
Abstract
Cystinuria is an autosomal inherited disorder of dibasic amino acid transport in the proximal tubule and small intestine, being responsible for 1–2 % of urinary stones in adults and up to 10 % in children. Recurrent stone formation and repeated need of surgical intervention are typical for patients suffering from cystinuria and are possibly leading to an impairment of renal function and quality of life. Thus, an appropriate strategy for stone removal as well as medical recurrence prevention is of great importance but is also demanding. Another most important factor is the patient’s compliance with conservative therapy. However, many of these patients, who require a lifelong therapy and regular follow-up, have problems achieving and maintaining a good adjustment. Traditionally, the disease was classified by the urine phenotype, measured by excretion of amino acids. Increased understanding of the underlying genetic characteristics has led to a revised classification based on the genotype. So far, two involved genes have been identified: SLC3A1 on chromosome 2 and SLC7A9 on chromosome 19. Their products form the bo,+-transporter, which transports dibasic amino acids in exchange for neutral amino acids. Conservative therapy aims at reducing the frequency of urological intervention and is based on two principles: to reduce the saturation of urine with cystine and to increase the solubility of cystine in the urine. Interventional therapy focuses on minimally invasive techniques. Despite the increased pathophysiological and genetic knowledge regarding cystinuria, no new treatment modalities could be derived from that during the last years.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Cystinuria
- Urolithiasis
- Prevention
- Treatment
- SLC3A1
- SLC7A9
- bo,+-transporter
- Autosomal recessive disorder
- Dibasic amino acid transport
Introduction
Cystinuria is an autosomal recessive inherited disorder of dibasic amino acid transport in the proximal tubule and small intestine, being responsible for 1–2 % of urinary stones in adults and up to 10 % in children [1]. Recurrent stone formation and repeated need of surgical intervention are typical for patients suffering from cystinuria and are possibly leading to an impairment of renal function and quality of life [2]. Thus, an appropriate strategy for stone removal as well as medical recurrence prevention is of great importance but is also demanding [3]. Despite the increased pathophysiological and genetic knowledge regarding cystinuria, no new treatment modalities could be derived from that during the last few years [4].
Epidemiology
The estimated global prevalence of cystinuria is 1:7,000 [5]. Considering the genetic transmission of the disease, it is not surprising that the reported prevalence differs remarkably from country to country. Since regular screening programs are quite rare, the prevalence shown in Table 92.1 is approximate. Some of these prevalence rates were derived from urine screening in newborns. Since maturation of SLC3A1 gene expression between midgestation and 4.5 years postnatal age may account for transient neonatal cystinuria [5], these newborn screenings may have resulted in false high findings.
Pathophysiology
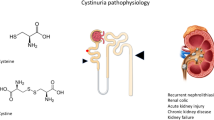
Cystinuria is characterized by a hyperexcretion of cystine and dibasic amino acids (lysine, ornithine, and arginine) into the urine [6, 7]. This is caused by an impaired transport of these amino acids across the apical membrane of epithelial cells of the renal proximal tubule and small intestine [4, 8, 9], thus hindering the reabsorption of the filtered amino acids (normally 98–99 %) [10]. Although all of these amino acids reach high concentrations in urine, only cystine is insoluble enough to form stones [1]. Due to this poor solubility, hyperexcretion and supersaturation lead to the development of cystine stones [11, 12]. The impairment of the intestinal transport does not result in malnutrition because the amino acids can be formed by metabolism or taken up by another transporter [13, 14].
The precipitation of characteristic hexagonal cystine crystals is influenced by urine pH with an acidic pH promoting it [15]. An alkaline pH > 8 leads to a threefold increase of cystine solubility [16]. Varying effect of pH on cystine solubility among patients suggests further influencing factors [17–19]. Besides known factors such as dietary intake of fluid, salt, and protein, other, yet unknown, genetic influences can be assumed to be contributory. This is supported by the observation that the proportion of cystine stone-producing mice increases from 40 % in the second filial generation of SLC7A9-knockout mice [20] to 85 % in the sixth filial generation [4]. Intake of dietary salt increases the cystine excretion. However, no prospective study has shown a decrease in stone activity resulting from sodium-restricted diets [17]. Another known factor is dietary intake of animal protein. Lowered intake of the cystine precursor methionine reduces urinary cystine excretion [21].
In patients with cystinuria, eventually, the cystine crystals will form stones with the corresponding typical clinical problems like obstruction, pain, or infection. Cystinuric patients are overrepresented among stone formers who have lost a kidney [22]. Histological examination of papillary biopsies of patients with cystine stones has shown cystine crystal-plugged ducts of Bellini, with the surrounding interstitium showing changes from inflammation to fibrosis. Also, many inner medullary collecting ducts will be seen to be dilated, some with and others without crystal plugs [22].
The main effector of cystine reabsorption in the kidney is the amino acid transport system bo,+, belonging to the family of heterodimeric amino acid transporters (HAT). These transporters, which consist of a heavy chain linked by a disulphide bridge to a range of light chains, prefer antiport mechanisms which lead to exchange of amino acids [14]. In the case of bo,+, the association of the light subunit called bo,+ AT (neutral and dibasic amino acid transporter) with the heavy chain called rBAT (related to basic amino transporter) forms the active transporter, which transports dibasic amino acids in exchange for neutral amino acids [14, 23]. Incorrect assembly of the subunits, caused by genetic mutations, can lead to clinical manifestation of cystinuria of varying severity, depending on the underlying mutation [24].
Genetics, Inheritance, and Classification
Positional genetics, genetic linkage studies, and mutational analysis helped to identify two genes whose mutations cause cystinuria [4]:
-
SLC3A1 on chromosome 2 (gene locus 2p16), coding for the heavy rBat subunit of the bo,+-transporter [25]
-
SLC7A9 on chromosome 19 (gene locus 19q12–13), coding for the light bo,+AT subunit of the bo,+-transporter [26]
So far, 133 mutations have been identified in SLC3A1 and 95 in SLC7A9. Reported mutations include nonsense, missense, splicing, frameshifts, and large sequence rearrangements [4]. Five hundred seventy-nine mutated alleles in SLC3A1 have been reported in patients from 23 countries, frequencies varying regionally, and 436 mutated SLC7A9 alleles in patients from 18 countries [4].
Cystinuria is usually considered an autosomal recessive disorder. Classification of the phenotype was traditionally achieved by identifying the urinary excretion pattern of cystine and dibasic amino acid acids of the obligate heterozygous parents [4]. Three phenotypes have been described, heterozygous relatives of patients with type I showing normal aminoaciduria, those of type II cystinuric patients having a high hyperexcretion (not matching that of a homozygous patient), and type III relatives having a moderate hyperexcretion (Table 92.2) [27].
Observation of the varying extent of amino acid excretion in patients who carry the same mutation has led to a revised classification to type I, non-type I (includes former type II and III), and mixed type (type I/non-type I). Due to rare formation of cystine calculi with concomitant variable hyperexcretion of cystine in non-type I heterozygotes, an autosomal dominant inheritance with incomplete penetration for the cystine lithiasis trait is assumed for non-type I cystinuria [4]. A lack of genotype-phenotype correlation in some cystinuria cases has led to the recommendation of a new classification based on the genotype [24, 28], defining patients with mutations in SLC3A1 as type A, patients with mutations in SLC7A9 as type B, and patients with mutations in both SLC3A1 and SLC7A9 as type AB.
Diagnostics
The average onset of the disease is during the second decade of life, with male patients having a more severe evolution and an earlier onset of symptoms [28]. Every younger stone-forming patient should be suspected to have cystine stones, especially if he or she is a member of a known cystinuric family. Diagnosis of cystinuria is possible even before the first stone has been passed spontaneously or extracted (Table 92.3). For patients with stones, a diagnosis of cystine stone can be confirmed by noting an increased urinary excretion of dibasic amino acids or by identifying mutations on both alleles of one of the involved genes. An excretion of >1,300 μmol/g creatinine of cystine or >5,900 μmol total dibasic amino acids also confirms the diagnosis in those without stones. Genotyping analyses can also be made in relatives of cystinuric patients.
Stone Analysis
Stone analysis should always be carried out after stone expulsion or extraction. Cystine stones have a pale amber color and a waxy appearance. They often present as staghorn stones at first diagnosis, due to their often rapid growth. Pure cystine stones are identified in 60–80 % of cases. Potential diagnostic error may occur due to partial analysis of the stone [13]. A distinction between rough and smooth subtypes of cystine stones has been made by electron microscopic evaluation. Smooth calculi have an irregular, interlacing crystal structure and are thereby more resistant to fragmentation than those of the rough subtype [29].
Radiological Appearance
Cystine stones are easily detectable by ultrasound. They appear poorly radio-opaque on plain film (Fig. 92.1). In case of large stone masses or a mixed stone composition, they may appear radio-opaque. In computed tomography (CT) cystine stones appear poorly attenuated, with CT-collimation and stone size influencing the attenuation [30]. The CT-based prediction of stone composition remains an interesting issue for research. Latest reports using a dual-energy multidetector CT with postprocessing techniques showed good results [31]. However, these findings are not yet generally transferred to routine clinical practice.

Cystine stones on plain film
Urine Analysis
Microscopic morning urine examination may reveal typical hexagonal cystine crystals (Fig. 92.2) that confirm the diagnosis. However, their absence does not exclude the diagnosis because they are only detectable in 20–39 % of urine specimens from cystinuric patients [1, 32]. Since cystine contains sulfur, the urine may have the characteristic smell of rotten eggs. Quantitative daily cystine excretion is determined by ion-exchange chromatography from collected urine. Twenty-four-hour urine collection is not only important for determination of cystine excretion but also for other metabolic abnormalities like hyperuricosuria, hypocitraturia, and hypercalciuria, which can be found in up to 45 % of cystinuric patients [33]. It should be performed regularly to plan and monitor the therapy. For a proper determination of cystine excretion, sodium bicarbonate should be added to the urine to achieve a pH above 7.5. In this way, cystine dissolution is promoted and false reporting of low cystine values can be avoided. Patients excreting 1,300 μmol of cystine per g creatinine are presumed to be homozygous and need further evaluation and treatment [34].

Cystine crystals
Conservative Management of Cystinuria
Recurrent stone formation causes repeated need of urological intervention with typical associated risks. Due to ongoing improvement of minimally invasive techniques, these are today’s preferential treatment modalities in case of existing stones. To reduce the frequency of intervention, conservative treatment plays a great role in the management of cystinuria. It is based on two main principles: to reduce the saturation of urine with cystine and to increase the solubility of cystine in urine. The former goal can be achieved by either lowering the excreted amount of cystine or by reducing the excreted cystine to better soluble cysteine or by increasing the total urine volume. Raising the urinary pH level entails an improved solubility of cystine.
Patient compliance is an important issue as patients often have problems to maintain the necessary high urinary flow, urine alkalinization, or the rigorous low-sodium, low-protein diets used to reduce cystine excretion [35]. Also, available pharmacological therapy often has adverse side effects that lower patient compliance with the therapy. Despite the increased pathophysiological and genetic knowledge regarding cystinuria, no methods to fix the transport-defect have been found as yet. Recently, an approach to retard cystine crystal growth was reported, by using new compounds in an in vitro environment [36]. However, it remains to be seen whether this can lead to an applicable in vivo therapy.
For all patients first-line therapy includes increased fluid intake, urine alkalinization, and low-salt diet. Other medical approaches are reserved for those patients who do not sufficiently respond to these measures.
Urine Dilution
The single most important factor for cystine-stone prevention is a constant hyperdiuresis to decrease the urine saturation with cystine. Up to pH 7 cystine solubility is approximately 250 mg/L (1 mmol/L); at a pH above 7.5, this is doubled to 500 mg/L (2 mmol/L); and at a pH above 8, it is tripled to 750 mg/L (3 mmol/L) [16]. Since a homozygous cystinuric patient excretes 600–1,400 mg of cystine per day, the needed urine volume ranges from at least 2.4–5.6 L. Usually, the total daily urine volume should be at least 3 L, thus making a daily intake of at least 4–4.5 L of fluid desirable. In children, fluid intake should be 1.5 L/m2 body surface. Ideally, this should be evenly distributed in equivalent doses across 24 h. Therefore, it is important to advise the patient to drink before going to bed to provoke nocturia, as well as to drink during the night after micturition [37, 38]. The European Association of Urology recommends an hourly intake of at least 150 mL in its 2011 guidelines update [39]. Urine neutral and alkalizing beverages like mineral water (preferably high in bicarbonate and low in sodium), herbal teas, and citrus juices are recommended. Dilution success can be evaluated by patient’s use of dipstick tests, keeping the urine specific gravity below 1.010 [40].
Dietary Prevention
Although it was shown that reduced intake of animal protein lowers the urinary excretion of cystine [21], such restrictions to the menu are not accepted by most patients and are therefore not generally recommended. Adults should follow a mixed common sense diet with relatively low protein. Protein restriction is not recommended in children [1].
Reduction of dietary intake of sodium chloride has been shown to reduce urinary cystine concentration significantly [41, 42]. Therefore, a reduction to less than 2 g/day has been recommended for adolescents and grown-ups [43]. A low-salt diet is also effective in children [44]. However, compliance with dietary restriction is often poor.
Urine Alkalinization
As described previously, the solubility of cystine increases with higher urine pH values. Therefore, continuous urine alkalinization is another important measure to improve cystine solubility. This can be achieved by either administering potassium citrate or sodium bicarbonate [45]. The latter is only recommended in cases with severe renal insufficiency because sodium increases the urinary cystine excretion. Therefore, potassium citrate is the first-line treatment for alkalinization in cystinuric patients. It should be given in 2–4 single doses of 3–10 mmol. Monitoring of the urine pH should be performed at least three times a day to keep a constant pH of at least 7.5. After starting with low doses, careful adjustment should be carried out until therapeutic pH values are reached. Monitoring of serum potassium levels is advisable [45].
There are reports that azetazolamide, a sulfonamide, can be used to reinforce the alkalizing effect of other pH-increasing therapy [46]. But then, azetazolamide is known to increase the risk of calcium phosphate stone formation and long-term data is not yet available. No general recommendation can be made at the moment.
Pharmacological Therapy
In case of a urinary cystine excretion above 3 mmol/day or if the aforementioned measures fail to prevent cystine stone recurrence, further pharmacological intervention is needed. Thiol-containing compounds can cleave the disulfide bond of cystine, forming mixed disulfides with the cysteine monomers that are as much as 50 times more soluble than cystine [47] (Fig. 92.3). Most widely studied and used thiol agents are D-penicillamine (DP) and α(alpha)-mercaptopropionylglycine (MPG, thiopronin) [48, 49]. In one study, MPG was shown to be 50 % more effective than DP [35]. Although both have the same side effect profile, MPG appears to have significantly lower frequency and severity of side effects [19], which is why it is the favored treatment option today. However, in a recent study, a good tolerance to DP was shown in a cohort of American children [50]. Typical side effects include rash, arthralgia, leukopenia, gastrointestinal intolerance, proteinuria, and nephrotic syndrome [43], thus occasionally limiting treatment success. It has been shown in a mouse model that the efficacy of MPG therapy is highly dependent on urinary pH [51] and should therefore always be based on a combination with urine alkalinization. The suggested initial MPG dose is 250 mg/day, which may be consecutively increased to a maximum dose of 2 g/day [39]. An early tachyphylaxia is possible. Treatment in children must be individualized as the required amount of drug is strictly dependent on body size [52]. The recommended dosage is 20–40 mg/kg body weight administered in two doses, which is important as cystine excretion is higher during the night [45].

Cystine – cysteine
Captopril, an angiotensin-converting-enzyme inhibitor, contains free sulfhydryl groups. Reported results regarding its therapeutic use in cystinuria are controversial [37, 48, 53–55]. Therefore, it remains a second-line therapeutic option, applicable when MPG therapy is unfeasible or unsuccessful [39].
Likewise, the use of ascorbic acid as a therapeutic agent for cystinuria is controversial [56, 57]. It has limited reductive power and is estimated to lower urinary cystine levels by about 20 % [56]. It was assumed that the positive effect on stone formation could be traced back to the fraction of sodium bicarbonate in the fizzy tablets used. High ascorbic acid supplementation may also increase urinary oxalate concentration and should therefore be avoided in patients with simultaneous calcium oxalate stone formation [58].
Many cystinuric patients form stones of mixed composition [29]. Hypercalciuria, hyperuricosuria, hyperoxaluria, hypocitraturia, or infections might accompany cystinuria and should be determined and treated if necessary.
Urological Intervention
Despite the prophylactic conservative treatment, recurrent stone formation is common in cystinuric patients, thus leading to a repeated need of urological intervention. Today, minimally invasive procedures are preferred in almost all cases. Indication criteria are generally the same as for any patient with urinary stones. The chosen technique does not influence stone recurrence, but being totally stone free after the procedure results in a longer time interval until stone recurrence [59].
Extracorporeal Shock Wave Lithotripsy
Cystine stones are usually considered to be relatively resistant to extracorporeal shock wave lithotripsy (SWL), which often leads to multiple treatments, especially in cases of larger stones. The classification in rough and smooth subtype in combination with a possible increasing clinical use of CT-based prediction of these subtypes could lead to a better selection of SWL-suited cystine stones. However, in regard to the low invasiveness and recurrent stone events in cystinuric patients, SWL remains the primary therapy option for all stone sites of the upper urinary tract and stone sizes below 1.5 cm, particularly in children [60–62]. Treatment success in case of lower pole stones might be limited due to remaining fragments, depending on anatomic characteristics (steep infundibular-pelvic angle, long lower pole, narrow infundibulum) [39, 63–66].
Ureterorenoscopy
Due to the development of new small semirigid and flexible scopes and the availability of the holmium:YAG laser, ureterorenoscopy (URS) offers a safe and efficient alternative for the treatment of cystine stones [67, 68], even in prepubertal children [69]. This applies especially to all ureteric sites and also to all localizations in the collecting system, although larger stone masses lead to long operative times, thus making a percutaneous approach or SWL more favorable. URS offers a good treatment opportunity for fragments refractory to prior SWL treatment.
Percutaneous Nephrolithotomy
For stones exceeding 1.5–2 cm or calyceal diverticular stones, percutaneous nephrolithotomy (PNL) is recommended and is also well suited for the treatment of cystine stones. Monotherapy (Fig. 92.4) with PNL is safe and efficient in the management of staghorn and complex renal calculi in one single hospital stay. It may be combined with SWL, so-called sandwich therapy, or with URS, even in one procedure [70], if necessary. Simultaneous PNL and URS (endoscopic combined intrarenal surgery, ECIRS) allows circumvention of multiple tract access in case of large and complex stone situations. The use of miniaturized instruments (Mini-Perc) as an alternative to conventional PNL [71, 72] has facilitated the successful application of PNL in the pediatric population [73].

Therapy overview
Conclusion
Cystinuria is responsible for 1–2 % of urinary stones in adults and up to 10 % of stones in children. The higher incidence reported in children might result from screening of newborn urines which detects transient cystinuria, which disappears as the SLC3A1 gene expression matures. Cystine stones are recurrent and impair renal function and the quality of life. The recurrent nature of the stone requires that definitive treatment be minimally invasive and active attempts be made to reduce recurrences by approaches that lower the saturation of urine with cystine and increasing the solubility of cystine in urine.
References
Knoll T, Zollner A, Wendt-Nordahl G, Michel MS, Alken P. Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment and follow-up. Pediatr Nephrol. 2005;20:19–24.
Assimos DG, Leslie SW, Ng C, Streem SB, Hart LJ. The impact of cystinuria on renal function. J Urol. 2002;168:27–30.
Tiselius HG. New horizons in the management of patients with cystinuria. Curr Opin Urol. 2010;20:169–73.
Chillarón J, Font-Llitjos M, Fort J, Zorzano A, Goldfarb DS, Nunes V, Palacin M. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010;6:424–34.
Boutros M, Vicanek C, Rozen R, Goodyer P. Transient neonatal cystinuria. Kidney Int. 2005;67(2):443–8.
Scriver CR, Beaudet AL, Sly WS, Valle D, Palacin M, Goodyer P, Nunes V, Gasparini P, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. p. 4909–32.
Palacin M, Borsani G, Sebastio G. The molecular bases of cystinuria and lysinuric protein intolerance. Curr Opin Genet Dev. 2001;11:328–35.
Rosenberg LE, Durant JL, Holland JM. Intestinal absorption and renal extraction of cystine and cysteine in cystinuria. N Engl J Med. 1965;273:1239–45.
Their S, Fox M, Segal S, Rosenberg LE. Cystinuria: in vitro demonstration of an intestinal transport defect. Science. 1964;143:482–4.
Goodyer P. The molecular basis of cystinuria. Nephron Exp Nephrol. 2004;98(2):e45–9.
Coe FL, Favus MJ, Pak CYC, Parks JH, Preminger GM, Tiselius HG, editors. Kidney stones: medical and surgical management. Philadelphia: Lippincott-Raven Publishers; 1996. p. 33–64.
Evan AP. Physiology and etiology of stone formation in the kidney and the urinary tract. Pediatr Nephrol. 2010;25:831–41.
Biyani SB, Cartledge JJ. Cystinuria – diagnosis and management. EAU-EBU Update Series. 2006;4:175–83.
Wagner CA, Lang F, Broeer S. Function and structure of heterodimeric amino acid transporters. Am J Physiol Cell Physiol. 2001;281:C1077–93.
Wagner CH, Mohebbi N. Urinary pH and stone formation. J Nephrol. 2010;23 Suppl 16:165–9.
Dent CE, Senior B. Studies on the treatment of cystinuria. Br J Urol. 1955;27(4):317–32.
Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int. 2006;69(6):1041–7.
Nakagawa Y, Asplin JR, Goldfarb DS, Parks JH, Coe FL. Clinical use of cystine supersaturation measurements. J Urol. 2000;164(5):1481–5.
Pak CY, Fuller C, Sakhaee K, Zerwekh JE, Adams BV. Management of cystine nephrolithiasis with alpha-mercaptopropionylglycin. J Urol. 1986;136:1003–8.
Feliubadaló L, Arbonés ML, Mañas S, Chillarón J, Visa J, Rodés M, et al. SLC7A9-deficient mice develop cystinuria non-I and cystine urolithiasis. Hum Mol Genet. 2003;12:2097–108.
Rodman JS, Blackburn P, Williams JJ, Brown A, Pospischil MA, Peterson CM. The effect of dietary protein on cystine excretion in patients with cystinuria. Clin Nephrol. 1984;22(6):273–8.
Evan AP, Coe FL, Lingeman JE, Shao Y, Matlaga BR, Kim SC, et al. Renal crystal deposits and histopathology in patients with cystine stones. Kidney Int. 2006;69:2227–35.
Feliubadalo L, Font M, Purroy J, Rosaud F, Estivill X, Nunes V, et al. Non-type I cystinuria caused by mutations in SLC7A9, encoding a subunit (bo,+AT) of rBAT. International Cystinuria Consortium. Nat Genet. 1999;23:52–7.
Font-Llitjos M, Jimenez-Vidal M, Bisceglia L, Di Perna M, De Sanctis L, Rousaud F, et al. New insights into cystinuria: 40 new mutations, genotype-phenotype correlation, and digenic inheritance causing partial phenotype. J Med Genet. 2005;42:58–68.
Calogne MJ, Gasparini P, Chillaron J, Chillon M, Gallucci M, Rosaud F, et al. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cysteine. Nat Genet. 1994;6:420–5.
Pras E, Kreiss Y, Frishberg Y, Prosen L, Aksentijevich I, Kastner DL. Refined mapping of the CSBU3 gene to a 1,8-Mb region on chromosome 19q13.1 using historical recombinants in Lybian Jewish cystinuria patients. Genomics. 1999;60:248–50.
Rosenberg LE, Downing S, Durant JL, Segal S. Cystinuria: biochemical evidence for three genetically distinct diseases. J Clin Invest. 1966;45:365–71.
Dello Strologo L, Pras E, Pontesilli C, Beccia E, Ricci-Barbini V, De Sanctis L, et al. Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classification. J Am Soc Nephrol. 2002;13:2547–53.
Bhatta KM, Prien Jr EL, Dretler SP. Cystine calculi – rough and smooth: a new clinical distinction. J Urol. 1989;142:937–40.
Saw KC, McAteer JA, Monga AG, Chua GT, Lingeman JE, Williams Jr JC. Helical CT of urinary calculi: effect of stone composition, stone size, and scan collimation. AJR Am J Roentgenol. 2000;175:329–32.
Boll DT, Patil NA, Paulson EK, Merkle EM, Simmons WN, Pierre SA, et al. Renal stone assessment with dual-energy multidetector CT and advanced postprocessing techniques: improved characterization of renal stone composition – pilot study. Radiology. 2009;250(3):813–20.
Daudon M, Cohen-Solal F, Barbey F, Gagnadoux MF, Knebelmann B, Jungers P. Cystine crystal volume determination: a useful tool in the management of cystinuric patients. Urol Res. 2003;31(3):207–11.
Sakhae K, Poindexter JR, Pak CY. The spectrum of metabolic abnormalities in patients with cystine nephrolithiasis. J Urol. 1989;141(4):819–21.
Guillen M, Corella D, Cabello ML, Garcia AM, Hernandez-Yago J. Reference values of urinary excretion of cystine and dibasic amino acids: classification of patients with cystinuria in the Valencian Community, Spain. Clin Biochem. 1999;32:25–30.
Pietrow PK, Auge BK, Weizer AZ, Delvecchio FC, Silverstein AD, Mathias B, et al. Durability of the medical management of cystinuria. J Urol. 2003;169(1):68–70.
Rimer JD, Zhihua A, Zhu Z, Lee MH, Goldfarb DS, Wesson JA, et al. Crystal growth inhibitors for the prevention of L-cystine kidney stones through molecular design. Science. 2010;330:337–41.
Monnens LA, Noordam K, Trijbels F. Necessary practical treatment of cystinuria at night. Pediatr Nephrol. 2000;14:1148–9.
Fjellstedt E, Denneberg T, Jeppson JO, Christensson A, Tiselius HG. Cystine analyses of separate day and night urine as a basis for the management of patients with homozygous cystinuria. Urol Res. 2001;29:303–10.
Türk C, Knoll T, Petrik A, Sarica K, Straub M, Seitz C. Guidelines on urolithiasis (update 2011) [online]. 2011. Link: http://www.uroweb.org/gls/pdf/18_Urolithiasis.pdf. Accessed on Aug 30.
Rutchik SD, Resnick MI. Cystine calculi: diagnosis and management. Urol Clin North Am. 1997;24:163.
Jaeger P, Portmann L, Saunders A, Rosenberg LE, Their SO. Anticystinuric effects of glutamine and of dietary sodium restriction. N Engl J Med. 1986;315:1120–3.
Jaeger P. Cystinuria: pathophysiology and treatment. Adv Nephrol Necker Hosp. 1989;18:107.
Ng CS, Streem SB. Contemporary management of cystinuria. J Endourol. 1999;13:647–51.
Rodriguez LM, Santos F, Malaga S, Martinez V. Effect of a low sodium diet on urinary elimination of cystine in cystinuric children. Nephron. 1995;71:416–8.
Fjellstedt E, Denneberg T, Jeppsson JO, Tiselius HG. A comparison of the effects of potassium citrate and sodium bicarbonate in the alkalinization of urine in homozygous cystinuria. Urol Res. 2001;29(5):295–302.
Sterret SP, Penniston KL, Wolf Jr JS, Nakada SY. Azetazolamide is an effect adjunct for urinary alkalinization in patients with uric acid and cystine stone formation recalcitrant to potassium citrate. Urology. 2008;72:278–81.
Lotz M, Potts Jr JT, Holland JM, Kiser WS, Bartter FC. D-penicillamine therapy in cystinuria. J Urol. 1966;95:257.
Chow GK, Streem SB. Medical treatment of cystinuria: results of contemporary clinical practice. J Urol. 1996;156:1576–8.
Harbar JA, Cusworth DC, Lawes LC, Wrong OM. Comparison of 2-mercaptopropionylglycine and D-Penicillamine in the treatment of cystinuria. J Urol. 1986;136:146–9.
DeBerardinis RJ, Coughlin 2nd CR, Kaplan P. Penicillamine therapy for pediatric cystinuria: experience from a cohort of American children. J Urol. 2009;180:2620–3.
Wendt-Nordahl G, Meister L, Michel S, Knoll T. Evaluation of alkalinization and chelating agent therapy for cystinuria in animal model [abstract #340]. Eur Urol. 2009;S8:205.
DelloStrologo L, Laurenzi C, Legato A, Pastore A. Cystinuria in children and young adults: success of monitoring free-cystine urine levels. Pediatr Nephrol. 2007;22(11):1869–73.
Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J Urol. 1995;154:164–6.
Perazella MA, Buller GK. Successful treatment of cystinuria with captopril. Am J Kidney Dis. 1993;21:504–7.
Michelakakis H, Delis D, Anastasiadou V, Bartsocas C. Ineffectiveness of captopril in reducing cystine excretion in cystinuric children. J Inherit Metab Dis. 1993;16:1042–3.
Birwe H, Schneeberger W, Hesse A. Investigations of the efficacy of ascorbic acid therapy in cystinuria. Urol Res. 1991;19(3):199–201.
Ragone R. Medical treatment of cystinuria with vitamin C. Am J Kidney Dis. 2000;35(5):1020.
Traxer O, Huet B, Poindexter JR, Pak CY, Pearle MS. Effect of ascorbic acid consumption on urinary stone risk factors. J Urol. 2003;170:397–401.
Chow GK, Streem SB. Contemporary urological intervention for cystinuric patients: immediate and long-term impact and implications. J Urol. 1998;160:341–4.
Kachel TA, Vijan SR, Dretler SP. Endourological experience with cystine calculi and a treatment algorithm. J Urol. 1991;145:25–8.
Brinkmann OA, Griehl A, Kuwertz-Broking E, Bulla M, Hertle L. Extracorporeal shock wave lithotripsy in children. Efficacy, complications and long-term follow-up. Eur Urol. 2001;39:591–7.
Muslumanoglu AY, Tefekli A, Sarilar O, Binbay M, Altunrende F, Ozkuvanci U. Extracorporeal shock wave lithotripsy as first line treatment alternative for urinary tract stones in children: a large retrospective analysis. J Urol. 2003;170:2405–8.
Argyropoulos AN, Tolley DA. Evaluation of outcome following lithotripsy. Curr Opin Urol. 2010;20(2):154–8.
Knoll T, Musial A, Trojan L, Ptashnyk T, Michel MS, Alken P, et al. Measurement of renal anatomy for prediction of lower-pole caliceal stone clearance: reproducibility of different parameters. J Endourol. 2003;17(7):447–51.
Preminger GM. Management of lower pole renal calculi: shock wave lithotripsy versus percutaneous nephrolithotomy versus flexible ureteroscopy. Urol Res. 2006;34(2):108–11.
Pearle MS, Lingeman JE, Leveillee R, Kuo R, Preminger GM, Nadler RB, et al. Prospective, randomized trial comparing shock wave lithotripsy and ureteroscopy for lower pole caliceal calculi 1 cm or less. J Urol. 2005;173(6):2005–9.
Ruggera L, Zanin M, Beltrami P, Zattoni F. Retrograde transureteral approach: a safe and efficient treatment for recurrent cystine renal stones. Urol Res. 2011;39(5):411–5.
Kourambas J, Munver R, Preminger GM. Ureterorenoscopic management of recurrent renal cystine calculi. J Endourol. 2000;14(6):489–92.
Schuster TG, Russell KY, Bloom DA, Koo HP, Faerber GJ. Ureteroscopy for the treatment of urolithiasis in children. J Urol. 2002;167(4):1813.
Scoffone CM, Cracco CM, Cossu M, Grande S, Poggio M, Scarpa RM. Endoscopic combined intrarenal surgery in Galdakao-modified supine Valdivia position: a new standard for percutaneous nephrolithotomy? Eur Urol. 2008;54(6):1393–403.
Lahme S, Zimmermanns V, Hochmuth A, Janitzki V. Minimally invasive PCNL (Mini-perc). Alternative treatment modality or replacement of conventional PCNL? Urologe A. 2008;47(5):563–8. [Article in German].
Lahme S, Bichler KH, Strohmaier WL, Götz T. Minimally invasive PCNL in patients with renal pelvic and calyceal stones. Eur Urol. 2001;40(6):619–24.
Schuster TK, Smaldone MC, Averch TD, Ost MC. Percutaneous nephrolithotomy in children. J Endourol. 2009;23(10):1699–705.
Oezalp I, Coskun T, Tokol S, Demircin G, Moench E. Inherited metabolic disorders in Turkey. J Inherit Metab Dis. 1990;13(5):732–8.
Tanzer F, Ozgur A, Bardakci F. Type I cystinuria and its genetic basis in a population of Turkish school children. Int J Urol. 2007;14(10):914–7.
Cabello ML, Guillen M. Pilot screening programme for cystinuria in the Valencian community. Eur J Epidemiol. 1999;15:681–4.
Dello Strologo L, Rizzoni G. Cystinuria. Acta Paediatr Suppl. 2006;95(452):31–3.
Turner B, Brown DA. Amino acid excretion in infancy and early childhood. A survey of 200,000 infants. Med J Aust. 1972;1(2):62–5.
Hyanek J. Cystinurie. Dědičné metabolické poruchy. Praha: Avicenum. 1991; 53–58.
Scriver CR, Clow CL, Reade TM, Goodyer P, Auray-Blais C, Giguere R, et al. Ontogeny modifies manifestations of cystinuria genes: implications for counseling. J Pediatr. 1985;106(3):411–6.
Ito H, Murakami M, Miyauchi T, Mori I, Yamaguchi K, Usui T, et al. The incidence of cystinuria in Japan. J Urol. 1983;129(5):1012–4.
Scriver CR, Beaudet AL, Sly WS, Valle D, Segal S, Thier SO, editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1995. p. 3581–601.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer-Verlag London
About this chapter
Cite this chapter
Jessen, J.P., Knoll, T. (2012). Management of Cystinuria. In: Talati, J., Tiselius, HG., Albala, D., YE, Z. (eds) Urolithiasis. Springer, London. https://doi.org/10.1007/978-1-4471-4387-1_92
Download citation
DOI: https://doi.org/10.1007/978-1-4471-4387-1_92
Published:
Publisher Name: Springer, London
Print ISBN: 978-1-4471-4383-3
Online ISBN: 978-1-4471-4387-1
eBook Packages: MedicineMedicine (R0)