
Abstract
Cystine stones are an uncommon form of kidney stones but carry high morbidity. They occur in patients with cystinuria, a rare genetic defect in tubular reabsorption of cationic amino acids leading to high amounts of cystine amino acids in urine. Inheritance is autosomal and two major genetic types have been identified. Cystine nephrolithiasis and related kidney diseases are usually the only clinically pertinent manifestation of this disorder.
These patients, mostly presenting in their teens, frequently have a high stone burden and high recurrence rate of kidney stones. Compared with the other more common forms of kidney stones, these patients require a much higher number of invasive procedures and are at higher risk for chronic kidney disease.
Diagnosis is made by stone analysis and measurement of very high cystine levels in urine. Medical management of these stones is successful at reducing the rate of recurrence and the need for surgical procedures but requires high compliance with dietary and pharmacological interventions as well as close follow-up. Cystine capacity is a new urine assay, which is used as a measure of cystine saturation in urine and is helpful in guiding the ongoing management of cystine stones.
The goal of therapy is to modify the urinary milieu to increase solubility of cystine and may include drugs that bind cystine in urine. Dietary management is critical with focus on increasing the urine volume and pH. Pharmacological therapy with urine alkalizing agents and cystine-binding thiol drugs (CBTDs) is often utilized. The two CBTDs, D-penicillamine and tiopronin, are effective agents but require close monitoring due to the need for dose titration and high incidence of adverse reactions.
Ongoing research may help develop newer therapeutic options in the future for this disease with high medical and psychosocial morbidity.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
FormalPara Key Points-
1.
Cystine stones are caused by cystinuria, a rare hereditary disorder with autosomal inheritance. Cystine nephrolithiasis and related kidney diseases are usually the only clinically pertinent manifestation of this disorder.
-
2.
Patients with cystinuria usually present with first kidney stone in their early teens although presentation may be as early as infancy or as late as the fifth decade.
-
3.
Diagnosis is made by stone analysis and measurement of very high cystine levels in urine.
-
4.
Cystine capacity, a new urine assay, is used as a measure of cystine saturation in urine and is helpful in guiding the ongoing management of cystine stones.
-
5.
High urine volume, urine alkalization, and cystine-binding drugs are the three pillars of medical and nutritional management.
-
6.
The two cystine-binding agents in clinical use are D-penicillamine and tiopronin, both of which have a high incidence of adverse reactions.
-
7.
Frequent follow-up of patients is needed to ensure compliance with burdensome dietary recommendations, to re-assess the need for management adjustment based on urine studies, and to look for adverse effects of the medications.
Introduction/Physiology
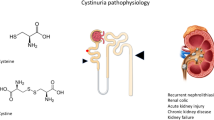
Cystine stones, as the name suggests, are formed by high concentration of the amino acid cystine in the urine. It is due to a rare genetic disorder called cystinuria , which occurs due to a defective transport mechanism in the tubular lumen allowing for a large amount of cystine and other cationic amino acids (cystine, ornithine, arginine, lysine (COAL)) to appear in the urine.
After glomerular filtration cystine normally undergoes near complete reabsorption in proximal tubule via a cystine transporter , with a fractional excretion of about 0.4% [1]. This can increase to more than 100% in patients with cystinuria (suggestive of tubular secretion) [2]. These large amounts of unreabsorbed cystine in the urine crystallize at usual human urine pH of 5.5–7.0 and cause stone formation. The other three amino acids (see above) are also present in high quantities in urine but do not precipitate at normal urine pH and thus lack clinical significance.
The cystine transporter consists of a heavy subunit, rBAT encoded by SLC3A1, and a light subunit, bo,+AT encoded by SLC7A9 . The light subunit is the actual transporter, whereas the heavy subunit is responsible for trafficking of the light subunit to the apical membrane of the cells [3, 4]. In addition to renal proximal tubule, this transporter is also expressed in the intestine where its malfunction is compensated by peptide transporter 1 (PEPT1) , which is not present in nephron tubular lumen. Thus cystine stones are the only clinically relevant feature of this disorder.
The normal excretion of cystine in urine, which is less than 30 mg/day, exceeds 400 mg/day in patients with cystinuria [5]. Patients with cystinuria usually present with first kidney stone in their early teens (median age of 12 years) although the age of presentation may be as early as infancy or as late as the fifth decade [6,7,8]. In fact, 80% of symptomatic patients experience their first stone episode by the end of the second decade [7]. Compared with other stone formers, patients with cystinuria tend to have stones earlier, more often, more bilateral, and more of the staghorn variant that require more urological interventions [8,9,10]. They also have a higher risk of chronic kidney disease (CKD) and lower quality of life compared with non-cystine stone formers [10,11,12]. The higher prevalence of CKD is, however, variable across different studies. In a 2002 study looking at the International Cystinuria Consortium (ICC) data , impairment of the renal function was noted in 17% of the patients [7]. In a 2015 study from France, whereas only 22.5% of the patients had glomerular filtration rate (GFR) > 90, 27% of the patients had GFR below 60, and in 0.6%, the GFR was below 15 [13]. In addition, cystinuria patients were almost five times more likely to have nephrectomy compared with patients with calcium oxalate stones; 14% vs. 3%, respectively [11].
In the past, cystinuria was classified phenotypically as type I–III or type I, and non-type I, based on the level of cystine excretion in parents, type I being the group where parents had normal cystine in urine and non-type I where parents had high cystine excretion. However, after identification of the genes behind cystinuria , it was found that this classification had poor correlation with the genotypes, and hence a genotypic classification has been adopted [7]. SLC3A1 and SLC7A9 are the two gene mutations which can be identified in the vast majority of patients. Patients with SLC3A1 and SLC7A9 mutations are called type A and type B, respectively. Rare patients with single mutated allele in both genes are called AB and usually have mild disease.
Prevalence, Inheritance
Cystinuria was first described in the 1800s [1] and was one of the disorders used by Sir Archibald Garrod in 1908 to illustrate the concept of inborn errors of metabolism [14]. The prevalence of cystinuria varies from 1/2500 in Libyan Jewish population to 1/100,000 in Swedish population [2]. The average prevalence is estimated to be about 1/7000 [15]. Nephrolithiasis and related complications are the only clinically significant manifestation of this disorder.
Up to 1% of adult stone formers and 5–10% of pediatric stone formers have cystinuria [13, 16]. A recent study also noted an increasing trend of cystine stones as a proportion of all stones in males [17]. About 6% of patients with cystinuria never present with urolithiasis [15].
Cystinuria has autosomal inheritance with SLC3A1 or type A showing to be autosomal recessive and SLC7A9 showing to be autosomal dominant with variable penetrance pattern [8]. Most patients are homozygotes of either of these gene mutations: 100% of homozygotes have high cystine excretion, and 94% of the homozygotes develop kidney stones in their lifetime [15]. Whereas heterozygotes of type A have normal urine cystine, heterozygotes of type B usually have higher than normal levels of urine cysteine in 86–90% of cases and kidney stone formation in 2–18% of cases [8, 15]. There is no good correlation between the concentration of amino acids in the urine to stone event frequency [7, 18].
Although in a large cohort [19] type A accounted for about 45% of the patients with cystinuria and type B for about 53%, this proportion varies among different cohorts; type AB constitutes 1.2–4% of the cases [20]. Type A and type B homozygotes have similar clinical manifestation [7] although males seem to be affected more severely for as of yet unclear reasons [21]. So far, 160 and 116 different mutations have been reported in SLC3A1 and SLC7A9 , respectively [20].
Diagnosis
The diagnosis of cystinuria is essentially based on high urine cystine levels. It should especially be suspected in younger patients with high stone burden and high recurrence.
In the office, examination of urine sediment for cystine crystals and sodium nitroprusside test may be helpful . Under microscopy, cystine crystals are colorless, refractory, and hexagonal and are pathognomonic of the disease. But they are identified in only 25% of patients [5]. Sodium nitroprusside test can identify urine cystine concentration of more than 75 mg/L (urine turns purple) and may be used as a screening test with sensitivity of about 70–80% and specificity of approximately 83–95% [22, 23].
All patients with suspected cystinuria should undergo a 24-hour urine collection to assess cystine excretion along with other metabolic factors that affect crystallization and stone formation. Of these, urine pH and volume are the two most important factors. Considering a relatively fixed total cystine excretion per day, the concentration halves as the volume of urine doubles. The solubility of cystine is highly affected by the urine pH as cystine is much more soluble in alkaline urine. As a rule, at pH of 7, cystine is soluble at concentration of 250 mg/L. This doubles to 500 mg/L at pH 7.5 and goes up further at pH of 8 [2, 24].
It is hard to determine cystine supersaturation based on cystine concentration and pH alone given high inter-patient variability. Therefore, cystine capacity, a new solid phase assay, has been developed to determine urine’s cystine saturation. In this assay, a known amount of solid phase cystine is added to urine. In supersaturated urine, cystine precipitates onto added crystals, thus the recovered solid phase after incubation will be more. A negative cystine capacity implies oversaturated urine and vice versa [25].
Cystine stones are less radio-opaque than oxalate, and X-ray may miss small stones. Computed tomography (CT) scan may help not only in identifying but also in managing small stones as it can differentiate “rough stones” from “smooth stones” based on the appearance and lower Hounsfield units in rough stones [1]; rough stones tend to be more fragile and amenable to lithotripsy. Renal ultrasound is a good noninvasive test to identify and monitor stone formation.
Genetic testing is not routinely performed as it does not affect the management or clinical course of the disease.
Management and Prevention
Cystinuria is a highly morbid disease given recurrent stone formation, requiring frequent procedures and pain management, and for increasing the risk of CKD development. If poorly managed, the risk of forming new stones is about 1/patient-year, requiring urological procedure every 3–4 years [24]. Although medical therapy is often met with poor compliance and fails to achieve the desired control, it certainly helps in reducing the number of stone events and procedures required [10, 24] by up to 78% and 52%, respectively [24].
Also, in patients with cystinuria the stone composition may be mixed [26]. Therefore, stone analysis (for composition) is an important part of medical evaluation and management of these patients.
The goal of medical management in cystinuria is to reduce the recurrence of stones and minimize the burden of urological procedures and chronic kidney disease. It is aimed at achieving low cystine concentration in urine and modifying the milieu to promote its solubility with a goal to achieve urine supersaturation of <1 or a positive cystine capacity.
High urine volume, urine alkalization, and cystine-binding drugs are the three pillars of medical and nutritional management.
Nutritional Management
-
(a)
High fluid intake is the cornerstone of nutritional management. The goal is to bring the cystine concentration to below 250 mg/L. A fluid intake of at least 3–4 L is recommended. Emphasis should be given on staying well hydrated at night and drinking water before bed and in the middle of the night.
-
(b)
Low protein (particularly of animal source) diet may help urine alkalization in addition to reducing cystine production by lowering the supply of its precursor methionine [27]. Children and adolescents require normal protein intake for their overall growth.
-
(c)
Low sodium diet can help reduce cystine concentration in urine, the mechanism of which is not clear [28].
-
(d)
Diet rich in fruits and vegetable is recommended as it may help with increasing the urine pH [29].
Medical Management
Urine Alkalization
Potassium citrate is the preferred choice to increase urine pH. Citrate may also reduce the risk of calcium phosphate stone formation associated with alkaline urine.
Sodium bicarbonate is another option but is less desirable due to its sodium load. Acetazolamide is used by some as the last resort, although it is poorly tolerated, has questionable efficacy, and may have adverse effect(s) on bone health due to metabolic acidosis.
High Urine Volume
This is achieved through high fluid intake, as discussed above. Tolvaptan has been proposed as an option in patients with low volume but comes with high expense, risk of liver disease, and unclear benefit [30, 31].
Cystine-Binding Thiol Drugs
Often patients are refractory to nutritional changes and urine alkalization and require cystine-binding thiol drugs (CBTDs). These drugs reduce the insoluble cystine to cysteine monomer and form complexes with it. This complex is about 50 times more soluble than cystine [1, 20], thus preventing crystallization and stone formation.
The two agents in clinical use are D-penicillamine and 2-meracptopropionylglycine (tiopronin ). D-penicillamine and tiopronin are similar agents with tiopronin having the advantage of fewer side effects; drug withdrawal of 69% vs. 31%, respectively [32]. Studies have consistently shown reduction in stone size and dissolution of stones with D-penicillamine and tiopronin . Compared with conservative management, treatment with one of these two drugs decreased stone events by 32–65% [33]. In one study, they reduced new stone formation or enlargement of existing stones from 1.6 per year to 0.5 per year [34]. In another study with median daily dose of 900 mg D-penicillamine or 750 mg tiopronin , the mean decrease in free cystine excretion was 32% (35% and 29%, respectively) [24].
The dose of D-penicillamine in adults ranges from 0.5 to 2 g/day (20–40 mg/kg per day in children) in 3–4 divided doses. Tiopronin initial dose is 250 mg/day, gradually increasing to 400–1200 mg/day in 3–4 divided doses with maximum dose of 2 g/day (10–15 mg/kg per day) [20].
Both agents have a high incidence of adverse reactions that limit their tolerability although the incidence is lower for tiopronin [1]. They include abnormal taste, fever, arthralgia, skin rash, leukopenia, and proteinuria among others [35]. In fact, D-penicillamine is known to cause proteinuria and nephrotic syndrome mainly due to membranous nephropathy and minimal change disease although other lesions including crescentic glomerulonephritis have been reported [36,37,38,39]. D-penicillamine causes vitamin B6 (pyridoxine) deficiency and requires supplementation of 50 mg/day of vitamin B6 [20]. Most adverse effects are dose-dependent and more common during the first year of therapy, hence the recommendation for gradual dose escalation. Of note, autoimmune and hypersensitivity-related side effects are not dose dependent and typically occur in patients with other autoimmune disorders [1].
Captopril is also a potential CBTD. Captopril has good cystine binding in-vitro, but it is of little clinical value given its low urinary concentration [25]. Multiple studies have produced contradictory results on its ability to improve cystine concentration in urine even at high doses of 75–150 mg/day [40, 41]. However, it may be utilized as a preferred anti-hypertensive agent.
Although pH does not appear to affect the maximum effect of CBTDs, it has been shown that it does affect the time by which they reach the maximum effect; the higher the pH, the faster the effect. This higher rate of action at higher pH is attributed to higher deprotonated (active) form of the drug. This may have clinical implications given that urine transition time through renal pelvis may be short and only a few minutes. Therefore, it is very important to remember that the effects of CBTDs and high urine pH are complimentary, and one should not be considered as a substitute for the other [42, 43].
Follow-Up
Compliance with strict nutritional, medical, and follow-up requirements is difficult, and studies have shown poor compliance with subsequent treatment failure [44, 45].
Frequent follow-up of patients is recommended to insure compliance with burdensome dietary recommendations, to re-assess the need for management change based on urine studies, and to look for adverse effects of the medications. A patient with severe disease should be followed up every 3 months with radiological studies [20]. Patient with mild disease and minimal or no stone activity can be followed every 6 months. A 24-hour urine analysis should be done at least annually and preferably more frequently. Isolated overnight collection may be necessary in refractory cases [46].
Management in specialty clinic may help reduce recurrence and the need for urological procedures [47].
Cystine stones do not occur post-transplant as the graft kidney does not have the genetic defect [48].
Genetic testing of siblings is not done routinely but may be helpful if they have not been diagnosed with cystinuria . Genetic counseling of siblings and spouse may be beneficial.
Future Direction
Structural mimics of cystine (cystine dimethyl ester (CDME) and cystine methyl ester (CME) ) are being studied for their potential to inhibit cystine crystallization and crystal growth by steric inhibition [49]. In one study, SLC3A1 knockout mice treated with CDME demonstrated smaller but more bladder stones [50].
SCL3A1 and SCL7A9 knock out mice and a proximal tubular cell line derived from normal human kidney mimicking cystinuria phenotype have been developed allowing for further investigation [1].
Atomic force microscopy is a new technique to study crystal growth and is being used to identify molecules which may inhibit crystal growth [51].
Formation of Rare Kidney Stone Consortium , which studies primary hyperoxaluria, cystinuria , adenine phosphoribosyl transferase (APRT) deficiency , and Dent disease , and the establishment of an International Cystinuria Registry may help in further understanding of the disease and to facilitate clinical trials [43].
References
Pereira DJ, Schoolwerth AC, Pais VM. Cystinuria: current concepts and future directions. Clin Nephrol. 2015;83(3):138–46.
Mattoo A, Goldfarb DS. Cystinuria. Semin Nephrol. 2008;28(2):181–91.
Fotiadis D, Kanai Y, Palacin M. The SLC3 and SLC7 families of amino acid transporters. Mol Asp Med. 2013;34(2–3):139–58.
Chillaron J, et al. Pathophysiology and treatment of cystinuria. Nat Rev Nephrol. 2010;6(7):424–34.
Fattah H, Hambaroush Y, Goldfarb DS. Cystine nephrolithiasis. Transl Androl Urol. 2014;3(3):228–33.
Lambert EH, et al. Analysis of 24-hour urine parameters as it relates to age of onset of cystine stone formation. J Endourol. 2010;24(7):1179–82.
Dello Strologo L, et al. Comparison between SLC3A1 and SLC7A9 cystinuria patients and carriers: a need for a new classification. J Am Soc Nephrol. 2002;13(10):2547–53.
Rhodes HL, et al. Clinical and genetic analysis of patients with cystinuria in the United Kingdom. Clin J Am Soc Nephrol. 2015;10(7):1235–45.
Cranidis AI, et al. Cystine stones: the efficacy of percutaneous and shock wave lithotripsy. Urol Int. 1996;56(3):180–3.
Worcester EM, et al. Reduced renal function and benefits of treatment in cystinuria vs other forms of nephrolithiasis. BJU Int. 2006;97(6):1285–90.
Assimos DG, et al. The impact of cystinuria on renal function. J Urol. 2002;168(1):27–30.
Modersitzki F, et al. Health-Related Quality of Life (HRQoL) in cystine compared with non-cystine stone formers. Urolithiasis. 2014;42(1):53–60.
Prot-Bertoye C, et al. CKD and its risk factors among patients with cystinuria. Clin J Am Soc Nephrol. 2015;10(5):842–51.
Scriver CR. Garrod’s Croonian lectures (1908) and the charter ‘inborn errors of metabolism’: albinism, alkaptonuria, cystinuria, and pentosuria at age 100 in 2008. J Inherit Metab Dis. 2008;31(5):580–98.
Eggermann T, et al. Clinical utility gene card for: cystinuria. Eur J Hum Genet. 2012;20(2):1–2.
Knoll T, et al. Cystinuria in childhood and adolescence: recommendations for diagnosis, treatment, and follow-up. Pediatr Nephrol. 2005;20(1):19–24.
Moses R, et al. Changes in stone composition over two decades: evaluation of over 10,000 stone analyses. Urolithiasis. 2015;43(2):135–9.
Stoller ML, et al. Acalculous cystinuria. J Endourol. 1997;11(4):233–8.
Cochat P, et al. Nephrolithiasis related to inborn metabolic diseases. Pediatr Nephrol. 2010;25(3):415–24.
Andreassen KH, et al. How should patients with cystine stone disease be evaluated and treated in the twenty-first century? Urolithiasis. 2016;44(1):65–76.
Masotti A, et al. Gender-related effects on urine L-cystine metastability. Amino Acids. 2014;46(2):415–27.
Finocchiaro R, et al. Usefulness of cyanide-nitroprusside test in detecting incomplete recessive heterozygotes for cystinuria: a standardized dilution procedure. Urol Res. 1998;26(6):401–5.
Giugliani R, Ferrari I, Greene LJ. An evaluation of four methods for the detection of heterozygous cystinuria. Clin Chim Acta. 1987;164(2):227–33.
Barbey F, et al. Medical treatment of cystinuria: critical reappraisal of long-term results. J Urol. 2000;163(5):1419–23.
Goldfarb DS, Coe FL, Asplin JR. Urinary cystine excretion and capacity in patients with cystinuria. Kidney Int. 2006;69(6):1041–7.
Rogers A, et al. Management of cystinuria. Urol Clin North Am. 2007;34(3):347–62.
Rodman JS, et al. The effect of dietary protein on cystine excretion in patients with cystinuria. Clin Nephrol. 1984;22(6):273–8.
Jaeger P, et al. Anticystinuric effects of glutamine and of dietary sodium restriction. N Engl J Med. 1986;315(18):1120–3.
Meschi T, et al. The effect of fruits and vegetables on urinary stone risk factors. Kidney Int. 2004;66(6):2402–10.
Torres VE, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–18.
de Boer H, Roelofsen A, Janssens PM. Antidiuretic hormone antagonist to reduce cystine stone formation. Ann Intern Med. 2012;157(6):459–60.
Pak CY, et al. Management of cystine nephrolithiasis with alpha-mercaptopropionylglycine. J Urol. 1986;136(5):1003–8.
Moe OW, Pearle MS, Sakhaee K. Pharmacotherapy of urolithiasis: evidence from clinical trials. Kidney Int. 2011;79(4):385–92.
Chow GK, Streem SB. Medical treatment of cystinuria: results of contemporary clinical practice. J Urol. 1996;156(5):1576–8.
Xu H, et al. Kidney stones: an update on current pharmacological management and future directions. Expert Opin Pharmacother. 2013;14(4):435–47.
Hall CL, et al. Natural course of penicillamine nephropathy: a long term study of 33 patients. Br Med J (Clin Res Ed). 1988;296(6629):1083–6.
Habib GS, et al. Penicillamine and nephrotic syndrome. Eur J Intern Med. 2006;17(5):343–8.
Ntoso KA, et al. Penicillamine-induced rapidly progressive glomerulonephritis in patients with progressive systemic sclerosis: successful treatment of two patients and a review of the literature. Am J Kidney Dis. 1986;8(3):159–63.
Tasic V, et al. Nephrotic syndrome occurring during tiopronin treatment for cystinuria. Eur J Pediatr. 2011;170(2):247–9.
Cohen TD, Streem SB, Hall P. Clinical effect of captopril on the formation and growth of cystine calculi. J Urol. 1995;154(1):164–6.
Michelakakis H, et al. Ineffectiveness of captopril in reducing cystine excretion in cystinuric children. J Inherit Metab Dis. 1993;16(6):1042–3.
Asplin DM, Asplin JR. The interaction of thiol drugs and urine pH in the treatment of cystinuria. J Urol. 2013;189(6):2147–51.
Sumorok N, Goldfarb DS. Update on cystinuria. Curr Opin Nephrol Hypertens. 2013;22(4):427–31.
Pietrow P, et al. Durability of the medical management of cystinuria. J Urol. 2003;169(1):68–70.
Pareek G, Steele TH, Nakada SY. Urological intervention in patients with cystinuria is decreased with medical compliance. J Urol. 2005;174(6):2250–2, discussion 2252.
Fjellstedt E, et al. Cystine analyses of separate day and night urine as a basis for the management of patients with homozygous cystinuria. Urol Res. 2001;29(5):303–10.
Haritopoulos K, et al. Impact of a metabolic stone clinic on management of patients with cystinuria: 5 years follow-up. Clin Ter. 2010;161(4):341–4.
Tuso P, et al. Cystinuria and renal transplantation. Nephron. 1993;63(4):478.
Sahota A, et al. Novel cystine ester mimics for the treatment of cystinuria-induced urolithiasis in a knockout mouse model. Urology. 2014;84(5):1249 e9–15.
Lee MH, et al. Cystine growth inhibition through molecular mimicry: a new paradigm for the prevention of crystal diseases. Curr Rheumatol Rep. 2015;17(5):33.
Rimer JD, et al. Crystal growth inhibitors for the prevention of L-cystine kidney stones through molecular design. Science. 2010;330(6002):337–41.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Agrawal, N., Zandi-Nejad, K. (2019). Cystine Stones. In: Han, H., Mutter, W., Nasser, S. (eds) Nutritional and Medical Management of Kidney Stones. Nutrition and Health. Humana, Cham. https://doi.org/10.1007/978-3-030-15534-6_12
Download citation
DOI: https://doi.org/10.1007/978-3-030-15534-6_12
Published:
Publisher Name: Humana, Cham
Print ISBN: 978-3-030-15533-9
Online ISBN: 978-3-030-15534-6
eBook Packages: MedicineMedicine (R0)