
Abstract
The innate immune system harbors a multitude of different receptor systems and cells that are constantly prepared to sense and eliminate invading microbial pathogens. Staphylococcus aureus enters the body on its exposed epithelial surfaces, e.g., on skin and mucosa. The initial interaction with epithelial cells is governed by Toll-like receptor (TLR)-2-mediated local production of soluble mediators, including cytokines, chemokines, and antimicrobial peptides. The overall goal is to achieve a steady state of immune mediators and colonizing bacteria. Following cell and tissue invasion clearance of bacteria depends on intracellular microbial sensors and subsequent activation of the inflammasomes. Tissue-resident mast cells and macrophages recruit neutrophils, macrophages, and NK cells. This inflammatory response supports the generation of IL-17 producing NKT, γδ T cells, and T helper cells. Local dendritic cells migrate to the lymph nodes and fine-tune the adaptive immune response. The scope of this chapter is to provide an overview on the major cell types and receptors involved in innate immune defense against S. aureus. By segregating the different stages of infection from epithelial barrier to intracellular and systemic infection, this chapter highlights the different qualities of the innate immune response to S. aureus at different stages of invasiveness.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
1 Introduction
In the last two decades, our understanding of the cellular and molecular mechanisms regulating innate immune defense has been refined. The researchers working in this field have not only cloned and described a broad variety of receptors responsible for the sensing of microbial pathogens but we have also learned how the different immune cell subsets contribute to immune defense and shape the effector function of the adaptive immunity. Despite all of these efforts, we have only started to understand how all of these cells and receptors integrate to mount a pathogen-specific protective immune response and which factors determine the differences in the outcome and quality of the immune response observed among individuals.
Numerous studies have dealt with the innate immune response against S. aureus. They have defined the receptors and cells required for innate immune protection against infection with this pathogen. It has even been postulated that clearance of S. aureus infection can solely rely on the innate immune system (Schmaler et al. 2011; Josefsson and Tarkowski 1999). By contrast, activation of adaptive immune cells in S. aureus infection has rather been associated with exacerbated inflammation and development of arthritis (Josefsson and Tarkowski 1999) and is most likely due to aberrant activation of B and T lymphocytes by S. aureus superantigens (reviewed in Broker et al. 2014). Nevertheless, a multitude of studies also highlighted that S. aureus also possesses a broad variety of virulence factors that promote its superb ability to adapt to a hostile environment and successfully evade the innate immune defense (reviewed elsewhere in this volume).
This chapter provides an overview on the innate immune mechanisms involved in physiological recognition and immune defense against S. aureus in carriers and local and systemic infections. Unfortunately, to date, there are no studies available that systemically compare the immune response to colonizing, invasive, and intracellularly persisting S. aureus strains. The authors have, therefore, defined different layers of antibacterial immune defense on a cellular (Fig. 1A) and a receptor level (Fig. 1B) and assigned these to the different stages and types of infection.

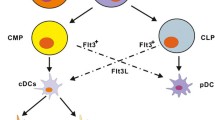
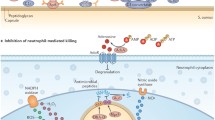
The different layers of innate immune defense. A: Antibacterial defense on a cellular level: 1. Epithelial barrier: keratinocytes and mucosal epithelial cells are the first to encounter bacteria. Bacteria are sensed by surface receptors and intracellular pattern recognition receptors (PRRs). These cells produce antimicrobial peptides (blue) and cytokines in response to stimulation of TLR2 or Nod-like receptors in response to S. aureus lipoproteins and/or peptidoglycan. 2. Tissue-resident cells: mast cells, tissue-resident macrophages, innate lymphoid cells (ILCs), plasmacytoid dendritic cells (pDCs), NKT, and innate B cell subsets present in the tissues are prepared to fight invading pathogens. They express a broad variety of scavenger receptors and PRR that enable bacterial recognition, phagocytosis, and release of chemokines and proinflammatory cytokines, which attract leukocytes from blood. 3. Transmigration of cells from blood: neutrophils, monocytes, and NK cells immigrate from blood and participate in phagocytosis and clearance of bacteria and abscess formation. 4. Lymph nodes: Cell migration from the infected site initiates the antigen-specific adaptive immune response. Dendritic cells migrate to lymph nodes and present antigen to T cells. Neutrophils and macrophages interact with B cells. B: Cellular levels of innate immune receptors. 1. Surface receptors on epithelial cells and leukocytes mediate the recognition of bacterial cell wall components. TLR2 recognizes staphylococcal lipoproteins and plays a central role in phagocyte activation. Scavenger receptors such as CD36 and SR-A recognize S. aureus and promote phagocytosis and bacterial clearance. 2. Endosomal TLRs recognize microbial nucleic acids. They are activated upon degradation of the bacterium and acidification of the phagosome. 3. Cytosolic receptors sense liberated bacterial degradation products such as peptidoglycan and microbial DNA. In infections with S. aureus, they activate the NLRP3, NLRC5, and AIM2 inflammasomes and promote caspase-1-dependent activation of IL-1β and IL-18, thus driving the formation of IL-17-producing T cells
2 The Encounter at the Epithelial Barrier
When encountering the body surfaces, S. aureus cells interact with and adhere to epithelial surfaces in skin and mucosa. The presence of adherent bacterial cells is sensed by epithelial surface receptors that recognize microbial components derived from the bacterial cell wall. These include wall teichoic acid (WTA), lipoteichoic acid (LTA), peptidoglycan (PG), and lipoproteins (Lpp). Additionally, secreted bacterial toxins such as alpha toxin, beta hemolysin, and Panton-Valentine leukocidin (PVL) are neutralized by preformed IgA present in the mucosa (Verkaik et al. 2009). Notably, to date, there is nearly no information available that allows a comparison of the epithelial and leukocyte responses upon initial encounter of the pathogen to those maintained in chronic colonization with S. aureus.
2.1 The Sentinel Function of Toll-like Receptor-2: Permitting Colonization and Preventing Invasion
The major surface receptor implicated in epithelial recognition of Gram-positive commensals such as S. aureus is Toll-like receptor (TLR)-2, a pattern recognition receptor (PRR) expressed on cells of epithelial, endothelial, and leukocyte origin. It senses di- and triacylated bacterial Lpp by forming heterodimers together with TLR6 and TLR1, respectively (Jin et al. 2007; Kang et al. 2009). Its engagement by S. aureus-derived TLR2 ligands contributes to the integrity of the epithelial barrier by supporting tight junctions and skin wound repair (Kuo et al. 2013). In vivo studies demonstrated that mice lacking TLR2 (or the central TLR adaptor molecules MyD88 and IRAK4) are highly susceptible to S. aureus infection (Takeuchi et al. 2000; Kielian et al. 2005; Yimin Kohanawa et al. 2013; Suzuki et al. 2002). In infection with this pathogen, these mice exhibit reduced cytokine responses and a higher bacterial burden in the affected organs. Well in line with these findings, patients with genetic mutations in the irak4 and myd88 loci display increased frequencies with pyogenic infections, among them S. aureus (reviewed in von Bernuth et al. 2012). However, the requirement for TLR2 in the human is less clear.
Staphylococcal Lpp account for approximately 2 % of the proteome and function as regulators of the iron transport through the cytoplasmic membrane (Schmaler et al. 2010; Babu et al. 2006; Maresso and Schneewind 2006). S. aureus expresses more than 50 lipoproteins, among them SitC or the tandem lipoproteins encoded in the pathogenicity island vSaα (Stoll et al. 2005; Hashimoto et al. 2006; Kurokawa et al. 2011; Nguyen et al. 2015). Maturation of Lpp involves several steps of posttranslational modification, the most important one being the transfer of a diacylglycerol group by the phosphatidyl glycerol diacylglyceryl transferase (lgt) (Sankaran et al. 1995; Sankaran and Wu 1995). This modification is essential for recognition via TLR2 (Stoll et al. 2005; Hashimoto et al. 2006; Kurokawa et al. 2011; Schmaler et al. 2009).
Activation of TLR2 on keratinocytes leads to the release of antimicrobial peptides (AMP) and proinflammatory cytokines and neutrophil-attracting chemokines (Niebuhr et al. 2010a; Wanke et al. 2011; Olaru and Jensen 2010). Well in line with a central role of TLR2 in the recognition of S. aureus in the skin, unresponsiveness of keratinocytes from patients with atopic dermatitis to TLR2 stimulation might contribute to colonization of affected skin with S. aureus (Niebuhr et al. 2011).
Similarly, expression of TLR2 protected against nasal colonization with S. aureus (Gonzalez-Zorn et al. 2005). Moreover, deficient secretion of nasal AMP and S. aureus-triggered downregulation of TLR2 have been proposed to support S. aureus nasal carriage (Gonzalez-Zorn et al. 2005; Nurjadi et al. 2013; Zanger et al. 2011; Quinn and Cole 2007) and adherence of S. aureus on bronchial epithelial cells that express low to absent levels of TLR2 (Mayer et al. 2007). Furthermore, recent reports proposed that S. aureus induces expression of TLR2 on salivary epithelial cells (Negrini et al. 2014) and triggers the release of the chemoattractant cytokine IL-8 from colonic epithelial cells in a TLR2-dependent manner (Kang et al. 2015), a finding important in light of the gastrointestinal tract serving as an important reservoir for colonizing S. aureus strains (Nowrouzian et al. 2011; Gries et al. 2005; Kato-Matsunaga and Okonogi 1996; Bhalla et al. 2007; Klotz et al. 2005).
Notably, sensing via TLR2 cannot distinguish between viable and dead bacteria. Furthermore, TLR2 activity is subject to strain variation, e.g., it varies depending on the proliferative and metabolic state (Hilmi et al. 2014). TLR2 recognition, therefore, exerts an immune stimulatory effect on epithelial cells and a broad range of immune cells, but it is not solely responsible for bacterial clearance or containment of adherent S. aureus on the epithelial surfaces. Not surprisingly, it was, thus, not possible to prove an association of single nucleotide polymorphisms in the tlr2 gene with either S. aureus carriage and/or associated rhinopolyposis (Sachse et al. 2010; Tewfik et al. 2008) or infection with S. aureus (El-Helou et al. 2011; Moore et al. 2004).
Keeping in mind that sensing of TLR2-active lipoproteins is coupled to bacterial proliferation and loss of cell wall integrity that uncover the cytoplasmic membrane (Hilmi et al. 2014; Wolf et al. 2011), the central role of TLR2 sensing might be attributed to two key determinants: Firstly, TLR2-dependent induction of AMP that degrade bacterial cells achieves a steady-state condition in the quantity of colonizing bacterial cells, thus making further recruitment and activation of phagocytes unnecessary: This principle has been described in drosophila but very probably is also valid for mammalian immune defense (Zaidman-Remy et al. 2006). Secondly, TLR2 exerts an important priming effect on epithelial cells and immune cells, thus preparing these cells to recognize invading S. aureus via intracellular sensors that subsequently induce its elimination by intracellular bacterial lysis in phagocytes or death of infected epithelial cells.
These events prepare the grounds for the polarization of T cell responses, most importantly the generation of Th17 cells in S. aureus skin infections, which are reviewed in Miller and Cho (2011). Nonetheless, S. aureus and diacylated TLR2 Lpp derived thereof also induce thymic stromal lymphopoietin (TSLP) (Takai et al. 2014; Vu et al. 2010), a cytokine expressed by human epidermal keratinocytes and mucosal epithelial cells that favors Th2 cell responses and blocks Th1/Th17 differentiation (Ziegler et al. 2013). In addition, S. aureus and TLR2 ligands have been implicated in the promotion of tolerance established via TLR2-dependent IL-10-mediated suppression of T cell responses induced by S. aureus or TLR2-dependent infiltration of colonized skin with myeloid suppressor cells (Skabytska et al. 2014; Chau et al. 2009).
Altogether, these findings highlight the central role of TLR2 in the recognition of S. aureus on epithelial cells from different body origins. Notably, the stimulatory function of TLR2 on epithelial cells is supplemented by its effects on professional phagocytes, which are discussed later in this chapter. The current findings further illustrate that TLR2 balances pro- and anti-inflammatory immune responses, thus permitting colonization but preventing infection.
2.2 Bacterial Invasion: Immune Defense Relies on Intracellular Sensors and Inflammasome Activation
Upon invasion of epithelial cells (or endosomal escape in professional phagocytes), cytosolic pattern recognition receptors (PRRs) secure immune detection of invading pathogens. In this context, recognition of S. aureus occurs via peptidoglycan binding by Nod receptors. The Nod1 receptor senses meso-diaminopimelic acid (mesoDAP) in PG, which is present in peptidoglycan of Gram-negative species; the Nod2 receptor recognizes the muramyl dipeptide present in peptidoglycan of Gram-positive and Gram-negative organisms (Girardin et al. 2003). Nod2−/− cells were irresponsive to S. aureus PG (Volz et al. 2010). Nevertheless, one report claimed that both Nod receptors contribute to the recognition of S. aureus (Kapetanovic et al. 2007). A central role of Nod2 in vivo was demonstrated in a skin infection model using in nod2-deficient mice (Hruz et al. 2009). In this study, production of α-toxin facilitated Nod2 recognition by promoting access to the cytosol through pore formation in the plasma membrane.
TLR2 stimulation represents an important costimulus for Nod2-dependent recognition of peptidoglycan and vice versa (Volz et al. 2010; Schaffler et al. 2014). Furthermore, staphylococcal lipoteichoic acid (LTA) enhances recognition of TLR2 ligands and PG by a, so far, ill-defined mechanism. Additionally, Nod2 ligand MDP enhances LTA-triggered induction of cyclooxygenase-2 in macrophages (Ahn et al. 2014), which increases bactericidal activity against S. aureus (Bernard and Gallo 2010).
Notably, recognition of PG is greatly facilitated by uncovering the minimal PG recognition motifs: PG digestion by PG recognition proteins (PGRP) facilitates Toll signaling in drosophila and phagocytosis of S. aureus in human cells (De Marzi et al. 2015; Garver et al. 2006). Moreover, PG digestion by lysozyme is an important prerequisite for efficient activation of the NLRP3 inflammasome (Shimada et al. 2010). However, the PG-hydrolyzing activity of the major autolysin (atl) and D-alanylated wall teichoic acid interferes with recognition of S. aureus PG by drosophila PGRP (Kurokawa et al. 2011; Atilano et al. 2011, 2014) and O-acetylation of muramic acid (oat) typically found in S. aureus makes PG resistant to the hydrolytic activity of lysozyme (Shimada et al. 2010; Bera et al. 2007). This results in a pathogenicity factor-mediated suppression of proinflammatory cytokine production and subsequent inflammasome activation, facilitates colonization of skin and mucosa, and enables prolonged intracellular persistence. Relevance of these mechanisms was further provided by studies on S. aureus mutants with minimized PG synthesis, i.e., absence of nonessential peptidoglycan binding proteins, which rendered S. aureus less virulent while more susceptible to antibiotics and resulted in better bacterial clearance in infection (Reed et al. 2015).
Subsequent to engagement of cytosolic PRR, S. aureus induces activation of the inflammasomes. These cytosolic molecular complexes mediate the activation of caspase-1, which triggers the release of biologically active IL-1β and IL-18 and cell death. Notably, next to TLR activation, inflammasome activation depends on the presence of ATP, which is required for the induction of an intracellular potassium flux (Franchi et al. 2007; Arlehamn et al. 2010; Petrilli et al. 2007). S. aureus establishes this intracellular potassium gradient via agr-dependent expression of pore-forming toxins, e.g., α-hemolysin, Panton-Valentine leucocidin (PVL), and phenol-soluble modulins; this results in NLRP3 (NACHT, leucine-rich repeat (LRR), and pyrin domain-containing protein 3)-dependent caspase-1 activation in a variety of cell types, ultimately leading to pyroptosis and secretion of IL-1β and IL-18 (Soong et al. 2015; Accarias et al. 2015; McGilligan et al. 2013; Holzinger et al. 2012; Perret et al. 2012; Craven et al. 2009; Munoz-Planillo et al. 2009; Chi et al. 2014; DuMont and Torres 2014; Queck et al. 2008; Bocker et al. 2001; Gurcel et al. 2006). By contrast, the absence of agr and/or pore-forming toxins promotes intracellular persistence and survival of S. aureus by inducing autophagy, a process counteracting NLRP3 and caspase-1 activation and associated pyroptosis and IL-1β production (Soong et al. 2015). Nevertheless, the role of autophagy and Nod/NLRP3 activation in immune recognition of intracellularly persisting metabolically inactive small colony variants has not been sufficiently addressed to date.
Moreover, an earlier study suggested that low MOIs of S. aureus promote IL-1β and IL-18 cleavage via activation of NLRC5 (Davis et al. 2011). In addition, a recent study suggested a specific role of the DNA-activated absent in melanoma-2 (AIM2) inflammasome in IL-1β-mediated bacterial immune defense in S. aureus CNS infection (Hanamsagar et al. 2014). To date, however, there is no evidence for involvement of the cytosolic RNA-sensing RIG-I-like receptors (RLR) in immune defense against S. aureus.
Notably, in murine S. aureus infection models, neutrophil recruitment, cutaneous abscess formation, and clearance of S. aureus in pneumonia were shown to be dependent on IL-1β levels (Labrousse et al. 2014; Cho et al. 2012; Miller et al. 2007). However, based on a murine model of α-toxin-dependent severe necrotizing pneumonia, pharmacological intervention with NLRP3 activation was also discussed to ameliorate the course of disease (Kebaier et al. 2012). Moreover, a study in patients with atopic dermatitis suggested that the Th2-derived cytokines IL-4, IL-5, and IL-13 counteract activation of S. aureus α-toxin-induced activation of NLRP3 and ASC (Niebuhr et al. 2014). This finding supported the hypothesis that susceptibility of Th2-prototypical DBA/2 mice to S. aureus infection could be attributed to the failure to activate the NLRP3 inflammasome, while S. aureus-resistant C57BL/6 mice with an inherent Th1-profile clear S. aureus by activating the inflammasome (Accarias et al. 2015).
2.3 Linking Inflammasomes to Protective T Cell Responses: The Role of NLRP3 in Th17 Differentiation
Interestingly, a recent study proposed that NLRP3/IL-1β-dependent recruitment of IL-17-producing γδ-T cells is required for recruitment of neutrophils to the infection site (Maher et al. 2013). Further in vitro studies showed that IL-1β and IL-23 support a T cell receptor and CD1d-dependent IL-17A response to heat-killed S. aureus in invariant NK T cells (Doisne et al. 2011). This T cell subset expresses a T cell receptor that recognizes lipid antigens in a CD1d-restricted manner and NK cell markers. It is typically found in barrier tissues where it assumes a protective role in colonization with S. aureus (Nieuwenhuis et al. 2009) and a murine S. aureus sepsis model (Kwiecinski et al. 2013). Alternatively, induction of Th17 cells was reported to be induced by dendritic cells; here, IL-1β production and IL-23 production were triggered by dual activation of surface TLRs (TLR2/4/5) and concomitant engagement of FcγRIIA by staphylococcal immune complexes with IgG (den Dunnen et al. 2012).
Taken together, these findings argue for the coexistence of redundant innate immune mechanisms for the induction of Th17-mediated immune defense. Future work is needed to understand whether certain mechanisms are more relevant in distinct disease entities.
3 Professional Phagocytes and Their Effector Functions
3.1 Phagocytosis: Linking Intracellular Lysis to Antigen Presentation
Engulfment, ingestion, and phagosomal degradation of microorganisms by processes such as acidification and enzyme digestion are essential for killing of the microbes and are required for activation of cells of the adaptive immune response via antigen presentation by MHCII molecules. This process is initiated by cellular recognition of S. aureus PAMPs and opsonins. One important opsonin is mannose-binding lectin (MBL), a collection that contributes to bacterial recognition and mediates activation of complement and innate immune cells. Its binding to S. aureus facilitates phagocytosis by enhancing opsonization by complement components and synergy with TLR2/6 ligands in the phagosome (Neth et al. 2000, 2002; Ip et al. 2008). Survival of mice lacking MBL is severely impaired after intraperitoneal injection of S. aureus (Shi et al. 2004). MBL has been shown to increase uptake of S. aureus via upregulation of the scavenger receptor SR-A (Ono et al. 2006). However, strain-specific differences in MBL binding have been attributed to differences in the surface carbohydrate structures (Shang et al. 2005).
It should, however, not be disregarded that the processes of internalization and phagosome maturation are important prerequisites for MyD88/TLR-dependent recognition of S. aureus (Wolf et al. 2011; Ip et al. 2010). Albeit TLR2 itself does not mediate phagocytosis, sensing of S. aureus and induction of the proinflammatory cytokine response only occur upon recruitment of TLR2 to the phagosome (Underhill et al. 1999). Degradation of bacteria in the phagosome facilitates recognition of PG via Nod receptors and, thus, activation of the inflammasome (Wolf et al. 2011; Shimada et al. 2010; Ip et al. 2010; Sokolovska et al. 2013; Kaplan et al. 2012). Interestingly, inflammasome-mediated activation of caspase-1 not only activates IL-1 and IL-18 precursors but also participates in the acidification of the phagosome (Sokolovska et al. 2013), which enables recognition of microbial nucleic acids by TLRs (Parcina et al. 2008, 2013).
Staphylococcal ribosomal (23S) RNA is recognized in a sequence-specific manner by TLR13 expressed in murine conventional CD8HIGH DC but not plasmacytoid dendritic cells (pDCs) (Oldenburg et al. 2012). This receptor is not expressed in humans, but staphylococcal RNA was recently shown to mediate human monocyte activation by TLR8 (Bergstrom et al. 2015). By contrast, in human B cells, pDC activation by S. aureus is blocked by inhibitory oligonucleotides blocking TLR7 and TLR9, the only TLRs expressed in these cell types (Parcina et al. 2008, 2013; Hornung et al. 2002).
Further support for a role of phagocytosis in cellular defense against S. aureus is provided by the importance of scavenger receptors in infection. These receptors mediate recognition and clearance of S. aureus and are typically expressed on phagocytes:
Most prominently, mice lacking CD36, a receptor expressed in macrophages, monocytes, endothelial, and epithelial cells, are highly susceptible to S. aureus infections (Hoebe et al. 2005; Stuart et al. 2005; Blanchet et al. 2014). Of note, mice with a double deficiency in CD36 and SR (scavenger receptor)-A, another scavenger receptor required for phagocytosis of S. aureus (Amiel et al. 2009), died of pneumonia but were protected from S. aureus peritonitis (Blanchet et al. 2014).
Interestingly, CD36 has been implicated in the recognition of phosphatidylserine on apoptotic cells (Fadok et al. 1998). The same receptor specifically recognizes S. aureus via the diacylglycerol moiety in LTA and acts as a coreceptor for TLR2 (Hoebe et al. 2005; Nilsen et al. 2008; Baranova et al. 2008). Supporting its association with TLR2, expression of CD36 was higher in patients with atopic dermatitis carrying a TLR2 polymorphism (R753Q) that leads to reduced TLR2 reactivity (Niebuhr et al. 2010b; Mrabet-Dahbi et al. 2008).
Moreover, PIR-B (paired immunoglobulin-like receptor) was also found to bind S. aureus LTA (Nakayama et al. 2012). A protective function in S. aureus pneumonia was found to be due to suppression of the inflammatory response (Banerjee et al. 2010).
Notably, CD36 and α5β1 integrin, the receptor for vitronectin, a plasma protein that binds to apoptotic cells, collaborate in apoptotic cell recognition (Fadok et al. 1998). S. aureus binds vitronectin via extracellular adherence protein (Eap) (Hussain et al. 2008). It is, therefore, likely that cells involved in apoptotic cell clearance also participate in the recognition of S. aureus. Well in line with the tolerogenic response induced by apoptotic cells, in murine peritonitis the presence of Eap on S. aureus had an anti-inflammatory effect that led to a reduction in infiltrating neutrophils (Chavakis et al. 2002).
3.2 Tissue-resident Phagocytes
3.2.1 Mast Cells: Well-prepared Guardians of Skin and Mucosa
Mast cells are important sentinels in the skin and mucosa. These phagocytic cells are prepared to kill bacteria with intracellular ROS or release extracellular traps and AMP. They further have the unique ability to rapidly release cytoplasmic granules storing preformed vasoactive and immune stimulatory mediators, e.g., histamine, TNF, and the proteases tryptase and chymase into the extracellular environment. In bacterial infections, they have mainly been implicated in defense against respiratory pathogens in pneumonia (reviewed in Urb and Sheppard 2012). Since they predominantly reside at sites where encounters with invading pathogens are to be expected due to constant contact with the exterior environment, it should not be over interpreted that mast cells were not required for bacterial clearance or host survival in an in vivo S. aureus peritonitis model (Ronnberg et al. 2014).
Mast cells contribute to the elimination of S. aureus by release of extracellular traps, proinflammatory cytokines, and AMP (Abel et al. 2011; Rocha-de-Souza et al. 2008; von Kockritz-Blickwede et al. 2008a). S. aureus-derived PG, LTA, protein A, and TLR2 ligands have been implicated in mast cell activation (Jawdat et al. 2006; Matsui and Nishikawa 2002, 2005; Terada et al. 2006; McCurdy et al. 2003). Degranulation was recently postulated to be specifically induced by S. aureus δ-toxin, which links colonization to allergic reactions in atopic dermatitis (Nakamura et al. 2013). However, recent studies highlight that S. aureus evades these bactericidal effects by invading mast cells via α5β1 integrin, a reaction demonstrated in mucosal tissue from nasal polyposis (Abel et al. 2011; Hayes et al. 2015).
3.2.2 Macrophages: Tissue-Specific Vigilants Balancing the Local Immune Response
Macrophages are professional phagocytes that are either resident in the tissue or develop from monocytes that enter the tissue from the blood vessels. Their major function is the clearance of invading pathogens from the site of infection and antigen presentation to T cells in the periphery and in the lymphatic organs. Tissue-resident macrophages are often highly specialized on their specific environment, e.g., marginal zone macrophages in the spleen, microglia in the brain, alveolar macrophages, and macrophages in the thymus are each equipped to fulfill their specific tasks.
Again, TLR2 plays a central role in the release of soluble mediators mediating bacterial defense and in bacterial killing, e.g., in macrophages, staphylococcal lipoproteins induce proinflammatory cytokines, nitric oxide (NO), and reactive oxygen species (ROS) (Nguyen et al. 2015; Kim et al. 2015; Nandi et al. 2015; Bishayi et al. 2014). Moreover, in brain abscesses of TLR2-deficient mice, secretion of proinflammatory cytokines and NO was abrogated, but IL-17 production was increased (Kielian et al. 2005). Furthermore, expression of S. aureus-binding scavenger receptor, lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) (Shimaoka et al. 2001), and murine macrophage scavenger receptor (MSR)-AI/II was increased in response to S. aureus and TLR2 ligands in microglia arguing for a specific role of TLR2 and these scavenger receptors in CNS infection (Kielian et al. 2005).
By contrast, a recent study claimed that neither TLR2 expression on macrophages nor proinflammatory cytokine production by macrophages is essential for immune defense against S. aureus (Yimin Kohanawa et al. 2013). Nevertheless, the importance of macrophages in antistaphylococcal immune defense was recently demonstrated in a murine post-influenza pneumonia model where resistance to S. aureus infection was achieved by GM-CSF-mediated influx of neutrophils and alveolar macrophages and mainly established by the production of reactive oxygen species (ROS) in macrophages (Subramaniam et al. 2014). In an additional study, MyD88-dependent activation of dermal macrophages and their interaction with neutrophils was shown to be required for control of neutrophil-mediated inflammation in cutaneous S. aureus infection (Feuerstein et al. 2015).
In addition, several studies reported binding and uptake of S. aureus cells in marginal zone (MZ) macrophages (Palecanda et al. 1999; van der Laan et al. 1999; Westerberg et al. 2008; Birjandi et al. 2011). It was further suggested that in vivo uptake was mediated by macrophage receptor with collagenous structure (MARCO) (Palecanda et al. 1999; van der Laan et al. 1999). This receptor might, therefore, play a central role in MZ macrophage-mediated removal of S. aureus from the bloodstream.
Notably, the currently available studies do not differentiate between pro- and anti-inflammatory monocyte and macrophage subsets. However, macrophage polarization is highly dependent on the microenvironment, e.g., GM-CSF, TLR2 ligands, and Fcγ receptors differentially affect macrophage function (reviewed in Martinez and Gordon 2014). Contradictory findings, might therefore, arise from the tissue- and/or mouse strain-specific milieu and the predominance of functionally distinct subpopulations.
3.3 Blood-Derived Phagocytes
Both epithelial cells and tissue-resident innate immune cells release chemokines and cytokines upon bacterial encounter. These soluble mediators provoke transmigration of cells from the blood circulation. Activation induces differentiation peripheral blood monocytes to inflammatory monocytes, macrophages, and dendritic cells that transmigrate into the tissue and participate in bacterial clearance.
3.3.1 Neutrophils: Recruited to Resolve Uncontrolled Spread of Infection
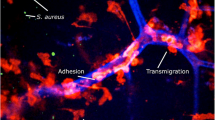
Neutrophils are recruited to the site of infection and represent the hallmark of early antistaphylococcal defense in the invaded tissue (reviewed in Rigby and DeLeo 2012). They further participate in abscess formation, an event that is supported by S. aureus-derived toxins and their ability to induce a specific form of programmed cell necrosis (necroptosis) (reviewed in Kobayashi et al. 2015; Greenlee-Wacker et al. 2015). Indeed, phagocytosis and subsequent intracellular lysis of S. aureus in neutrophils are often prevented by staphylococcal virulence factors, and infected neutrophils containing viable bacteria evade efferocytosis by macrophages via an upregulation of CD47 expression, the phosphatidylserine “don’t eat me” signal (Greenlee-Wacker et al. 2014).
Nevertheless, neutrophils are essential for antistaphylococcal innate immune defense, a fact that was demonstrated in mice depleted of neutrophils (Kohler et al. 2011; Robertson et al. 2008; Verdrengh and Tarkowski 1997) but can also be derived from increased susceptibility to S. aureus seen in neutropenic patients and in patients with genetic defects in neutrophil function, i.e., chronic granulomatosis disease (CGD) (Hartl et al. 2008; McNeil 2014) and mouse models thereof (Kohler et al. 2011; Pizzolla et al. 2012). Furthermore, increased mortality in S. aureus septicemia after depletion of complement was mainly attributed to insufficient recruitment of neutrophils (Sakiniene et al. 1999). In addition, in mice, MyD88-dependent IL-1 and IL-18 signalings are required for mobilization of neutrophils and resolution of staphylococcal skin abscesses (Miller et al. 2006) and post-burn infection (Kinoshita et al. 2011). Therefore, neutrophils remain central cells in immune defense against S. aureus and other compensatory mechanisms such as the release of bactericidal antimicrobial peptides most likely govern neutrophil-dependent first-line immune defense against S. aureus.
Despite their synthesis in many different types of phagocytes, e.g., monocytes, macrophages, and DC, the highest levels of defensins as HNP1-3 are expressed by neutrophils (Ryu et al. 2014). AMP, such as cathelicidin (LL-37) and α-defensin, contribute to bacterial killing and degradation in the phagosome (Jann et al. 2009), neutralize toxins (Cardot-Martin et al. 2015), induce the production of cytokines (Chaly et al. 2000), exert chemotactic activity (Yang et al. 2001), and facilitate the formation of neutrophil extracellular traps (NETs) (Neumann et al. 2014). However, again, S. aureus virulence factors shield the pathogen from AMP. This occurs through AMP-inactivating enzymes (Braff et al. 2007; Jin et al. 2004; Sieprawska-Lupa et al. 2004) and modification of charge of surface structures, e.g., via D-alanylation of WTA and LTA (Collins et al. 2002; Peschel et al. 1999; Simanski et al. 2013).
A recent study further highlights that in vivo recruitment of neutrophils to the site of infection occurs in two waves, e.g., initially from the blood, and secondly, they are mobilized from the bone marrow (Kamenyeva et al. 2015). The authors propose that in S. aureus infection, neutrophils enter the lymph node medulla and interfollicular areas where they interact with B cells to interfere with antibody production (Kamenyeva et al. 2015). The significance of these findings remains unclear but is well compatible with a previously described anti-inflammatory role of neutrophils in staphylococcal arthritis (Verdrengh and Tarkowski 1997). Concomitant with the release of neutrophil myeloid-related proteins (MRP) -8 and -14 into the bronchoalveolar fluid exerts an additional protective effect in pneumonia by mediating transmigration of neutrophils, this phenomenon might be necessary for resolution of inflammation (Achouiti et al. 2015).
3.4 Dendritic Cells: Orchestrating the Adaptive Immune Response in Tissue and Lymph Nodes
3.4.1 Myeloid Dendritic Cells: Expert Control of T Cell Responses
Dendritic cells regulate the T cell response in infection via presentation of antigen and concomitant expression of T cell polarizing costimulatory ligands and release of cytokines and other soluble mediators.
Myeloid dendritic cells (mDCs) are specialized cells whose major function is to present antigen to T cells after migration to the local lymph nodes. They play a key role in the clearance of S. aureus, and their depletion increases mortality in an in vivo model of bacteremia (Schindler et al. 2012). Furthermore, Jin et al. highlighted that mDCs expressing the C-type lectin BDCA1+ are highly responsive to S. aureus and support differentiation of T cells into IFNγ-producing CD4+ (Th1) and CD8+ (Tc1) cells (Jin et al. 2014). Interestingly, this property was attributed to high expression levels for TLR2 and the scavenger receptor SR-A whose expression levels were low on mDCs with a BDCA3+ CD16+ phenotype.
Another study differentiated the effects of S. aureus Lpp and PG on monocytes and on in vitro generated monocyte-derived dendritic cells: While PI3K-dependent production of anti-inflammatory IL-10 is induced on monocytes, production of Th1/Th17 polarizing cytokines IL-12 and IL-23 is triggered in monocyte-derived dendritic cells (Frodermann et al. 2011). Well in line with this report, it was demonstrated that IRAK4 reverts IL-10-dominated tolerogenic MyD88-dependent signaling in S. aureus- or TLR2/4-stimulated human monocytes from PKB/Akt/mTOR-dependent IL-10 to Th1-promoting IL-12 secretion (Over et al. 2013). However, release of phenol-soluble modulins by S. aureus results in the loss of T cell priming capacity and the induction of T regulatory cells (Schreiner et al. 2013).
In the skin, S. aureus is phagocytosed by Langerhans cells, a specialized DC subset found in dermis (Reis e Sousa et al. 1993). Furthermore, in a murine atopic dermatitis model, dual activation of DC via TLR2 ligands and IL-4 induced the progression of self-limited Th2-mediated dermatitis to chronic cutaneous inflammation (Kaesler et al. 2014).
Myeloid-derived suppressor cells, on the contrary, suppress T cell responses and thereby contribute to the persistence of S. aureus in chronic infections (Tebartz et al. 2015). They further halt monocyte and macrophage-mediated clearance of S. aureus biofilm (Heim et al. 2014), and once induced by TLR2/6 ligands are recruited to S. aureus colonized skin where they suppress T cell responses to the pathogen (Skabytska et al. 2014).
3.4.2 Plasmacytoid Dendritic Cells and Type I Interferons: Fine-Tuning of Innate and Adaptive Immune Responses
Plasmacytoid dendritic cells are considered to be DC of lymphoid origin. They represent the major producers of systemically active interferon-α in the human body and have been implicated in tolerance to oral antigens (Goubier et al. 2008). They are present in the blood, in the lymphatic organs, and probably all peripheral tissues. S. aureus induces pDC-derived secretion of IFN-α, TNF, and IL-6 (Parcina et al. 2008; Michea et al. 2013). However, depending on the tissue environment, soluble mediators such as prostaglandins and TGF-β alter pDC-derived cytokine secretion panels and their function (Michea et al. 2013; Bekeredjian-Ding et al. 2009; Contractor et al. 2007). Their role in infection, particularly S. aureus infection, was extensively reviewed in Bekeredjian-Ding et al. (2014).
One of their most important characteristics is the expression of Fcγ receptor IIA and Fcε receptor that exert positive and negative regulatory effects, respectively, on pDC activation and release of IFN-α. Therefore, pDC most likely orchestrate the secondary immune response to S. aureus (Parcina et al. 2008), and their presumptive role in first-line immune defense is limited to invasion by protein A-bearing S. aureus strains and pDC-mediated support of polyclonal B cell activation (Parcina et al. 2013).
Additionally, pDC might be involved regulation of the antistaphylococcal immune response by type I interferon. In vivo, induction of IFN-α production in pDC by TLR9 ligand CpG ODN was protective against S. aureus pneumonia in a hemorrhagic shock model (Roquilly et al. 2010) and IFN-α-mediated resistance of host cells against S. aureus alpha toxin (Lizak and Yarovinsky 2012). Similarly, IFN-β increased clearance of S. aureus from murine BMDC and human monocytes in vitro and in an cutaneous infection model in vivo (Kaplan et al. 2012), and deficiency in IFN-α/β receptor or TLR9 expression resulted in improved clearance of bacteria from mice with S. aureus pneumonia (Parker and Prince 2012). However, IFN-β production was associated with increased inflammation and cellular necrosis in murine skin infection (Kaplan et al. 2012).
Notably, several studies showed that IFN-α- and IFN-α-inducing TLR7/9 agonists suppressed the formation of Th17 cells under healthy conditions, in infection, and autoimmune disease (Cui et al. 2014; Hirohata et al. 2010; Liu et al. 2011; Meyers et al. 2006; Vultaggio et al. 2011). The inhibitory effect of IFN-α on Th17 responses can be attributed to the induction of the IL-17-suppressing cytokine IL-27 (reviewed in Goriely et al. 2009). In accordance with these findings, lack of IL-27 receptor expression increases Th17 cell numbers and decreases the bacterial burden in post-influenza S. aureus pneumonia (Robinson et al. 2015).
Moreover, a recent report showed that S. aureus-induced expression of osteopontin in DC was responsible for increased Th17 induction (Salvi et al. 2013). Upregulation of osteopontin occurred upon TLR2 activation and concomitant absence of IFN-β induction, which was observed with Gram-negative bacteria after engagement of LPS/TLR4 and subsequent recruitment of TRIF (Salvi et al. 2013). Notably, S. aureus-induced IFN-β release is further prevented by lysozyme resistance of S. aureus and its resistance to degradation in the phagosome (Kaplan et al. 2012). Nevertheless, protective effects of IFN-I-mediated suppression of Th17 responses on DC have also been described: In a murine EAE model, IFN-I suppressed the expression of an intracellular translational isoform of osteopontin (iOPN) in myeloid DC; this enabled IL-27 synthesis and, in turn, decreased Th17-mediated inflammation, which ultimately slowed down the progression of EAE (Shinohara et al. 2008; Guo et al. 2008).
Furthermore, generation of monocyte-derived dendritic cells in the presence of IL-27 resulted in increased suppression of intracellular growth of S. aureus, upregulation of MHC II, and increased IL-12 secretion that shifted the T cell response to a Th1 phenotype (Jung et al. 2015). IL-27 further induced the synthesis of IFN-α and IFNλ1 in PBMC and DC derived from healthy donors, thus promoting an autoregulatory negative feedback loop (Cao et al. 2014). Thus, intertwined regulation of IFN-α and IL-17 determines the polarization of CD4+ T cell responses, the efficacy of bacterial clearance, and the degree of inflammation in autoimmune disease and infection. A fine-tuned balancing of these two cytokines is, therefore, most likely very critical for the resolution of S. aureus infection and immunopathology.
4 The Last Frontiers Before Adaptive Immunity
4.1 Innate Immune B Cells: Rapid Supply of Antibacterial Antibodies
In murine peritonitis and sepsis, rapid accumulation of IgM-secreting plasmablasts is observed within 48 h after bacterial challenge (Martin et al. 2001). The cells responsible for this early IgM secretion are B cell subsets that express B cell receptors that recognize thymus-independent (TI) antigens, e.g., bacterial PAMPs such as capsular polysaccharides, LPS, and phosphocholine and phosphatidylserine, which are also present on apoptotic cells. Together with PRR, stimulation targeting of these B cell receptors elicits B cell proliferation and differentiation to plasmablast in a T cell-independent manner. Release of these antibacterial IgM molecules (also called natural IgM) enables bacterial opsonization and subsequent complement activation. It has further been postulated that cellular uptake of S. aureus immune complexes with IgM is promoted by an Fcα/μ receptor (Shibuya and Honda 2006).
Among these, specialized B cell subsets are CD5-positive B1a and CD5-negative B1b cells that reside in the peritoneal and pleural cavities in mice. B1b cells are capable of phagocytosis of S. aureus, intracellular bacterial killing, and antigen presentation to T cells (Gao et al. 2012). Albeit this response is weaker than in macrophages, it underlines the phylogenetic relationship of innate immune B cells and macrophages.
Notably, S. aureus targets VH3+ B cells via protein A, which results in long-term depletion of the B1a and MZ B cell pools (Goodyear and Silverman 2004; Viau et al. 2005). In the presence of pDC and costimulation via TLR2-active Lpp and endosomal TLRs, protein A-activated B cells are not directly harmed by programmed cell death but undergo differentiation to IL-10-secreting B regulatory cells and IgM-producing plasmablasts (Parcina et al. 2013; Bekeredjian-Ding et al. 2007). This process enables IL-10-mediated suppression of T cell responses before ultimately resulting in cell death due to the physiologically short life span of plasmablasts and, again, extinction of these B cell subsets.
4.2 Natural Killer Cells: Neglected Sensors for Intracellular Persisting S. aureus?
NK cells have mainly been studied in viral infection and malignancy. They are attracted to the infected tissue and enter by transmigration from the bloodstream. Their ability to recognize infected and damaged cells independent of MHC-restricted antigen presentation of antibodies makes them very flexible and invaluable cells for the rapid defense against invading pathogens. Upon encounter of suspicious cells, the release of IFNγ and granules containing cytotoxic proteases (granzymes) and perforine induces apoptosis of the encountered cell.
Although resistance Rag2-IL2Rγ−/− C57BL/6 mice against S. aureus infection suggested that NK cells are not essential for innate immune defense against S. aureus (von Kockritz-Blickwede et al. 2008b), the interaction of natural killer cells with alveolar macrophages was shown to be beneficial in murine S. aureus pneumonia models (Zhao et al. 2014; Small et al. 2008). Similarly, upon exposure to S. aureus interaction with monocytes was required for activation of human NK cells in vitro (Haller et al. 2002). A protective role of NK cells was further described in the development of S. aureus arthritis (Nilsson et al. 1999). Detection of NK cells in human joint infections caused by chronically persistent S. aureus, indeed, argues for a potential role of NK cells at the site of infection (Wagner et al. 2006). Nevertheless, the obvious role of NK cells in the detection intracellularly persisting S. aureus and the elimination of infected cells have not yet been demonstrated.
4.3 Innate Lymphoid Cells: Confinement of S. aureus to Its Niche?
In tissues exposed to the environment and colonized by commensals, innate lymphoid cells (ILCs) prevent bacterial translocation beyond the epithelial barrier, trigger mucosal IgA production, and function as important regulators of immune homeostasis (reviewed in Philip and Artis 2013; Tait Wojno and Artis 2012; Diefenbach et al. 2014). Notably, commensals trigger the development of ILC, and ILCs phenotypically and functionally adapt to changes in the composition of the local microbiome (Tait Wojno and Artis 2012). NK cell receptors (NCR) such as NKp44/46 and expression of TLR2 and its coreceptors enable binding and sensing of the local microbiota (Philip and Artis 2013).
Three different classes of murine ILC have been defined by the expression of prototypical transcription factors and T helper cell-like cytokine secretion profiles, e.g., Tbet+ ILC (group 1) produce IFNγ, GATA3+ ILC (group 2) secrete IL-5, IL-9, and IL-13 and RORγt+ ILC (group 3) release IL-17A, IFNγ, and IL-22 (Diefenbach et al. 2014; Robinette et al. 2015). Notably, NK cells have recently been allocated to group 1 ILC (Philip and Artis 2013; Robinette et al. 2015; Monticelli et al. 2012). In the human, similar ILC subsets have been described, including different group 2-like ILCs in the respiratory tract and skin or different CD127+ group 3-like ILCs in tonsils, appendix, and Peyer’s patches (Tait Wojno and Artis 2012; Monticelli et al. 2012; Mjosberg and Eidsmo 2014). Nevertheless, despite their presence at the main sites of S. aureus colonization, at present there is no available information on their role in preventing invasion. We can only speculate that in chronic carriers, ILC might confine S. aureus to its niche, thus preventing systemic immune responses.
5 Conclusion
A broad variety of receptor systems cooperates in sensing S. aureus and regulating the host immune response to this pathogen (see Table 1 for summary). TLR2-dependent recognition of S. aureus by epithelial cells limits the spread of colonizing S. aureus on the skin and mucosal surfaces. Upon cell and tissue invasion activation of resident innate immune cells by S. aureus fosters a proinflammatory environment that attracts neutrophils, NK cells, and monocytes from the blood. Notably, degradation of bacteria in the phagosomes is essential for bacterial recognition via TLR, activation of the inflammasomes, and subsequent bacterial clearance. However, evolution has selected S. aureus strains that are resistant to these processes.
At present, we know that innate immune cells and PRR also participate in the resolution of S. aureus infections. However, the exact mechanisms involved remain to be investigated and are potentially exploited by S. aureus to promote tolerance. Finally, future work is needed to clarify the role of innate immunity and, in particular, NK cells in the recognition of intracellular persisting S. aureus and the role of ILC in immune homeostasis in chronic carriage.
References
Abel J, Goldmann O, Ziegler C, Holtje C, Smeltzer MS, Cheung AL, Bruhn D, Rohde M, Medina E (2011) Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. J Innate Immun 3(5):495–507. doi:10.1159/000327714
Accarias S, Lugo-Villarino G, Foucras G, Neyrolles O, Boullier S, Tabouret G (2015) Pyroptosis of resident macrophages differentially orchestrates inflammatory responses to Staphylococcus aureus in resistant and susceptible mice. Eur J Immunol 45(3):794–806. doi:10.1002/eji.201445098
Achouiti A, Vogl T, Van der Meer AJ, Stroo I, Florquin S, de Boer OJ, Roth J, Zeerleder S, van’t Veer C, de Vos AF, van der Poll T (2015) Myeloid-related protein-14 deficiency promotes inflammation in staphylococcal pneumonia. Eur Respir J. doi:10.1183/09031936.00183814
Ahn KB, Jeon JH, Baik JE, Park OJ, Kang SS, Yun CH, Park JH, Han SH (2014) Muramyl dipeptide potentiates staphylococcal lipoteichoic acid induction of cyclooxygenase-2 expression in macrophages. Microbes Infect 16(2):153–160. doi:10.1016/j.micinf.2013.10.018
Amiel E, Alonso A, Uematsu S, Akira S, Poynter ME, Berwin B (2009) Pivotal advance: Toll-like receptor regulation of scavenger receptor-A-mediated phagocytosis. J Leukoc Biol 85(4):595–605. doi:10.1189/jlb.1008631
Arlehamn CS, Petrilli V, Gross O, Tschopp J, Evans TJ (2010) The role of potassium in inflammasome activation by bacteria. J Biol Chem 285(14):10508–10518. doi:10.1074/jbc.M109.067298
Atilano ML, Pereira PM, Vaz F, Catalao MJ, Reed P, Grilo IR, Sobral RG, Ligoxygakis P, Pinho MG, Filipe SR (2014) Bacterial autolysins trim cell surface peptidoglycan to prevent detection by the Drosophila innate immune system. Elife 3:e02277. doi:10.7554/eLife.02277
Atilano ML, Yates J, Glittenberg M, Filipe SR, Ligoxygakis P (2011) Wall teichoic acids of Staphylococcus aureus limit recognition by the drosophila peptidoglycan recognition protein-SA to promote pathogenicity. PLoS Pathog 7(12):e1002421. doi:10.1371/journal.ppat.1002421
Babu MM, Priya ML, Selvan AT, Madera M, Gough J, Aravind L, Sankaran K (2006) A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J Bacteriol 188(8):2761–2773. doi:188/8/2761 [pii] 10.1128/JB.188.8.2761-2773.2006
Banerjee A, Stevenaert F, Pande K, Haghjoo E, Antonenko S, Gorman DM, Sathe M, McClanahan TK, Pierce R, Turner SP, Bigler ME, Phillips JH, Heyworth PG (2010) Modulation of paired immunoglobulin-like type 2 receptor signaling alters the host response to Staphylococcus aureus-induced pneumonia. Infect Immun 78(3):1353–1363. doi:10.1128/iai.00969-09
Baranova IN, Kurlander R, Bocharov AV, Vishnyakova TG, Chen Z, Remaley AT, Csako G, Patterson AP, Eggerman TL (2008) Role of human CD36 in bacterial recognition, phagocytosis, and pathogen-induced JNK-mediated signaling. J Immunol 181(10):7147–7156
Bekeredjian-Ding I, Greil J, Ammann S, Parcina M (2014) Plasmacytoid dendritic cells: neglected regulators of the immune response to Staphylococcus aureus. Front Immunol 5:238. doi:10.3389/fimmu.2014.00238
Bekeredjian-Ding I, Inamura S, Giese T, Moll H, Endres S, Sing A, Zahringer U, Hartmann G (2007) Staphylococcus aureus protein A triggers T cell-independent B cell proliferation by sensitizing B cells for TLR2 ligands. J Immunol 178(5):2803–2812
Bekeredjian-Ding I, Schafer M, Hartmann E, Pries R, Parcina M, Schneider P, Giese T, Endres S, Wollenberg B, Hartmann G (2009) Tumour-derived prostaglandin E and transforming growth factor-beta synergize to inhibit plasmacytoid dendritic cell-derived interferon-alpha. Immunology 128(3):439–450. doi:10.1111/j.1365-2567.2009.03134.x
Bera A, Biswas R, Herbert S, Kulauzovic E, Weidenmaier C, Peschel A, Gotz F (2007) Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J Bacteriol 189(1):280–283. doi:JB.01221-06 [pii] 10.1128/JB.01221-06
Bergstrom B, Aune MH, Awuh JA, Kojen JF, Blix KJ, Ryan L, Flo TH, Mollnes TE, Espevik T, Stenvik J (2015) TLR8 senses Staphylococcus aureus RNA in human primary monocytes and macrophages and induces IFN-beta production via a TAK1-IKKbeta-IRF5 signaling pathway. J Immunol. doi:10.4049/jimmunol.1403176
Bernard JJ, Gallo RL (2010) Cyclooxygenase-2 enhances antimicrobial peptide expression and killing of Staphylococcus aureus. J Immunol 185(11):6535–6544. doi:10.4049/jimmunol.1002009
Bhalla A, Aron DC, Donskey CJ (2007) Staphylococcus aureus intestinal colonization is associated with increased frequency of S. aureus on skin of hospitalized patients. BMC Infect Dis 7:105. doi:10.1186/1471-2334-7-105
Birjandi SZ, Ippolito JA, Ramadorai AK, Witte PL (2011) Alterations in marginal zone macrophages and marginal zone B cells in old mice. J Immunol 186(6):3441–3451. doi:10.4049/jimmunol.1001271 jimmunol.1001271 [pii]
Bishayi B, Bandyopadhyay D, Majhi A, Adhikary R (2014) Possible role of Toll-like receptor-2 in the intracellular survival of Staphylococcus aureus in murine peritoneal macrophages: involvement of cytokines and anti-oxidant enzymes. Scand J Immunol 80(2):127–143. doi:10.1111/sji.12195
Blanchet C, Jouvion G, Fitting C, Cavaillon JM, Adib-Conquy M (2014) Protective or deleterious role of scavenger receptors SR-A and CD36 on host resistance to Staphylococcus aureus depends on the site of infection. PLoS ONE 9(1):e87927. doi:10.1371/journal.pone.0087927
Bocker U, Manigold T, Watson JM, Singer MV, Rossol S (2001) Regulation of Staphylococcus aureus-mediated activation of interleukin-18 in peripheral blood mononuclear cells. Eur Cytokine Netw 12(4):631–638
Braff MH, Jones AL, Skerrett SJ, Rubens CE (2007) Staphylococcus aureus exploits cathelicidin antimicrobial peptides produced during early pneumonia to promote staphylokinase-dependent fibrinolysis. J Infect Dis 195(9):1365–1372. doi:10.1086/513277
Broker BM, Holtfreter S, Bekeredjian-Ding I (2014) Immune control of Staphylococcus aureus—regulation and counter-regulation of the adaptive immune response. Int J Med Microbiol 304(2):204–214. doi:10.1016/j.ijmm.2013.11.008
Cao Y, Zhang R, Zhang W, Zhu C, Yu Y, Song Y, Wang Q, Bai L, Liu Y, Wu K, Wu J (2014) IL-27, a cytokine, and IFN-lambda1, a type III IFN, are coordinated to regulate virus replication through type I IFN. J Immunol 192(2):691–703. doi:10.4049/jimmunol.1300252
Cardot-Martin E, Casalegno JS, Badiou C, Dauwalder O, Keller D, Prevost G, Rieg S, Kern WV, Cuerq C, Etienne J, Vandenesch F, Lina G, Dumitrescu O (2015) alpha-Defensins partially protect human neutrophils against Panton-Valentine leukocidin produced by Staphylococcus aureus. Lett Appl Microbiol. doi:10.1111/lam.12438
Chaly YV, Paleolog EM, Kolesnikova TS, Tikhonov II, Petratchenko EV, Voitenok NN (2000) Neutrophil alpha-defensin human neutrophil peptide modulates cytokine production in human monocytes and adhesion molecule expression in endothelial cells. Eur Cytokine Netw 11(2):257–266
Chau TA, McCully ML, Brintnell W, An G, Kasper KJ, Vines ED, Kubes P, Haeryfar SM, McCormick JK, Cairns E, Heinrichs DE, Madrenas J (2009) Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat Med 15(6):641–648
Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT (2002) Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med 8(7):687–693. doi:10.1038/nm728
Chi CY, Lin CC, Liao IC, Yao YC, Shen FC, Liu CC, Lin CF (2014) Panton-Valentine leukocidin facilitates the escape of Staphylococcus aureus from human keratinocyte endosomes and induces apoptosis. J Infect Dis 209(2):224–235. doi:10.1093/infdis/jit445
Cho JS, Guo Y, Ramos RI, Hebroni F, Plaisier SB, Xuan C, Granick JL, Matsushima H, Takashima A, Iwakura Y, Cheung AL, Cheng G, Lee DJ, Simon SI, Miller LS (2012) Neutrophil-derived IL-1beta is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog 8(11):e1003047. doi:10.1371/journal.ppat.1003047
Collins LV, Kristian SA, Weidenmaier C, Faigle M, Van Kessel KP, Van Strijp JA, Gotz F, Neumeister B, Peschel A (2002) Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J Infect Dis 186(2):214–219. doi:10.1086/341454
Contractor N, Louten J, Kim L, Biron CA, Kelsall BL (2007) Cutting edge: Peyer’s patch plasmacytoid dendritic cells (pDCs) produce low levels of type I interferons: possible role for IL-10, TGFbeta, and prostaglandin E2 in conditioning a unique mucosal pDC phenotype. J Immunol 179(5):2690–2694
Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA (2009) Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 4(10):e7446. doi:10.1371/journal.pone.0007446
Cui F, Meng J, Luo P, Chen P (2014) IFN- alpha blocks IL-17 production by peripheral blood mononuclear cells in patients with chronic active hepatitis B Infection. BMC Infect Dis 14:55. doi:10.1186/1471-2334-14-55
Davis BK, Roberts RA, Huang MT, Willingham SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ, Accavitti-Loper MA, Duncan JA, Ting JP (2011) Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol 186(3):1333–1337. doi:10.4049/jimmunol.1003111
De Marzi MC, Todone M, Ganem MB, Wang Q, Mariuzza RA, Fernandez MM, Malchiodi EL (2015) Peptidoglycan recognition protein-peptidoglycan complexes increase monocyte/macrophage activation and enhance the inflammatory response. Immunology. doi:10.1111/imm.12460
den Dunnen J, Vogelpoel LT, Wypych T, Muller FJ, de Boer L, Kuijpers TW, Zaat SA, Kapsenberg ML, de Jong EC (2012) IgG opsonization of bacteria promotes Th17 responses via synergy between TLRs and FcgammaRIIa in human dendritic cells. Blood 120(1):112–121. doi:10.1182/blood-2011-12-399931
Diefenbach A, Colonna M, Koyasu S (2014) Development, differentiation, and diversity of innate lymphoid cells. Immunity 41(3):354–365. doi:10.1016/j.immuni.2014.09.005
Doisne JM, Soulard V, Becourt C, Amniai L, Henrot P, Havenar-Daughton C, Blanchet C, Zitvogel L, Ryffel B, Cavaillon JM, Marie JC, Couillin I, Benlagha K (2011) Cutting edge: crucial role of IL-1 and IL-23 in the innate IL-17 response of peripheral lymph node NK1.1- invariant NKT cells to bacteria. J Immunol 186(2):662–666. doi:10.4049/jimmunol.1002725
DuMont AL, Torres VJ (2014) Cell targeting by the Staphylococcus aureus pore-forming toxins: it’s not just about lipids. Trends Microbiol 22(1):21–27. doi:10.1016/j.tim.2013.10.004 S0966-842X(13)00202-3 [pii]
El-Helou O, Berbari EF, Brown RA, Gralewski JH, Osmon DR, Razonable RR (2011) Functional assessment of Toll-like receptor 2 and its relevance in patients with Staphylococcus aureus infection of joint prosthesis. Hum Immunol 72(1):47–53. doi:10.1016/j.humimm.2010.10.001
Fadok VA, Warner ML, Bratton DL, Henson PM (1998) CD36 is required for phagocytosis of apoptotic cells by human macrophages that use either a phosphatidylserine receptor or the vitronectin receptor (alpha v beta 3). J Immunol 161(11):6250–6257
Feuerstein R, Seidl M, Prinz M, Henneke P (2015) MyD88 in macrophages is critical for abscess resolution in staphylococcal skin infection. J Immunol 194(6):2735–2745. doi:10.4049/jimmunol.1402566 jimmunol.1402566 [pii]
Franchi L, Kanneganti TD, Dubyak GR, Nunez G (2007) Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282(26):18810–18818. doi:10.1074/jbc.M610762200
Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J (2011) A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis 204(2):253–262. doi:10.1093/infdis/jir276
Gao J, Ma X, Gu W, Fu M, An J, Xing Y, Gao T, Li W, Liu Y (2012) Novel functions of murine B1 cells: active phagocytic and microbicidal abilities. Eur J Immunol 42(4):982–992. doi:10.1002/eji.201141519
Garver LS, Wu J, Wu LP (2006) The peptidoglycan recognition protein PGRP-SC1a is essential for Toll signaling and phagocytosis of Staphylococcus aureus in Drosophila. Proc Natl Acad Sci U S A 103(3):660–665. doi:10.1073/pnas.0506182103
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ (2003) Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278(11):8869–8872. doi:10.1074/jbc.C200651200
Gonzalez-Zorn B, Senna JP, Fiette L, Shorte S, Testard A, Chignard M, Courvalin P, Grillot-Courvalin C (2005) Bacterial and host factors implicated in nasal carriage of methicillin-resistant Staphylococcus aureus in mice. Infect Immun 73(3):1847–1851. doi:73/3/1847 [pii] 10.1128/IAI.73.3.1847-1851.2005
Goodyear CS, Silverman GJ (2004) Staphylococcal toxin induced preferential and prolonged in vivo deletion of innate-like B lymphocytes. Proc Natl Acad Sci U S A 101(31):11392–11397. doi:10.1073/pnas.0404382101 0404382101 [pii]
Goriely S, Cavoy R, Goldman M (2009) Interleukin-12 family members and type I interferons in Th17-mediated inflammatory disorders. Allergy 64(5):702–709. doi:10.1111/j.1398-9995.2009.02039.x
Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, Trinchieri G, Kaiserlian D (2008) Plasmacytoid dendritic cells mediate oral tolerance. Immunity 29(3):464–475. doi:S1074-7613(08)00372-5 [pii] 10.1016/j.immuni.2008.06.017
Greenlee-Wacker M, DeLeo FR, Nauseef WM (2015) How methicillin-resistant Staphylococcus aureus evade neutrophil killing. Curr Opin Hematol 22(1):30–35. doi:10.1097/moh.0000000000000096
Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM (2014) Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol 192(10):4709–4717. doi:10.4049/jimmunol.1302692
Gries DM, Pultz NJ, Donskey CJ (2005) Growth in cecal mucus facilitates colonization of the mouse intestinal tract by methicillin-resistant Staphylococcus aureus. J Infect Dis 192(9):1621–1627. doi:JID34054 [pii] 10.1086/491737
Guo B, Chang EY, Cheng G (2008) The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J Clin Invest 118(5):1680–1690. doi:10.1172/jci33342
Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG (2006) Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126(6):1135–1145. doi:10.1016/j.cell.2006.07.033
Haller D, Serrant P, Granato D, Schiffrin EJ, Blum S (2002) Activation of human NK cells by staphylococci and lactobacilli requires cell contact-dependent costimulation by autologous monocytes. Clin Diagn Lab Immunol 9(3):649–657
Hanamsagar R, Aldrich A, Kielian T (2014) Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J Neurochem 129(4):704–711. doi:10.1111/jnc.12669
Hartl D, Lehmann N, Hoffmann F, Jansson A, Hector A, Notheis G, Roos D, Belohradsky BH, Wintergerst U (2008) Dysregulation of innate immune receptors on neutrophils in chronic granulomatous disease. J Allergy Clin Immunol 121(2):375–382 e379. doi:10.1016/j.jaci.2007.10.037
Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, Krikae F, Kirikae T, Gotz F (2006) Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol 177(5):3162–3169
Hayes SM, Howlin R, Johnston DA, Webb JS, Clarke SC, Stoodley P, Harries PG, Wilson SJ, Pender SL, Faust SN, Hall-Stoodley L, Salib RJ (2015) Intracellular residency of Staphylococcus aureus within mast cells in nasal polyps: a novel observation. J Allergy Clin Immunol. doi:10.1016/j.jaci.2014.12.1929
Heim CE, Vidlak D, Scherr TD, Kozel JA, Holzapfel M, Muirhead DE, Kielian T (2014) Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol 192(8):3778–3792. doi:10.4049/jimmunol.1303408
Hilmi D, Parcina M, Stollewerk D, Ostrop J, Josten M, Meilaender A, Zaehringer U, Wichelhaus TA, Bierbaum G, Heeg K, Wolz C, Bekeredjian-Ding I (2014) Heterogeneity of host TLR2 stimulation by Staphylocoocus aureus isolates. PLoS One 9(5):e96416. doi:10.1371/journal.pone.0096416 PONE-D-13-55098 [pii]
Hirohata S, Shibuya H, Tejima S (2010) Suppressive influences of IFN-alpha on IL-17 expression in human CD4+ T cells. Clin Immunol 134(3):340–344. doi:10.1016/j.clim.2009.11.012
Hoebe K, Georgel P, Rutschmann S, Du X, Mudd S, Crozat K, Sovath S, Shamel L, Hartung T, Zahringer U, Beutler B (2005) CD36 is a sensor of diacylglycerides. Nature 433(7025):523–527. doi:nature03253 [pii] 10.1038/nature03253
Holzinger D, Gieldon L, Mysore V, Nippe N, Taxman DJ, Duncan JA, Broglie PM, Marketon K, Austermann J, Vogl T, Foell D, Niemann S, Peters G, Roth J, Loffler B (2012) Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J Leukoc Biol 92(5):1069–1081. doi:10.1189/jlb.0112014
Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G (2002) Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168(9):4531–4537
Hruz P, Zinkernagel AS, Jenikova G, Botwin GJ, Hugot JP, Karin M, Nizet V, Eckmann L (2009) NOD2 contributes to cutaneous defense against Staphylococcus aureus through alpha-toxin-dependent innate immune activation. Proc Natl Acad Sci U S A 106(31):12873–12878. doi:10.1073/pnas.0904958106
Hussain M, Haggar A, Peters G, Chhatwal GS, Herrmann M, Flock JI, Sinha B (2008) More than one tandem repeat domain of the extracellular adherence protein of Staphylococcus aureus is required for aggregation, adherence, and host cell invasion but not for leukocyte activation. Infect Immun 76(12):5615–5623. doi:10.1128/iai.00480-08
Ip WK, Sokolovska A, Charriere GM, Boyer L, Dejardin S, Cappillino MP, Yantosca LM, Takahashi K, Moore KJ, Lacy-Hulbert A, Stuart LM (2010) Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J Immunol 184(12):7071–7081. doi:10.4049/jimmunol.1000110
Ip WK, Takahashi K, Moore KJ, Stuart LM, Ezekowitz RA (2008) Mannose-binding lectin enhances Toll-like receptors 2 and 6 signaling from the phagosome. J Exp Med 205(1):169–181. doi:10.1084/jem.20071164
Jann NJ, Schmaler M, Kristian SA, Radek KA, Gallo RL, Nizet V, Peschel A, Landmann R (2009) Neutrophil antimicrobial defense against Staphylococcus aureus is mediated by phagolysosomal but not extracellular trap-associated cathelicidin. J Leukoc Biol 86(5):1159–1169. doi:10.1189/jlb.0209053
Jawdat DM, Rowden G, Marshall JS (2006) Mast cells have a pivotal role in TNF-independent lymph node hypertrophy and the mobilization of Langerhans cells in response to bacterial peptidoglycan. J Immunol 177(3):1755–1762
Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A (2004) Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J Immunol 172(2):1169–1176
Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130(6):1071–1082
Jin JO, Zhang W, Du JY, Yu Q (2014) BDCA1-positive dendritic cells (DCs) represent a unique human myeloid DC subset that induces innate and adaptive immune responses to Staphylococcus aureus Infection. Infect Immun 82(11):4466–4476. doi:10.1128/iai.01851-14
Josefsson E, Tarkowski A (1999) Staphylococcus aureus-induced inflammation and bone destruction in experimental models of septic arthritis. J Periodontal Res 34(7):387–392
Jung JY, Roberts LL, Robinson CM (2015) The presence of interleukin-27 during monocyte-derived dendritic cell differentiation promotes improved antigen processing and stimulation of T cells. Immunology 144(4):649–660. doi:10.1111/imm.12417
Kaesler S, Volz T, Skabytska Y, Koberle M, Hein U, Chen KM, Guenova E, Wolbing F, Rocken M, Biedermann T (2014) Toll-like receptor 2 ligands promote chronic atopic dermatitis through IL-4-mediated suppression of IL-10. J Allergy Clin Immunol 134(1):92–99. doi:10.1016/j.jaci.2014.02.017 S0091-6749(14)00267-X [pii]
Kamenyeva O, Boularan C, Kabat J, Cheung GY, Cicala C, Yeh AJ, Chan JL, Periasamy S, Otto M, Kehrl JH (2015) Neutrophil recruitment to lymph nodes limits local humoral response to Staphylococcus aureus. PLoS Pathog 11(4):e1004827. doi:10.1371/journal.ppat.1004827
Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO (2009) Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity 31(6):873–884
Kang SS, Noh SY, Park OJ, Yun CH, Han SH (2015) Staphylococcus aureus induces IL-8 expression through its lipoproteins in the human intestinal epithelial cell, Caco-2. Cytokine. doi:10.1016/j.cyto.2015.04.017
Kapetanovic R, Nahori MA, Balloy V, Fitting C, Philpott DJ, Cavaillon JM, Adib-Conquy M (2007) Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus-activated macrophages. Infect Immun 75(2):830–837. doi:10.1128/iai.01199-06
Kaplan A, Ma J, Kyme P, Wolf AJ, Becker CA, Tseng CW, Liu GY, Underhill DM (2012) Failure to induce IFN-beta production during Staphylococcus aureus infection contributes to pathogenicity. J Immunol 189(9):4537–4545. doi:10.4049/jimmunol.1201111
Kato-Matsunaga N, Okonogi K (1996) Gastrointestinal colonization by methicillin-resistant Staphylococcus aureus in immunosuppressed mice. Infect Immun 64(10):4231–4235
Kebaier C, Chamberland RR, Allen IC, Gao X, Broglie PM, Hall JD, Jania C, Doerschuk CM, Tilley SL, Duncan JA (2012) Staphylococcus aureus alpha-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis 205(5):807–817. doi:10.1093/infdis/jir846
Kielian T, Haney A, Mayes PM, Garg S, Esen N (2005) Toll-like receptor 2 modulates the proinflammatory milieu in Staphylococcus aureus-induced brain abscess. Infect Immun 73(11):7428–7435. doi:73/11/7428 [pii] 10.1128/IAI.73.11.7428-7435.2005
Kim NJ, Ahn KB, Jeon JH, Yun CH, Finlay BB, Han SH (2015) Lipoprotein in the cell wall of Staphylococcus aureus is a major inducer of nitric oxide production in murine macrophages. Mol Immunol 65(1):17–24. doi:10.1016/j.molimm.2014.12.016 S0161-5890(14)00358-7 [pii]
Kinoshita M, Miyazaki H, Ono S, Inatsu A, Nakashima H, Tsujimoto H, Shinomiya N, Saitoh D, Seki S (2011) Enhancement of neutrophil function by interleukin-18 therapy protects burn-injured mice from methicillin-resistant Staphylococcus aureus. Infect Immun 79(7):2670–2680. doi:10.1128/iai.01298-10
Klotz M, Zimmermann S, Opper S, Heeg K, Mutters R (2005) Possible risk for re-colonization with methicillin-resistant Staphylococcus aureus (MRSA) by faecal transmission. Int J Hyg Environ Health 208(5):401–405. doi:10.1016/j.ijheh.2005.05.004
Kobayashi SD, Malachowa N, DeLeo FR (2015) Pathogenesis of Staphylococcus aureus abscesses. Am J Pathol 185(6):1518–1527. doi:10.1016/j.ajpath.2014.11.030
Kohler J, Breitbach K, Renner C, Heitsch AK, Bast A, van Rooijen N, Vogelgesang S, Steinmetz I (2011) NADPH-oxidase but not inducible nitric oxide synthase contributes to resistance in a murine Staphylococcus aureus Newman pneumonia model. Microbes Infect 13(11):914–922. doi:10.1016/j.micinf.2011.05.004
Kuo IH, Carpenter-Mendini A, Yoshida T, McGirt LY, Ivanov AI, Barnes KC, Gallo RL, Borkowski AW, Yamasaki K, Leung DY, Georas SN, De Benedetto A, Beck LA (2013) Activation of epidermal toll-like receptor 2 enhances tight junction function: implications for atopic dermatitis and skin barrier repair. J Invest Dermatol 133(4):988–998. doi:10.1038/jid.2012.437
Kurokawa K, Gong JH, Ryu KH, Zheng L, Chae JH, Kim MS, Lee BL (2011) Biochemical characterization of evasion from peptidoglycan recognition by Staphylococcus aureus D-alanylated wall teichoic acid in insect innate immunity. Dev Comp Immunol 35(8):835–839. doi:10.1016/j.dci.2011.03.001
Kwiecinski J, Rhost S, Lofbom L, Blomqvist M, Mansson JE, Cardell SL, Jin T (2013) Sulfatide attenuates experimental Staphylococcus aureus sepsis through a CD1d-dependent pathway. Infect Immun 81(4):1114–1120. doi:10.1128/iai.01334-12
Labrousse D, Perret M, Hayez D, Da Silva S, Badiou C, Couzon F, Bes M, Chavanet P, Lina G, Vandenesch F, Croisier-Bertin D, Henry T (2014) Kineret(R)/IL-1ra blocks the IL-1/IL-8 inflammatory cascade during recombinant Panton Valentine Leukocidin-triggered pneumonia but not during S. aureus infection. PLoS ONE 9(6):e97546. doi:10.1371/journal.pone.0097546
Liu X, Yang P, Wang C, Li F, Kijlstra A (2011) IFN-alpha blocks IL-17 production by peripheral blood mononuclear cells in Behcet’s disease. Rheumatology (Oxford) 50(2):293–298. doi:10.1093/rheumatology/keq330
Lizak M, Yarovinsky TO (2012) Phospholipid scramblase 1 mediates type i interferon-induced protection against staphylococcal alpha-toxin. Cell Host Microbe 11(1):70–80. doi:10.1016/j.chom.2011.12.004 S1931-3128(11)00405-7 [pii]
Maher BM, Mulcahy ME, Murphy AG, Wilk M, O’Keeffe KM, Geoghegan JA, Lavelle EC, McLoughlin RM (2013) Nlrp-3-driven interleukin 17 production by gammadeltaT cells controls infection outcomes during Staphylococcus aureus surgical site infection. Infect Immun 81(12):4478–4489. doi:10.1128/iai.01026-13
Maresso AW, Schneewind O (2006) Iron acquisition and transport in Staphylococcus aureus. Biometals 19(2):193–203. doi:10.1007/s10534-005-4863-7
Martin F, Oliver AM, Kearney JF (2001) Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 14(5):617–629. doi:S1074-7613(01)00129-7 [pii]
Martinez FO, Gordon S (2014) The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6:13. doi:10.12703/p6-13
Matsui K, Nishikawa A (2002) Lipoteichoic acid from Staphylococcus aureus induces Th2-prone dermatitis in mice sensitized percutaneously with an allergen. Clin Exp Allergy 32(5):783–788
Matsui K, Nishikawa A (2005) Percutaneous application of peptidoglycan from Staphylococcus aureus induces an increase in mast cell numbers in the dermis of mice. Clin Exp Allergy 35(3):382–387. doi:10.1111/j.1365-2222.2005.02190.x
Mayer AK, Muehmer M, Mages J, Gueinzius K, Hess C, Heeg K, Bals R, Lang R, Dalpke AH (2007) Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J Immunol 178(5):3134–3142
McCurdy JD, Olynych TJ, Maher LH, Marshall JS (2003) Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol 170(4):1625–1629
McGilligan VE, Gregory-Ksander MS, Li D, Moore JE, Hodges RR, Gilmore MS, Moore TC, Dartt DA (2013) Staphylococcus aureus activates the NLRP3 inflammasome in human and rat conjunctival goblet cells. PLoS ONE 8(9):e74010. doi:10.1371/journal.pone.0074010
McNeil JC (2014) Staphylococcus aureus—antimicrobial resistance and the immunocompromised child. Infect Drug Resist 7:117–127. doi:10.2147/idr.s39639
Meyers JA, Mangini AJ, Nagai T, Roff CF, Sehy D, van Seventer GA, van Seventer JM (2006) Blockade of TLR9 agonist-induced type I interferons promotes inflammatory cytokine IFN-gamma and IL-17 secretion by activated human PBMC. Cytokine 35(5–6):235–246. doi:10.1016/j.cyto.2006.09.001
Michea P, Vargas P, Donnadieu MH, Rosemblatt M, Bono MR, Dumenil G, Soumelis V (2013) Epithelial control of the human pDC response to extracellular bacteria. Eur J Immunol 43(5):1264–1273. doi:10.1002/eji.201242990
Miller LS, Cho JS (2011) Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 11(8):505–518. doi:10.1038/nri3010
Miller LS, O’Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL (2006) MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity 24(1):79–91. doi:10.1016/j.immuni.2005.11.011
Miller LS, Pietras EM, Uricchio LH, Hirano K, Rao S, Lin H, O’Connell RM, Iwakura Y, Cheung AL, Cheng G, Modlin RL (2007) Inflammasome-mediated production of IL-1beta is required for neutrophil recruitment against Staphylococcus aureus in vivo. J Immunol 179(10):6933–6942
Mjosberg J, Eidsmo L (2014) Update on innate lymphoid cells in atopic and non-atopic inflammation in the airways and skin. Clin Exp Allergy 44(8):1033–1043. doi:10.1111/cea.12353
Monticelli LA, Sonnenberg GF, Artis D (2012) Innate lymphoid cells: critical regulators of allergic inflammation and tissue repair in the lung. Curr Opin Immunol 24(3):284–289. doi:10.1016/j.coi.2012.03.012
Moore CE, Segal S, Berendt AR, Hill AV, Day NP (2004) Lack of association between Toll-like receptor 2 polymorphisms and susceptibility to severe disease caused by Staphylococcus aureus. Clin Diagn Lab Immunol 11(6):1194–1197. doi:10.1128/cdli.11.6.1194-1197.2004
Mrabet-Dahbi S, Dalpke AH, Niebuhr M, Frey M, Draing C, Brand S, Heeg K, Werfel T, Renz H (2008) The Toll-like receptor 2 R753Q mutation modifies cytokine production and Toll-like receptor expression in atopic dermatitis. J Allergy Clin Immunol 121(4):1013–1019. doi:10.1016/j.jaci.2007.11.029
Munoz-Planillo R, Franchi L, Miller LS, Nunez G (2009) A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol 183(6):3942–3948. doi:jimmunol.0900729 [pii] 10.4049/jimmunol.0900729
Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, Villaruz AE, Cheung GY, McGavin MJ, Travers JB, Otto M, Inohara N, Nunez G (2013) Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature 503(7476):397–401. doi:10.1038/nature12655
Nakayama M, Kurokawa K, Nakamura K, Lee BL, Sekimizu K, Kubagawa H, Hiramatsu K, Yagita H, Okumura K, Takai T, Underhill DM, Aderem A, Ogasawara K (2012) Inhibitory receptor paired Ig-like receptor B is exploited by Staphylococcus aureus for virulence. J Immunol 189(12):5903–5911. doi:10.4049/jimmunol.1201940
Nandi A, Dey S, Biswas J, Jaiswal P, Naaz S, Yasmin T, Bishayi B (2015) Differential induction of inflammatory cytokines and reactive oxygen species in murine peritoneal macrophages and resident fresh bone marrow cells by acute staphylococcus aureus infection: contribution of toll-like receptor 2 (TLR2). Inflammation 38(1):224–244. doi:10.1007/s10753-014-0026-8
Negrini TC, Arthur RA, Waeiss RA, Carlosa IZ, Srinivasan M (2014) Salivary epithelial cells as model to study immune response against cutaneous pathogens. Clin Transl Sci 7(1):48–51. doi:10.1111/cts.12113
Neth O, Jack DL, Dodds AW, Holzel H, Klein NJ, Turner MW (2000) Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect Immun 68(2):688–693
Neth O, Jack DL, Johnson M, Klein NJ, Turner MW (2002) Enhancement of complement activation and opsonophagocytosis by complexes of mannose-binding lectin with mannose-binding lectin-associated serine protease after binding to Staphylococcus aureus. J Immunol 169(8):4430–4436
Neumann A, Berends ET, Nerlich A, Molhoek EM, Gallo RL, Meerloo T, Nizet V, Naim HY, von Kockritz-Blickwede M (2014) The antimicrobial peptide LL-37 facilitates the formation of neutrophil extracellular traps. Biochem J 464(1):3–11. doi:10.1042/bj20140778
Nguyen MT, Kraft B, Yu W, Demicrioglu DD, Hertlein T, Burian M, Schmaler M, Boller K, Bekeredjian-Ding I, Ohlsen K, Schittek B, Gotz F (2015) The nuSaalpha specific lipoprotein like cluster (lpl) of S. aureus USA300 contributes to immune stimulation and invasion in human cells. PLoS Pathog 11(6):e1004984. doi:10.1371/journal.ppat.1004984 PPATHOGENS-D-15-00227 [pii]
Niebuhr M, Baumert K, Heratizadeh A, Satzger I, Werfel T (2014) Impaired NLRP3 inflammasome expression and function in atopic dermatitis due to Th2 milieu. Allergy 69(8):1058–1067. doi:10.1111/all.12428
Niebuhr M, Baumert K, Werfel T (2010a) TLR-2-mediated cytokine and chemokine secretion in human keratinocytes. Exp Dermatol 19(10):873–877. doi:10.1111/j.1600-0625.2010.01140.x
Niebuhr M, Langnickel J, Sigel S, Werfel T (2010b) Dysregulation of CD36 upon TLR-2 stimulation in monocytes from patients with atopic dermatitis and the TLR2 R753Q polymorphism. Exp Dermatol 19(8):e296–e298. doi:10.1111/j.1600-0625.2009.00989.x
Niebuhr M, Heratizadeh A, Wichmann K, Satzger I, Werfel T (2011) Intrinsic alterations of pro-inflammatory mediators in unstimulated and TLR-2 stimulated keratinocytes from atopic dermatitis patients. Exp Dermatol 20(6):468–472. doi:10.1111/j.1600-0625.2011.01277.x
Nieuwenhuis EE, Matsumoto T, Lindenbergh D, Willemsen R, Kaser A, Simons-Oosterhuis Y, Brugman S, Yamaguchi K, Ishikawa H, Aiba Y, Koga Y, Samsom JN, Oshima K, Kikuchi M, Escher JC, Hattori M, Onderdonk AB, Blumberg RS (2009) Cd1d-dependent regulation of bacterial colonization in the intestine of mice. J Clin Invest 119(5):1241–1250. doi:10.1172/jci36509
Nilsen NJ, Deininger S, Nonstad U, Skjeldal F, Husebye H, Rodionov D, von Aulock S, Hartung T, Lien E, Bakke O, Espevik T (2008) Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J Leukoc Biol 84(1):280–291
Nilsson N, Bremell T, Tarkowski A, Carlsten H (1999) Protective role of NK1.1+ cells in experimental Staphylococcus aureus arthritis. Clin Exp Immunol 117(1):63–69
Nowrouzian FL, Dauwalder O, Meugnier H, Bes M, Etienne J, Vandenesch F, Lindberg E, Hesselmar B, Saalman R, Strannegard IL, Aberg N, Adlerberth I, Wold AE, Lina G (2011) Adhesin and superantigen genes and the capacity of Staphylococcus aureus to colonize the infantile gut. J Infect Dis 204(5):714–721. doi:10.1093/infdis/jir388
Nurjadi D, Herrmann E, Hinderberger I, Zanger P (2013) Impaired beta-defensin expression in human skin links DEFB1 promoter polymorphisms with persistent Staphylococcus aureus nasal carriage. J Infect Dis 207(4):666–674. doi:10.1093/infdis/jis735
Olaru F, Jensen LE (2010) Staphylococcus aureus stimulates neutrophil targeting chemokine expression in keratinocytes through an autocrine IL-1alpha signaling loop. J Invest Dermatol 130(7):1866–1876. doi:10.1038/jid.2010.37
Oldenburg M, Kruger A, Ferstl R, Kaufmann A, Nees G, Sigmund A, Bathke B, Lauterbach H, Suter M, Dreher S, Koedel U, Akira S, Kawai T, Buer J, Wagner H, Bauer S, Hochrein H, Kirschning CJ (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337(6098):1111–1115. doi:10.1126/science.1220363 science.1220363 [pii]
Ono K, Nishitani C, Mitsuzawa H, Shimizu T, Sano H, Suzuki H, Kodama T, Fujii N, Fukase K, Hirata K, Kuroki Y (2006) Mannose-binding lectin augments the uptake of lipid A, Staphylococcus aureus, and Escherichia coli by Kupffer cells through increased cell surface expression of scavenger receptor A. J Immunol 177(8):5517–5523
Over B, Ziegler S, Foermer S, Weber AN, Bode KA, Heeg K, Bekeredjian-Ding I (2013) IRAK4 turns IL-10+ phospho-FOXO+ monocytes into pro-inflammatory cells by suppression of protein kinase B. Eur J Immunol 43(6):1630–1642. doi:10.1002/eji.201243217
Palecanda A, Paulauskis J, Al-Mutairi E, Imrich A, Qin G, Suzuki H, Kodama T, Tryggvason K, Koziel H, Kobzik L (1999) Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J Exp Med 189(9):1497–1506
Parcina M, Miranda-Garcia MA, Durlanik S, Ziegler S, Over B, Georg P, Foermer S, Ammann S, Hilmi D, Weber KJ, Schiller M, Heeg K, Schneider-Brachert W, Gotz F, Bekeredjian-Ding I (2013) Pathogen-triggered activation of plasmacytoid dendritic cells induces IL-10-producing B cells in response to Staphylococcus aureus. J Immunol 190(4):1591–1602. doi:10.4049/jimmunol.1201222
Parcina M, Wendt C, Goetz F, Zawatzky R, Zahringer U, Heeg K, Bekeredjian-Ding I (2008) Staphylococcus aureus-induced plasmacytoid dendritic cell activation is based on an IgG-mediated memory response. J Immunol 181(6):3823–3833. doi:181/6/3823 [pii]
Parker D, Prince A (2012) Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J Immunol 189(8):4040–4046. doi:10.4049/jimmunol.1201055
Perret M, Badiou C, Lina G, Burbaud S, Benito Y, Bes M, Cottin V, Couzon F, Juruj C, Dauwalder O, Goutagny N, Diep BA, Vandenesch F, Henry T (2012) Cross-talk between Staphylococcus aureus leukocidins-intoxicated macrophages and lung epithelial cells triggers chemokine secretion in an inflammasome-dependent manner. Cell Microbiol 14(7):1019–1036. doi:10.1111/j.1462-5822.2012.01772.x
Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274(13):8405–8410
Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14(9):1583–1589. doi:10.1038/sj.cdd.4402195
Philip NH, Artis D (2013) New friendships and old feuds: relationships between innate lymphoid cells and microbial communities. Immunol Cell Biol 91(3):225–231. doi:10.1038/icb.2013.2
Pizzolla A, Hultqvist M, Nilson B, Grimm MJ, Eneljung T, Jonsson IM, Verdrengh M, Kelkka T, Gjertsson I, Segal BH, Holmdahl R (2012) Reactive oxygen species produced by the NADPH oxidase 2 complex in monocytes protect mice from bacterial infections. J Immunol 188(10):5003–5011. doi:10.4049/jimmunol.1103430
Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M (2008) RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus aureus. Mol Cell 32(1):150–158. doi:10.1016/j.molcel.2008.08.005 S1097-2765(08)00537-6 [pii]
Quinn GA, Cole AM (2007) Suppression of innate immunity by a nasal carriage strain of Staphylococcus aureus increases its colonization on nasal epithelium. Immunology 122(1):80–89
Reed P, Atilano ML, Alves R, Hoiczyk E, Sher X, Reichmann NT, Pereira PM, Roemer T, Filipe SR, Pereira-Leal JB, Ligoxygakis P, Pinho MG (2015) Staphylococcus aureus survives with a minimal peptidoglycan synthesis machine but sacrifices virulence and antibiotic resistance. PLoS Pathog 11(5):e1004891. doi:10.1371/journal.ppat.1004891
Reis e Sousa C, Stahl PD, Austyn JM (1993) Phagocytosis of antigens by Langerhans cells in vitro. J Exp Med 178(2):509–519
Rigby KM, DeLeo FR (2012) Neutrophils in innate host defense against Staphylococcus aureus infections. Semin Immunopathol 34(2):237–259. doi:10.1007/s00281-011-0295-3
Robertson CM, Perrone EE, McConnell KW, Dunne WM, Boody B, Brahmbhatt T, Diacovo MJ, Van Rooijen N, Hogue LA, Cannon CL, Buchman TG, Hotchkiss RS, Coopersmith CM (2008) Neutrophil depletion causes a fatal defect in murine pulmonary Staphylococcus aureus clearance. J Surg Res 150(2):278–285. doi:10.1016/j.jss.2008.02.009
Robinette ML, Fuchs A, Cortez VS, Lee JS, Wang Y, Durum SK, Gilfillan S, Colonna M (2015) Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat Immunol 16(3):306–317. doi:10.1038/ni.3094
Robinson KM, Lee B, Scheller EV, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF (2015) The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res 16(1):10. doi:10.1186/s12931-015-0168-8
Rocha-de-Souza CM, Berent-Maoz B, Mankuta D, Moses AE, Levi-Schaffer F (2008) Human mast cell activation by Staphylococcus aureus: interleukin-8 and tumor necrosis factor alpha release and the role of Toll-like receptor 2 and CD48 molecules. Infect Immun 76(10):4489–4497. doi:10.1128/iai.00270-08
Ronnberg E, Johnzon CF, Calounova G, Garcia Faroldi G, Grujic M, Hartmann K, Roers A, Guss B, Lundequist A, Pejler G (2014) Mast cells are activated by Staphylococcus aureus in vitro but do not influence the outcome of intraperitoneal S. aureus infection in vivo. Immunology 143(2):155–163. doi:10.1111/imm.12297
Roquilly A, Gautreau L, Segain JP, de Coppet P, Sebille V, Jacqueline C, Caillon J, Potel G, Lejus C, Josien R, Asehnoune K (2010) CpG-ODN and MPLA prevent mortality in a murine model of post-hemorrhage-Staphyloccocus aureus pneumonia. PLoS One 5(10):e13228. doi:10.1371/journal.pone.0013228 e13228 [pii]
Ryu S, Song PI, Seo CH, Cheong H, Park Y (2014) Colonization and infection of the skin by S. aureus: immune system evasion and the response to cationic antimicrobial peptides. Int J Mol Sci 15(5):8753–8772. doi:10.3390/ijms15058753 ijms15058753 [pii]
Sachse F, Becker K, Rudack C (2010) Incidence of staphylococcal colonization and of the 753Q Toll-like receptor 2 variant in nasal polyposis. Am J Rhinol Allergy 24(1):e10–e13. doi:10.2500/ajra.2010.24.3416
Sakiniene E, Bremell T, Tarkowski A (1999) Complement depletion aggravates Staphylococcus aureus septicaemia and septic arthritis. Clin Exp Immunol 115(1):95–102
Salvi V, Scutera S, Rossi S, Zucca M, Alessandria M, Greco D, Bosisio D, Sozzani S, Musso T (2013) Dual regulation of osteopontin production by TLR stimulation in dendritic cells. J Leukoc Biol 94(1):147–158. doi:10.1189/jlb.0412194
Sankaran K, Gupta SD, Wu HC (1995) Modification of bacterial lipoproteins. Methods Enzymol 250:683–697. doi:0076-6879(95)50105-3 [pii]
Sankaran K, Wu HC (1995) Bacterial prolipoprotein signal peptidase. Methods Enzymol 248:169–180
Schaffler H, Demircioglu DD, Kuhner D, Menz S, Bender A, Autenrieth IB, Bodammer P, Lamprecht G, Gotz F, Frick JS (2014) NOD2 stimulation by Staphylococcus aureus-derived peptidoglycan is boosted by Toll-like receptor 2 costimulation with lipoproteins in dendritic cells. Infect Immun 82(11):4681–4688. doi:10.1128/iai.02043-14
Schindler D, Gutierrez MG, Beineke A, Rauter Y, Rohde M, Foster S, Goldmann O, Medina E (2012) Dendritic cells are central coordinators of the host immune response to Staphylococcus aureus bloodstream infection. Am J Pathol 181(4):1327–1337. doi:10.1016/j.ajpath.2012.06.039
Schmaler M, Jann NJ, Ferracin F, Landolt LZ, Biswas L, Götz F, Landmann R (2009) Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J Immunol 182(11):7110–7118. doi:182/11/7110 [pii] 10.4049/jimmunol.0804292
Schmaler M, Jann NJ, Ferracin F, Landmann R (2011) T and B cells are not required for clearing Staphylococcus aureus in systemic infection despite a strong TLR2-MyD88-dependent T cell activation. J Immunol 186(1):443–452. doi:10.4049/jimmunol.1001407
Schmaler M, Jann NJ, Gotz F, Landmann R (2010) Staphylococcal lipoproteins and their role in bacterial survival in mice. IntJMedMicrobiol 300(2–3):155–160. doi:S1438-4221(09)00111-8 [pii]; 10.1016/j.ijmm.2009.08.018 [doi]
Schreiner J, Kretschmer D, Klenk J, Otto M, Buhring HJ, Stevanovic S, Wang JM, Beer-Hammer S, Peschel A, Autenrieth SE (2013) Staphylococcus aureus phenol-soluble modulin peptides modulate dendritic cell functions and increase in vitro priming of regulatory T cells. J Immunol 190(7):3417–3426. doi:10.4049/jimmunol.1202563
Shang SQ, Chen GX, Shen J, Yu XH, Wang KY (2005) The binding of MBL to common bacteria in infectious diseases of children. J Zhejiang Univ Sci B 6(1):53–56. doi:10.1631/jzus.2005.B0053
Shi L, Takahashi K, Dundee J, Shahroor-Karni S, Thiel S, Jensenius JC, Gad F, Hamblin MR, Sastry KN, Ezekowitz RA (2004) Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J Exp Med 199(10):1379–1390. doi:10.1084/jem.20032207
Shibuya A, Honda S (2006) Molecular and functional characteristics of the Fcalpha/muR, a novel Fc receptor for IgM and IgA. Springer Semin Immunopathol 28(4):377–382. doi:10.1007/s00281-006-0050-3
Shimada T, Park BG, Wolf AJ, Brikos C, Goodridge HS, Becker CA, Reyes CN, Miao EA, Aderem A, Gotz F, Liu GY, Underhill DM (2010) Staphylococcus aureus evades lysozyme-based peptidoglycan digestion that links phagocytosis, inflammasome activation, and IL-1beta secretion. Cell Host Microbe 7(1):38–49. doi:10.1016/j.chom.2009.12.008
Shimaoka T, Kume N, Minami M, Hayashida K, Sawamura T, Kita T, Yonehara S (2001) LOX-1 supports adhesion of Gram-positive and Gram-negative bacteria. J Immunol 166(8):5108–5114
Shinohara ML, Kim JH, Garcia VA, Cantor H (2008) Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: role of intracellular osteopontin. Immunity 29(1):68–78. doi:10.1016/j.immuni.2008.05.008
Sieprawska-Lupa M, Mydel P, Krawczyk K, Wojcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, Shafer W, McAleese F, Foster T, Travis J, Potempa J (2004) Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob Agents Chemother 48(12):4673–4679. doi:48/12/4673 [pii] 10.1128/AAC.48.12.4673-4679.2004
Simanski M, Glaser R, Koten B, Meyer-Hoffert U, Wanner S, Weidenmaier C, Peschel A, Harder J (2013) Staphylococcus aureus subverts cutaneous defense by D-alanylation of teichoic acids. Exp Dermatol 22(4):294–296. doi:10.1111/exd.12114
Skabytska Y, Wolbing F, Gunther C, Koberle M, Kaesler S, Chen KM, Guenova E, Demircioglu D, Kempf WE, Volz T, Rammensee HG, Schaller M, Rocken M, Gotz F, Biedermann T (2014) Cutaneous innate immune sensing of Toll-like receptor 2-6 ligands suppresses T cell immunity by inducing myeloid-derived suppressor cells. Immunity 41(5):762–775. doi:10.1016/j.immuni.2014.10.009
Small CL, McCormick S, Gill N, Kugathasan K, Santosuosso M, Donaldson N, Heinrichs DE, Ashkar A, Xing Z (2008) NK cells play a critical protective role in host defense against acute extracellular Staphylococcus aureus bacterial infection in the lung. J Immunol 180(8):5558–5568
Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, Tanne A, Vanaja SK, Moore KJ, Fitzgerald KA, Lacy-Hulbert A, Stuart LM (2013) Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol 14(6):543–553. doi:10.1038/ni.2595
Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, Brown A, Lauren C, Dowd M, West E, Horst B, Planet P, Prince A (2015) Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. MBio 6(2). doi:10.1128/mBio.00289-15
Stoll H, Dengjel J, Nerz C, Gotz F (2005) Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73(4):2411–2423
Stuart LM, Deng J, Silver JM, Takahashi K, Tseng AA, Hennessy EJ, Ezekowitz RA, Moore KJ (2005) Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol 170(3):477–485. doi:jcb.200501113 [pii] 10.1083/jcb.200501113
Subramaniam R, Barnes PF, Fletcher K, Boggaram V, Hillberry Z, Neuenschwander P, Shams H (2014) Protecting against post-influenza bacterial pneumonia by increasing phagocyte recruitment and ROS production. J Infect Dis 209(11):1827–1836. doi:10.1093/infdis/jit830
Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC (2002) Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature 416(6882):750–756. doi:10.1038/nature736
Tait Wojno ED, Artis D (2012) Innate lymphoid cells: balancing immunity, inflammation, and tissue repair in the intestine. Cell Host Microbe 12(4):445–457. doi:10.1016/j.chom.2012.10.003
Takai T, Chen X, Xie Y, Vu AT, Le TA, Kinoshita H, Kawasaki J, Kamijo S, Hara M, Ushio H, Baba T, Hiramatsu K, Ikeda S, Ogawa H, Okumura K (2014) TSLP expression induced via Toll-like receptor pathways in human keratinocytes. Methods Enzymol 535:371–387. doi:10.1016/b978-0-12-397925-4.00021-3
Takeuchi O, Hoshino K, Akira S (2000) Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol 165(10):5392–5396
Tebartz C, Horst SA, Sparwasser T, Huehn J, Beineke A, Peters G, Medina E (2015) A major role for myeloid-derived suppressor cells and a minor role for regulatory T cells in immunosuppression during Staphylococcus aureus infection. J Immunol 194(3):1100–1111. doi:10.4049/jimmunol.1400196
Terada M, Tsutsui H, Imai Y, Yasuda K, Mizutani H, Yamanishi K, Kubo M, Matsui K, Sano H, Nakanishi K (2006) Contribution of IL-18 to atopic-dermatitis-like skin inflammation induced by Staphylococcus aureus product in mice. Proc Natl Acad Sci U S A 103(23):8816–8821. doi:10.1073/pnas.0602900103
Tewfik MA, Bosse Y, Hudson TJ, Vallee-Smejda S, Al-Shemari H, Desrosiers M (2008) Assessment of Toll-like receptor 2 gene polymorphisms in severe chronic rhinosinusitis. J Otolaryngol Head Neck Surg 37(4):552–558
Urb M, Sheppard DC (2012) The role of mast cells in the defence against pathogens. PLoS Pathog 8(4):e1002619. doi:10.1371/journal.ppat.1002619 PPATHOGENS-D-12-00120 [pii]
Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A (1999) The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature 401(6755):811–815. doi:10.1038/44605
van der Laan LJ, Dopp EA, Haworth R, Pikkarainen T, Kangas M, Elomaa O, Dijkstra CD, Gordon S, Tryggvason K, Kraal G (1999) Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J Immunol 162(2):939–947
Verdrengh M, Tarkowski A (1997) Role of neutrophils in experimental septicemia and septic arthritis induced by Staphylococcus aureus. Infect Immun 65(7):2517–2521
Verkaik NJ, de Vogel CP, Boelens HA, Grumann D, Hoogenboezem T, Vink C, Hooijkaas H, Foster TJ, Verbrugh HA, van Belkum A, van Wamel WJ (2009) Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J Infect Dis 199(5):625–632
Viau M, Longo NS, Lipsky PE, Zouali M (2005) Staphylococcal protein a deletes B-1a and marginal zone B lymphocytes expressing human immunoglobulins: an immune evasion mechanism. J Immunol 175(11):7719–7727
Volz T, Nega M, Buschmann J, Kaesler S, Guenova E, Peschel A, Rocken M, Gotz F, Biedermann T (2010) Natural Staphylococcus aureus-derived peptidoglycan fragments activate NOD2 and act as potent costimulators of the innate immune system exclusively in the presence of TLR signals. FASEB J 24(10):4089–4102. doi:10.1096/fj.09-151001
von Bernuth H, Picard C, Puel A, Casanova JL (2012) Experimental and natural infections in MyD88- and IRAK-4-deficient mice and humans. Eur J Immunol 42(12):3126–3135. doi:10.1002/eji.201242683
von Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M, Medina E (2008a) Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 111(6):3070–3080. doi:10.1182/blood-2007-07-104018
von Kockritz-Blickwede M, Rohde M, Oehmcke S, Miller LS, Cheung AL, Herwald H, Foster S, Medina E (2008b) Immunological mechanisms underlying the genetic predisposition to severe Staphylococcus aureus infection in the mouse model. Am J Pathol 173(6):1657–1668. doi:10.2353/ajpath.2008.080337
Vu AT, Baba T, Chen X, Le TA, Kinoshita H, Xie Y, Kamijo S, Hiramatsu K, Ikeda S, Ogawa H, Okumura K, Takai T (2010) Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J Allergy Clin Immunol 126(5):985–993, 993 e981-983. doi:10.1016/j.jaci.2010.09.002
Vultaggio A, Nencini F, Pratesi S, Maggi L, Guarna A, Annunziato F, Romagnani S, Parronchi P, Maggi E (2011) The TLR7 ligand 9-benzyl-2-butoxy-8-hydroxy adenine inhibits IL-17 response by eliciting IL-10 and IL-10-inducing cytokines. J Immunol 186(8):4707–4715. doi:10.4049/jimmunol.1002398
Wagner C, Iking-Konert C, Hug F, Stegmaier S, Heppert V, Wentzensen A, Hansch GM (2006) Cellular inflammatory response to persistent localized Staphylococcus aureus infection: phenotypical and functional characterization of polymorphonuclear neutrophils (PMN). Clin Exp Immunol 143(1):70–77. doi:10.1111/j.1365-2249.2005.02963.x
Wanke I, Steffen H, Christ C, Krismer B, Gotz F, Peschel A, Schaller M, Schittek B (2011) Skin commensals amplify the innate immune response to pathogens by activation of distinct signaling pathways. J Invest Dermatol 131(2):382–390. doi:10.1038/jid.2010.328
Westerberg LS, de la Fuente MA, Wermeling F, Ochs HD, Karlsson MC, Snapper SB, Notarangelo LD (2008) WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function. Blood 112(10):4139–4147. doi:10.1182/blood-2008-02-140715 blood-2008-02-140715 [pii]
Wolf AJ, Arruda A, Reyes CN, Kaplan AT, Shimada T, Shimada K, Arditi M, Liu G, Underhill DM (2011) Phagosomal degradation increases TLR access to bacterial ligands and enhances macrophage sensitivity to bacteria. J Immunol 187(11):6002–6010. doi:10.4049/jimmunol.1100232
Yang D, Chertov O, Oppenheim JJ (2001) Participation of mammalian defensins and cathelicidins in anti-microbial immunity: receptors and activities of human defensins and cathelicidin (LL-37). J Leukoc Biol 69(5):691–697
Yimin Kohanawa M, Zhao S, Ozaki M, Haga S, Nan G, Kuge Y, Tamaki N (2013) Contribution of toll-like receptor 2 to the innate response against Staphylococcus aureus infection in mice. PLoS ONE 8(9):e74287. doi:10.1371/journal.pone.0074287
Zaidman-Remy A, Herve M, Poidevin M, Pili-Floury S, Kim MS, Blanot D, Oh BH, Ueda R, Mengin-Lecreulx D, Lemaitre B (2006) The Drosophila amidase PGRP-LB modulates the immune response to bacterial infection. Immunity 24(4):463–473
Zanger P, Nurjadi D, Vath B, Kremsner PG (2011) Persistent nasal carriage of Staphylococcus aureus is associated with deficient induction of human beta-defensin 3 after sterile wounding of healthy skin in vivo. Infect Immun 79(7):2658–2662. doi:10.1128/iai.00101-11
Zhao H, Li W, Gao Y, Li J, Wang H (2014) Exposure to particular matter increases susceptibility to respiratory Staphylococcus aureus infection in rats via reducing pulmonary natural killer cells. Toxicology 325:180–188. doi:10.1016/j.tox.2014.09.006
Ziegler SF, Roan F, Bell BD, Stoklasek TA, Kitajima M, Han H (2013) The biology of thymic stromal lymphopoietin (TSLP). Adv Pharmacol 66:129–155. doi:10.1016/b978-0-12-404717-4.00004-4
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Bekeredjian-Ding, I., Stein, C., Uebele, J. (2015). The Innate Immune Response Against Staphylococcus aureus . In: Bagnoli, F., Rappuoli, R., Grandi, G. (eds) Staphylococcus aureus. Current Topics in Microbiology and Immunology, vol 409. Springer, Cham. https://doi.org/10.1007/82_2015_5004
Download citation
DOI: https://doi.org/10.1007/82_2015_5004
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-72061-6
Online ISBN: 978-3-319-72063-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)