
Abstract
Rheumatologic diseases in children compromise a small but sometimes perplexing portion of patients admitted to the Pediatric Intensive Care Unit. Systemic Lupus Erythematosus (SLE) is the most variable and indiscriminate vasculitis affecting children. SLE may cause devastating injury to virtually any organ system and therefore must often be included in the differential diagnosis of critically ill children. Juvenile Idiopathic Arthritis (JIA) does not commonly require admission to the ICU in itself, but is associated with macrophage activation syndrome (MAS). MAS is a spectrum of hemophagocytic lymphohistiocytosis (HLH) which often calls for intensive care and aggressive therapies. Henoch-Schoenlein Purpura is a small vessel vasculitis affecting children commonly associated with intussusception, but may also give rise to significant cardioplulmonary complications. Kawasaki Disease is the most common vasculitis in children and the recently described Kawasaki Disease Shock Syndrome may require invasive monitoring and therapies. Antiphospholipid Antibody Syndrome, Goodpasture Disease, and Wegener’s Granulomatosis while not common, give rise to complications which also may necessitate critical care. The following chapter discusses these disease entities and other rheumatologic illness affecting children and common complications arising from them which may indicate critical care. Included in the discussion are the most widely accepted diagnostic approaches and therapies, and supporting evidence is described.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Systemic Lupus Erythematosus
- Juvenile Idiopathic Arthritis
- Macrophage Activation Syndrome
- Kawasaki Disease
- Kawasaki Disease Shock Syndrome
- Antiphospholipid Antibody Syndrome
- Infliximab
- Henoch-Schoenlein Purpura
- Goodpasture Disease
- Churg-Strauss Syndrome
- Juvenile Dermatomyositis
- Systemic Sclerosis
Introduction
In the general pediatric population, rheumatologic diseases vary greatly in their features and presentation. Thus, despite their infrequency, these disorders often must be included in the differential diagnosis when evaluating a critically ill child. It has been estimated that between 5 and 10,000 children within the United States carry the diagnosis of systemic lupus erythematosus [1], the most common connective tissue disease. In addition, 30,000–50,000 children suffer from juvenile idiopathic arthritis (JIA) [2], the most common rheumatologic disease. The most common vasculitis in childhood, Henoch-Schonlein Purpura (HSP), has an estimated incidence of nine per 100,000 children [3, 4]. More rare rheumatologic diseases affecting children include antiphospholipid antibody syndrome (APS), juvenile dermatomyositis (JDM), Goodpasture disease, Wegener granulomatosis, Churg-Strauss syndrome, and scleroderma. The pediatric intensivist cares for rheumatology patients for varying reasons, related to the primary disease process, adverse effects of treatment for the underlying disease, and/or illnesses that are complicated by the underlying disease [5]. In the following sections, we will examine selected rheumatologic diseases affecting children, and will review their presentation, pathophysiology, complications, and management.
Systemic Lupus Erythematosus (SLE)
Systemic lupus erythematosus (SLE) (Table 35.1) is the prototypic, and most variable rheumatologic disease described to date, affecting all organ systems. SLE affects patients of all ethnicities and ages, but 15–20 % of patients are diagnosed in the first two decades of life [8]. SLE is extremely variable in presentation, organ system involvement, and disease progression [9, 10]. Patients with SLE are hospitalized in the intensive care setting for a wide array of diagnoses. One retrospective study of adult SLE patients requiring intensive care unit admission found that the primary diagnosis was infection in 41 % of admissions (the majority of which were diagnosed with septicemia), renal disease in 21 %, cardiovascular disease in 16 %, and coagulopathy (both thrombotic and hemorrhagic) in 14 % [11].
The pathogenesis of SLE differs depending on which tissues are examined and is best described in renal disease, which shows inflammation, cellular proliferation, and immune complex deposition affecting the glomerular basement membrane (GBM). Deposition of circulating immune complexes or de novo synthesis of immune complexes on the GBM activates the complement cascade, leading to cellular damage, cellular proliferation, and matrix deposition [12]. Tissue classification of lupus nephritis is complex secondary to the variability of disease expression among patients and among various glomeruli within one tissue biopsy [12]. The World Health Organization (WHO) has established a classification scheme based on light microscopy, immunofluorescence, and electron microscopy, which divides lupus nephritis into six classes. Findings range from class I with normal glomeruli by light microscopy, immunofluorescence, and electron microscopy, to class VI where >90 % of observed glomeruli are sclerotic [12].
The cardiovascular system, including both the heart and blood vessels, shows pathologic changes due to the ongoing autoimmune process as well. Inflammation is seen in the pericardium, myocardium, and endocardium. Immunofluorescence studies have shown complement and immune complex deposition within the myocardial vessels as well as the myocardium itself [13].
CNS Complications of SLE
Approximately 20–30 % of children will develop CNS involvement, or neuropsychiatric lupus (NPS), during the course of their SLE, with 75–85 % of these developing during the first year [14, 15]. The term neuropsychiatric lupus describes 19 clinical situations seen in SLE patients, which may be the consequence of ongoing CNS vasculitis, peripheral nervous system involvement, or stroke due to associated antiphospholipid antibodies [16, 17]. Symptoms range from headache or memory impairment to psychosis, paralysis, seizures, and coma. When considering serious or life-threatening CNS lupus only (including seizures, stroke, major cognitive disorder, chorea, psychosis, major depression, and acute confusional state) Sibbit and colleagues found that 76 % of the patients in their series were affected during the 6-year study period [18]. A similar case series from Spinosa and colleagues reports nearly 62 % of patients with SLE diagnosed before age 16 years had NPS syndromes as defined by the American College of Rheumatology during their 14 year study period [19]. CNS disease has been reported as the initial presentation of SLE in 16–28 % of pediatric patients [15, 20, 21]. However, before CNS symptoms are attributed to the SLE, other entities such as infection, mass, or hemorrhage must be evaluated thoroughly.
Seizures
Seizure has been reported as the most common serious CNS complication of pediatric SLE, occurring in up to 51–61 % of patients [15, 18, 20, 22]. Seizures may be the initial presentation of disease [15, 18, 20, 22] and may portend a more severe disease course when present at diagnosis of SLE [23]. Seizures associated with SLE are usually easily controlled with standard antiepileptic therapy and remit with control of SLE, although status epilepticus has been reported [24]. Mecarelli and colleagues reported a case of SLE induced status epilepticus requiring barbiturate coma [25].
Cerebral Vein Thrombosis
Cerebral vein thrombosis (CVT), usually seen in association with antiphospholipid antibodies, has been associated with pediatric SLE more commonly than in adults [8]. The presence of a CVT in pediatric patients should be considered when there is a severe unremitting headache, especially when the patient has known antiphospholipid antibodies [26]. Head CT images are often normal in the presence of a CVT, but abnormalities are sometimes identified. The most common CT finding is the delta sign which appears as a central dark area in the torcula, corresponding to thrombus, surrounded by enhancing contrast representing flowing blood [27]. With early consideration of the diagnosis, imaging, and treatment, reported outcomes have been favorable [26]. Delays in diagnosis and/or therapy have been associated with progressive neurological deficits, and death [26].
Supportive medical and neurologic care, including hydration and seizure control, are the cornerstones of therapy for CVT in children [27]. Anticoagulation is considered by some to be appropriate therapy, but remains controversial, and the most appropriate agent remains unclear. Many studies have shown the safe use of anticoagulation in adult and pediatric patients with sinovenous thrombosis, but none has clearly shown significant benefit [28, 29]. In the acute phase, intravenous infusions of heparin have been most widely used [28, 30], but safe and effective use of low-molecular weight heparin has also been reported in pediatric patients without evidence of preceding intracranial hemorrhage [31–33]. Continued or intensified therapy for the underlying vasculitis is also appropriate [14]. Data regarding the use of thrombolytics in children with CVT are sparse. Studies regarding the use of thrombolytics for other forms of thromboses in children have shown effectiveness but with a low therapeutic index [34].
Stroke
Stroke has been reported in 3.4–12 % of pediatric patients with SLE [15, 20, 22] and in 20–40 % of patients with neuropsychiatric lupus [20, 22]. Strokes seen in pediatric SLE may be arterial ischemic strokes or sequelae of cerebral vein thrombosis. Most reports of stroke with pediatric SLE identify the concurrent presence of antiphospholipid antibodies (anti-cardiolipin antibody or lupus anticoagulant) [30]. Treatment of stroke in SLE patients is largely supportive, aiming at maintaining normothermia, hydration, and normal hemodynamics [30]. Anticoagulation is becoming accepted practice although the ideal agent remains controversial, but should be considered in these patients. Patients with antiphospholipid antibodies, particularly lupus anticoagulant, have a high risk of recurrent cerebrovascular disease [30, 35, 36] and therefore may be more likely to benefit from anticoagulation. Therapy should also include ongoing or intensified immunosuppressive therapy provided the absence of a contraindication [14].
Psychosis, Delirium, and Depression
Although children who present with depression, psychosis, delirium, or movement disorders secondary to SLE often have normal CSF findings and non-focal CT and MRI studies, discrete lesions can sometimes be seen on CT and MRI [37]. If abnormalities are seen on MRI, they are usually diffuse gray and white matter lesions that dramatically improve or resolve with aggressive SLE treatment [38]. There are also reports of single photon emission computed tomography (SPECT) scan abnormalities in the parietal and frontal lobes [39], as well as diffuse small areas of decreased uptake [40]. Other than possibly supporting the diagnosis of neuropsychiatric lupus, the clinical utility of SPECT scan abnormalities remains unclear as they usually persist despite clinical improvement [8, 39–41] and may be present in pediatric SLE patients without clinical CNS disease [40]. Favorable outcome of neuropsychiatric SLE in both pediatric and adult patients has been reported after treatment with intravenous methylprednisolone and cyclophosphamide [16, 38, 42, 43].
Pulmonary Complications of SLE
Pulmonary involvement in adults with SLE is common, with infection being the most frequent complication [44]. The incidence of pulmonary involvement in children with SLE is difficult to estimate as the literature is limited primarily to case reports and small series; however published figures place the incidence anywhere between 5 and 67 % during the course of the disease, and acute lupus pneumonitis may be the first manifestation of disease [45]. Pulmonary hypertension, diffuse interstitial disease, pulmonary hemorrhage, pneumothorax, acute lupus pneumonitis, and shrinking lungs have all complicated SLE in pediatric patients [45].
SLE Pneumonitis
Acute lupus pneumonitis is difficult to clinically differentiate from an infectious process, and must be a diagnosis of exclusion [44, 45]. Mortality in adult patients with acute lupus pneumonitis is approximately 50 % [44]. Presenting symptoms include fever, dyspnea, tachypnea, hypoxia, cough, and occasionally hemoptysis [45]. On blood gas analysis, hypoxemia and a respiratory alkalosis are often seen [44]. Initial therapy should consist of corticosteroids; cyclophosphamide, plasmapheresis, or intravenous immunoglobulin may be added if there is no clinical response [44, 46]. One must be cognizant of the fact that treatment with cyclophosphamide has been linked with an increased risk for opportunistic infections and mortality [11, 47].
Pulmonary Hemorrhage
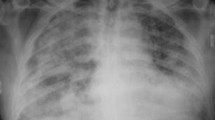
Pulmonary hemorrhage in SLE is an uncommon, but often deadly occurrence, with mortality approaching 80 % [45]. Symptoms associated with pulmonary hemorrhage are similar to those seen in pneumonitis, with cough, dyspnea, tachypnea, and hypoxia seen most commonly, in association with a sudden drop in hematocrit. Hemoptysis is an unreliable indicator of the presence or absence of pulmonary hemorrhage [44, 45]. Chest radiography will usually reveal patchy alveolar infiltrates, particularly in the lower lobes; however, infiltrates are not universally present [44]. Reported therapies consist of pulse methylprednisolone with or without cytotoxic agents [45, 48]. Respiratory failure associated with SLE and pulmonary hemorrhage requires aggressive ventilatory and hemodynamic support, and extracorporeal life support has been used successfully in conjunction with ongoing immunosuppressive therapy [49].
Cardiac Complications of SLE
SLE Pericarditis
Pericarditis is the most common cardiac complication of pediatric SLE; throughout the disease process, pericarditis is seen in up to 25 % of SLE patients, and can rarely be the first manifestation of disease [50, 51]. In adult patients, pericarditis is present in 6–45 % of SLE patients, and in autopsy series, as many as 60–80 % of patients have pericardial lesions [52]. Pericardial tamponade is a rare, life-threatening, but usually early complication of SLE [52], occurring in less than 4 % of pediatric patients [50, 53].
Pericardial fluid in SLE serositis is an inflammatory exudate consisting of fibrinous debris and inflammatory cells, which can mimic bacterial pericarditis [54]. An ANA titer of >1:160 in the pericardial fluid has been shown to be a sensitive, but not specific, indicator of underlying SLE [55]. While the presence of lupus erythematosus cells (phagocytic cells which have ingested the nucleus of another cell) in a pericardial aspirate has been shown to have high sensitivity and specificity [54, 55].
Therapy for SLE pericarditis is guided by severity. In pediatric SLE, pericarditis is usually mild, and effusions small [50]. In this setting, treatment is aimed at the underlying disease and consists of monitoring, steroids, antimalarials, and other immune modulators [50, 56]. The presence of a large pericardial effusion is an indication for ICU monitoring, and may warrant aspiration, particularly when present with diminished cardiac function or cardiovascular instability.
SLE Endocarditis
Verrucous endocarditis (Libman-Sacks endocarditis) describes the valvar vegetations seen in association with SLE. Most commonly these involve the mitral valve, but also involve the aortic, pulmonic, and tricuspid valves in order of decreasing incidence [50]. The lesions consist of deposits of immune complexes, cellular debris, and fibrin [57, 58]. In autopsy series, these nodular lesions have been found uniformly in SLE patients [59]. However, lesions detectable by echocardiography occur generally in older adolescents and adults [50]. Rarely, heart failure from mitral insufficiency can be the clinical presentation in SLE, and require mitral valve replacement in childhood [60].
SLE Myocarditis and Cardiomyopathy
Although cardiac processes in association with SLE are widely recognized, clinically apparent acute myocarditis is a rare complication of SLE, particularly in infants and children [61, 62]. Even so, acute myocarditis has been reported as the initial presentation of SLE [63]. The literature regarding lupus-associated myocarditis is limited owing to the rarity of this clinical entity. Therefore, there is no consensus on the best management of SLE myocarditis [62]. The literature contains reports of poor responses of SLE myocarditis to steroid therapy [62, 64]; however, James et al. [62] state that further immunosuppression seems a reasonable approach in light of the underlying autoimmune process. In the adult literature, high dose intravenous corticosteroids are the norm, with anecdotal reports of other immunosuppressive agents (azathioprine and cyclophosphamide) and intravenous immune globulin providing some benefit [13, 65]. Data from Johns Hopkins suggest that the mortality of myocarditis associated with SLE is greater than that of primary myocarditis [66].
Infectious Complications of SLE
The most frequent complication, reason for admission to the ICU, and cause of death among SLE patients is infection [11, 67–72]. The rate of infectious complications in these patients is increased by therapy for the underlying disease [71, 73, 74], although SLE intrinsically increases the risk of serious infection [70, 73]. Localized infections are usually related to the underlying disease, while systemic infections are generally caused by immunosuppression from therapy, in particular corticosteroids [70].
Glucocorticoid therapy has multiple effects on immunity, including suppressing phagocytic function, cell-mediated immunity, and humoral immunity [70, 74]. As a result, the offending agents in infections related to glucocorticoid therapy are diverse. Defective phagocytic function places the patient at risk for gram-positive, gram-negative, and fungal infections [71, 74]. Ineffective cell-mediated immunity places the patient at risk for organisms such as Mycobacterium, Listeria monocytogenes, Salmonella, and Nocardia, as well as Histoplasma, Coccidiodes, and Cryptococcus [74]. Protozoal (Pneumocystis, Toxoplasma, and Strongyloides) and viral (cytomegalovirus, Epstein-Barr virus, and Varicella-zoster virus) infections are also related to defective cell-mediated immunity [74]. Multiple studies have reported that patients maintained on high dose corticosteroids, particularly more than 20 mg per day, are at increased risk for serious infections [75, 76]. In fact, one series reported that 90 % of infected patients on more than 40 mg per day of prednisone were bacteremic [76]. Although even lower doses have been shown to increase the risk [75].
Bacterial pathogens account for more than 90 % of infections in SLE patients [70], and the most frequently isolated organisms are S. aureus and enteric gram-negative bacteria [70, 71, 74]. Gram-negative sepsis or bacteremia was the cause of death in 32 % of SLE-related deaths in one series of 544 patients [71]. Patients with SLE have been found to be at increased risk for pneumococcal infections secondary to functional hyposplenism, hypocomplementemia, impaired chemotaxis, and defects in opsonization, all secondary to SLE itself [75].
Renal Complications of SLE
Two-thirds of children and adolescents with SLE will develop nephritis during the course of their illness, and in 90 % of these, the renal disease is present within 1 year of diagnosis [8], with diffuse proliferative glomerulonephritis being the most common form. Unfortunately this form of nephritis is the form most likely to progress to end-stage renal disease and/or death [8]. In patients with SLE admitted to the intensive care unit, renal failure has been associated with increased mortality [67].
Juvenile Idiopathic Arthritis (JIA)
Juvenile idiopathic arthritis (JIA) (Table 35.2), formerly known as juvenile rheumatoid arthritis (JRA), is a common rheumatic disease of childhood, responsible for significant morbidity. The underlying cause of JIA remains unclear, but an underlying immunogenetic susceptibility is likely required to react to an external stimulus thereby inducing disease [77]. Approximately 113/100,000 children currently carry the diagnosis of JIA, with an annual incidence of approximately 13.9/100,000 children 15 years old or younger [77].
Three types, or onset forms of JIA are currently described:, oligoarthritis (pauciarticular), polyarthritis, and systemic onset disease [77]. While oligoarthritis generally affects the large joints of the lower extremities, polyarthritis affects both large and small joints, often with 20–40 individual joints involved [77]. Systemic onset disease is characterized by a cyclical fever which may exceed 39 °C, in conjunction with a faint salmon-colored evanescent macular rash [77]. Hepatomegaly, splenomegaly, and lymphadenopathy are present with arthritis. Serositis in the form of pericardial effusion may be present as well [77].
Although the overall mortality of JIA is low, when comparing affected individuals to non-affected individuals, the diagnosis of JIA does impart an increase in age-adjusted mortality [2]. Approximately two-thirds of deaths attributable to JIA are in patients with systemic onset JIA, which compromises only 10–20 % of all patients with JIA [2]. Deaths due to JIA are most likely secondary to infections, cardiac complications, and macrophage activation syndrome [2].
Infectious Complications of JIA
Similar to SLE, infections in patients with JIA are usually a complication of therapy for the underlying disease. The disease modifying drugs used most commonly in JRA include steroids and methotrexate. Other medications, including azathioprine, cyclosporine, and etanercept, have also been used. Although these medications have the potential for significant improvement in the daily function of patients with JIA, several of these medications carry risks of serious infectious complications. As discussed previously, steroid therapy has multiple effects on immunity, and sustained therapy, particularly at high doses, is associated with increased infection rates. With other disease modifying drugs available, steroids are generally reserved for severe systemic onset disease or life threatening complications of JIA [2].
Anti-TNF therapy is the newest class of medications currently being used for the treatment of JIA. Etanercept (Enbrel; Amgen Inc.) and infliximab (Remicade; Centocor Inc.) are two such medications. Etanercept is a human fusion protein of Fc IgG1 and the p75 TNF receptor which has been FDA approved for use in rheumatoid arthritis and psoriatic arthritis. Its use has proven to provide improvement in patient functionality and pain, as well as slowed disease progression [79]. In children, etanercept has been shown to be well tolerated and effective in the treatment of JIA, particularly those with pauciarticular or polyarticular JIA [80]. Placebo controlled trials found no increase in the rate of serious infections between treatment and control groups [81, 82]. However, post-marketing reports of serious bacterial infections have been published. Patients who are prescribed etanercept with concurrent steroid therapy are at particular risk [83] and in patients with active sepsis etanercept has been shown to increase mortality [84].
Infliximab (Remicade; Centocor Inc.) is a monoclonal anti-TNF-α antibody which has also shown beneficial in the management of inflammatory arthritis [79], but is not currently labeled for use in JIA. In placebo controlled studies evaluating anti-TNF therapy, several studies have shown a similar incidence of serious bacterial infections (life-threatening or requiring hospitalization) to that of placebo [81, 82]. In post-marketing reports, anti-TNF therapy, infliximab in particular, has been associated with development of active tuberculosis. This most likely represents activation of a latent infection and patients should be screened prior to initiation of infliximab therapy [85].
Cardiac Complications of JIA
Pericarditis is a common complication of JIA, and may be present at diagnosis, or precede the development of arthritis [86], although usually not clinically significant [78]. Pericarditis occurs in as many as 30 % of patients, and in autopsy series, as many as 45 % [87]. Pericardial effusions seen with JIA are usually small and clinically insignificant, but cardiac tamponade requiring pericardiocentesis or pericadiectomy has been reported [88, 89]. Non-steroidal anti-inflammatory medications and intensification of anti-inflammatory therapy are usually effective [88, 90].
Valvar disease is a well known, but uncommon complication of JRA [91, 92]. The aortic valve is the most commonly involved; however, the mitral valve may also be diseased [93]. In a recent study of patients with HLA-B27-associated juvenile arthritis, 10 % were found to have aortic regurgitation of varying degrees after a mean of 3 years of illness, compared with none of the healthy control patients [93]. Generally, the severity of valvar disease correlates with articular disease and the degree of aortic regurgitation may be such that valve replacement is warranted [92]. Recently, the presence of rheumatoid nodules in the diseased valve has been described in a patient without other rheumatoid nodules [92]. Myocarditis is a commonly cited but rarely seen complication of JIA. It occurs with much less frequency than pericarditis, but carries higher mortality and severe sequelae. Patients who recover may be left with residual dilated cardiomyopathy [94].
Macrophage Activation Syndrome
Macrophage activation syndrome (MAS), also known as hemophagocytic syndrome, is an uncommon life-threatening complication of JIA (usually systemic JIA), brought about by an infection, medications (gold salts, NSAID’s, or methotrexate), or autologous stem cell transplant [2, 95, 96]. MAS is a form of hemophagocytic lymphohistiocytosis (HLH) characterized by sudden onset of sustained fever, generalized lymphadenopathy, hepatosplenomegaly, and coagulopathy [2] (Table 35.3). Encephalopathy, respiratory distress or failure, and/or renal failure may develop [95, 98] with a mortality rate between 11 and 60 % [99, 100].
Laboratory studies which may suggest or support the diagnosis of MAS are a falling ESR, pancytopenia, elevated transaminases, increased triglyceride levels, elevated serum ferritin, and studies suggestive of a consumptive coagulopathy (elevated D-Dimers, PT, and PTT, decreased fibrinogen, and fibrin split products) [2, 98]. Bone marrow aspirates in MAS provide pathognomonic findings consisting of well differentiated macrophage proliferation with active phagocytosis of hematopoetic elements of the marrow [98]. Similar infiltration may also be seen in organs such as the liver and spleen [98, 100].
Differentiation of MAS from systemic JIA is important as therapy should be initiated early to prevent sequelae and death. The fever pattern seen with systemic JIA is usually cyclical with daily or twice daily spikes, whereas with MAS, the fever is high and unremitting [95, 98]. In MAS, the ESR falls precipitously, as opposed to systemic JIA, where it will be elevated. Also with MAS, there is a sudden drop in all hematologic cell lines, in contrast to systemic JIA, which is commonly accompanied by leukocytosis and thrombocytosis [95, 98]. Hypertriglyceridemia is another feature present in MAS which is not generally associated with systemic JIA [95].
First line therapy of MAS includes withdrawal of NSAID’s, disease-modifying antirheumatic drugs and/or immunosuppressives, and early initiation of high dose steroid therapy (methylprednisolone). Despite initiating therapy, the disease may still progress [99]. Because cyclosporine has been useful in a familial form of HLH, it has been investigated in MAS as well. Mouy and colleagues [95] reported the use of cyclosporine in five children with systemic JIA and MAS with good outcome, and recommended its use with high dose parenteral steroids. Other authors recommend cyclosporine for MAS as well, particularly in patients unresponsive to steroids alone [99].
The prevailing current treatment protocols contain steroids and cyclosporine, but other disease modifying agents are gaining evidence for effective use in JIA and MAS. Etanercept (Enbrel; Immunex), a TNF-α blocker, has been used in JIA with good disease control, and its use for MAS has been reported with good outcome [101]. Others, however, have reported a possible link between etanercept therapy and the development of MAS [102]. Anakinra (Kineret; Amgen), an IL-1 receptor antagonist, is becoming widely used for maintenance therapy and has been reported as a treatment of MAS, capable of inducing rapid disease [103, 104]. Tocilizumab (Actemra; Roche), and IL-6 receptor antibody has been shown safe, effective, and well-tolerated in a phase III randomized, placebo-controlled trial in children with JIA and may prove to be an effective therapy in MAS complicating JIA [105, 106]. Tocilizumab is currently pending FDA approval is only available for compassionate use in the United States.
Henoch-Schoenlein Purpura (HSP)
Henoch-Schoenlein Purpura (HSP) (Table 35.4), the most common cause of purpura in children with normal platelet counts, is an idiopathic vasculitis syndrome involving the small vessels of the skin, gut, and glomeruli most commonly seen following a viral or streptococcal infection [3, 107, 108]. One study has shown a stronger correlation between an elevated serum Bartonella henselae antibody titer and HSP than with elevated ASO or anti-DNase B [109] titers, but this correlation is as yet unsubstantiated. HSP nephritis may represent a variant of IgA nephropathy [108].
Serious complications of HSP are uncommon, but include neurologic (seizure, intracranial hemorrhage, coma) [110–113], gastrointestinal (intussusception, obstruction, perforation, hemorrhage) [114], and renal (nephritis) [108]. There are two reported cases of myocardial necrosis associated with HSP, although there is some question as to whether another underlying diagnosis may have been present in at least one of the two cases [115, 116].
CNS Complications of HSP
Most believe that the incidence of neurologic involvement, from headaches to intracranial hemorrhages, with HSP lies somewhere between 2 and 10 %, but estimates have been as high as 20 % [110, 117]. More than 50 % of patients reported to have had neurologic complications had seizures, and most of these were generalized [110]. The etiology of the neurologic complications is unclear. Multiple possible causes, such as hypertensive encephalopathy, uremic encephalopathy, steroids, cytotoxic drugs, cerebral vasculitis and electrolyte imbalances are often simultaneously present in HSP [110]. Magnetic resonance imaging of patients with HSP who manifest seizures and/or encephalopathy has shown lesions consistent with demyelination in the posterior parieto-temporal region that resolve with clinical improvement [118–120].
Intracranial hemorrhage has been reported in less than ten patients [121, 122]. Most of the reported cases are intraparenchymal and parieto-temporal in location [121]. It is thought that in most cases, the vasculitis itself leads to a friability of vessels, allowing the hemorrhage. However, studies have linked HSP to a decrease of factor XIII, possibly contributing to hemorrhage. One recent report documented a low factor XIII level in a patient with HSP complicated by intracerebral hemorrhage [123].
Pulmonary Complications of HSP
A rare, but striking and potentially fatal complication of HSP is pulmonary hemorrhage. A recent retrospective analysis reported two of 136 patients with HSP experienced pulmonary hemorrhage [124]. Without rapid diagnosis and institution of treatment, pulmonary hemorrhage can progress to death [113, 125]. A review of the literature demonstrates that mortality in patients with HSP complicated by pulmonary hemorrhage approaches 50 % [113], but other authors suggest that mortality may depend on age, with lower mortality seen in younger children [126]. The treatment regimen reported by many authors for pulmonary hemorrhage in HSP includes aggressive ventilatory support, glucocorticoids, and occasionally cyclophosphamide, although no controlled trial has shown clear benefit of immunosuppressives [113, 127].
Gastrointestinal Complications of HSP
Intussusception is the most common, and best described, abdominal complication of HSP. The lead point is edematous bowel wall secondary to intramural vasculitis and hemorrhage [128]. Unlike spontaneous intussusception, which is usually ileocolic, intussusception in HSP usually involves entirely small bowel. This distinction is important as contrast enema is not useful when the pathology is proximal to the ileocecal valve. Bowel perforation is rare, and has decreased dramatically with earlier diagnosis [128]. Gastrointestinal hemorrhage has been reported as a common abdominal complication of HSP, occurring in approximately 50 % of patients. The bleeding is usually minimal and self-limited, but may occasionally be massive and lead to hypovolemic shock [128–131].
Nephritis in HSP
Nepritis is the most common serious, long-term complication of HSP, affecting 85–90 % of children within 1 month of diagnosis and nearly all children within 6 months to varying degrees [132]. Rarely does nephritis warrant PICU admission, however patients with nephrotic range proteinuria may require admission for correction of fluid and electrolyte abnormalities. Standard therapy consists of glucocorticoids, but those resistant to steroids and more severely ill have been treated with ACE inhibitors, urokinase, plasmapheresis, cyclosporine A, cyclophosphamide, azathioprine, and mycophenolate in various regimens [133, 134]. The outcome of HSP nephritis is generally good with 1–2 % of all patients with HSP nephritis developing chronic renal disease [132] however, those with more severe disease may be at increased risk for the development of chronic renal disease and even end stage renal disease.
Kawasaki Disease
First described in Japan in 1967, Kawasaki disease (Table 35.5) is an acute, self-limited vasculitis affecting infants and children of all races. It is characterized by high fever, conjunctival injection, erythema of the oral mucosal, strawberry tongue, peripheral edema, cervical lymphadenopathy, and variable rash [135, 136]. Kawasaki disease (KD) affects approximately 3,000–4,000 children annually in the United States [135, 136]. The incidence of KD is approximately 112/100,000 in Japanese children <5 years old. In contrast, children <5 years old of Asian or Pacific island descent have the highest annual incidence in the United States at approximately 32.5/100,000 children [135]. Many features of KD (such as the presence of epidemics and seasonality, the age group affected, fever, self-limited course, and rash) suggest an infectious etiology, although none has been proven [135, 136].
The vascular inflammation seen with KD affects all vessels, but is more pronounced in the medium arteries, particularly the coronary arteries [136]. Inflammation and infiltration affects all layers of the arterial wall and results in destruction of the elastic lamina. Aneurysmal dilatation is a result of this destruction, and is not limited to the coronary arteries, having been reported in many muscular arteries throughout the body, including the axillary, celiac, femoral, iliac, mesenteric, and renal arteries [135]. Ultimately local scarring leads to stenosis of the affected vessel [136]. KD will lead to coronary abnormalities in 15–25 % of children if left untreated [135]. Nearly all deaths attributable to KD are a direct result of cardiac involvement [135].
Myocarditis, pericardial effusions, coronary rupture, cardiac tamponade, and myocardial infarction secondary to coronary thrombosis or stenosis may complicate KD [58, 135–138]. Myocarditis is common and likely universal. Diminished ventricular contractility is often seen on the initial echocardiogram [135]. The dysfunction is usually mild and improves after intravenous immune globulin, but may be severe [58, 135]. Pericardial effusions are common, seen on the initial echocardiogram in up to 30 % of patients with KD, but are generally small and clinically insignificant [58].
Giant coronary artery aneurysms (greater than 8 mm), seen in only 1 % of appropriately treated children, carry a mortality rate of approximately 4 % usually secondary to obstructive disease [58, 139]. Rupture of an aneurysm can occur and is rapidly fatal secondary to cardiac tamponade. There are reports of three children who have survived cardiac arrest after coronary rupture. All three had surgical intervention at the bedside and were taken to the operating room emergently for definitive coronary bypass grafting. One patient subsequently required heart transplantation [139, 140]. Macrophage activation syndrome (discussed previously) has also been reported in the context of Kawasaki disease [141].
Kawasaki Disease Shock Syndrome
Shock as a presentation of KD has become more widely recognized in recent years and has been described as Kawasaki Disease Shock Syndrome [142]. Clinically KDSS may be difficult to distinguish from toxic shock syndrome (TSS). Both are characterized by fever, desquamating rash, and mucous membrane erythema. In contrast, shock is a hallmark of TSS, but historically not considered a feature of KD, and coronary artery abnormalities often associated with KD are not seen in TSS. Several recent case reports and series have described KDSS and report that KDSS is more resistant to IVIG, more likely to require steroid or infliximab therapy, more likely to have a delayed diagnosis, and more likely to have coronary artery dilatation than KD without shock. KDSS has also been more commonly seen with female gender, lower platelet counts, higher band counts, and higher C-reactive protein measurements than KD without shock [142–144]. KDSS requires a vigilance to the diagnosis, particularly in the absence of early coronary artery abnormalities. IVIG should be considered early even if the diagnosis of TSS remains a question, as delay in treatment is associated with increased risk of coronary artery aneurysms.
Kawasaki Disease Management
Management of Kawasaki disease consists initially of supportive care, IVIG (2 g/kg), and high dose aspirin (80–100 mg/kg/day divided q 6 h). The aspirin dose is usually decreased to 3–5 mg/kg/day after the 14th day of illness [136]. Most experts agree that patients who fail to respond to the initial dose of IVIG should receive a second infusion of 2 g/kg, and steroid therapy should be reserved for failures after two doses of IVIG [135]. Although the exact mechanism of action is unknown, IVIG has consistently shown benefit if given within the first 10 days of illness [135]. With IVIG and aspirin, approximately 5 % of children with Kawasaki disease will develop transient coronary dilatation and only 1 % develop giant aneurysms [135]. A trial of dexamethasone with IVIG found no difference in the number of patients who developed aneurysms of the coronary arteries, but reported more rapid clinical improvement (defervescence) and decrease in C-reactive protein (CRP) [145].
Monoclonal antibodies have recently been used as a part of the therapy for Kawasaki disease. Abciximab (Reopro®, Eli Lilly and Company), a chimeric human/mouse monoclonal antibody directed against the platelet glycoprotein IIa/IIIb receptor, has been used to inhibit platelet aggregation in patients with giant aneurysms secondary to Kawasaki disease. Short-term outcome was reported as favorable in a case report with coronary thrombi [146]. In 15 patients with 50 coronary aneurysms, a trial of standard therapy (IVIG 2 g/kg once with aspirin 80–100 mg/kg/day) versus standard therapy with abciximab administered 24–48 h after IVIG found greater aneurysmal regression in the abciximab group [147]. However, current evidence lacks sufficient strength to recommend the addition of abciximab to first-line therapy.
Infliximab (Remicade®, Centocor) has been used in children at various centers for refractory Kawasaki disease. Reports of infliximab’s efficacy are mixed, with some centers reporting benefit while others reporting no effect [148]. A recent retrospective analysis of 17 patients with refractory Kawasaki disease treated with infliximab reported rapid defervescence, often after a single dose [149]. A recent randomized prospective trial of infliximab therapy (5 mg/kg) versus a second infusion of IVIG in refractory KD found similar efficacy and safety between the two groups [150], but as the authors identified, several patients in the second dose IVIG group also received infliximab which may reduce any differences between treatment groups. With the growing body of supporting evidence infliximab is being used more commonly as a second teir therapy for KD [151]. As with abciximab, current evidence lacks the strength to recommend infliximab as initial therapy over IVIG.
Antiphospholipid Antibody Syndrome
Antiphospholipid antibody syndrome (Table 35.6) or antiphospholipid syndrome (APS) is characterized by thromboembolic disease or immune thrombocytopenia caused by antibody-mediated platelet activation in the presence of antiphospholipid antibodies (aPL) [153]. Several aPL have been identified to date [154], with lupus anticoagulant (LA) and anticardiolipin (aCL) antibodies felt to be the most clinically relevant [153, 154]. In adults, the thromboembolic disease may be worsened by atherosclerotic disease and is accompanied by recurrent abortions [155]. APS remains a rare entity in pediatric patients that is often diagnosed after a clinical event prompts further investigation [153].
Primary APS refers to the above clinical situation with the presence of aPL antibodies on two occasions at least 6 weeks apart and the absence of a comorbid condition associated with aPL antibodies (such as SLE). Secondary APS describes a scenario with the presence of aPL antibodies, in the setting of vasculitis (SLE, JRA, HSP, among others), certain chronic infections (Lyme disease), or certain drug exposures (quinide, penicillin) [153].
Several authors have reported the presence of aPL antibodies in asymptomatic individuals. A study of over 700 presumably healthy adult volunteer blood donors in Indiana found that 8.1 % were positive for aPL antibodies [154]. In healthy children, less data is available, and reported frequencies range from 2 to 82 %, depending on testing methods, cutoff values used, and populations studied [156]. Experts estimate that the actual frequency most likely lies in the 5–10 % range [156].
In pediatric patients, a strong association between antiphospholipid antibodies and SLE has been found, and APS occurs most frequently as secondary APS with SLE in children [156]. Also of note is that children who present with what appears initially to be primary APS, may later develop SLE [156]. Similarly, some authors have reported a high prevalence of aCL antibodies in patients with JRA, unlike with adult rheumatoid arthritis [157]. Interestingly, aCL antibodies have been found to be the cause of false positive VDRL tests in some patients with SLE [158]. In a study of 57 Brazilian children and adolescents with SLE, investigators found that 63 % of the patients tested positive for aPL antibodies on at least one occasion during the 22 month study period [155]. The investigators found that SLE remission was achieved less often in patients testing positive for aPL antibodies as well [155]. Other investigators have found a correlation between neuropsychiatric manifestations in pediatric SLE and positive testing for aPL antibodies [159].
Most complications of APS are thrombotic, and no major differences have been found between primary and secondary APS [154]. APS, much like heparin-induced thrombocytopenia, predisposes to both venous and arterial thromboses, but deep venous thrombosis remains the most common in both adult [154] and pediatric series [156], affecting 29–55 % of patients. Subsequent pulmonary embolus may occur in up to half of these patients [154]. Antiphospholipid antibodies have been associated with the first arterial ischemic stroke in children as well as adults [160, 161]. Whether aPL increase the risk of recurrent acute arterial stroke or TIA, however, remains unclear [162]. Sinovenous thrombosis has also been associated with aPL antibodies [32]. A recent study from the Canadian Pediatric Ischemic Stroke Registry found that the most common prothrombotic disorder identified in infants and children with sinovenous thrombosis was the presence of aCL antibodies (10 of 123 patients) [32].
Catastrophic antiphospholipid syndrome has been used to describe the minority of patients who present with acute and widespread vascular occlusions throughout the body [154]. This may be the initial presentation of APS in as many as 46 % of patients [163]. Diagnostic criteria include evidence of three or more organ system involvement, development of manifestations simultaneously or within less than 1 week, histopathologic evidence of small vessel occlusion, and laboratory confirmation of aPL antibodies [164]. The presence of all four of the above criteria indicates definite catastrophic APS, with the presence of only three or only two organ system involvement indicating “probable” catastrophic APS [164]. These patients often rapidly deteriorate and mortality has been reported to be near 50 % secondary to multiorgan failure [154, 163].
Optimal management of catastrophic APS is not known. Therapy should be aimed at supporting the involved organ systems, and suppressing progression of the microvascular disease [165]. The majority of patients receive a combination of several treatments that varies by institution [164]. A consensus statement from the Catastrophic Antiphospholipid Syndrome Registry Project Group published in 2003 states that patients with clinically suspected catastrophic APS should be managed with anticoagulation and “high doses” of steroids with plasma exchange and/or IVIG [164]. A recent report from the Catastophic Antiphospholipid Syndrome Registry found a higher recovery rate in patients treated with anticoagulants, plus steroids, plus plasma exchange, and some with IVIG [163]. Other authors have recommended reservation of plasma exchange for patients who do not respond to anticoagulation and steroids [165]. If such treatment fails, patients should be considered for cyclophosphamide, prostacyclin, and/or fibrinolytics. Defibrotide, an investigatory thrombomodulator, may prove a useful adjunct in recalcitrant catastrophic APS [164].
Goodpasture Disease
Goodpasture disease (Table 35.7) is a rare autoimmune disease characterized by the presence of autoantibodies directed against the GBM. These circulating antibodies may attach to GBM, alveolar basement membrane (ABM), or both depending on the individual. Why the GBM, ABM, or both are affected in different individuals remains unclear [168]. Approximately 10 % of patients with anti-GBM antibodies will have pulmonary disease alone, 20–40 % will have renal disease alone, and 60–80 % will have both, termed Goodpasture’s disease or Goodpasture’s syndrome [168]. Unlike systemic vasculitides, the ESR is seldom significantly elevated. ANA and RF are usually negative as well [168].
In patients with pulmonary disease, almost all will have hemoptysis at some point during their disease [168]. Alveolar bleeding is usually minute in volume, however massive bleeding may occur leading to respiratory failure and death. Massive pulmonary hemorrhage is the most common cause of death in patients with anti-GBM disease [168]. As discussed briefly with HSP, management of severe pulmonary hemorrhage consists of general supportive measures, aggressive ventilatory support, and immunosuppressives. Extra corporeal life support (ECLS) may be required in extreme cases.
Central nervous system involvement has been reported in two adolescents with Goodpasture’s disease. The patients presented with generalized seizures and abnormalities consistent with vasculitis on MRI. Both patients improved with intensification of immunosuppressive therapy and plasmapheresis [169].
Wegener’s Granulomatosis
Wegener’s granulomatosis (WG) is an antineutrophil cytoplasm antibody positive necrotizing granulomatous vasculitis affecting the lower and upper respiratory tracts, often associated with renal disease [170]. WG predominately affects adults; Hoffman and colleagues found that 15 % of WG patients seen at their referral center over a 24 year period were under age 19 [171], while other authors have reported much lower rates in children and adolescents [170, 172]. Children usually present with fever, anorexia, weight loss, cough, hemoptysis, and pain (chest pain, myalgia, arthralgia) [172]. Sinusitis, proteinuria, and hematuria are found nearly universally during initial evaluation [172].
Pulmonary disease with WG is rapidly progressive in children. Four of the ten patients in a Swedish study required admission to the PICU as a result of pulmonary dysfunction, and two required mechanical ventilation [172]. Pulmonary hemorrhage has been reported during the course of the disease, with severity enough to warrant mechanical ventilation, and the successful use of ECMO has been reported [49]. Isolated subglottic stenosis has also been associated with WG, more commonly in children than adults, which may require dilatation [173].
Therapy for WG consists of supportive medical care and immunosuppression [172]. A high dose pulse steroid (methylprednisolone) is given acutely, followed by an oral regimen. Usually cyclophosphamide is given in conjunction with steroids, and plasma exchange has been used [172]. In adults, steroids plus cyclophosphamide is considered standard of care [173]. With widespread use, steroid and cyclophosphamide therapy has changed the prognosis from universally fatal to quite good. Adult patients are able achieve disease remission in more than 90 % of cases, and relapse is declining with newer maintenance treatment protocols [174].
Churg-Strauss Syndrome
Churg Strauss Syndrome (CSS) is an antineutrophil cytoplasm antibody positive small vessel vasculitis much like Wegener’s Granulomatosis, which usually presents with fever, pulmonary infiltrates, and respiratory distress in a patient with asthma [173]. Among pediatric patients, CSS is most commonly seen in adolescent patients, but remains rare. In 1984, Lanham published clinical criteria, which require only asthma, peripheral eosinophilia (>1.5 × 109/L), and evidence of systemic vasculitis involving two or more extrapulmonary organs to make the diagnosis of CSS [175]. However, the diagnostic criteria for CSS, according to the American College of Rheumatology, requires the presence of asthma, blood eosinophilia (>10 %), mono- or polyneuropathy, infiltrates on CXR, paranasal sinus abnormalities, and extravascular eosinophils on blood vessel biopsy [176]. Published reports have implicated a causative role for leukotriene modifiers in CSS, while others have found no such link [177]. A post-marketing analysis confirmed an association between leukotriene modifier therapy (alone or in combination with other asthma therapies) and CSS, but could not speculate on causality [178].
Cardiac Complications of CSS
Pericarditis or cardiomyopathy and congestive heart failure may also be present at diagnosis of CSS [173, 179, 180]. Cardiomyopathy secondary to coronary artery vasculitis is common in adult CSS patients, occurring in 22–26 % of patients, and corresponds to advanced disease [179].
Pulmonary Complications of CSS
The literature contains multiple reports of serious pulmonary hemorrhage in patients with CSS [181, 182]. Without treatment CSS is fatal within months, secondary to pulmonary disease. Widespread use of corticosteroids has significantly improved survival and become accepted as first-line therapy [183]. While some authors consider a combination of corticosteroids with another immuno-suppressive (cyclophosphamide) to be first-line [179], others reserve cyclophosphamide for those patients who fail corticosteroids alone or have severe disease [183].
Gastrointestinal Complications of CSS
While abdominal complaints are common, serious abdominal involvement is rare. Small bowel perforation and peritonitis in association with CSS has been reported [184].
Juvenile Dermatomyositis (JDM)
Juvenile dermatomyositis (JDM) (Table 35.8) is a rare autoimmune inflammatory muscle disease with an incidence of 1.7–3.1 per million children, affecting twice as many females as males [185–188]. JDM is characterized by proximal muscle weakness, heliotrope rash, and elevated serum levels of skeletal muscle enzymes such as creatinine phosphokinase (CPK), aldolase, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and lactate dehydrogenase (LDH) [186, 189]. Muscle biopsy specimens reveal complement-mediated microvascular endothelial damage, which leads to local hypoperfusion and perifascicular atrophy [187]. Overall mortality rate among children with JDM is approximately 1.5 % [185].
CNS Complications of JDM
Although extremely rare, CNS complications of JDM are serious and life threatening. CNS involvement likely represents manifestation of severe, uncontrolled disease [188, 190]. Reported manifestations include visual disturbances, seizures, pseudoseizures, psychosis, and brainstem infarction [186, 190]. Seizures are likely the most common manifestation, but are much less common in JDM than in other vasculitides such as SLE [186]. Immunosuppressive therapy has been used with these complications, and has improved symptoms [190].
Pulmonary Complications of JDM
The most common pulmonary manifestations of JDM are those secondary to muscle weakness. Asymptomatic restrictive lung disease has been reported in up to 40 % of JDM patients at presentation [188]. Decreased ventilatory capacity may occur in up to 78 % of JDM patients throughout the course of the illness secondary to respiratory muscle weakness and decreased chest wall compliance [186]. Dysphagia secondary to esophageal dysmotility and pharyngeal weakness is a common symptom in JDM [186, 188], and may lead to pathologic aspiration severe enough to require parenteral nutrition and tracheostomy [186, 191].
Interstitial lung disease (ILD) is a well known complication of dermatomyositis, occurring in up to 65 % of adults [189, 192]. In children, interstitial lung disease has been less well described in association with JDM, however, it has been associated with anti-Jo-1 antibody positive patients [188], and may be detected early as a decrease in diffusion capacity [188]. Unfortunately, JDM-associated interstitial lung disease is often severe and rapidly progressive, with a high mortality [192]. Interstitial lung disease in children with JDM has been reported to occur early in the illness, particularly in patients whom the biochemical markers of ongoing disease (AST, ALT, aldolase, etc.) do not normalize with steroid therapy [189, 192]. Interstitial lung disease associated with DM has been treated with cyclosporine in both adults and children with promising results [192, 193]. Currently, however, the pediatric literature is limited to a few case reports and small series [192]. Cyclosporine is generally reserved for steroid resistant ILD or rapidly progressive ILD [189, 192].
Cardiac Complications of JDM
Clinically important cardiac involvement is rare; however, reports have been published of fatal cardiac vasculitis and cardiac tamponade with JDM [186]. Non-specific ECG abnormalities and pericarditis have also been reported in conjunction with JDM, although all of these are primarily associated with adult DM [188].
Systemic Sclerosis
Juvenile scleroderma is an autoimmune disease of unknown etiology characterized by hard skin with an onset before the age of 16 years. Two broad categories have been defined, morphea (localized skin sclerosis), and systemic sclerosis (skin sclerosis with diffuse internal organ involvement) [194]. Systemic sclerosis (SSc) is rare in children, with only 10–17 % of patients with SSc patients accounted for by those less than 20 years of age [194, 195].
Mononuclear cell infiltration is a key component of the lesions of scleroderma. Cytokine liberation by these mononuclear cells leads to immune activation, increased collagen production, and endothelial damage. Ultimately fibrosis occurs and microvascular disease results in localized tissue ischemia [194].
Management of SSc has been challenging, and no therapy as yet has been shown to have significant benefit without significant risk [194, 196]. Methotrexate trials have had mixed results and steroids have generally been ineffective. Stem cell transplantation has been studied in adults, with disease improvement in 70 % of patients, but 19 % of patients had further disease progression and 17 % died from complications of the transplant itself [194]. One series describing the use of etanercept in ten adults with SSc published in abstract form showed no clinically important improvements [196].
Cardiopulmonary Complicationsof Systemic Sclerosis
Clinically insignificant cardiac disease is common in SSc, but severe cardiopulmonary involvement may be a major cause of morbidity and mortality among SSc patients [194, 195]. Involvement may come in the form of myocardial fibrosis leading to cardiomyopathy or arrhythmia, valvar insufficiency, or pericardial disease [194, 197]. Systolic [198] and diastolic [197, 199] dysfunction have been reported with SSc. Pulmonary hypertension with or without pulmonary fibrosis is another well-known complication of SSc [194, 200].
Autopsy series including both children and adults have shown myocardial fibrosis to be common in patients with SSc [201]. Myocardial fibrosis may lead to cardiomyopathy and arrhythmias including atrioventricular block, supraventricular tachycardia, and ventricular tachycardia [194, 197]. Frank cardiomyopathy is uncommon in pediatric systemic sclerosis, although dilated cardiomyopathy may be more common in children who have myositis in addition to systemic sclerosis [202]. Rokicki reported finding diastolic dysfunction among SSc patients without cardiac symptoms, as well as increased left ventricular mass without overt hypertrophy [197].
Steroid therapy has proven generally ineffective for the management of SSc, except in early muscle involvement or the edematous phase of cutaneous disease, and may be associated with renal crisis [194]. Nevertheless, given the autoimmune component of this disease, authors have reported the use of glucocorticoids in SSc cardiomyopathy [203]. In 32 adult patients with scleroderma and secondary myocardial dysfunction, prednisolone treatment was shown to increase LVEF, particularly in patients with the SSc form [198].
Subclinical pericardial disease has been reported as a common occurrence in autopsy series of SSc patients, found in 33–72 % of specimens [201]. Clinical pericardial involvement has been reported in 7–20 % of patients during the disease course [201]. However, Rokicki and colleagues evaluated 43 children with various forms of scleroderma and found no pericardial disease by clinical exam or echocardiography [197]. Rokiciki implied that pericardial disease may be a component of adult SSc as opposed to juvenile SSc [197].
Conduction abnormalities may occur in up to 60 % of adult patients with SSc, particularly in patients with concurrent myositis [204]. These patients are at high risk for sudden death; Follansbee and colleagues found retrospectively that 12 of 25 adult patients with SSc and myositis experienced sudden cardiac deaths [204]. Conduction system disease is often asymptomatic with the first manifestations being slight resting tachycardia and a decrease in heart rate variability attributable to dysautonomia [205].
Pulmonary fibrosis with pulmonary vascular disease has been reported as the most common cause of death among adult patients with SSc [200]. In adults, these two entities generally occur in simultaneously. While this may be the case in children, isolated pulmonary vascular disease is more common in young patients with scleroderma, in particular with SSc [194]. Unfortunately, this isolated pulmonary vascular disease has a much worse prognosis [194]; also, for reasons that remain unclear, pulmonary arterial hypertension (PAH) in scleroderma patients carries a higher risk of death than similarly affected patients with primary pulmonary hypertension [206]. In SSc patients as a whole, PAH likely has a prevalence of 12–15 %, but values as high as 50 % have been published [207–209]. The pulmonary vascular disease associated with SSc is similar in pathology and management to primary pulmonary hypertension [206, 209, 210]. Pathologic evaluation shows prominent intimal proliferation, medial hypertrophy, and adventitial fibrosis [206].
Prostacyclin analogues iloprost [Ventavis®, CoTherix] [211], epoprostenol [Flolan®, GlaxoSmithKline] [209], and treprostinil [Remodulin®, United Therapeutics] [212] have all been evaluated in adults with pulmonary hypertension secondary to the scleroderma. Improvements in exercise tolerance were seen with all; however the greatest improvement in pulmonary hemodynamics was seen with epoprostenol. Epoprostenol is currently considered the “gold standard” for management of pulmonary hypertension unresponsive to acute vasodilators [213]. Further studies with treprostinil at optimal doses may show similar efficacy with greater ease of administration (subcutaneous infusion) and elimination of the need for indwelling venous catheters. Systemic sclerosis patients with pulmonary hypertension which does not respond to prostacyclin analogues have been treated with concurrent burst cyclophosphamide with marked improvement in hemodynamic measurements [214].
Endothelin-1 is a potent vasoactive peptide which acts through two receptors, “A” and “B”, and has been implicated in various forms of pulmonary hypertension. Bosentan (Tracleer®, Actelion Pharmaceuticals) is the first orally available non-selective endothelin receptor antagonist approved for use in severe pulmonary hypertension. Channick and colleagues [215] reported significant improvements in pulmonary hemodynamics and exercise tolerance in adults with scleroderma and pulmonary arterial hypertension after 3 months of bosentan therapy. The same group reported after 1 year of treatment that these improvements had been sustained with ongoing bosentan therapy [216]. Initial monotherapy for pulmonary hypertension complicating SSc in adults is generally endothelin blockade [210] and successful use of endothelin blockade in pediatric SSc associated pulmonary hypertension has been reported [217].
In adult patients with systemic sclerosis and severe lung disease unresponsive to medical management, lung transplantation has been performed. Several case series have found that the outcomes of patients with scleroderma who undergo lung transplantation for severe pulmonary disease (both with and without pulmonary hypertension) have similar outcomes to individuals with primary lung disease in the absence of rheumatologic disease [218–220].
Scleroderma Renal Crisis
Scleroderma renal crisis is the abrupt onset of severe hypertension and rapidly worsening oliguric renal failure occurring in the setting of scleroderma [221]. It is usually accompanied by left ventricular failure and microangiopathic hemolytic anemia [221]. The pathophysiology is as yet unclear; however it likely involves vascular disease of the small arteries and arterioles of the kidney. This vascular disease causes decreased perfusion, which in turn leads to angiotensin-II mediated hypertension [221]. Some authors have implicated the recent addition of high dose steroids or non-steroidal anti-inflammatory medications in the development of scleroderma renal crisis [221].
Scleroderma renal crisis occurs in 14–18 % of adult patients with SSc during the course of their disease [221, 222]. With a mortality rate approaching 35 % despite aggressive antihypertensive management [221, 222], Scleroderma renal crisis is the most common cause of death in adult patients with SSc. Survivors usually have permanent renal damage, and may be dialysis dependent [221]. Management consists of aggressive antihypertensive therapy, and the use of ACE inhibitors (specifically captopril) has proven to improve survival in adult patients [221].
Behcet’s Disease
Behcet’s disease (BD) is a multisystem vasculitis of unknown etiology characterized by recurrent oral ulcers, genital ulcers, and uveitis [223, 224]. A positive pathhergy test (formation of a papule or pustule 48 h after skin pin-prick or intradermal saline injection) supports the diagnosis of BD but is not required [223]. In Japanese and Middle Eastern patients, HLA-B51 has been associated with BD, although this association does not hold true for Western patients [223, 224]. Behcet’s disease is uncommon in childhood, generally diagnosed in adults in the third decade of life, and is most prevalent in Japan, Turkey, Iran, and Mediterranean countries [223].
Systemic involvement with BD includes nearly all organ systems, and a wide range of severity. Central nervous involvement associated with BD includes headaches, aseptic meningitis, mild intracranial hypertension [225], organic psychiatric disturbances, seizures, hemiparesis, and sinovenous thrombosis [223]. Pulmonary involvement may include pleuritis [225], hemoptysis, parenchymal infarction, and pulmonary vasculitis with inflammatory aneurysms [223, 226]. Rupture of pulmonary artery aneurysms has resulted in hemorrhage and death [223, 226]. Cardiac involvement can include myocarditis, arrhythmia [223], myocardial fibrosis, or endocardial fibrosis [226]. Arterial and venous vascular thromboses are associated with BD, although more commonly in adult patients [225]. Multiple thromboses may be present and can lead to death [223]. Gastrointestinal manifestations of BD include colicky abdominal pain, diarrhea, bloody stools, and ulcerative colitis [223, 225]. Supportive medical care with corticosteroids is the mainstay of therapy for acute exacerbations or complications of BD [227].
Conclusion
Children with rheumatologic diseases require admission to the PICU for a multitude of diagnoses, affecting all organ systems. In addition to the broad spectrum of disease, the limited number of pediatric trials makes caring for these patients challenging. Supportive care remains the cornerstone of therapy in these patients while disease specific therapies (Table 35.9) continue to evolve. Continuing research into the pathophysiology of these diseases and new therapies will improve patient care and patient outcomes.
References
King KK, Kornreich HK, Bernstein BH, et al. The clinical spectrum of systemic lupus erythematosus in childhood. Arthritis Rheum. 1977;20:287.
Schneider R, Passo MH. Juvenile rheumatoid arthritis. Rheum Dis Clin N Am. 2002;28:503–30.
Miller ML, Pachman LM. Vasculitis syndromes. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson textbook of pediatrics. 17th ed. Philadelphia: Saunders; 2004. p. 826–31.
Schärer K, Krmar R, Querfeld U, et al. Clinical outcome of Schönlein-Henoch purpura nephritis in children. Pediatr Nephrol. 1999;13(9):816–23.
Janssen NM, Karnad DP, Guntupalli KK. Rheumatologic disease in the intensive care unit : epidemiology, clinical approach, management, and outcome. Crit Care Clin. 2002;18:729–48.
Hahn BH, Karpouzas GA, Tsa BP. Pathogenesis of systemic lupus erythematosus. In: Harris ED, Budd RC, Genovese MC, Firestein GS, Sargent JS, Sledge CB, editors. Kelley’s textbook of rheumatology. 7th ed. Philadelphia: Saunders; 2005. p. 1174–201.
Edworthy SM. Clinical manifestations of systemic lupus erythematosus. In: Harris ED, Budd RC, Genovese MC, Firestein GS, Sargent JS, Sledge CB, editors. Kelley’s textbook of rheumatology. 7th ed. Philadelphia: Saunders; 2005. p. 1201–25.
Klein-Gitelman M, Reiff A, Silverman ED. Systemic lupus erythematosus in childhood. Rhem Dis Clin N Am. 2002;28:561–77.
Iqbal S, Sher MR, Good RA, et al. Diversity in presenting manifestations of systemic lupus erythematosus in children. J Pediatr. 1999;135:500–5.
Font J, Cervera R, Ramos-Casals M, et al. Clusters of clinical and immunologic features in systemic lupus erythematosus: analysis of 600 patients from a single center. Sem Arth Rheum. 2004;33:217–30.
Williams FMK, Chinn S, Hughes GRV, et al. Critical illness in systemic lupus erythematosus and antiphospholipid syndrome. Ann Rheum Dis. 2002;66:414–21.
Appel GB, Radhakrishnan J, D’Agati VD. Secondary glomerular disease. In: Brenner BM, editor. Brenner and Rector’s: the kidney. 7th ed. St. Louis: W.B. Saunders; 2004. p. 1382–97.
Wijetunga M, Rockson S. Myocarditis in systemic lupus erythematosus. Am J Med. 2002;113:419–23.
Olfat M, Al-Mayouf SM, Muzaffer MA. Pattern of neuropsychiatric manifestations and outcome in juvenile systemic lupus erythematosus. Clin Rhematol. 2004;23:395–9.
Hussain HMI, Loh WF, Sofiah A. Childhood cerebral lupus in an oriental population. Brain Dev. 1999;21:229–35.
Butani L, Makker SP. Life-threatening extrarenal lupus in children despite improvement in serologic findings. Rhematol Int. 2003;23:93–5.
ACRAd Hoc Committee. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42(4):599–608.
Sibbitt Jr WL, Brandt JR, Johnson CR, et al. The incidence and prevalence of neuropsychiatric syndromes in pediatric onset systemic lupus erythematosus. J Rheumatol. 2002;29:1536–42.
Spinosa MJ, Bandeira M, Liberalesso PBN, et al. Clinical, laboratory and neuroimage findings in juvenile systemic lupus erythematosus presenting involvement of the nervous system. Arq Neuropsiquiatr. 2007;65(2-B):433–9.
Quintero-Del-Rio AI, Miller V. Neurologic symptoms in children with systemic lupus erythematosus. J Child Neurol. 2000;15:803–7.
Steinlin MI, Blaser SI, Gilday DL, et al. Neurologic manifestations of pediatric systemic lupus erythematosus. Pediatr Neurol. 1995;13:191–7.
Parikh S, Swaiman KF, Kim Y. Neurologic characteristics of childhood lupus erythematosus. Pediatr Neurol. 1995;13:198–201.
Rood MJ, ten Cate R, van Suijlekom-Smit LW, et al. Childhood-onset systemic lupus erythematosus: clinical presentation and prognosis in 31 patients. Scand J Rheumatol. 1999;28:222–6.
Eren M, Baskin E, Cila A, et al. Mannitol treatment in central nervous system lupus. Clin Rheumatol. 2001;20:160–1.
Mecarelli O, de Feo MR, Accornero N, et al. Systemic lupus erythematosus and myoclonic epileptic manifestations. Ital J Neurol Sci. 1999;20:129–32.
Uziel Y, Laxer RM, Blaser S, et al. Cerebral vein thrombosis in childhood systemic lupus erythematosus. J Pediatr. 1995;126:722–7.
Carvalho KS, Garg BP. Cerebral venous thrombosis and venous malformations in children. Neurol Clin N Am. 2002;20:1061–77.
Einhaupl KM, Villringer A, Meister W, et al. Heparin treatment in sinus venous thrombosis. Lancet. 1991;338:597–600.
Sébire G, Tabarki B, Saunders DE, et al. Cerebral venous sinus thrombosis in children: risk factors, presentation, diagnosis and outcome. Brain. 2005;128:477–89.
Carlin TM, Chanmugam A. Stroke in children. Emerg Med Clin N Am. 2002;20:671–85.
de Bruijn SFTM, Stam J, Cerebral Venous Sinus Thrombosis Study Group. Randomized, placebo-controlled trial of anticoagulant treatment with low-molecular-weight heparin for cerebral sinus thrombosis. Stroke. 1999;30:484–8.
DeVeber G, Andrew M, Adams C, et al. Cerebral sinovenous thrombosis in children. N Engl J Med. 2001;345:417–23.
Dix D, Andrew M, Marzinotto V, et al. The use of low molecular weight heparin in pediatric patients: a prospective cohort study. J Pediatr. 2000;136:439–45.
Gupta AA, Leaker M, Andrew M, et al. Safety and outcomes of thrombolysis with tissue plasminogen activator for treatment of intravascular thrombosis in children. J Pediatr. 2001;139:682–8.
Lanthier S, Carmant L, David M, et al. Stroke in children: the coexistence of multiple risk factors predicts poor outcome. Neurology. 2000;54:371–8.
Avcin T, Benseler SM, Tyrrell PN, et al. A followup study of antiphospholipid antibodies and associated neuropsychiatric manifestations in 137 children with systemic lupus erythematosus. Arthritis Rheum. 2008;59(2):206–13.
Steens SC, Bosma GP, Cate RT, et al. A neuroimaging follow up study of a patient with juvenile central nervous system systemic lupus erythematosus. Ann Rheum Dis. 2003;62:583–6.
Gieron MA, Khoromi S, Campos A. MRI changes in the central nervous system in a child with lupus erythematosus. Pediatr Radiol. 1995;25:184–5.
Turkel SB, Miller JH, Reiff A. Case series: neuropsychiatric symptoms with pediatric systemic lupus erythematosus. J Am Acad Child Adolesc Psychiatry. 2001;40:482–5.
Russo R, Gilday D, Laxer RM, et al. Single photon emission computed tomography scanning in childhood systemic lupus erythematosus. J Rheumatol. 1998;25:576–82.
Reiff A, Miller J, Shaham B, et al. Childhood central nervous system lupus; longitudinal assessment using single photon emission computed tomography. J Rheumatol. 1997;24:2461–5.
Baca V, Lavalle C, García R, et al. Favorable response to intravenous methylprednisolone and cyclophosphamide in children with severe neuropsychiatric lupus. J Rheumatol. 1999;26:432–9.
Stojanovich L, Stojanovich R, Kostich V, et al. Neuropsychiatric lupus favorable response to low dose i.v. cyclophosphamide and prednisolone (pilot study). Lupus. 2003;12:3–7.
Raj R, Murin S, Matthay RA, et al. Systemic lupus erythematosus in the intensive care unit. Crit Care Clin. 2002;18:781–803.
Ciftçi E, Yalçinkaya F, Ince E, et al. Pulmonary involvement in childhood-onset systemic lupus erythematosus: a report of five cases. Rheumatology. 2004;43:587–91.
Chetan G, Mahadevan S, Sulanthung K, Narayanan P. Intravenous immunoglobulin therapy of lupus pneumonitis. Indian J Pediatr. 2007;74(11):1032–3.
Hellmann DB, Petri M, Whiting-O’Keefe Q. Fatal infections in systemic lupus erythematosus: the role of opportunistic organisms. Medicine. 1987;66:341–8.
Vijatov-Djuriic G, Stojanovic V, Konstantinidis N, Konstantinidis G. Systemic lupus erythematosus complicated with pulmonary hemorrhage in a a17-year-old female. Lupus. 2010;18:1561–4.
Kolovos NS, Schuerer DJ, Moler FW, et al. Extracorporeal life support for pulmonary hemorrhage in children: a case series. Crit Care Med. 2002;30:577–80.
Fitch JA. Emergency and critical care issues in pediatric rheumatology. Rheum Dis Clin N Am. 1997;23:439–60.
Barton EN, Chung EE, Smikle MF, et al. Asymptomatic cardiac involvement in systemic lupus erythematosus. West Indian Med J. 1995;44:14–5.
McDonnell JK. Cardiac disease and the skin. Dermatol Clin. 2002;20:503–11.
Malcić I, Senecić I, Dasović A, Radonić M. Incipient pericardial tamponade as the first symptom of systemic lupus erythematosus in 2 children. Reumatizam. 1995;42:19–22.
Longo MJ, Remetz MS. Cardiovascular manifestations of systemic autoimmune diseases. Clin Chest Med. 1998;19:793–808.
Wang DY, Yang PC, Yu WL, et al. Comparison of different diagnostic methods for lupus pleuritis and pericarditis: a prospective three-year study. J Formos Med Assoc. 2000;99(5):375–80.
DeSilva TN. Management of collagen vascular diseases in childhood. Dermatol Clin. 1998;16:579–92.
Pisetsky DS. Systemic lupus erythematosus. In: Klippel JH, editor. Primer on the rheumatic diseases. 12th ed. Atlanta: Arthritis Foundation; 2001. p. 329–34.
Sondheimer HM, Lorts A. Cardiac involvement in inflammatory disease: systemic lupus erythematosus, rheumatic fever, and Kawasaki disease. Adolesc Med. 2001;12:69–78.
Nadorra RL, Landing BH. Pulmonary lesions in childhood onset systemic lupus erythematosus: analysis of 26 cases, and summary of literature. Pediatr Pathol. 1987;7:1–18.
Durand I, Blaysat G, Chauvaud S, et al. Extensive fibrous endocarditis as first manifestation of systemic lupus erythematosus. Arch Fr Pediatr. 1993;50:685–8.
Walsh DS, Farley MF, Beard JS, et al. Systemic lupus erythematosus: nephritis, dilated cardiomyopathy, and extensive cutaneous depigmentation responsive to hydroxychloroquine. J Am Acad Dermatol. 1995;33:828–30.
James KB, Ratliff N, Starling R, et al. Inflammatory cardiomyopathy. The controversy of diagnosis and management. Rheum Dis Clin N Am. 1997;23:333–43.
Frustaci A, Gentiloni N, Caldarulo M. Acute myocarditis and left ventricular aneurysm as presentations of systemic lupus erythematosus. Chest. 1996;109:282–4.
Gottenberg JE, Roux S, Assayag P, et al. Specific cardiomyopathy in lupus patients: report of three cases. Joint Bone Spine. 2004;71:66–9.
Naarendorp M, Kerr LD, Khan AS, et al. Dramatic improvement of left ventricular function after cytotoxic therapy in lupus patients with acute cardiomyopathy: report of 6 cases. J Rheumatol. 1999;26:2257–60.
Pulerwitz TC, Cappola TP, Felker GM, et al. Mortality in primary and secondary myocarditis. Am Heart J. 2004;147:746–50.
Ansell SM, Bedhesi S, Ruff B, et al. Study of critically ill patients with systemic lupus erythematosus. Crit Care Med. 1996;24:981–4.
Huicochea Grobet ZL, Berrón R, Ortega Martell JA, et al. Survival up to 5 and 10 years of Mexican pediatric patients with systemic lupus erythematosus. Overhaul of 23 years experience. Allergol Immunopathol (Madr). 1996;24:36–8.
Godeau B, Mortier E, Roy PM, et al. Short and long-term outcomes from patients with systemic rheumatoid disease admitted to intensive care units: a prognostic study of 181 patients. J Rheumatol. 1997;24:1317–23.
Bouza E, Garcia-Lechuz J, Munoz P. Infections in systemic lupus erythematosus and rheumatoid arthritis. Inf Dis Clin N Am. 2001;15:335–61.
Kim WU, Min JK, Lee SH, et al. Causes of death in Korean patients with systemic lupus erythematosus: a single center retrospective study. Clin Exp Rheumatol. 1999;17:539–45.
Chen Y, Yang Y, Lin Y, et al. Risk of infection in hospitalized children with systemic lupus erythematosus: a 10 year follow-up. Clin Rheumatol. 2004;23:235–8.
Stuck AE, Minder CE, Frey FJ. Risk of infectious complications in patients taking glucocorticoseteroids. Rev Infect Dis. 1989;11:954–63.
Segal BH, Sneller MC. Infectious complications of immunosuppressive therapy in patients with rheumatic diseases. Rheum Dis Clin N Am. 1997;23:219–37.
Petri M. Infection in systemic lupus erythematosus. Rheum Dis Clin N Am. 1998;24:423–56.
Staples PJ, Gerding DN, Decker JL, et al. Incidence of infection in systemic lupus erythematosus. Arthritis Rheum. 1974;17:1–10.
Miller ML, Cassidy JT. Juvenile rheumatoid arthritis. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson textbook of pediatrics. 17th ed. Philadelphia: Saunders; 2004. p. 799–805.
Cassidy JT. Juvenile rheumatoid arthritis. In: Harris ED, Budd RC, Genovese MC, Firestein GS, Sargen JS, Sledge CB, editors. Kelley’s textbook of rheumatology. 7th ed. Philadelphia: Saunders; 2005. p. 1579–96.
Mease PJ. Disease-modifying antirheumatic drug therapy for spondyloarthropathies: advances in treatment. Curr Opin Rheumatol. 2003;15:205–12.
Quartier P, Taupin P, Bordeaut F, et al. Efficacy of etanercept for the treatment of juvenile idiopathic arthritis according to the onset type. Arthritis Rheum. 2003;48:1093–101.
Weinblatt ME, Kremer JM, Bankhurst AD, et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. N Engl J Med. 1999;340:253–9.
Kroesen S, Widmer AF, Tyndall A, Hasler P. Serious bacterial infections in patients with rheumatoid arthritis under anti-TNF-alpha therapy. Rheumatology. 2003;42:617–21.
Culy CR, Keating GM. Etanercept: an updated review of its use in rheumatoid arthritis, psoriatic arthritis, and juvenile rheumatoid arthritis. Drugs. 2002;62:2493–537.
Fisher CJ, Agosti JM, Opal SM, et al. Treatment of septic shock with the tumor necrosis factor receptor: Fc fusion protein. N Engl J Med. 1996;334:1697–702.
Keane J, Gerson S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor α-neutralizing agent. N Engl J Med. 2001;345:1098–104.
Bernstein D. Diseases of the myocardium and pericardium. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson textbook of pediatrics. 17th ed. Philadelphia: Saunders; 2004. p. 1580.
Park MK. Cardiac involvement in systemic diseases. In: Park MK, editor. Pediatric cardiology for practitioners. 4th ed. St. Louis: Mosby Inc; 2002. p. 327.
Yancey CL, Doughty RA, Cohlan BA, Athreya BH. Pericarditis and cardiac tamponade in juvenile rheumatoid arthritis. Pediatrics. 1981;68:369–73.
Alukal MK, Costello PB, Green FA. Cardiac tamponade in systemic juvenile rheumatoid arthritis requiring emergency pericardiectomy. J Rheumatol. 1984;11:222–5.
Majeed HA, Kvasnicka J. Juvenile rheumatoid arthritis with cardiac tamponade. Ann Rheum Dis. 1978;37:273–6.
Ozer S, Alehan D, Ozme S, Bakkaloglu A, Soylemezoglu O. Mitral and aortic insufficiency in polyarticular juvenile rheumatoid arthritis. Pediatr Cardiol. 1994;15:151–3.
Bultink IEM, Lems WF, Dijkmans BAC, van Soesbergen RM, Lindeman J. Severe aortic regurgitation in RF positive polyarticular JIA. Ann Rheum Dis. 2002;61:282–3.
Huppertz HI, Voigt I, Müller-Scholden J, Sandhage K. Cardiac manifestations in patients with HLA B27-associated juvenile arthritis. Pediatr Cardiol. 2000;21:141–7.
Oguz D, Ocal B, Ertan U, Narin H, Karademir S, Senocak F. Left ventricular diastolic functions in juvenile rheumatoid arthritis. Pediatr Cardiol. 2000;21:374–7.
Mouy R, Stephan J, Pillet P, Haddad E, Hubert P, Prieur A. Efficacy of cyclosporine A in the treatment of macrophage activation syndrome in juvenile arthritis: report of five cases. J Pediatr. 1996;129:750–4.
Ravelli A, Caria MC, Buratti S, Malattia C, Temporini F, Martini A. Methotrexate as a possible trigger of macrophage activation syndrome. J Rheumatol. 2001;28:865–7.
Davi S, Consolaro A, Guseinova D, et al. An international consensus survey of diagnostic criteria for macrophage activation syndrome in systemic juvenile idiopathic arthritis. J Rheumatol. 2011;38(4):764–8.
Grom AA, Passo M. Macrophage activation syndrome in systemic juvenile rheumatoid arthritis. J Pediatr. 1996;129:630–2.
Silva CAA, Silva CHM, Robazzi TCMV, et al. Macrophage activation syndrome associated with systemic juvenile idiopathic arthritis. J Pediatr (Rio J). 2004;80:517–22.
Billiau AD, Roskams T, Van Damme-Lombaerts R, Matthys P, Wouters C. Macrophage activation syndrome: characteristic findings on liver biopsy illustrating the key role of activated, INF-γ-producing lymphocytes and IL-6 and TNF-α-producing macrophages. Blood. 2005;105:1648–51.
Sampath P, Bove K, Dickens D, Lovell D, Grom A. Etanercept in the treatment of macrophage activation syndrome. J Rheumatol. 2001;28:2120–4.
Ramanan AV, Schneider R. Macrophage activation syndrome following initiation of etanercept in a child with systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2003;30:401–3.
Miettunen PM, Narendran A, Jayanthan A, et al. Successful treatment of severe paediatric rheumatic disease associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford). 2011;50:417–9.
Bruk N, Suttorp M, Kabus M, et al. Rapid and sustained remission of systemic juvenile idiopathic artheritis associated macrophage activation syndrome through treatment with anakinra and corticosteroids. J Clin Rheumatol. 2011;17:23–7.
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of toclizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomized, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371:998–1006.
Yokota S, Kishimoto T. Tocilizumab: molecular intervention therapy in children with systemic juvenile idiopathic arthritis. Exp Rev Clin Immunol. 2010;6(5):7335–743.
Gershoni-Baruch R, Broza Y, Brik R, et al. Prevalence and significance of mutations in the familial Mediterranean fever gene in Henoch-Schönlein purpura. J Pediatr. 2003;143:658–61.
Rai A, Nast C, Adler S. Henoch-Schönlein purpura nephritis. J Am Soc Nephrol. 1999;10(12):2637–44.
Ayoub EM, McBride J, Schmiederer M, et al. Role of Bartonella henselae in the etiology of Henoch-Schönlein purpura. Pediatr Infect Dis J. 2002;21:28–31.
Belman AL, Leicher CR, Moshe SL, et al. Neurologic manifestations of Schoenlein-Henoch purpura: report of three cases and review of the literature. Pediatrics. 1985;74:687–92.
Sokol DK, McIntyre JA, Short RA, et al. Henoch-Schönlein purpura and stroke: antiphosphatidyl-ethanolamine antibody in CSF and serum. Neurology. 2000;55:1379–81.
Nadeau SE. Neurologic manifestations of systemic vasculitis. Neurol Clin. 2002;20:123–50.
Paller AS, Kelly K, Sethi RM. Pulmonary hemorrhage: an often fatal complication of Henoch-Schoenlein Purpura. Pediatr Dermatol. 1997;14:299–302.
Esaki M, Matsumoto T, Nakamura S, et al. GI involvement in Henoch-Schönlein purpura. Gastrointest Endosc. 2002;56:920–3.
Agraharkar M, Gokhale S, Le L, et al. Cardiopulmonary manifestations of Henoch-Schönlein purpura. Am J Kidney Dis. 2000;35:319–22.
Robson WL, Leung AK. Henoch-Schönlein purpura and myocardial necrosis. Am J Kidney Dis. 2000;36(1):225–6.
Fielding RE, Hawkis CP, Hand MF, et al. Seizures complicating adult Henoch–Schonlein purpura. Nephrol Dial Transplant. 1998;13:761–2.
Ha TS, Cha SH. Cerebral vasculitis in Henoch-Schönlein purpura: a case report with sequential magnetic resonance imaging. Pediatr Nephrol. 1996;10:634–6.
Woolfenden AR, Hukin J, Poskitt KJ, et al. Encephalopathy complicating Henoch-Schönlein purpura: reversible MRI changes. Pediatr Neurol. 1998;19:74–7.
Chen CL, Chiou YH, Wu CY, et al. Cerebral vasculitis in Henoch-Schönlein purpura: a case report with sequential magnetic resonance imaging changes and treated with plasmapheresis alone. Pediatr Nephrol. 2000;15:276–8.
Chiaretti A, Caresta E, Piastra M, et al. Cerebral hemorrhage in Henoch Schoenlein syndrome. Child Nerv Syst. 2002;18:365–7.
Ng CC, Huang SC, Huang LT. Henoch-Schönlein purpura with intracerebral hemorrhage: case report. Pediatr Radiol. 1996;26:276–7.
Imai T, Okada H, Nanba M, et al. Henoch-Schönlein purpura with intracerebral hemorrhage. Brain Dev. 2002;24:115–7.
Nadrous HF, Yu AC, Specks U, Ryu JH. Pulmonary involvement in Henoch-Schönlein purpura. Mayo Clin Proc. 2004;79:1151–7.
Wright WK, Krous HF, Griswold WR, et al. Pulmonary vasculitis with hemorrhage in anaphylactoid purpura. Pediatr Pulmonol. 1994;17:269–71.
Vats KR, Vats A, Kim Y, et al. Henoch-Schönlein purpura and pulmonary hemorrhage: a report and literature review. Pediatr Nephrol. 1999;13:530–4.
Al-Harbi NN. Henoch-Schönlein nephritis complicated with pulmonary hemorrhage but treated successfully. Pediatr Nephrol. 2002;17:762–4.
Choong CK, Beasley SW. Intra-abdominal manifestations of Henoch-Schoenlein purpura. J Paediatr Child Health. 1998;34:405–9.
Lippl F, Huber W, Werner M, et al. Life-threatening gastrointestinal bleeding due to a jejunal lesion of Henoch-Schönlein purpura. Endoscopy. 2001;33:811–3.
Chen SY, Kong MS. Gastrointestinal manifestations and complications of Henoch-Schönlein purpura. Chang Gung Med J. 2004;27:175–81.
Chang WL, Yang YH, Lin YT, Chiang BL. Gastrointestinal manifestations in Henoch-Shonlein purpura: a review of 261 patients. Acta Paediatr. 2004;93:1427–31.
Bogdanovic R. Henoch-Schonlein purpura nephritis in children: risk factors, prevention and treatment. Acta Paeditrica. 2009;98:1882–9.
Ninchoji T, Kaito H, Nozu K, et al. Treatment strategies for Henoch-Schonelein purpura nephritis by histological and clinical severity. Pediatr Nephrol. 2011;26:563–9.
Nikibakhsh AA, Mahmoodzadeh H, Karamyyar M, et al. Treatment of complicated Henoch-Schonlein purpura with mycophenolate mofetil: a retrospective case series report. Int J Rheum. 2010;2010:254316. Epub 2010 Jun 15.
Newburger JW, Takahashi M, Gerber A, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114:1708–33.
Rowley AH, Shulman ST. Kawasaki disease. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson textbook of pediatrics. 17th ed. Philadelphia: Saunders; 2004. p. 823–6.
Wilson N, Heaton P, Calder L, Nicholson R, Stables S, Gavin R. Kawasaki disease with severe cardiac sequelae: lessons from recent New Zealand experience. J Paediatr Child Health. 2004;40:524–9.
Hunsaker DM, Hunsaker 3rd JC, Adams KC, Noonan JA, Ackermann DM. Fatal Kawasaki disease due to coronary aneurysm rupture with massive cardiac tamponade. J Ky Med Assoc. 2003;101:233–8.
Mok GC, Sung RY, Yam M, Arifi AA, Lam WW, Fok TF. A child with Kawasaki disease who survived after rupture of a coronary artery aneurysm. Eur J Pediatr. 2003;162:634–6.
Hwong TM, Arifi AA, Wan IYP, et al. Rupture of a giant coronary artery aneurysm due to Kawasaki disease. Ann Thorac Surg. 2004;78:693–5.
Muise A, Tallett SE, Silverman ED. Are children with Kawasaki disease and prolonged fever at risk for macrophage activation syndrome? Pediatrics. 2003;112:e495–7.
Kanegaye JT, Wilder MS, Molkara D, et al. Recognition of a Kawasaki disease shock syndrome. Pediatrics. 2009;123(5):e783–9.
Dominguez SR, Friedman K, Seewald R, et al. Kawaski disease in a pediatric intensive care unit: a case-control study. Pediatrics. 2008;122(4):e786–90.
Thabet F, Bafaqik H, Al-Mohaimeed S, et al. Shock: an unusual presentation of Kawasaki disease. Eur J Pediatr. 2011;170:941–3.
Jibiki T, Terai M, Kurosak T, et al. Efficacy of intravenous immune globulin therapy combined with dexamethasone for the initial treatment of acute Kawasaki disease. Eur J Pediatr. 2004;163:229–33.
Etheridge SP, Tani LY, Minich LL, Revenaugh JR. Platelet glycoprotein IIb/IIIa receptor blockade therapy for large coronary aneurysms and thrombi in Kawasaki disease. Cathet Cardiovasc Diagn. 1998;45:264–8.
Williams RV, Wilke VM, Tani LY, Minich LL. Does abciximab enhance regression of coronary aneurysms resulting from Kawasaki disease? Pediatrics. 2002;109:E4.
Freeman AF, Shulman ST. Refractory Kawasaki disease. Pediatr Infect Dis J. 2004;23:463–4.
Burns JC, Mason WH, Hauger SB, et al. Infliximab treatment for refractory Kawasaki syndrome. J Pediatr. 2005;146:662–7.
Burns JC, Best BM, Mejias A, et al. Infliximab treatment of intravenous immunoglobulin-resistant Kawasaki disease. J Pediatr. 2008;153(6):833–8.
Rowley AH, Shulman ST. Recent advances in the understanding and management of Kawasaki disease. Curr Infect Dis Rep. 2010;12(2):96–102.
Ravelli A, Martini A. Antiphospholipid syndrome. Pediatr Clin N Am. 2005;52:469–91.
Barlas S, Tansel T. Antiphospholipid syndrome in a child: an insight into the pathology, identification, and means of cure. J Pediatr Surg. 2004;39:1280–2.
McIntyre JA, Wagenknecht DR, Waxman DW. Frequency and specificities of antiphospholipid antibodies in volunteer blood donors. Immunobiology. 2003;207:59–63.
Campos LMA, Kiss MHB, D’Amico EA, Silva CAA. Antiphospholipid antibodies in 57 children and adolescents with systemic lupus erythematosus. Rev Hosp Clin Fac Med S Paulo. 2003;58:157–62.
Avcin T, Cimaz R, Meroni PL. Recent advances in antiphospholipid antibodies and antiphospholipid syndromes in pediatric populations. Lupus. 2002;11:4–10.
von Sheven E, Athreya BH, Rose CD, Goldsmith DP, Morton L. Clinical characteristics of antiphospholipid antibody syndrome in children. J Pediatr. 1996;129:339–45.
Levine JS, Branch DW, Rauch J. Medical progress: the antiphospholipid syndrome. N Engl J Med. 2002;346:752–63.
Ravelli A, Caporali R, Di Fucia G, Zonta L, Montecucco C, Martini A. Anticardiolipin antibodies in pediatric systemic lupus erythematosus. Arch Pediatr Adolesc Med. 1994;148:398–402.
Kenet G, Sadetzki S, Murad H, et al. Facto V Leiden and antiphospholipid antibodies are significant risk factors for ischemic stroke in children. Stroke. 2000;31:1283–8.
Angelini L, Ravelli A, Caporali R, Rumi V, Nardocci N, Martini A. High prevalence of antiphospholipid antibodies in children with idiopathic cerebral ischemia. Pediatrics. 1994;94:500–3.
Lanthier S, Kirkham FJ, Mitchell LG, et al. Increased anticardiolipin antibody IgG titers do not predict recurrent stroke or TIA in children. Neurology. 2004;62:194–200.
Cervera R, Bucciarelli S, Plasin MA, et al. Catastrophic antiphospholipid syndrome (CAPS): descriptive analysis of a series of 280 patients from the “CAPS Registry”. J Autoimmun. 2009;32(3–4):240–5.
Asherson RA, Cervera R, de Groot PG, et al. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530–4.
Westney GE, Harris EN. Catastrophic antiphospholipid syndrome in the intensive care unit. Crit Care Clin. 2002;18:805–17.
Shah MK, Hugghis SY. Characteristics and outcomes of patients with Goodpasture’s syndrome. South Med J. 2002;95:1411–8.
Rodriguez W, Hanania N, Guy E, Guntupalli J. Pulmonary-renal syndromes in the intensive care unit. Crit Care Clin. 2002;18:881–95.
Ball JA, Young Jr KR. Pulmonary manifestations of Goodpasture’s syndrome. Antiglomerular basement membrane disease and related disorders. Clin Chest Med. 1998;19:777–91.
Gittins N, Basu A, Eyre J, Gholkar A, Moghal N. Cerebral vasculitis in a teenager with Goodpasture’s syndrome. Nephrol Dial Transplant. 2004;19:3168–71.
Calabrese LH, Duna G. Antineutrophil cytoplasmic antibody-associated vasculitis. In: Harris Jr ED, Budd RC, Genovese MC, Firestein GS, Sargent JS, Sledge CB, editors. Kelley’s textbook of rheumatology. 7th ed. St. Louis: Saunders; 2005. p. 1362–70.
Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488–98.
Stegmayr B, Gothefors L, Malmer B, Muller Wiefel DM, Nilsson K, Sundelin B. Wegener granulomatosis in children and young adults. A case study of ten patients. Pediatr Nephrol. 2000;14:208–13.
Sundel R, Szer I. Vasculitis in childhood. Rheum Dis Clin N Am. 2002;28:625–54.
Langford CA, Taler-Williams C, Barron KS, Sneller MC. A staged approach to the treatment of Wegener’s granulomatosis: induction of remission with glucocorticoids and daily cyclophosphamide switching to methotrexate for remission maintenance. Arthritis Rheum. 1999;42:2666–73.
Lanham JG, Elkon K. Systemic vasculitis with asthma and eosinophilia: a clinical approach to the Churg-Strauss syndrome. Medicine. 1984;63:65–81.
Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990;33:1094–100.
Keogh KA, Ulrich SU. Churg-Strauss syndrome: clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am J Med. 2003;115:284–90.
DuMouchel W, Smith ET, Beasley R, et al. Association of asthma therapy and Churg-Strauss syndrome: an analysis of post marketing surveillance data. Clin Ther. 2004;26:1092–104.
Lilly CM, Churg A, Lazarovich M, et al. Asthma therapies and Churg-Strauss syndrome. J Allergy Clin Immunol. 2002;109:S1–19.
Berlioz M, Triolo V, Sirvent N, et al. Churg-Strauss syndrome revealed by acute abdominal pain. Pediatr Pulmonol. 2001;32:92–4.
Clutterbuck EJ, Pusey CD. Severe alveolar hemorrhage in Churg-Strauss syndrome. Eur J Respir Dis. 1987;71:158–63.
Lai RS, Lin SL, Lai NS, et al. Churg-Strauss syndrome presenting with pulmonary capillaritis and diffuse alveolar hemorrhage. Scand J Rheumatol. 1998;27:230–2.
Goetzl EJ, Luce JM. Eosinophilic lung diseases. In: Murray JF, Nadel JA, editors. Textbook of respiratory medicine. 3rd ed. Philadelphia: Saunders; 2000. p. 1769–71.
Sharma MC, Safaya R, Sidhu BS. Perforation of small intestine caused by Churg-Strauss syndrome. J Clin Gastroenterol. 1996;23:232–5.
Pachman LM. Juvenile dermatomyositis: immunogenetics, pathophysiology, and disease expression. Rheum Dis Clin N Am. 2002;28:579–602.
Ramanan AV, Feldman B. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin N Am. 2002;28:833–57.
Chitnis T, Khoury SJ. Immunologic neuromuscular disorders. J Allergy Clin Immunol. 2003;111:S659–68.
Compeyrot-Lacassagne S, Feldman BM. Inflammatory myopathies in children. Pediatr Clin N Am. 2005;52:493–520.
Strange C, Highland KB. Interstitial lung disease in the patient who has connective tissue disease. Clin Chest Med. 2004;25:549–59.
Elst EF, Kamphuis SS, Prakken BJ, et al. Case report: severe central nervous system involvement in juvenile dermatomyositis. J Rheumatol. 2003;30:2059–63.
Iester A, Puleo MG, Buzzanca C, Debbia C, Callegarini L, Alpigiani MG. A case of organic psychosis in dermatomyositis. Etiopathogenetic hypothesis. Minerva Pediatr. 1994;46:565–8.
Kobayashi I, Yamada M, Takahashi Y, et al. Interstitial lung disease associated with juvenile dermatomyositis: clinical features and efficacy of cyclosporine A. Rheumatology. 2003;43:371–4.
Miyake S, Ohtani Y, Sawada M, et al. Usefulness of cyclosporine A on rapidly progressive interstitial pneumonia in dermatomyositis. Sarcoidosis Vasc Diffuse Lung Dis. 2002;19:128–33.
Zulian F. Scleroderma in children. Pediatr Clin N Am. 2005;52:521–45.
Foeldvari I, Zhavanial M, Birdi N, et al. Favorable outcome in 135 children with juvenile systemic sclerosis: results of a multi-national survey. Rheumatology. 2000;39:556–9.
Ellman MH, MacDonald PA, Hayes FA. Etanercept as treatment for diffuse scleroderma: a pilot study. Arthritis Rheum. 2000;43:S392.
Rokicki W, Dukalska M, Rubisz-Brzezinska J, Gasior Z. Circulatory system in children with localized scleroderma. Pediatr Cardiol. 1997;18:213–7.
Antoniades L, Sfikakis PP, Mavrikakis M. Glucocorticoid effects on myocardial performance in patients with systemic sclerosis. Clin Exp Rheumatol. 2001;19:431–7.
Isaeva LA, Deliagin VM, Bazhenova LK. Main manifestations of carditis in diffuse connective tissue diseases in children. Cor Vasa. 1988;30:211–7.
Hesselstrand R, Scheja A, Akesson A. Mortality and causes of death in a Swedish series of systemic sclerosis patients. Ann Rheum Dis. 1998;57:682–6.
Byers RJ, Marshall DA, Freemont AJ. Pericardial involvement in systemic sclerosis. Ann Rheum Dis. 1997;56:393–4.
Quartier P, Bonnet D, Fournet JC, et al. Severe cardiac involvement in children with systemic sclerosis and myositis. J Rheumatol. 2002;29:1767–73.
Moore EC, Cohen F, Farooki Z, Chang CH. Focal scleroderma and severe cardiomyopathy. Patient report and brief review. Am J Dis Child. 1991;145:229–31.
Follansbee WP, Zerbe TR, Medsger Jr TA. Cardiac and skeletal muscle disease in systemic sclerosis (scleroderma): a high risk association. Am Heart J. 1993;125:194–203.
Wranicz J, Zielinska M, Cygankiewicz I, Dziankowska-Bartkowiak B, Sysa-Jedrzejowska A. Early cardiovascular involvement in patients with systemic sclerosis (SSc). Med Sci Monit. 2002;8:CR78–82.
Kawut SM, Taichman DB, Archer-Chicko CL, Palevsky HI, Kimmel SE. Hemodynamics and survival in patients with pulmonary arterial hypertension related to systemic sclerosis. Chest. 2003;123:344–50.
Denton CP, Black CM. Pulmonary hypertension in systemic sclerosis. Rheum Dis Clin N Am. 2003;29:335–49.
MacGregor AJ, Canavan R, Knight C, et al. Pulmonary hypertension in systemic sclerosis: risk factors for progression and consequences for survival. Rheumatology. 2001;40:453–9.
Badesch DB, Tapson VF, McGoon MD, et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. Ann Intern Med. 2000;132:425–34.
Opitz C, Klein-Weigel PF, Riemekasten G. Sestemic Sclerosis – a systematic overview: part 2 – immunosuppression, treatment of SSC-associated vasculopathy, and treatment of pulmonary arterial hypertension. Vasa. 2011;40(1):20–30.
de la Mata J, Gomez-Sanchez MA, Aranzana M, Gomez-Reino JJ. Long-term iloprost infusion therapy for severe pulmonary hypertension in patients with connective tissue diseases. Arthritis Rheum. 1994;37:1528–33.
Oudiz RJ, Schilz RJ, Barst RJ, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126:420–7.
Rosenzweig EB, Widlitz AC, Barst RJ. Pulmonary arterial hypertension in children. Pediatr Pulmonol. 2004;38:2–22.
Cossio M, Menon Y, Wilson W, deBoisblanc BP. Life-threatening complications of systemic sclerosis. Crit Care Clin. 2002;18:819–39.
Channick RN, Simonneau G, Sitbon O, et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomized placebo-controlled study. Lancet. 2001;358:1119–23.
Sitbon O, Badesch DB, Channick RN, et al. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary arterial hypertension. Chest. 2003;124:247–54.
Shimizu M, Hashida Y, Ueno K, et al. Successful treatment with bosentan for pulmonary hypertension and reduced peripheral circulation in juvenile systemic sclerosis. Pediatr Cardiol. 2011;32:1040–2.
Levine SM, Anzueto A, Peters JI, Calhoon JH, Jenkinson SG, Bryan CL. Single lung transplantation in patients with systemic disease. Chest. 1994;105:837–41.
Pigula FA, Griffith BP, Zenati MA, Dauber JH, Yousem SA, Keenan RJ. Lung transplantation for respiratory failure resulting from systemic disease. Ann Thorac Surg. 1997;64:1630–4.
Rosas V, Conte JV, Yang SC, et al. Lung transplantation and systemic sclerosis. Ann Transplant. 2000;5:38–43.
Walker JG, Ahern MJ, Smith MD, et al. Scleroderma renal crisis: poor outcome despite aggressive antihypertensive treatment. Intern Med J. 2003;33:216–20.
Steen VD, Medseger Jr TA. Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum. 2000;43:2437–44.
Kone-Paut I, Yurdakul S, Barbari S, et al. Clinical features of Behcet’s disease in children: an international collaborative study of 86 cases. J Pediatr. 1998;132:721–5.
Dedeoglu F, Sundel RP. Vasculitis in children. Pediatr Clin N Am. 2005;52:547–72.
Krause I, Uziel Y, Guedj D, et al. Childhood Behcet’s disease clinical features and comparison with adult onset disease. Rheumatology. 1999;38:457–62.
Cohle SD, Colby T. Fatal hemoptysis from Behcet’s disease in a child. Cardiovasc Pathol. 2002;11:296–9.
Barrit R, Shannon KM. Autoimmune and inflammatory disorders. In: Goetz CG, editor. Textbook of clinical neurology. 2nd ed. Philadelphia: Saunders; 2003. p. 1124–6.
Abernathy DR, Arnold GJ, Azarnoff D, et al., editors. Mosby’s drug consult. 15th ed. St. Louis: Mosby; 2002.
Beresford MW, Cleary AG, Sills JA, Couriel J, Davidson JE. Cardio-pulmonary involvement in juvenile systemic lupus erythematosus. Lupus. 2005;14:152–8.
Harel L, Mukamel M, Brik R, Blau H, Straussberg R. Peripheral neuropathy in pediatric SLE. Pediatr Neurol. 2002;27:53–6.
Marie I, Hachulla E, Cherin P, et al. Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum. 2002;47:614–22.
Hajj-Ali RA, Chamande S, Calabrese LH, Arroliga AC. Central nervous system vasculitis in the intensive care unit. Crit Care Clin. 2002;18:897–914.
Haas JP, Metzler M, Ruder H, Waldherr R, Boswald M, Rupprecht T. An unusual manifestation of Wegener’s granulomatosis in a 4-year-old girl. Pediatr Neurol. 2002;27:71–4.
Wallace CA, French JW, Kahn SJ, Sherry DD. Initial intravenous gammaglobulin treatment failure in Kawasaki disease. Pediatrics. 2000;105:e78.
Yekeler E, Tunaci A, Tunaci M, Kamali S, Gul A, Ancunas B. Successful medical treatment of abdominal aortic aneurysms in a patient with Behcet’s disease: imaging findings. Australas Radiol. 2005;49:182–4.
Schnabel A, Reuter M, Biederer J, Richter C, Gross WL. Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum. 2003;32:273–84.
Frankel SK, Sullivan EJ, Brown K. Vasculitis: Wegener granulomatosis, Churg-Strauss syndrome, microscopic polyangiitis, polyarteritis nodosa, and Takaysu arteritis. Crit Care Clin. 2002;18:855–79.
Besbas N, Ozyurek E, Balkanci S, et al. Behcet’s disease with severe arterial involvement in a child. Clin Rheumatol. 2002;21:176–9.
Mitsui Y, Mitsui M, Urakami R, Kihara M, Takahashi M, Kusunoki S. Behcet disease presenting with neurological complications immediately after conversion from conventional cyclosporine A to microemulsion formulation. Intern Med. 2005;44:149–52.
Reiff A, Takei S, Sadeghi S, et al. Etanercept therapy in children with treatment-resistant uveitis. Arthritis Rheum. 2001;44:1411–5.
Suzuki Kurokawa M, Suzuki N. Behcet’s disease. Clin Exp Med. 2004;4:10–20.
Sarwar H, McGrath Jr H, Espinoza LR. Successful treatment of long-standing neuro-Behcet’s disease with infliximab. J Rheumatol. 2005;32:181–3.
Lindstedt EW, Baarsma GS, Kuijpers RWAM, van Hagen PM. Anti-TNF-α therapy for sight threatening uveitis. Br J Ophthalmol. 2005;89:533–6.
Wechsler B, Sable-Fourtassou R, Bodaghi B, et al. Infliximab in refractory uveitis due to Behcet’s disease. Clin Exp Rheumatol. 2004;22:S14–6.
Erkan D, Cervera R, Asherson RA. Catastrophic antiphospholipid syndrome: where do we stand? Arthritis Rheum. 2003;48:3320–7.
Shibata M, Kibe T, Ishikawa T, et al. Diffuse central nervous system lupus involving white matter, basal ganglia, thalami and brainstem. Brain Dev. 1999;21:337–40.
Vieira JP, Ortet O, Barata D, Abranches M, Gomes JM. Lupus myelopahy in a child. Pediatr Neurol. 2002;27:303–6.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag London
About this chapter
Cite this chapter
Martin, S.W., Anderson, M.R. (2014). Rheumatologic Disorders in the PICU. In: Wheeler, D., Wong, H., Shanley, T. (eds) Pediatric Critical Care Medicine. Springer, London. https://doi.org/10.1007/978-1-4471-6416-6_35
Download citation
DOI: https://doi.org/10.1007/978-1-4471-6416-6_35
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-6415-9
Online ISBN: 978-1-4471-6416-6
eBook Packages: MedicineMedicine (R0)