-Goffer RS, Bachelard HS, Morris PG (1990) Cerebral metabolism of acetate and glucose studied by 13C-n.m.r. spectroscopy. A technique for investigating metabolic compartmentation in the brain. Biochem J 266:133–139
Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS (2006) Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab 26:1285–1297
Bak LK, Waagepetersen HS, Melø TM, Schousboe A, Sonnewald U (2007) Complex glutamate labeling from [U-13C]glucose or [U-13C]lactate in co-cultures of cerebellar neurons and astrocytes. Neurochem Res 32:671–680
Bakken IJ, Johnsen SF, White LR, Unsgard G, Aasly J, Sonnewald U (1997) NMR spectroscopy study of the effect of 3-nitropropionic acid on glutamate metabolism in cultured astrocytes. J Neurosci Res 47:642–649
Barany M, Arus C, Chang YC (1985) Natural-abundance 13C NMR of brain. Magn Reson Med 2:289–295
Baxter CF, Roberts E (1961) Elevation of gamma-aminobutyric acid in brain: selective inhibition of gamma-aminobutyric-alpha-ketoglutaric acid transaminase. J Biol Chem 236:3287–3294
Beckmann N, Turkalj I, Seelig J, Keller U (1991) 13C NMR for the assessment of human brain glucose metabolism in vivo. Biochemistry 30:6362–6366
Benjamin AM (1981) Control of glutaminase activity in rat brain cortex in vitro: influence of glutamate, phosphate, ammonium, calcium and hydrogen ions. Brain Res 208:363–377
Blüml S, Moreno A, Hwang JH, Ross BD (2001) 1-13C glucose magnetic resonance spectroscopy of pediatric and adult brain disorders. NMR Biomed 14:19–32
Blüml S, Moreno-Torres A, Shic F, Nguy CH, Ross BD (2002) Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed 15:1–5
Bradford HF (1995) Glutamate, GABA and epilepsy. Prog Neurobiol 47:477–511
Castillo J, Davalos A, Naveiro J, Noya M (1996) Neuroexcitatory amino acids and their relation to infarct size and neurological deficit in ischemic stroke. Stroke 27:1060–1065
Cerdan S, Kunnecke B, Seelig J (1990) Cerebral metabolism of [1,2-13C2]acetate as detected by in vivo and in vitro 13C NMR. J Biol Chem 265:12916–12926
Cerdán S, Rodrigues TB, Sierra A, Benito M, Fonseca LL, Fonseca CP, García-Martín ML (2006) The redox switch/redox coupling hypothesis. Neurochem Int 48:523–530
Choi DW (1994) Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res 100:47–51
Choi IY, Gruetter R (2003) In vivo 13C NMR assessment of brain glycogen concentration and turnover in the awake rat. Neurochem Int 43:317–322
Cremer JE, Lucas HM (1971) Sodium pentobarbitone and metabolic compartments in rat brain. Brain Res 35:619–621
de Lanerolle NC, Lee TS (2005) New facets of the neuropathology and molecular profile of human temporal lobe epilepsy. Epilepsy Behav 10:190–203
Eid T, Thomas MJ, Spencer DD, Runden-Pran E, Lai JC, Malthankar GV, Kim JH, Danbolt NC, Ottersen OP, de Lanerolle NC (2004) Loss of glutamine synthetase in the human epileptogenic hippocampus: possible mechanism for raised extracellular glutamate in mesial temporal lobe epilepsy. Lancet 363:28–37
Eloqayli H, Dahl CB, Gotestam KG, Unsgard G, Hadidi H, Sonnewald U (2003) Pentylenetetrazole decreases metabolic glutamate turnover in rat brain. J Neurochem 85:1200–1207
Erecinska M, Nelson D, Daikhin Y, Yudkoff M (1996) Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem 67:2325–2334
Fan TWM (1996) Metabolite profiling by one and two-dimensional NMR analysis of complex mixture. Prog Nucl Magn Reson Spectrosc 528:161–219
Fan TW-M, Lane AN (2008) Structure-based profiling of metabolites and isotopomers by NMR. Prog Nucl Magn Reson Spectrosc 52:69–117
Freo U, Ori C (2004) Effects of anesthesia and recovery from ketamine racemate and enantiomers on regional cerebral glucose metabolism in rats. Anesthesiology 100:1172–1178
Fujibayashi Y, Waki A, Wada K, Ueno M, Magata Y, Yonekura Y, Konishi J, Takeda T, Yokoyama A (1994) Differential aging pattern of cerebral accumulation of radiolabeled glucose and amino acid in the senescence accelerated mouse (SAM), a new model for the study of memory impairment. Biol Pharm Bull 17:102–105
Gaitonde MK, Dahl DR, ElliotT KA (1965) Entry of glucose carbon into amino acids of rat brain and liver in vivo after injection of uniformly 14-C-labelled glucose. Biochem J 94:345–352
Garcia-Espinosa MA, Rodrigues TB, Sierra A, Benito M, Fonseca C, Gray HL, Bartnik BL, Garcia-Martin ML, Ballesteros P, Cerdan S (2004) Cerebral glucose metabolism and the glutamine cycle as detected by in vivo and in vitro 13C NMR spectroscopy. Neurochem Int 45:297–303
Gibson GE, Sheu KF, Blass JP (1998) Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm 105:855–870
Griffin JL, Rae C, Radda GK, Matthews PM (1999) Delayed labelling of brain glutamate after an intra-arterial [13C]glucose bolus: evidence for aerobic metabolism of guinea pig brain glycogen store. Biochim Biophys Acta 1450:297–307
Gruetter R, Novotny EJ, Boulware SD, Rothman DL, Mason GF, Shulman GI, Shulman RG, Tamborlane WV (1992) Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci USA 89:1109–1112. Erratum in: Proc Natl Acad Sci USA 1992 Dec 15;89(24):12208
Haberg A, Qu H, Bakken IJ, Sande LM, White LR, Haraldseth O, Unsgard G, Aasly J, Sonnewald U (1998a) In vitro and ex vivo 13C-NMR spectroscopy studies of pyruvate recycling in brain. Dev Neurosci 20:389–398
Haberg A, Qu H, Haraldseth O, Unsgard G, Sonnewald U (1998b) In vivo injection of [1-13C]glucose and [1,2-13C]acetate combined with ex vivo 13C nuclear magnetic resonance spectroscopy: a novel approach to the study of middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab 18:1223–1232
Haberg A, Qu H, Saether O, Unsgard G, Haraldseth O, Sonnewald U (2001) Differences in neurotransmitter synthesis and intermediary metabolism between glutamatergic and GABAergic neurons during 4 hours of middle cerebral artery occlusion in the rat: the role of astrocytes in neuronal survival. J Cereb Blood Flow Metab 21:1451–1463
Haberg A, Qu H, Hjelstuen MH, Sonnewald U (2006) Glutamate and GABA metabolism in transient and permanent middle cerebral artery occlusion in rat: importance of astrocytes for neuronal survival. Neurochem Int 48:531–540
Haberg A, Qu H, Hjelstuen MH, Sonnewald U (2007) Effect of the pyrrolopyrimidine lipid peroxidation inhibitor U-101033E on neuronal and astrocytic metabolism and infarct volume in rats with transient middle cerebral artery occlusion. Neurochem Int 50:932–940
Haberg AK, Qu H, Sonnewald U (2009) Acute changes in intermediary metabolism in cerebellum and contralateral hemisphere following middle cerebral artery occlusion in rat. J Neurochem 109(Suppl 1):174–181
Hansen TD, Warner DS, Todd MM, Vust LJ, Trawick DC (1988) Distribution of cerebral blood flow during halothane versus isoflurane anesthesia in rats. Anesthesiology 69:332–337
Harada M, Okuda C, Sawa T, Murakami T (1992) Cerebral extracellular glucose and lactate concentrations during and after moderate hypoxia in glucose- and saline-infused rats. Anesthesiology 77:728–734
Hassel B, Sonnewald U (1995a) Selective inhibition of the tricarboxylic acid cycle of GABAergic neurons with 3-nitropropionic acid in vivo. J Neurochem 65:1184–1191
Hassel B, Sonnewald U (1995b) Glial formation of pyruvate and lactate from TCA cycle intermediates: implications for the inactivation of transmitter amino acids? J Neurochem 65:2227–2234
Hassel B, Sonnewald U, Fonnum F (1995) Glial-neuronal interactions as studied by cerebral metabolism of [2-13C]acetate and [1-13C]glucose: an ex vivo 13C NMR spectroscopic study. J Neurochem 64:2773–2782
Hassel B, Bachelard H, Jones P, Fonnum F, Sonnewald U (1997) Trafficking of amino acids between neurons and glia in vivo. Effects of inhibition of glial metabolism by fluoroacetate. J Cereb Blood Flow Metab 17:1230–1238
Henry PG, Tkac I, Gruetter R (2003) 1H-localized broadband 13C NMR spectroscopy of the rat brain in vivo at 9.4 T. Magn Reson Med 50:684–692
Henry PG, Adriany G, Deelchand D, Gruetter R, Marjanska M, Oz G, Seaquist ER, Shestov A, Ug˘urbil K (2006) In vivo 13C NMR spectroscopy and metabolic modeling in the brain: a practical perspective. Magn Reson Imaging 24:527–539
Hertz E, Yu ACH, Shahar A, Juurlink BHJ, Schousboe A (1989a) Preparation of primary cultures of mouse cortical neurons. In: A dissection and tissue culture manual for the nervous system. Alan R. Liss, New York, pp 183–186
Hertz L, Juurlink BHJ, Hertz E, Fosmark H, Schousboe A (1989b) Preparation of primary cultures of mouse (rat) astrocytes. In: A dissection and tissue culture manual for the nervous system. Alan R. Liss, New York, pp 105–108
Hoffman WE, Edelman G, Kochs E, Werner C, Segil L, Albrecht RF (1991) Cerebral autoregulation in awake versus isoflurane-anesthetized rats. Anesth Analg 73:753–757
Hogstad S, Svenneby G, Torgner IA, Kvamme E, Hertz L, Schousboe A (1988) Glutaminase in neurons and astrocytes cultured from mouse brain: kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem Res 13:383–388
Hosford DA (1995) Models of primary generalized epilepsy. Curr Opin Neurol 8:121–125
Hyder F, Brown P, Nixon TW, Behar KL (2003) Mapping cerebral glutamate 13C turnover and oxygen consumption by in vivo NMR. Adv Exp Med Biol 530:29–39
Karelson G, Ziegler A, Kunnecke B, Seelig J (2003) Feeding versus infusion: a novel approach to study the NAA metabolism in rat brain. NMR Biomed 16:413–423
Kauppinen RA, Pirttilä TR, Auriola SO, Williams SR (1994) Compartmentation of cerebral glutamate in situ as detected by 1H/13C n.m.r. Biochem J 298:121–127
Kondziella D, Bidar A, Urfjell B, Sletvold O, Sonnewald U (2002) The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: behavior and metabolism. Neurochem Int 40:413–418
Kondziella D, Hammer J, Sletvold O, Sonnewald U (2003) The pentylenetetrazole-kindling model of epilepsy in SAMP8 mice: glial-neuronal metabolic interactions. Neurochem Int 43:629–637
Kvamme E, Roberg B, Torgner IA (2000) Phosphate-activated glutaminase and mitochondrial glutamine transport in the brain. Neurochem Res 25:1407–1419
Laake JH, Slyngstad TA, Haug FM, Ottersen OP (1995) Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J Neurochem 65:871–881
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22:1523–1531
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Magistretti PJ, Pellerin L, Rothman DL, Shulman RG (1999) Energy on demand. Science 283:496–497
Mason GF, Gruetter R, Rothman DL, Behar KL, Shulman RG, Novotny EJ (1995) Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J Cereb Blood Flow Metab 15:12–25
Mason GF, Petersen KF, de Graaf RA, Shulman GI, Rothman DL (2007) Measurements of the anaplerotic rate in the human cerebral cortex using 13C magnetic resonance spectroscopy and [1-13C] and [2-13C] glucose. J Neurochem 100:73–86
McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66: 386–393
McKenna MC, Dienel GA, Sonnewald U, Waagepetersen HS, Schousboe A (2011) Energy metabolism of the brain. In: Siegel G, Albers W, Brady S, Price D (eds) Basic neurochemistry, vol 8, Elsevier, London, pp 201–226, ISBN 978-0-12-374947-5
Melø TM, Nehlig A, Sonnewald U (2005) Metabolism is normal in astrocytes in chronically epileptic rats: a 13C NMR study of neuronal-glial interactions in a model of temporal lobe epilepsy. J Cereb Blood Flow Metab 25:1254–1264
Melø TM, Bastholm IA, Sonnewald U, Nehlig A (2007) Astrocytes may play a role in the etiology of absence epilepsy: a comparison between immature GAERS not yet expressing seizures and adults. Neurobiol Dis 28:227–235
Memezawa H, Minamisawa H, Smith ML, Siesjo BK (1992) Ischemic penumbra in a model of reversible middle cerebral artery occlusion in the rat. Exp Brain Res 89:67–78
Minchin MC, Beart PM (1975a) Compartmentation of amino acid metabolism in the rat dorsal root ganglion; a metabolic and autoradiographic study. Brain Res 83:437–449
Minchin MC, Beart PM (1975b) Compartmentation of amino acid metabolism in the rat posterior pituitary. J Neurochem 24:881–884
Moreno A, Blüml S, Hwang JH, Ross BD (2001a) Alternative 1-13C glucose infusion protocols for clinical 13C MRS examinations of the brain. Magn Reson Med 46:39–48
Moreno A, Ross BD, Blüml S (2001b) Direct determination of the N-acetyl-L-aspartate synthesis rate in the human brain by 13C MRS and [1-13C]glucose infusion. J Neurochem 771:347–350
Muller TB, Haraldseth O, Jones RA, Sebastiani G, Godtliebsen F, Lindboe CF, Unsgard G (1995) Combined perfusion and diffusion-weighted magnetic resonance imaging in a rat model of reversible middle cerebral artery occlusion. Stroke 26:451–457
Muller B, Qu H, Garseth M, White LR, Aasly J, Sonnewald U (2000) Amino acid neurotransmitter metabolism in neurones and glia following kainate injection in rats. Neurosci Lett 279:169–172
Nehlig A, Wittendorp-Rechenmann E, Lam CD (2004) Selective uptake of [14 C]2-deoxyglucose by neurons and astrocytes: high-resolution microautoradiographic imaging by cellular 14 C-trajectography combined with immunohistochemistry. J Cereb Blood Flow Metab 24:1004–1014
Obrenovitch TP (1996) Origins of glutamate release in ischaemia. Acta Neurochir Suppl 66:50–55
Olstad O, Qu H, Sonnewald U (2007) Glutamate is preferred over glutamine for intermediary metabolism in cultured cerebellar neurons. J Cereb Blood Flow Metab 50:1004–1013
Ottersen OP, Storm-Mathisen J (1984) Glutamate- and GABA-containing neurons in the mouse and rat brain, as demonstrated with a new immunocytochemical technique. J Comp Neurol 229:374–392
Oz G, Berkich DA, Henry PG, Xu Y, LaNoue K, Hutson SM, Gruetter R (2004) Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J Neurosci 24:11273–11279
Oz G, Seaquist ER, Kumar A, Criego AB, Benedict LE, Rao JP, Henry PG, Van De Moortele PF, Gruetter R (2007) Human brain glycogen content and metabolism: implications on its role in brain energy metabolism. Am..J Physiol. Endocrinol Metab 292:E946–51
Pan JW, de Graaf RA, Petersen KF, Shulman GI, Hetherington HP, Rothman DL (2002) [2,4-13C2 ]-beta-Hydroxybutyrate metabolism in human brain. J Cereb Blood Flow Metab 22:890–898
Pascual JM, Carceller F, Roda JM, Cerdán S (1998) Glutamate, glutamine, and GABA as substrates for the neuronal and glial compartments after focal cerebral ischemia in rats. Stroke 29:1048–1056, discussion 1056–1057
Pellerin L, Bouzier-Sor AK, Aubert A, Serres S, Merle M, Costala R, Magistretti PJ (2007) Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55:1251–1262
Peng L, Zhang X, Hertz L (1994) High extracellular potassium concentrations stimulate oxidative metabolism in a glutamatergic neuronal culture and glycolysis in cultured astrocytes but have no stimulatory effect in a GABAergic neuronal culture. Brain Res 663:168–172
Petroff OA, Errante LD, Rothman DL, Kim JH, Spencer DD (2002) Glutamate-glutamine cycling in the epileptic human hippocampus. Epilepsia 43:703–710
Qu H, Haberg A, Haraldseth O, Unsgard G, Sonnewald U (2000a) 13C MR spectroscopy study of lactate as substrate for rat brain. Dev Neurosci 22:429–436
Qu H, Waagepetersen HS, van Hengel M, Wolt S, Dale O, Unsgard G, Sletvold O, Schousboe A, Sonnewald U (2000b) Effects of thiopental on transport and metabolism of glutamate in cultured cerebellar granule neurons. Neurochem Int 37:207–215
Qu H, Konradsen JR, van HM, Wolt S, Sonnewald U (2001a) Effect of glutamine and GABA on [U-13C]glutamate metabolism in cerebellar astrocytes and granule neurons. J Neurosci Res 66:885–890
Qu H, van der GM, Le MT, Sonnewald U (2001b) The effect of thiopental on glutamate metabolism in mouse cerebellar astrocytes in vitro. Neurosci Lett 304:141–144
Qu H, Eloqayli H, Müller B, Aasly J, Sonnewald U (2003) Glial-neuronal interactions following kainate injection in rats. Neurochem Int 42:101–106
Rae C, Moussa Cel-H, Griffin JL, Parekh SB, Bubb WA, Hunt NH, Balcar VJA (2006) Metabolomic approach to ionotropic glutamate receptor subtype function: a nuclear magnetic resonance in vitro investigation. J Cereb Blood Flow Metab 26:1005–1017
Rossi DJ, Brady JD, Mohr C (2007) Astrocyte metabolism and signaling during brain ischemia. Nat Neurosci 10:1377–1386
Rothman SM, Olney JW (1986) Glutamate and the pathophysiology of hypoxic–ischemic brain damage. Ann Neurol 19:105–111
Rothman DL, Howseman AM, Graham GD, Petroff OA, Lantos G, Fayad PB, Brass LM, Shulman GI, Shulman RG, Prichard JW (1991) Localized proton NMR observation of [3-13C]lactate in stroke after [1-13C]glucose infusion. Magn Reson Med 21:302–307
Rothman DL, Novotny EJ, Shulman GI, Howseman AM, Petroff OA, Mason G, Nixon T, Hanstock CC, Prichard JW, Shulman RG (1992) 1H-[13C] NMR measurements of [4-13C]glutamate turnover in human brain. Proc Natl Acad Sci USA 89:9603–9606
Sato E, Inoue A, Kurokawa T, Ishibashi S (1994) Early changes in glucose metabolism in the cerebrum of senescence accelerated mouse: involvement of glucose transporter. Brain Res 637:133–138
Schousboe A, Wu JY, Roberts E (1973) Purification and characterization of the 4-aminobutyrate–2, ketoglutarate transaminase from mouse brain. Biochemistry 12:2868–2873
Schousboe A, Meier E, Drejer J, Hertz L (1989) Preparation of primary cultures of mouse (rat) cerebellar granule cells. In: A dissection and tissue culture manual for the nervous system. Alan R. Liss, New York, pp 183–186
Schroeder H, Becker A, Schroeder U, Hoellt V (1999) 3 H-L-glutamate binding and 3 H-D-aspartate release from hippocampal tissue during the development of pentylenetetrazole kindling in rats. Pharmacol Biochem Behav 62:349–352
Schwartz-Bloom RD, Sah R (2001) gamma-Aminobutyric acid(A) neurotransmission and cerebral ischemia. J Neurochem 77:353–371
Serres S, Bouyer JJ, Bezancon E, Canioni P, Merle M (2003) Involvement of brain lactate in neuronal metabolism. NMR Biomed 16:430–439
Serres S, Bezancon E, Franconi JM, Merle M (2004) Ex Vivo Analysis of Lactate and Glucose Metabolism in the Rat Brain under Different States of Depressed Activity. J Biol Chem 279:47881–47889
Shank RP, Leo GC, Zielke HR (1993) Cerebral metabolic compartmentation as revealed by nuclear magnetic resonance analysis of D-[1-13C]glucose metabolism. J Neurochem 61:315–323
Shic F, Ross B (2003) Automated data processing of [1H-decoupled] 13C MR spectra acquired from human brain in vivo. J Magn Reson 162:259–268
Sibson NR, Dhankhar A, Mason GF, Behar KL, Rothman DL, Shulman RG (1997) In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glutamine cycling. Proc Natl Acad Sci USA 94:2699–2704
Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG (2001) In vivo 13C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during. J Neurochem 2001(76):975–989
Sommer W (1880) Erkrankung des Ammonshorns als aetiologisches Moment der Epilepsie. Arch Psychiatr Nervenkr 10:631–675
Sonnewald U, Rae C (2010) Pyruvate carboxylation in different model systems studied by 13C MRS. Neurochem Res 35(12):1916–21
Sonnewald U, Westergaard N, Krane J, Unsgard G, Petersen SB, Schousboe A (1991) First direct demonstration of preferential release of citrate from astrocytes using [13C]NMR spectroscopy of cultured neurons and astrocytes. Neurosci Lett 128:235–239
Sonnewald U, Petersen SB, Krane J, Westergaard N, Schousboe A (1992) 1H NMR study of cortex neurons and cerebellar granule cells on microcarriers and their PCA extracts: lactate production under hypoxia. Magn Reson Med 23:166–171
Sonnewald U, Westergaard N, Petersen SB, Unsgard G, Schousboe A (1993a) Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem 61:1179–1182
Sonnewald U, Westergaard N, Schousboe A, Svendsen JS, Unsgard G, Petersen SB (1993b) Direct demonstration by [13C]NMR spectroscopy that glutamine from astrocytes is a precursor for GABA synthesis in neurons. Neurochem Int 22:19–29
Sonnewald U, White LR, Odegard E, Westergaard N, Bakken IJ, Aasly J, Unsgard G, Schousboe A (1996) MRS study of glutamate metabolism in cultured neurons/glia. Neurochem Res 21:987–993
Sonnewald U, Olstad E, Qu H, Babot Z, Cristofòfol R, Suñol C, Schousboe A, Waagepetersen H (2004) First direct demonstration of extensive GABA synthesis in mouse cerebellar neuronal cultures. J Neurochem 91:796–803
Sonnewald U, Schousboe A, Waagepetersen HW (2011) 13C NMR spectroscopy and mass spectrometry analysis of intermediary metabolism in cultured neural cells in Neuromethods. W Walz (Series ed), Aschner M (Volume Eds), Sunol C, Price A. Springer Science-Business Media, New York, pp 403–414, ISBN 978-1-61779-076-8, http://issuu.com/asartehutiimhotep/docs/cell-culturing-techniques
Stephen LJ, Brodie MJ (2000) Epilepsy in elderly people. Lancet 355:1441–1446
Swanson RA, Shiraishi K, Morton MT, Sharp FR (1990) Methionine sulfoximine reduces cortical infarct size in rats after middle cerebral artery occlusion. Stroke 21:322–327
Sze PY (1979) L-Glutamate decarboxylase. Adv Exp Med Biol 123:59–78
Takagi K, Ginsberg MD, Globus MY, Dietrich WD, Martinez E, Kraydieh S, Busto R (1993) Changes in amino acid neurotransmitters and cerebral blood flow in the ischemic penumbral region following middle cerebral artery occlusion in the rat: correlation with histopathology. J Cereb Blood Flow Metab 13:575–585
Taylor A, McLean M, Morris P, Bachelard H (1996) Approaches to studies on neuronal/glial relationships by 13C-MRS analysis. Dev Neurosci 18:434–442
Tirilazad International Steering Committee (2000) Tirilazad mesylate in acute ischemic stroke: A systematic review. Stroke 31:2257–2265
Torp R, Arvin B, Le PE, Chapman AG, Ottersen OP, Meldrum BS (1993) Effect of ischaemia and reperfusion on the extra- and intracellular distribution of glutamate, glutamine, aspartate and GABA in the rat hippocampus, with a note on the effect of the sodium channel blocker BW1003C87. Exp Brain Res 96:365–376
Tyson RL, Gallagher C, Sutherland GR (2003) 13C-Labeled substrates and the cerebral metabolic compartmentalization of acetate and lactate. Brain Res 992:43–52
Vrba R (1962) Glucose metabolism in rat brain in vivo. Nature 195:663–665
Waagepetersen HS, Bakken IJ, Larsson OM, Sonnewald U, Schousboe A (1998) Metabolism of lactate in cultured GABAergic neurons studied by 13C nuclear magnetic resonance spectroscopy. J Cereb Blood Flow Metab 18:109–117
Waagepetersen HS, Qu H, Schousboe A, Sonnewald U (2001) Elucidation of the quantitative significance of pyruvate carboxylation in cultured cerebellar neurons and astrocytes. J Neurosci Res 66:763–770
Waagepetersen H, Melø T, Schousboe A, Sonnewald U (2004) Homeostasis of neuroactive amino acids in cultured cerebellar and neocortical neurons is influenced by environmental cues. J Neurosci Res (in press)
Waagepetersen HS, Qu H, Sonnewald U, Shimamoto K, Schousboe A (2005a) Role of glutamine and neuronal glutamate uptake in glutamate homeostasis and synthesis during vesicular release in cultured glutamatergic neurons. Neurochem Int 47:92–102
Waagepetersen HS, Melø TM, Schousboe A, Sonnewald U (2005b) Homeostasis of neuroactive amino acids in cultured cerebellar and neocortical neurons is influenced by environmental cues. J Neurosci Res 79:97–105
Waagepetersen HS, Qu H, Sonnewald U, Schousboe A (2009) Energy and amino acid neurotransmitter metabolism in astrocytes. In: Parpura V, Haydon PG, (eds), Astrocytes in (patho)physiology of the nervous system, Springer, Boston, pp 177–200
Waniewski RA, Martin DL (1998) Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci 18:5225–5233
Westergaard N, Fosmark H, Schousboe A (1991a) Metabolism and release of glutamate in cerebellar granule cells cocultured with astrocytes from cerebellum or cerebral cortex. J Neurochem 56:59–66
Westergaard N, Sonnewald U, Petersen SB, Schousboe A (1991b) Characterization of microcarrier cultures of neurons and astrocytes from cerebral cortex and cerebellum. Neurochem Res 16:919–923
Westergaard N, Larsson OM, Jensen B, Schousboe A (1992) Synthesis and release of GABA in cerebral cortical neurons co-cultured with astrocytes from cerebral cortex or cerebellum. Neurochem Int 20:567–575
Westergaard N, Drejer J, Schousboe A, Sonnewald U (1996) Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia 17:160–168
Whitehead KJ, Rose S, Jenner P (2004) Halothane anesthesia affects NMDA-stimulated cholinergic and GABAergic modulation of striatal dopamine efflux and metabolism in the rat in vivo. Neurochem Res 29:835–842
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this chapter
Cite this chapter
Haberg, A., Sonnewald, U., Hammer, J., Melø, T., Eloqayli, H. (2012). 13C NMR Spectroscopy as a Tool in Neurobiology. In: Choi, IY., Gruetter, R. (eds) Neural Metabolism In Vivo. Advances in Neurobiology, vol 4. Springer, Boston, MA. https://doi.org/10.1007/978-1-4614-1788-0_8
Download citation
DOI: https://doi.org/10.1007/978-1-4614-1788-0_8
Published:
Publisher Name: Springer, Boston, MA
Print ISBN: 978-1-4614-1787-3
Online ISBN: 978-1-4614-1788-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)