
Abstract
Humans are well equipped to defend themselves against bacteria. The innate immune system employs diverse mechanisms to recognize, control and initiate a response that can destroy millions of different microbes. Microbes that evade the sophisticated innate immune system are able to escape detection and could become pathogens. The pathogens Streptococcus pneumoniae and Staphylococcus aureus are particularly successful due to the development of a wide variety of virulence strategies for bacterial pathogenesis and they invest significant efforts towards mechanisms that allow for neutrophil evasion. Neutrophils are a primary cellular defense and can rapidly kill invading microbes, which is an indispensable function for maintaining host health. This review compares the key features of Streptococcus pneumoniae and Staphylococcus aureus in epidemiology, with a specific focus on virulence mechanisms utilized to evade neutrophils in bacterial pathogenesis. It is important to understand the complex interactions between pathogenic bacteria and neutrophils so that we can disrupt the ability of pathogens to cause disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Epidemiology of Staphylococcus aureus and Streptococcus pneumoniae
Both S. pneumoniae and S. aureus colonize in the human upper respiratory tract and typically have a rather commensal lifestyle. Occasionally, the immune response may be dampened or incapacitated; this weakness allows for S. pneumoniae and S. aureus to act as pathogens that contribute towards the development of potentially fatal diseases such as pneumonia, meningitis, endocarditis, toxic-shock syndrome, bacteremia and soft-tissue/skin infections (Klevens et al. 2006; Deleo et al. 2010). Alarmingly, despite serious attempts to eradicate it, S. aureus has remained a leading cause of healthcare and community-associated infections in the western world over the past decade. Furthermore, S. pneumoniae is the most common cause of community-acquired pneumonia worldwide, accounting for an estimated 3.5 million deaths worldwide (Black et al. 2010). At present, approximately 1.6 million people die from pneumococcal diseases each year, placing pneumonia and meningitis as the most prevalent invasive pneumococcal diseases (IPD). In western countries, the main burden of pneumococcal disease is among adults over the age of 50 and provides evidence that the elderly are a rapidly expanding population with increased risk for infection (Hussain et al. 2005).
Among the many risk factors associated with staphylococcal diseases, colonization with S. aureus has the strongest correlation. At present, approximately 20-30% of the healthy population carries S. aureus (Weidenmaier et al. 2012). Colonization with S. pneumoniae is most frequently detected in young children but transient nasopharyngeal pneumococcal carriage is common among all ages. The most at risk for development of pneumococcal diseases are at both extremities of life, i.e., children under the age of 4 years and adults over 50 years of age. As with S. pneumoniae, carriage of S. aureus serves as the first step to infection as well as the frequent source of transmission between individuals (Wertheim et al. 2005). Although carriage of these organisms is situated in the same niche, the upper respiratory tract, colonization in children with S. pneumoniae shows a strong negative association with S. aureus carriage (Regev-Yochay et al. 2004; Bogaert et al. 2004).
Competition is considered the most common interaction between different organisms occupying the same niche but host factors also contribute to the creation of a living environment that is suitable for the bacterium. Several different mechanisms were recently reviewed that could account for the inverse correlation in the nasopharyngeal space between S. aureus and S. pneumoniae, including the composition of the microbiome, host immune responses, production of bactericidal hydrogen peroxide (H202) or pilus by S. pneumoniae (Reiss-Mandel and Regev-Yochay 2016). Importantly, the effects of this negative correlation is bimodal, in that S. aureus factors could also hamper pneumococcal growth. In support of this, Cremers et al. reported that with controlled inoculation of healthy human adults with S. pneumoniae, colonization could not be established in individuals with staphylococcal-dominated nasopharyngeal microbiomes (Cremers et al. 2014). In fact, several staphylococcal peptides have been shown to directly kill S. pneumoniae (Cogen et al. 2010). However, the exact molecular mechanism for this negative interaction has yet to be determined and is further complicated by the fact that experimental murine models do not support colonization or transmission studies with S. aureus (Weidenmaier et al. 2012; Baur et al. 2014).
The main difference between the two pathogens is that the most common pneumococcal diseases do not contribute to pneumococcal transmission, suggesting that the virulence characteristics of the pneumococcus are adaptations that increase its persistence within a host during colonization (Weiser 2010; Musher 2003). On the contrary, skin infections caused by community-associated methicillin-resistant S. aureus (CA-MRSA) have been shown to enhance transmissibility of the pathogen (Coronado et al. 2007; Fontanilla et al. 2010). Although MRSA infections were historically restricted to the healthcare system (Barrett et al. 1968), in the past two decades the epidemiology shifted to community-associated (CA-)MRSA strains that have rapidly emerged and initiated infections in previously healthy individuals (Vandenesch et al. 2003; Li et al. 2009). Enhanced expression of virulence factors, such as the toxins alpha toxin (hemolysin, Hla), phenol- soluble modulins (PSMs) and or Panton–Valentine leucocidin (PVL), may have a contributory role in the epidemiological transition (Rigby and DeLeo 2012). In addition, a recent study found that wall teichoic acid (WTA) production is involved in the transition to CA-MRSA because the CA-MRSA strains that exhibited more WTA content had advanced abscess formation compared to low WTA-producing strains (Wanner et al. 2017). The enhanced virulence potential of CA-MRSA is, in part, related to the ability of these strains to evade killing by human neutrophils, thereby making them more prone to cause disease.
Antibiotic resistance drives the evolution of S. pneumoniae and S. aureus to become important pathogens, which is further complicated by the empty pipeline for the development of novel therapeutics (Chambers and DeLeo 2009). High antibiotic usage within the healthcare system has driven significant selective pressure for gaining antibiotic resistance. Strains for both S. pneumoniae and S. aureus have been isolated that are resistant for almost every antibiotic introduced into clinical practice (Laxminarayan et al. 2013; McGuinness et al. 2017; Charpentier and Tuomanen 2000). Resistance is mediated through horizontal gene transfer, a process that allows for rapid exchange of resistance genes and virulence factors either through natural competence for S. pneumoniae or through (pro)phage transduction and plasmid exchange for MRSA (Barlow 2009). For instance, complete vancomycin resistance can develop in MRSA through uptake of vancomycin-resistant enterococci plasmids followed by an integration of the vancomycin resistance genes into one of their resident plasmids (Zhu et al. 2010). The implementation of effective vaccination provides an important therapeutic strategy and has decreased the concern that S. pneumonia has become increasingly difficult to treat with antibiotics. Although vaccination is crucial to limiting over-use of antibiotics and resistance, to date, only a limited number of the known 93 polysaccharide capsule serotypes are included in the current vaccines. The selection of serotypes included in the 7-, 10- and 13-valent pneumococcal conjugate vaccines is based upon the frequency with which these serotypes caused IPD prior to conjugate vaccine introduction (Poland 1999). The introduction of the pneumococcal conjugate vaccines in national vaccination schemes has significantly reduced the incidence of pneumococcal disease caused by the vaccine serotypes but, alarmingly, non-vaccine serotypes are being isolated from both pediatric and adult patients with IPD with higher frequencies (Weinberger et al. 2011; Miller et al. 2011). There is currently no vaccine available for S. aureus, despite ample attempts by a multitude of pharmaceutical companies. As such, S. aureus remains high on the WHO priority list of most dangerous pathogens. Altogether, this underscores the urgent need for development of antimicrobials that are effective in the treatment of pneumococcal and staphylococcal infections.
Neutrophil response to infection
Polymorphnuclear cells or neutrophils are the major cellular defense against S. aureus and S. pneumoniae infections. Neutrophils are the first to arrive at the local infectious nidus and migrate out of the vasculature in an attempt to eradicate the pathogen through an armamentarium of defenses including protease release, production of reactive oxygen species (ROS) and antimicrobial peptides/proteins. Neutrophils are essential towards combating Gram-positive pathogens and patients with deficiencies in neutrophil function, or with reduced neutrophil numbers due to chemotherapy are particularly susceptible to S. aureus infections. Neutropenia, chronic granulomatosis disease (CGD) and leukocyte adhesion deficiency are all associated with severe bacterial infections and can commonly be attributed to Gram-positive bacteria as the causative organism (Bogomolski-Yahalom and Matzner 1995; Boxer and Morganroth 1987). However, S. pneumoniae and other catalase-negative organisms are not frequently associated with infection in CGD patients. In Chedaik–Higashi syndrome, patients have a reduced capacity to produce antimicrobials via neutrophil granules and show hampered degranulation. Furthermore, neutrophils isolated from Chedaik–Higashi syndrome patients had a diminished capacity to kill S. pneumonia and may be an explanation of the recurrent infections characteristic of the disorder (Ganz et al. 1988; Root et al. 1972) In addition, splenectomized or asplenic patients have a 50-fold increased risk of developing an IPD and a 50–70% mortality rate is associated with these cases (Di Sabatino et al. 2011). Recently, populations of immature and mature neutrophils from the spleen were recognized as key components in the local eradication of S. pneumonia (Deniset et al. 2017). This converging evidence suggests that neutrophils can utilize multiple mechanisms to eliminate S. aureus and S. pneumonia. It is important to understand the mechanisms utilized by neutrophils to combat these pathogens, so that we may have a better understanding of human health and capitalize on these strategies for improved therapeutic techniques.
Neutrophils develop and mature in bone marrow and are released en masse into peripheral circulation. Approximately 60% of human blood leukocytes are comprised of neutrophils and these cells flood the vasculature during a rapid response to infected tissue. Circulating neutrophils are directed to the infected tissue by chemotactic signals that could be provided by bacteria or released by host cells. The innate immune system provides a rapid response and elicits an almost immediate anticipation of neutrophils towards chemotactic signals that promote migration and induce inflammation. During the translation process, growing bacteria produce n-formylated peptides, which promote chemotactic migration of the neutrophil towards the nidus of invading bacteria. Other important host-derived chemotactic molecules, such as interleukin 8 (IL8), chemokine ligand 1 (CXCL1 or GROα), leukotriene B4 (LTB4) and complement component C5a, are used by neutrophils to find their way into areas of infection. Further recruitment is propagated with participation of resident cells such as macrophages and endothelial cells, which produce additional chemotactic signals to mount a proper cellular response in the infected tissue.
Neutrophil recruitment out of the circulation is mediated by the leukocyte recruitment cascade, whereby a complex series of cellular interactions and signaling events dictate the neutrophil dynamics of infiltration into infected tissue (Fig. 1). The leukocyte recruitment cascade can be divided into four distinct stages: neutrophil rolling and/or tethering on endothelial cells, firm adhesion and crawling of the neutrophils, transmigration, and subsequent chemotactic migration. The first step to recruitment requires circulating phagocytes to be slowed down near the site of infection. Activated endothelial cells rapidly express P- and E-selectins, which under sheer force of the blood flow are able to interact with glycoprotein P-selectin glycoprotein ligand-1 (PSGL-1) on the neutrophil surface. The carbohydrate–protein interactions result in initial tethering and subsequent rolling of neutrophils along the endothelial wall (Moore et al. 1995). The second step is the progression from rolling to complete arrest of the neutrophils on the endothelial lining. Neutrophils use integrin-dependent interactions via leukocyte adhesion molecules, such as clusters of differentiation molecule 11a (CD11a/CD18; LFA-1) and CD11b/CD18 (Mac-1), to firmly adhere to intercellular adhesion molecule 1 (ICAM-1) molecules on the endothelial cells (Diamond et al. 1990). The β2 integrins expressed on the cellular surface can increase their affinity for ICAM-1 through inside–out signaling following activation by proinflammatory mediators displayed on the endothelium. Once the neutrophil begins crawling along the endothelial cells, the neutrophil will prepare for transmigration with cytoskeletal rearrangement and β2 integrins clustering to alter morphology. Transmigration from the endothelium into infected tissue occurs through the endothelial junction or transcellularly and is facilitated by complex interactions between the receptors CD31 and CD11b/CD18 and junction adhesion molecules A–C, CD47 and CD44 (Phillipson et al. 2006). The leukocyte recruitment cascade described above is the generally held view of recruitment of neutrophils into the skin or muscle. Recruitment of neutrophils to other organs or vascular beds such as the lungs and liver do not require selectins, indicating that alternative adhesion molecules may be involved (McDonald et al. 2008; Yipp et al. 2012).

Neutrophil extravasation. Spinning disk intravital microscopy image showing a skin postcapillary venule (blue; CD31) with neutrophils (red; Ly6G) in the process of recruitment to staphylococcal infection. The skin of a wild-type C57BL/6 mouse was infected with S. aureus (strain MW2-GFP (Surewaard and Kubes 2017)) and the intravital microscopy (IVM) image was taken 2 h later. It captured neutrophils at different stages of migration: rolling cells, adhering neutrophils, cells that extravasated out of the blood vessel and chemotactic neutrophils towards S. aureus
When the neutrophil is internalized across the endothelial barrier, it is guided towards the site of infection by various chemo-attractants and becomes activated by inflammatory stimulants. Staphylococci and pneumococci directly release molecules that are recognized by sensors of the innate immune system called pattern recognition receptors (PRRs), such as Toll-like receptors (TLR) or chemoattractant receptors of the G-protein-coupled receptor (GPCR) family. Importantly, all PRRs recognize microbe-associated, evolutionarily conserved structures known as pathogen-associated molecular patterns (PAMPs). TLRs are the most studied group of PRRs and are responsible for the distinction between endogenous immune stimuli compared to exogenous pathogens, which constitute the basis of the host-immune surveillance. Ligands of the TLR are of relevance for staphylococcal and pneumococcal infections and include an incredibly diverse range of agonists including: bacterial lipoproteins (TLR1, TLR2, TLR6), bacterial CpG-rich DNA (TLR9), pneumolysin (TLR4) (Kawai and Akira 2010; Koppe et al. 2012). In general, activated TLRs initiate intracellular signal transduction cascades that enhance phagocytosis and induce cytokine production. However, activation of TLRs alone does not stimulate neutrophil migration, nor is it sufficient for phagocytosis. The GPCR family of transmembrane-bound receptors is crucial for mediating directed migration along a chemotactic gradient (Bestebroer et al. 2010a). The main chemo-attractants secreted by bacteria are PSMs and N-formylated proteins/peptides that are recognized by the formyl peptide receptors (FPRs) (Le et al. 2001; Wang et al. 2007; Kretschmer et al. 2010). One of the most potent host-derived chemo-attractants is the small complement fragment C5a and the less-potent relative, C3a. The complement fragments are generated during the activation of the complement cascade. Along with chemokines such as IL8, LTB4 and GROα, which are released by tissue resident cells, all these signals engage specific GPCRs leading to polarization, priming or activation. All theses chemotactic factors are integrated by signal transduction cascades in the neutrophil resulting in directed migration towards the site of infection (Mullaly and Kubes 2006; Phillipson and Kubes 2011).
The main mechanism by which neutrophils destroy pathogens is a process known as phagocytosis. Neutrophil phagocytosis starts with the recognition and binding of bacteria, followed by their ingestion. Complement and immunoglobulins serve as activated serum proteins that coat the extracellular surface of the pathogen and greatly enhance phagocytose, through a process called opsonization. The complement system is normally inactive and is constitutively present at high concentrations in the serum or interstitial fluid. When a pathogen is recognized through PRRs or specific IgGs, there is an activation of the proteolytic complement cascade, which directs the deposition of complement on the microbial surface. Neutrophils display leukocyte Fc gamma receptors (FcγR) and the complement receptors (CR); these receptors recognize IgG or activated complement fragment C3b on opsonized bacteria. Upon cross-linking by ligand binding, the CRs and FcγR mediate the initial uptake, or phagocytosis of the pathogen. Internalization of the pathogen initiates a series of vesicular transport events along with fusion and fission of neutrophil granules to develop the phagolysosome. As a result of granule fusion, the contents are released into the lumen of the phagolysosome and there is subsequent activation of proteases or enzymes. Phagocytosis also initiates signaling complexes that are responsible for the generation of ROS and acidification of the phagosome (Kinchen and Ravichandran 2008).
The NADPH oxidase is responsible for ROS production in neutrophils and functions by pumping electrons across the phagosomal membrane to produce O2−. This superoxide anion is almost instantly converted to hydrogen peroxide (H2O2) by superoxide dismutase. Unique to neutrophils, myeloperoxidase can catalyze a reaction of H2O2 with chloride to form the microbiocidal hypochlorous acid (HOCl). In addition, other secondary reactions generate hydroxyl radical, chloramines, hydroperoxyl radical and singlet oxygen, all very potent antimicrobial compounds (Babior 1999). In addition to oxidant-dependent killing mechanisms, neutrophils also undergo degranulation, a process whereby neutrophil granules fuse with phagosomes. As a result of granule fusion, the contents are released into the lumen of the phagolysosome and there is subsequent activation of proteases or enzymes. Degranulation will enrich the antimicrobial milieu of the phagolysosomal lumen, as these granules are packed with antimicrobial proteins such as: bactericidal/permeability-increasing protein, lysozyme, defensins and lactoferrin and the serine proteases, neutrophil elastase, proteinase 3 and cathepsin G (de Leeuw et al. 2010; Amulic et al. 2012).
Next to phagocytosis and intracellular killing by neutrophils, a new antimicrobial strategy has been described in which neutrophils actively release their DNA together with several cytosolic and granular proteins. These so-called neutrophil extracellular traps (NETs) prevent further dissemination by trapping pathogens (Brinkmann et al. 2004). NETs have been shown to bind and kill microbes in vitro and have been found in various disease models in vivo (McDonald et al. 2012). Importantly, neutrophils lack the ability to directly catch bacteria out of the bloodstream; therefore, release of NETs can assist in the capture of invading pathogens from the circulation (McDonald et al. 2012). A follow-up study from the same group used intravital microscopy to show that, in vivo, a neutrophil could release DNA without cell lysis or death, a process referred to as “vital NETosis” (Yipp et al. 2012). While the potential role of NETs in the innate immune system appear promising and may be of vital importance, there is currently a knowledge gap in our understanding of the molecular signaling events that underlie NET formation, that will require further investigation.
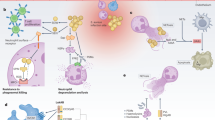
Taken together, neutrophils are extremely capable of recognizing and destroying bacterial pathogens. Many pathogens, such as S. aureus and S. pneumoniae, have co-evolved with the human innate immune system. Therefore, it is not surprising that these pathogens have an abundant repertoire of factors aimed at evasion of the innate system, which are used to thwart host defense and cause disease. The secreted and surface-attached molecules produced by S. aureus and S. pneumoniae have the potential to evade and/or disrupt almost the entire antimicrobial capacity of neutrophils. The remainder of this review will highlight recent findings and the most important strategies of these pathogens to circumvent the activation, recognition, uptake and killing by neutrophils (Fig. 2).

Immune evasion factors targeting neutrophils from Staphylococcus aureus and Streptococcus pneumoniae. Depicted are different stages in neutrophil responses towards pathogenic bacteria. The first column represents the detection of invading microorganisms by neutrophils and recruitment to the site of infection. In the gray boxes, the bacterial evasion strategies with bacterial evasion proteins/compounds in white, bacterial toxins in red and the host’s molecular targets in black. Details for S. pneumoniae are grouped on the top and information for S. aureus is grouped on the bottom. The first column represents strategies for evasion of neutrophil activation/recruitment and toxins that lyse neutrophils. Pneumococcal phosphorylcholine (ChoP) esterase (CbpE) depletes platelet-activating factor (PAF) for molecular mimicry resulting in decreased neutrophil activation and pneumococcal killing. Zinc metalloproteinase C (ZmpC) is utilized by S. pneumoniae and staphylococcal superantigen-like protein 5 (SSL5) by S. aureus to inhibit p-selectin glycoprotein 1 (PSGL-1), by either degradation of the glycoprotein or by antagonism, respectively. Staphylococcal extracellular adherence protein (EAP) binds and blocks intracellular adhesion molecule 1 (ICAM-1). Many G-protein-coupled receptors are required for activation and are inhibited by S.aureus; formyl-peptide receptors 1 (FPR1/2) are inhibited by formyl peptide receptor-like 1 inhibitor (FLIPr) and FLIPr-like (FLIPr-L). Chemotaxis inhibitory protein of S. aureus (CHIPS) inhibits the complement receptors C5aR and FPR1. Staphopain (ScpA) blocks signaling from the chemokine receptor CXCR2. SSL6 blocks cluster of differentiation 47 (CD47). Secreted staphylococcal toxins bind specific receptors on neutrophils to promote cell lysis. The bi-component pore-forming leukocidins (Luk) include LukAB that utilizes CD11b or macrophage antigen 1 (Mac-1) to facilitate lysis. LukED and γ-haemolysin CD (HlgCD) use both CXCR1 and CXCR2 to mediate neutrophil lysis. Additionally, C5aRs on neutrophils are used by HlgAB and Panton–Valentine leucocidin (PVL). Alpha toxin or hemolysin alpha (Hla) can lyse neutrophils through a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) and phenol-soluble modulins (PSM) lyse neutrophils independently of a receptor but are recognized at sub-lytic concentrations by FPR2. Pneumococal toxin pneumolysin (Ply) uses cholesterol in the plasma membrane to form pores; this is a host–receptor-independent process. Ply is recognized by neutrophils through Toll-like receptor 4 (TLR4). The second column represents molecules that inhibit opsonization and phagocytosis. The capsular polysaccharide (CPS) protects the pneumococcus from opsonization with complement and immunoglobulin (Ig), thereby preventing phagocytosis. Pneumococcal IgA protese, staphylococcal protein A (SpA), staphylococcal binder of IgG and SSL7 all function on immunoglobulins to interfere directly with Ig opsonization or prevent the classical complement pathway. Pneumococcal surface protein A (PspA) can interfere with complement opsonization by blocking complement reactive protein (CRP) and or activating factor B. PspC, transcription elongation factor (Tuf) and staphylococcal protein SdrE all recruit factor H to their surface to prevent further complement opsonization. Pneumococcal endopeptidase (PepO) and staphylokinase SAK interact with plasminogen and activation of this zymogen leads to reduced opsonization with C3. Pneumococcal α-Enolase (Eno) recruits another negative regulator of complement, C4 binding protein (C4BP). Staphylococcal complement inhibitor (SCIN), extracellular fibrinogen-binding protein (Efb) and extracellular complement-binding protein (Ecb) inhibit C3 or C5 convertases; and aureolysin cleaves the complement factor C3, which all compromise opsonization. Clumping factor A (ClfA) and Efb both recruit fibrinogen to the surface to cloak the surface from recognition; Efb needs to additionally interact with surface-bound C3 for this mechanism. Anchored collagen adhesin (Cna) inhibits complement component 1q (C1q) and FLIPr and FLIPr-L both block Fcγ receptors (FcγR) to IgG mediated to prevent phagocytosis. The last column represents molecules that inhibit bacterial killing by neutrophils. Pneumococcal pyruvate oxidase (SpxB), glutathione reductase (gor), pneumococcal NADH oxidase (nox), pneumococcal superoxide dismutase (SodA), thiol peroxidase (TpxD) and staphylococcal staphyloxanthin/superoxide dismutase (SodA/SodM), catalase KatG and alkylhydroperoxide reductase (AhpC) are all anti-oxidants that reduce oxidative stress caused by phagosomal reactive oxygen species (ROS) generation. Staphylococcal peroxidase inhibitor (SPIN) inhibits MPO and thereby prevents damage from hydrogen peroxide. CPS and aureolysin hamper antimicrobial peptides (AMPs) from functioning. Additionally, the Dlt operon mediates D-alanyl esterification of teichoic acids and MprF modifies phosphatidylglycerol with alanine or lysine, to protect staphylococci from AMPs. Eap, EapH1 and EapH2 inhibit neutrophil serine proteases and OatA O-acetylates peptidoglycan, preventing it from degradation by the lysozyme
Evasion of recognition
Capsule
Perhaps the best-studied virulence factor of S. pneumonia is its capsular polysaccharide (CPS). Over 90 different pneumococcal capsular serotypes exist and all differ in their polysaccharide chemistry and antigenicity. The CPS functions to minimize or inhibit recognition by the host, achieving this feature by hiding and/or modifying the bacterial surface. This strategy critically relies on the production of CPS to form the bacterial capsule, which will protect the pneumococcus against phagocytic clearance by blocking the deposition of immunoglobulins (Ig) and complement on the pneumococcal cell surface (Abeyta et al. 2003; Hostetter 1986). Furthermore, the CPS can decrease trapping by NETs, compounding the complexity of evasion strategies utilized by these pathogens (Wartha et al. 2007). Pneumococcal strains that produce CPS in vitro are more virulent in vivo (Mac and Kraus 1950), although it should be noted that the degree of encapsulation does not strongly impact nasopharyngeal colonization (Hammerschmidt et al. 2005). Not surprisingly, the pneumococcus spends significant effort in producing CPS and is overall recognized as its main virulence factor.
There is an emerging body of evidence that staphylococcal strains also have some degree of encapsulation. To date, eight different CPS were described with serotype 5 and 8, which account for the majority of the clinical isolates. The staphylococcal capsule has been shown to be a virulence factor in animal models of bacteremia, surgical wound infection, arthritis and renal or skin abscess formation. Although, there is a clear role of the CPS in animal virulence, only 40% of all circulating S. aureus strains express a capsule and expression is limited to the stationary phase in humans (Bagnoli et al. 2012). Notwithstanding, all the endemic USA300 CA-MRSA strains that presently account for the majority of infections lack encapsulation, providing evidence that staphylococcal strains without a capsule are fully virulent.
Anti-opsonic properties
S. pneumoniae and S. aureus can express several surface and secreted proteins, which hamper opsonization. Phagocytosis of S. pneumoniae and S. aureus can be averted by anti-opsonic properties released directly from the pathogens. The list of pneumococcal anti-opsonic molecules is rapidly growing; however, they remain pale in comparison to the arsenal of staphylococcal anti-opsonic proteins (Rigby and DeLeo 2012; Serruto et al. 2010). In the last decade, over 15 different secreted or surface-bound molecules that inhibit serum complement or IgG-mediated uptake of staphylococci have been described. Great reviews on complement evasion by S. pneumoniae (Andre et al. 2017) and for S. aureus (Zipfel and Skerka 2014) have recently been published and only the most recently discovered molecules will be mentioned in this review.
The complement system is incredibly important for opsonization and, at the core of this proteolytic cascade, enzymatic convertase complexes are formed. These complexes mediate the cleavage of C3, which is essential for opsonizing microbes with complement components C3b and iC3b, resulting in enhanced phagocytosis. Likewise, activation of the complement system generates C5a, a potent neutrophil chemoattractant. The secreted staphylococcal metalloprotease, aureolysin and pneumococcal endopeptidase (PepO) recruit plasminogen to promote complement activation and rapid consumption of complement components around the bacterium. Protein catabolism of complement can lead to consequential inhibition of complement activation on the bacterial surface targets (Laarman et al. 2011; Agarwal et al. 2015). Aureolysin and PepO inhibit complement activation via C3, the central molecule in the complement system. Aureolysin resembles cobra venom factor and can efficiently activate C3 in a nonspecific manner, thereby inhibiting phagocytosis and killing of bacteria by neutrophils (Laarman et al. 2011). Staphylokinase can bind and activate plasminogen on the surface of bacteria, which results in cleavages of opsonins and reduced phagocytosis in vitro (Rooijakkers et al. 2005a). Staphylococcal complement inhibitor (SCIN) and homologs SCIN-B and SCIN-C are released to prevent directed migration of neutrophils and opsonophagocytic killing of staphylococci. SCIN stabilizes surface-bound C3 convertases, thereby impairing the enzymatic activity of the convertases, preventing subsequent complement opsonization and C5a release (Rooijakkers et al. 2005b). Very similar to SCIN, extracellular fibrinogen-binding protein (Efb) and extracellular complement-binding protein (Ecb), bind and inhibit convertases targeting only the alternative pathway and C5 convertases but not lectin or classical pathway convertases (Jongerius et al. 2010). Moreover, Efb can crosslink C3b with fibrinogen on the staphylococcal surface to cloak itself from recognition by FcγR or CR and subsequent uptake by neutrophils (Ko et al. 2013). Although SCIN and homologs are human-specific and thus cannot be tested effectively in animal infection models, Efb has been shown to inhibit phagocytosis of S. aureus in murine infection models (Jongerius et al. 2012). Pneumococcal surface protein A also interferes with the deposition of C3 molecules on the pneumococcal cell wall by inhibiting the binding of CRP to the phosphocholine moieties on the cell wall, which inhibits complement-mediated opsonization. Not surprisingly, complement factor H binding seems to be a common strategy of complement evasion as staphylococcal SrdE, pneumococcal PspC and the transcription elongating factor, Tuf, all bind to complement factor H. Once bound to complement factor H, complement convertases begin to decay and function to prevent further bacterial opsonization (Bergmann and Hammerschmidt 2006; Akong-Moore et al. 2012; Mohan et al. 2014). In addition, the pneumococcal protein Enolase was reported to bind to C4 binding protein (C4BP), consequently coating the surface of pneumococci with this complement regulatory molecule.
The pneumococcus can express IgA protease, allowing the specific cleavage of the most prominent Ig subclass in the human airway and thereby limits the humoral response on mucosal surfaces (Poulsen et al. 1996; Senior et al. 2000). Unlike S. pneumoniae, S. aureus does not harbor an IgA protease; however, SSL7 binds immunoglobulin A (IgA) and complement C5, thereby inhibiting IgA–FcαRI binding and serum killing of bacteria (Langley et al. 2005). Furthermore, SSL7 can prevent the C5a-induced phagocytosis of S. aureus and oxidative burst in an in vitro whole-blood inflammation model (Bestebroer et al. 2010b). S. aureus can modulate IgG responses through staphylococcal protein A (SpA). SpA is expressed by all clinical S. aureus isolates and has potent immunomodulatory properties. SpA has two distinct binding activities for human and animal immunoglobulins with different functions in immunology. The first discovered function of SpA was binding to the Fcγ-domain of nonspecific IgG (in the wrong orientation), thereby blocking opsonophagocytosis of staphylococci by neutrophils. In addition, SpA binding to the Fab domain and cross-linking of IgM promotes B cell superantigen activity. Elegant work from the Schneewind laboratory demonstrated that, during S. aureus infection in humans, SpA increases in clonal non-specific IgM while IgG has no benefit in host protection (Pauli et al. 2014). However, vaccination with SpA, mutated in the Ig-binding domains, raised neutralizing antibodies specific for S. aureus, promoted opsonophagocytic clearance and protected mice against lethal staphylococcal infection (Kim et al. 2010). Next to SpA, S. aureus has an additional IgG binding protein with similar functions. Sbi binds the Fcγ-domain of IgG and this protein can also form a stable tripartite complex with C3 and FH, both pathways resulting in the inhibition of complement and IgG-mediated opsonization (Haupt et al. 2008).
Inhibition of neutrophil recruitment
Neutrophils are typically the first leukocyte to be recruited during acute inflammation and are crucial for clearing infection; therefore, inhibiting neutrophil recruitment is another strategy by S. aureus to mediate immune evasion. Neutrophils are signaled by specific chemokine or anaphylactic toxin receptors and bacteria can specifically target these receptors to block the initiation of the inflammatory response and host immune defenses. Extracellular adherence protein (Eap), chemotaxis-inhibiting protein of S. aureus (CHIPS), staphopain A (scpA), FPR2 inhibitory protein (FLIPr), its homolog FLIPr-L and staphylococcal superantigen-like (SSL)3–5 are all small secreted staphylococcal proteins that interfere with neutrophil activation or recruitment, as recently reviewed (Thammavongsa et al. 2015; Spaan et al. 2013a). These proteins are unique to S. aureus. S. pneumoniae has a few other proteins directly targeting cellular receptors involved in neutrophil recruitment or activation. The pneumococcus uses molecular mimicy to exploit the host’s inability to recognize self-derived molecular structures by displaying the host-derived small molecule phosphorylcholine (ChoP) on its surface. Decoration of pneumococcal cell-wall components with ChoP contribute to fitness and have an important role in inhibiting bacterial opsonization. On top of that, the generation of ChoP turned out to be an immune evasion strategy itself. The recently discovered ChoP esterase (also known as CbpE) is a pneumococcal enzyme bound to the cell-wall and utilizes platelet-activating factor (PAF) to generate ChoP. Generation of ChoP functions to deplete PAF from the airway lumen and renders the neutrophils ineffective in proper activation and killing of S. pneumoniae (Hergott et al. 2015). Another mechanism that the pneumococcus utilizes to evade neutrophil responses is through the surface protein ZmpC. ZmpC targets the leukocyte adhesion molecule P-selectin glycoprotein ligand-1 (PSGL-1) and thereby hampers initial tethering and rolling of leukocytes on endothelial cells. Infection of mice with ZmpC-producing strain TIGR4 in the model of pneumococcal pneumonia decreased neutrophil infiltration into the lungs compared to animals infected with an isogenic zmpC knock-out strain. Targeting PSGL-1 appears to be another virulence mechanism shared by S. aureus and S. pneumoniae as S. aureus can secrete the PSGL-1 inhibitor, SSL5 (Bestebroer et al. 2007). Furthermore, Gram-negative bacteria secrete proteases, e.g., ImpA of Pseudomonas aeruginosa and serine protease autotransporters of Enterobacteriaceae that cleave multiple glycoproteins including PSGL-1, thereby contributing to bacterial pathogenesis (Bardoel et al. 2012; Ruiz-Perez et al. 2011). All of these proteins, except for ImpA, target PSGL-1 and depend on proper glycosylation of the receptor. The importance of glycosylation in the activation of PSGL-1 has been shown using ligand treatment with neuraminidase to cleave the glycosidic linkages, resulting in diminished functional properties of this receptor. A novel function for PSGL-1 has recently been described whereby neutrophils use this receptor for phagocytosis of S. pneumoniae by recognition of CPS or autolysin (LytA) (Ramos-Sevillano et al. 2016). It remains to be elucidated whether PSGL-1 inhibitors also influence the uptake of bacteria by neutrophils; however, this may prove to be a promising direction for novel therapeutics.
Killing of neutrophils
One common potent virulence strategy is the secretion of toxins or cytolysins. Pneumolysin (Ply) of S. pneumoniae is a cholesterol-dependent pore-forming toxin, which is conserved in all pneumococcal isolates. Ply is generally appreciated as a virulence factor and its contribution to disease has been described in multiple experimental models of infection (Kadioglu et al. 2008). Using in vivo models of acute pneumonia, Ply was shown to be essential for the survival of S. pneumoniae in the respiratory tract (Kadioglu et al. 2002). Furthermore, Ply is required for bacterial dissemination from the lungs to other organs via the bloodstream (Orihuela et al. 2004) and immunization against Ply protects against S. pneumoniae infection (Alexander et al. 1994). Intriguingly, secretion of Ply by S. pneumoniae occurs solely upon autolysis, as Ply lacks a Gram-positive secretion signal. This strategy may seem odd because the bacterium has to undergo autolysis before the toxin is released; however, pneumococcal strains deficient in either LytA or puryvate oxidase (SpxB) genes involved in apoptotic-like death of pneumococcal cells were outcompeted in an in vivo model of nasopharyngeal colonization. These findings suggest that release of virulence proteins from dead pneumococcal cells contributes to ultimate survival of pneumococcus within the host (Regev-Yochay et al. 2007).
S. aureus is characterized by the ability to secrete many toxins that can lyse host cells, contribute to development of abscesses or kill neutrophils that are attempting to engulf and destroy bacteria. A few of the toxins that play a role in bacterial pathogenesis are α-toxin (or α-hemolysin, Hla), β-hemolysin, the PSMs and the bi-component toxins or leukocidins. The leukocidins are pore-forming cytotoxins that help the bacteria invade host cells. A few leukocidins released by S. aureus are PVL, γ-hemolysins (HlgAB and HlgCB), leukocidin ED (LukED), leukocidin GH (LukGH or LukAB) and leukocidin (LukMF) (Rigby and DeLeo 2012). Attempts have been made to characterize the role of each toxin involved in staphylococcal pathogenesis; however, many of these investigations are technically hampered by the species specificity of the various toxins (Loffler et al. 2010). Although many of these toxins were discovered nearly a century ago, investigations into host specificity have only recently been pursued. Interest in this area was sparked by the identification of the cellular receptor for Hla, rapidly followed by the elucidation of the leukocydin receptors. The Hla toxin functions by binding to a disintegrin and metalloprotease domain-containing protein 10 (ADAM10) receptor on host cells, which initiates the assembly into a heptameric pore resulting in subsequent lysis of the target cell (Inoshima et al. 2011). Many cell types express ADAM10, including endothelial and epithelial cells, platelets and monocytes. In mouse models for lethal pneumonia, bacteraemia and skin infections, mice administered S. aureus with isogenic Hla mutants are hampered in disease severity (Bubeck Wardenburg et al. 2007, 2008). Interestingly, human neutrophils appear to be fairly resistant to Hla, an observation that could be explained by low ADAM10 expression (Powers et al. 2015; Seilie and Bubeck Wardenburg 2017). Discovery of the targets for staphylococcal toxins has provided a cohesive explanation for cellular tropisms and a molecular basis for the species specificity.
The leukocidins are secreted by S. aureus and can kill target cells in minute concentrations (~1 nM) in vitro. When leukocidin binds to receptors on myeloid cells and erythrocytes, these toxins assemble from two different subunits (F and S) into an octameric pore structure followed by lysis of the host cell. In general, the majority of receptors that are targeted by bi-component leukocidins belong to the complement and chemokine family of receptors. For example, PVL targets C5aRs and LukED targets CCR5, CXCR1 and CXCR2 (Spaan et al. 2013b; Francis et al. 2012; Reyes-Robles et al. 2013). LukAB uniquely targets CD11b, a subunit of the Mac-1 integrin (DuMont et al. 2013) (see Fig. 2 for all the specific cell cellular targets of the bicomponent leukocidins, while more detailed information can be found in a recent comprehensive review by Spaan et al. 2017).
Staphylococcal toxin PSMs are highly expressed cytolytic peptides that are only found in staphylococcus species. PSMs are small, amphipathic α-helical peptides of approximately 20–25 (α-type) and 44 (β-type) amino acids. Two main immunomodulatory functions for these toxins have been proposed, the first of which is the attraction of phagocytes at nanomolar concentrations and cytolytic activity at micromolar concentrations (Wang et al. 2007). The second immunomodulatory function of PSMs is due to the amphipathic helical structure that can facilitate lysis of neutrophils, peripheral blood mononuclear cells and erythrocytes. Although PSMs peptides can lyse multiple cell types or even liposomes, it has been proposed that neutrophils are particularly susceptible to PSMs in infection because they will sense and migrate towards the PSM-producing bacteria (Wang et al. 2007; Kretschmer et al. 2010). The PSMα peptides have the highest biological potency and have a clear role in pathogenesis; however, the role of cytolysis with specific PSMα peptides remains unknown. In human serum, binding of lipoproteins has been shown to diminish the cytolytic capacity of PSMs (Surewaard et al. 2012). While these peptides may continue to exhibit cytolytic ability in serum-excluded extracellular environments, such as in a skin abscess, evidence that such activity is diminished in serum suggests that cytolytic PSMs also contribute to pathogenesis in an intracellular environment. Indeed, several studies have shown that PSMα peptides can facilitate neutrophil killing after phagocytosis (Surewaard et al. 2013; Geiger et al. 2012).
Another functional aspect of PSMs is the property to attract neutrophils, which seems contradictory, as other staphylococcal immune evasion molecules, such as CHIPS, FLIPr and FLIPr-L, are employed to prevent the influx of neutrophils during certain stages of staphylococcal disease. This strategy is successful because the expression of these immune evasion molecules is regulated under different conditions compared to the PSM genes. For PSMs, production is strictly controlled by the Agr quorum-sensing system (Queck et al. 2008), whereas most immune evasion molecules, such as CHIPs, FLIPr and FLIPr-L, are under control of the two-component SeaSeR system. For instance, FLIPr is expressed upon contact with neutrophil granule contents (Rooijakkers et al. 2006; Malachowa et al. 2011). PSM expression is low at early stages of infection when it may be advantageous for the bacteria to remain unrecognized by the innate immune system and FLIPr and FLIPr-L may help to inhibit the activity of any residual PSMs produced by the bacteria. At the height of infection, S. aureus may switch to an Agr-dependent toxin-based survival strategy and evasion of detection may no longer be feasible. Given that S. aureus is notoriously hard to kill by neutrophils, attraction to the site of infection may possibly be advantageous for the bacterium at certain stages of disease and may be involved in abscess formation. This strategy may be useful to disseminate infection to other tissues and organs through intracellular transport where neutrophils abet the bacterium by acting as a ‘Trojan horse’ (Thwaites and Gant 2011).
Analogous to PSMs, Ply has also been described to act in a pro-inflammatory nature. In a recent study, it was shown that S. pneumoniae expressing a mutated Ply protein lacks cytolytic activity and was more virulent than a pneumococcus in which the ply gene was deleted. This pro-inflammatory action appears to be specific for TLR4, because the non-cytolytic mutant has been reported to activate TLR4-dependent responses (Malley et al. 2003). Despite this convincing evidence, it is important to note that there is an opposing opinion in the field that Ply and many other proposed ligands for TLRs are based on artifact (Hajjar et al. 2001). In fact, initially PSMs were wrongfully characterized as TLR2 ligands due to contaminating lipopeptides in the purification procedure. Elucidation of a co-crystal structure would help further our understanding of how TLR4 may be structurally associated in complex with or without myeloid differentiation factor 2 (MD2) and the respective pneumolysin.
Intracellular survival
Once S. aureus has entered the cell, it has the capacity to survive and even replicate intracellularly within endothelial cells, epithelial cells, osteoclasts and in ‘professional’ phagocytes such as neutrophils and macrophages (Gresham et al. 2000; Koziel et al. 2009; Tuchscherr et al. 2011; Rogers 1956). When S. pneumoniae is phagocytosed, it is readily eliminated, although some recent reports suggest invasion and survival in ‘non-professional’ phagocytes such as epithelial and endothelial cells and specialized glial cells, such as the olfactory ensheathing cells (Macedo-Ramos et al. 2011, 2016; Uchiyama et al. 2009; Zhang et al. 2000). This process may involve translocation of S. pneumoniae to the central nervous system, where it can lead to pathogenesis of disorders such as meningitis.
Neutrophils can release serine proteases (e.g., elastase, cathepsin G and proteinase 3) into the phagolysosome, which are believed to facilitate the killing of kill S. pneumoniae. However, it has been suggested that several enzymes will function to facilitate survival with exposure to oxidative stress by acting as anti-oxidants. S. pneumoniae can encounter ROS stress either from neutrophils or from the endogenously produced H2O2. Interestingly, the pneumococcus has the remarkable ability to produce up to 2 mM of H202, which is largely mediated through the catalytic activity of pyruvate oxidase (SpxB). The Spx activity converts pyruvate to acetyl phosphate, CO2 and H2O2 using oxygen. Protection of the pneumococcus from oxidative stress is mediated through glutathione reductase (GR), pneumococcal NADH oxidase (NOX), pneumococcal SOD (SodA), thiol peroxidase (TpxD) and the SpxB gene. Evidence suggests that common mechanisms may be involved in protecting the neutrophil from ROS-induced damage (Yesilkaya et al. 2013). In addition, several putative auto-transporters may be involved in the resistance of S. pneumoniae to antimicrobial peptides such as the Cathelicidin LL-37 (Majchrzykiewicz et al. 2010).
Even better than S. pneumoniae, S. aureus has multiple mechanisms in place to protect against oxygen-dependent microbicidal killing. In fact, the characteristic golden pigment, Staphyloxanthin (aureus is Latin for golden), functions as an antioxidant against ROS. Staphylococcal superoxide dismutases (SodA and SodM), catalase (KatA) and alkyl hydroperoxide reductase (AhpCF) confer further resistance by direct elimination of ROS produced in the phagolysosome (Beavers and Skaar 2016). In addition, the H2O2 molecules are shuttled by the MPO–halide system in neutrophils to produce the potent bactericidal HOCl, which contributes to killing of ingested S. aureus (McGuinness et al. 2016). Recent work has shown that S. aureus produces a molecule that targets this reaction by specific inhibition of human MPO. The staphylococcal peroxidase inhibitor (SPIN) potently inhibits the peroxidation reaction mediated by MPO and bacteria. When the gene encoding SPIN has been deleted, decreased survival compared with wild-type bacteria after phagocytosis by neutrophils was observed (de Jong et al. 2017).
Antimicrobial peptides and/or proteins and proteases elicit oxygen-independent killing mechanisms in neutrophils, for which S. aureus has evolved mechanisms to prevent killing. The S. aureus peptidoglycan is completely resistant to lysozyme degradation, through O-acetylation of peptidoglycan, a process catalyzed by the enzyme OatA. S. aureus can hamper neutrophil killing by modification of the cell-wall components such as d-alanylation of teichoic acids (dlt operon) and incorporation of lysyl-phosphatidylglycerol in the plasma membrane, thereby decreasing overall negative charge of the bacterial surface leading to decrease susceptibility to antimicrobial peptides (Bera et al. 2005; Herbert et al. 2007; Peschel et al. 1999).
Outlook
With the rise of antibiotic resistant strains of both S aureus and S pneumoniae, there is an urgent need for new classes of antibiotics. Vaccination is an attractive option as an alternative strategy, because it prevents the risk of infection. For the pneumococcus, well-defined polysaccharide vaccines are available that allow efficient clearance of S. pneumoniae by the innate immune system and that have significantly decreased the burden of disease with the included serotypes. Currently, the most commonly used vaccines consist of purified polysaccharides from 7 to 13 (conjugated CPS) or from 23 (pure polysaccharide) serotypes. Vaccination with pure polysaccharide vaccines is protective against IPD in adults whereas conjugated vaccines protect against disease and eradicate carriage in all age groups including infants (Black et al. 2000; Hammitt et al. 2006). A potential drawback of CPS vaccines is limited serotype coverage, as it may lead to strains not carried by the vaccine replacing the otherwise colonized niche (Weinberger et al. 2011), highlighting the need for constant inclusion of additional new CPS serotypes in the vaccine. Perhaps, inclusion of other surface and virulence factors such as pneumolysin, pillus or ZmpC may significantly boost the vaccine efficacy. Although this method presents an enormous technical challenge to design a vaccine that includes all CPS serotypes for all strains that cause disease in humans, it could possibly eradicate pneumococcal disease.
In contrast, there currently is no vaccine available for S. aureus. Although several attempts have been made, most vaccine candidates failed in clinical trials despite being protective against staphylococcal infection in animal models. A possible explanation for failure in trials is the lack of correlation between uptake of the bacteria by neutrophils and their subsequent destruction. Vaccination efficacy is highly dependent on the uptake and killing by neutrophils; however, staphylococcal isolates can survive and even proliferate inside neutrophils. Intracellular survival is, in part, due to the ability of S. aureus to resist the effects of neutrophil-derived ROS and AMPs because neutrophils undergo rapid lysis after phagocytosis by S. aureus (Surewaard et al. 2013; Gresham et al. 2000; Voyich et al. 2005). Therefore, the pronounced capacity to kill phagocytes after uptake, a hallmark of virulent S. aureus such as CA-MRSA, may explain why some attempts to develop traditional S. aureus vaccines have failed. Although several new molecules that target staphylococci in this intracellular compartment have recently been identified, development and clinical testing will need to be carried out before it will become a therapeutic option available for clinical practice (Surewaard et al. 2016; Lehar et al. 2015). Lastly, vaccinations may also be less effective in the presence of immune evasion factors (e.g., SpA, Sbi, SCIN, CHIPS Efb, Ecb, FLIPr, FLIPr-L and SSLs) that hamper the attraction of the neutrophil and opsonization by complement/IgG, which prevents phagocytic uptake by neutrophils and results in decreased effectiveness for traditional vaccines.
The success of S. aureus and S. pneumoniae as ‘professional’ pathogens can largely be attributed to the vast repertoire of capsular serotypes, immune evasion proteins, and toxins that hamper host immunity. Neutrophils make significant contributions to innate defenses and are armed to control microbial infection but fail to have sufficient defense mechanisms to eradicate S. aureus. While significant advances have been made towards understanding the molecular details of the interaction between these pathogens and the neutrophil, many questions remain and provide a bright horizon for future research in this area. The clinical impact of the disorders caused by S. aureus and S. pneumoniae highlight the importance of gaining a complete understanding of the antimicrobial defenses in the neutrophil and the mechanisms that are employed to evade defenses, which is necessary to develop newly targeted therapeutics that can render bacteria more susceptible to phagocyte attack and control infection.
References
Abeyta M, Hardy GG, Yother J (2003) Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus Pneumoniae. Infect Immun 71:218–225
Agarwal V, Talens S, Grandits AM, Blom AM (2015) A novel interaction between complement inhibitor C4b-binding protein and Plasminogen that enhances Plasminogen activation. J Biol Chem 290:18333–18342
Akong-Moore K, Chow OA, von Köckritz-Blickwede M, Nizet V (2012) Influences of chloride and hypochlorite on Neutrophil extracellular trap formation. PLoS ONE 7:e42984
Alexander JE, Lock RA, Peeters CC, Poolman JT, Andrew PW, Mitchell TJ, Hansman D, Paton JC (1994) Immunization of mice with pneumolysin toxoid confers a significant degree of protection against at least nine serotypes of Streptococcus Pneumoniae. Infect Immun 62:5683–5688
Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A (2012) Neutrophil function: from mechanisms to disease. Annu Rev Immunol 30:459–489
Andre GO, Converso TR, Politano WR, Ferraz LFC, Ribeiro ML, Leite LCC, Darrieux M (2017) Role of Streptococcus Pneumoniae proteins in evasion of complement-mediated immunity. Front Microbiol 8:224
Babior BM (1999) NADPH oxidase: An update. Blood 93:1464–1476
Bagnoli F, Bertholet S, Grandi G (2012) Inferring reasons for the failure of Staphylococcus Aureus vaccines in clinical trials. Front Cell Infect Microbiol 2:16
Bardoel BW, Hartsink D, Vughs MM, de Haas CJ, van Strijp JA, van Kessel KP (2012) Identification of an immunomodulating metalloprotease of Pseudomonas Aeruginosa (IMPa). Cell Microbiol 14:902–913
Barlow M (2009) What antimicrobial resistance has taught us about horizontal gene transfer. Methods Mol Biol 532:397–411
Barrett FF, McGehee RF Jr, Finland M (1968) Methicillin-resistant Staphylococcus Aureus at Boston City Hospital. Bacteriologic and epidemiologic observations. N Engl J Med 279:441–448
Baur S, Rautenberg M, Faulstich M, Grau T, Severin Y, Unger C, Hoffmann WH, Rudel T, Autenrieth IB, Weidenmaier C (2014) A nasal epithelial receptor for Staphylococcus Aureus WTA governs adhesion to epithelial cells and modulates nasal colonization. PLoS Pathog 10:e1004089
Beavers WN, Skaar EP (2016) Neutrophil-generated oxidative stress and protein damage in Staphylococcus Aureus. Pathog Dis 74:ftw060
Bera A, Herbert S, Jakob A, Vollmer W, Gotz F (2005) Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus Aureus. Mol Microbiol 55:778–787
Bergmann S, Hammerschmidt S (2006) Versatility of pneumococcal surface proteins. Microbiology 152:295–303
Bestebroer J, Poppelier MJ, Ulfman LH, Lenting PJ, Denis CV, van Kessel KP, van Strijp JA, de Haas CJ (2007) Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood 109:2936–2943
Bestebroer J, De Haas CJ, Van Strijp JA (2010a) How microorganisms avoid phagocyte attraction. FEMS Microbiol Rev 34:395–414
Bestebroer J, Aerts PC, Rooijakkers SH, Pandey MK, Kohl J, van Strijp JA, de Haas CJ (2010b) Functional basis for complement evasion by staphylococcal superantigen-like 7. Cell Microbiol 12:1506–1516
Black RE, Cousens S, Johnson HL, Lawn JE, Rudan I, Bassani DG, Jha P, Campbell H, Walker CF, Cibulskis R, Eisele T, Liu L, Mathers C (2010) Global, regional, and national causes of child mortality in 2008: a systematic analysis. Lancet 375:1969–1987
Black S, Shinefield H, Fireman B, Lewis E, Ray P, Hansen JR, Elvin L, Ensor KM, Hackell J, Siber G, Malinoski F, Madore D, Chang I, Kohberger R, Watson W, Austrian R, Edwards K (2000) Efficacy, safety and immunogenicity of heptavalent pneumococcal conjugate vaccine in children. Northern California Kaiser Permanente vaccine study center group. Pediatr Infect Dis J 19:187–195
Bogaert D, van Belkum A, Sluijter M, Luijendijk A, de Groot R, Rumke HC, Verbrugh HA, Hermans PW (2004) Colonisation by Streptococcus Pneumoniae and Staphylococcus Aureus in healthy children. Lancet 363:1871–1872
Bogomolski-Yahalom V, Matzner Y (1995) Disorders of neutrophil function. Blood Rev 9:183–190
Boxer LA, Morganroth ML (1987) Neutrophil function disorders. Dis Mon 33:681–780
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535
Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O (2007) Poring over pores: alpha-hemolysin and Panton-valentine leukocidin in Staphylococcus Aureus pneumonia. Nat Med 13:1405–1406
Bubeck Wardenburg J, Palazzolo-Ballance AM, Otto M, Schneewind O, DeLeo FR (2008) Panton-valentine leukocidin is not a virulence determinant in murine models of community-associated methicillin-resistant Staphylococcus Aureus disease. J Infect Dis 198:1166–1170
Chambers HF, DeLeo FR (2009) Waves of resistance: Staphylococcus Aureus in the antibiotic era. Nat Rev Microbiol 7:629–641
Charpentier E, Tuomanen E (2000) Mechanisms of antibiotic resistance and tolerance in Streptococcus Pneumoniae. Microbes Infect 2:1855–1864
Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, Torpey JW, Otto M, Nizet V, Kim JE, Gallo RL (2010) Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus Epidermidis, a normal resident of the skin. J Investig Dermatol 130:192–200
Coronado F, Nicholas JA, Wallace BJ, Kohlerschmidt DJ, Musser K, Schoonmaker-Bopp DJ, Zimmerman SM, Boller AR, Jernigan DB, Kacica MA (2007) Community-associated methicillin-resistant Staphylococcus Aureus skin infections in a religious community. Epidemiol Infect 135:492–501
Cremers AJ, Zomer AL, Gritzfeld JF, Ferwerda G, van Hijum SA, Ferreira DM, Shak JR, Klugman KP, Boekhorst J, Timmerman HM, de Jonge MI, Gordon SB, Hermans PW (2014) The adult nasopharyngeal microbiome as a determinant of pneumococcal acquisition. Microbiome 2:44
de Jong NWM, Ramyar KX, Guerra FE, Nijland R, Fevre C, Voyich JM, McCarthy AJ, Garcia BL, van Kessel KPM, van Strijp JAG, Geisbrecht BV, Haas P-JA (2017) Immune evasion by a staphylococcal inhibitor of myeloperoxidase. Proc Natl Acad Sci U S A 114:9439–9444
de Leeuw E, Li C, Zeng P, Diepeveen-de Buin M, Lu WY, Breukink E, Lu W (2010) Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett 584:1543–1548
Deleo FR, Otto M, Kreiswirth BN, Chambers HF (2010) Community-associated meticillin-resistant Staphylococcus Aureus. Lancet 375:1557–1568
Deniset JF, Surewaard BG, Lee WY, Kubes P (2017) Splenic Ly6Ghigh mature and Ly6Gint immature neutrophils contribute to eradication of S. Pneumoniae. J Exp Med 214:1333–1350
Di Sabatino A, Carsetti R, Corazza GR (2011) Post-splenectomy and hyposplenic states. Lancet 378:86–97
Diamond MS, Staunton DE, de Fougerolles AR, Stacker SA, Garcia-Aguilar J, Hibbs ML, Springer TA (1990) ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J Cell Biol 111:3129–3139
DuMont AL, Yoong P, Day CJ, Alonzo F 3rd, McDonald WH, Jennings MP, Torres VJ (2013) Staphylococcus Aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A 110:10794–10799
Fontanilla JM, Kirkland KB, Talbot EA, Powell KE, Schwartzman JD, Goering RV, Parsonnet J (2010) Outbreak of skin infections in college football team members due to an unusual strain of community-acquired methicillin-susceptible Staphylococcus Aureus. J Clin Microbiol 48:609–611
Francis A III, Lina K, Stephen AR, Tamara R-R, Ashley LD, David GM, Nathaniel RL, Derya U, Victor JT (2012) CCR5 is a receptor for Staphylococcus Aureus leukotoxin ED. Nature 493:51–55
Ganz T, Metcalf JA, Gallin JI, Boxer LA, Lehrer RI (1988) Microbicidal/cytotoxic proteins of neutrophils are deficient in two disorders: Chediak-Higashi syndrome and "specific" granule deficiency. J Clin Invest 82:552–556
Geiger T, Francois P, Liebeke M, Fraunholz M, Goerke C, Krismer B, Schrenzel J, Lalk M, Wolz C (2012) The stringent response of Staphylococcus aureus and its impact on survival after Phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog 8:e1003016
Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP (2000) Survival of Staphylococcus Aureus inside neutrophils contributes to infection. J Immunol 164:3713–3722
Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB (2001) Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble Modulin. J Immunol 166:15–19
Hammerschmidt S, Wolff S, Hocke A, Rosseau S, Müller E, Rohde M (2005) Illustration of pneumococcal polysaccharide capsule during adherence and invasion of epithelial cells. Infect Immun 73:4653–4667
Hammitt LL, Bruden DL, Butler JC, Baggett HC, Hurlburt DA, Reasonover A, Hennessy TW (2006) Indirect effect of conjugate vaccine on adult carriage of Streptococcus Pneumoniae: an explanation of trends in invasive pneumococcal disease. J Infect Dis 193:1487–1494
Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, Skerka C, Zipfel PF (2008) The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement factor H and C3b. PLoS Pathog 4:e1000250
Herbert S, Bera A, Nerz C, Kraus D, Peschel A, Goerke C, Meehl M, Cheung A, Götz F (2007) Molecular basis of resistance to Muramidase and cationic antimicrobial peptide activity of Lysozyme in staphylococci. PLoS Pathog 3:e102
Hergott CB, Roche AM, Naidu NA, Mesaros C, Blair IA, Weiser JN (2015) Bacterial exploitation of phosphorylcholine mimicry suppresses inflammation to promote airway infection. J Clin Invest 125:3878–3890
Hostetter MK (1986) Serotypic variations among virulent pneumococci in deposition and degradation of covalently bound C3b: implications for phagocytosis and antibody production. J Infect Dis 153:682–693
Hussain M, Melegaro A, Pebody RG, George R, Edmunds WJ, Talukdar R, Martin SA, Efstratiou A, Miller E (2005) A longitudinal household study of Streptococcus Pneumoniae nasopharyngeal carriage in a UK setting. Epidemiol Infect 133:891–898
Inoshima I, Inoshima N, Wilke GA, Powers ME, Frank KM, Wang Y, Bubeck Wardenburg J (2011) A Staphylococcus Aureus Pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med 17:1310–1314
Jongerius I, Garcia BL, Geisbrecht BV, van Strijp JA, Rooijakkers SH (2010) Convertase inhibitory properties of staphylococcal extracellular complement-binding protein. J Biol Chem 285:14973–14979
Jongerius I, von Kockritz-Blickwede M, Horsburgh MJ, Ruyken M, Nizet V, Rooijakkers SH (2012) Staphylococcus Aureus virulence is enhanced by secreted factors that block innate immune defenses. J Innate Immun 4:301–311
Kadioglu A, Taylor S, Iannelli F, Pozzi G, Mitchell TJ, Andrew PW (2002) Upper and lower respiratory tract infection by Streptococcus Pneumoniae is affected by pneumolysin deficiency and differences in capsule type. Infect Immun 70:2886–2890
Kadioglu A, Weiser JN, Paton JC, Andrew PW (2008) The role of Streptococcus Pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol 6:288–301
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 11:373–384
Kim HK, Cheng AG, Kim H-Y, Missiakas DM, Schneewind O (2010) Nontoxigenic protein a vaccine for methicillin-resistant Staphylococcus Aureus infections in mice. J Exp Med 207:1863–1870
Kinchen JM, Ravichandran KS (2008) Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol 9:781–795
Klevens RM, Edwards JR, Tenover FC, McDonald LC, Horan T, Gaynes R (2006) Changes in the epidemiology of methicillin-resistant Staphylococcus Aureus in intensive care units in US hospitals, 1992-2003. Clin Infect Dis 42:389–391
Ko YP, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, Spaan AN, van Kessel KP, Hook M, Rooijakkers SH (2013) Phagocytosis escape by a Staphylococcus Aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog 9:e1003816
Koppe U, Suttorp N, Opitz B (2012) Recognition of Streptococcus Pneumoniae by the innate immune system. Cell Microbiol 14:460–466
Koziel J, Maciag-Gudowska A, Mikolajczyk T, Bzowska M, Sturdevant DE, Whitney AR, Shaw LN, DeLeo FR, Potempa J (2009) Phagocytosis of Staphylococcus Aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS ONE 4:e5210
Kretschmer D, Gleske AK, Rautenberg M, Wang R, Koberle M, Bohn E, Schoneberg T, Rabiet MJ, Boulay F, Klebanoff SJ, van Kessel KA, van Strijp JA, Otto M, Peschel A (2010) Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus Aureus. Cell Host Microbe 7:463–473
Laarman AJ, Ruyken M, Malone CL, van Strijp JA, Horswill AR, Rooijakkers SH (2011) Staphylococcus Aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J Immunol 186:6445–6453
Langley R, Wines B, Willoughby N, Basu I, Proft T, Fraser JD (2005) The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol 174:2926–2933
Laxminarayan R, Duse A, Wattal C, Zaidi AK, Wertheim HF, Sumpradit N, Vlieghe E, Hara GL, Gould IM, Goossens H, Greko C, So AD, Bigdeli M, Tomson G, Woodhouse W, Ombaka E, Peralta AQ, Qamar FN, Mir F, Kariuki S, Bhutta ZA, Coates A, Bergstrom R, Wright GD, Brown ED, Cars O (2013) Antibiotic resistance-the need for global solutions. Lancet Infect Dis 13:1057–1098
Le Y, Oppenheim JJ, Wang JM (2001) Pleiotropic roles of formyl peptide receptors. Cytokine Growth Factor Rev 12:91–105
Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Hiroshi Morisaki J, Kim J, Park S, Darwish M, Lee B-C, Hernandez H, Loyet KM, Lupardus P, Fong R, Yan D, Chalouni C, Luis E, Khalfin Y, Plise E, Cheong J, Lyssikatos JP, Strandh M, Koefoed K, Andersen PS, Flygare JA, Wah Tan M, Brown EJ, Mariathasan S (2015) Novel antibody–antibiotic conjugate eliminates intracellular S. Aureus. Nature 527:323–328
Li M, Diep BA, Villaruz AE, Braughton KR, Jiang X, DeLeo FR, Chambers HF, Lu Y, Otto M (2009) Evolution of virulence in epidemic community-associated methicillin-resistant Staphylococcus Aureus. Proc Natl Acad Sci U S A 106:5883–5888
Loffler B, Hussain M, Grundmeier M, Bruck M, Holzinger D, Varga G, Roth J, Kahl BC, Proctor RA, Peters G (2010) Staphylococcus Aureus panton-valentine leukocidin is a very potent cytotoxic factor for human neutrophils. PLoS Pathog 6:e1000715
Mac LC, Kraus MR (1950) Relation of virulence of pneumococcal strains for mice to the quantity of capsular polysaccharide formed in vitro. J Exp Med 92:1–9
Macedo-Ramos H, Campos FS, Carvalho LA, Ramos IB, Teixeira LM, De Souza W, Cavalcante LA, Baetas-da-Cruz W (2011) Olfactory ensheathing cells as putative host cells for Streptococcus Pneumoniae: evidence of bacterial invasion via mannose receptor-mediated endocytosis. Neurosci Res 69:308–313
Macedo-Ramos H, Ruiz-Mendoza S, Mariante RM, Guimaraes EV, Quadros-de-Souza LC, Paiva MM, Ferreira EO, Pinto TC, Teixeira LM, Allodi S, Baetas-da-Cruz W (2016) Streptococcus Pneumoniae resists intracellular killing by olfactory ensheathing cells but not by microglia. Sci Rep 6:36813
Majchrzykiewicz JA, Kuipers OP, Bijlsma JJ (2010) Generic and specific adaptive responses of Streptococcus Pneumoniae to challenge with three distinct antimicrobial peptides, bacitracin, LL-37, and nisin. Antimicrob Agents Chemother 54:440–451
Malachowa N, Whitney AR, Kobayashi SD, Sturdevant DE, Kennedy AD, Braughton KR, Shabb DW, Diep BA, Chambers HF, Otto M, DeLeo FR (2011) Global changes in Staphylococcus Aureus gene expression in human blood. PLoS ONE 6:e18617
Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT (2003) Recognition of pneumolysin by toll-like receptor 4 confers resistance to pneumococcal infection. Proc Natl Acad Sci U S A 100:1966–1971
McDonald B, McAvoy EF, Lam F, Gill V, de la Motte C, Savani RC, Kubes P (2008) Interaction of CD44 and hyaluronan is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids. J Exp Med 205:915–927
McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P (2012) Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12:324–333
McGuinness WA, Kobayashi SD, DeLeo FR (2016) Evasion of Neutrophil killing by Staphylococcus Aureus. Pathogens 5:32
McGuinness WA, Malachowa N, DeLeo FR (2017) Vancomycin resistance in Staphylococcus Aureus. Yale J Biol Med 90:269–281
Miller E, Andrews NJ, Waight PA, Slack MP, George RC (2011) Herd immunity and serotype replacement 4 years after seven-valent pneumococcal conjugate vaccination in England and Wales: an observational cohort study. Lancet Infect Dis 11:760–768
Mohan S, Hertweck C, Dudda A, Hammerschmidt S, Skerka C, Hallstrom T, Zipfel PF (2014) Tuf of Streptococcus Pneumoniae is a surface displayed human complement regulator binding protein. Mol Immunol 62:249–264
Moore KL, Patel KD, Bruehl RE, Fugang L, Johnson DA, Lichenstein HS, Cummings RD, Bainton DF, McEver RP (1995) P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J Cell Biol 128:661–671
Mullaly SC, Kubes P (2006) The role of TLR2 in vivo following challenge with Staphylococcus Aureus and prototypic ligands. J Immunol 177:8154–8163
Musher DM (2003) How contagious are common respiratory tract infections? N Engl J Med 348:1256–1266
Orihuela CJ, Gao G, Francis KP, Yu J, Tuomanen EI (2004) Tissue-specific contributions of pneumococcal virulence factors to pathogenesis. J Infect Dis 190:1661–1669
Pauli NT, Kim HK, Falugi F, Huang M, Dulac J, Henry Dunand C, Zheng NY, Kaur K, Andrews SF, Huang Y, DeDent A, Frank KM, Charnot-Katsikas A, Schneewind O, Wilson PC (2014) Staphylococcus Aureus infection induces protein A-mediated immune evasion in humans. J Exp Med 211:2331–2339
Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Götz F (1999) Inactivation of the dlt operon in Staphylococcus Aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274:8405–8410
Phillipson M, Kubes P (2011) The neutrophil in vascular inflammation. Nat Med 17:1381–1390
Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P (2006) Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med 203:2569–2575
Poland GA (1999) The burden of pneumococcal disease: the role of conjugate vaccines. Vaccine 17:1674–1679
Poulsen K, Reinholdt J, Kilian M (1996) Characterization of the Streptococcus Pneumoniae immunoglobulin A1 protease gene (iga) and its translation product. Infect Immun 64:3957–3966
Powers ME, Becker RE, Sailer A, Turner JR, Bubeck Wardenburg J (2015) Synergistic action of Staphylococcus Aureus Alpha-toxin on platelets and myeloid lineage cells contributes to lethal sepsis. Cell Host Microbe 17:775–787
Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Sturdevant DE, Ricklefs SM, Li M, Otto M (2008) RNAIII-independent target gene control by the agr quorum-sensing system: insight into the evolution of virulence regulation in Staphylococcus Aureus. Mol Cell 32:150–158
Ramos-Sevillano E, Urzainqui A, de Andres B, Gonzalez-Tajuelo R, Domenech M, Gonzalez-Camacho F, Sanchez-Madrid F, Brown JS, Garcia E, Yuste J (2016) PSGL-1 on leukocytes is a critical component of the host immune response against invasive pneumococcal disease. PLoS Pathog 12:e1005500
Regev-Yochay G, Dagan R, Raz M et al (2004) Association between carriage of streptococcus pneumoniae and staphylococcus aureus in children. JAMA 292:716–720
Regev-Yochay G, Trzcinski K, Thompson CM, Lipsitch M, Malley R (2007) SpxB is a suicide gene of Streptococcus Pneumoniae and confers a selective advantage in an in vivo competitive colonization model. J Bacteriol 189:6532–6539
Reiss-Mandel A, Regev-Yochay G (2016) Staphylococcus Aureus and Streptococcus Pneumoniae interaction and response to pneumococcal vaccination: myth or reality? Hum Vaccin Immunother 12:351–357
Reyes-Robles T, Alonzo F 3rd, Kozhaya L, Lacy DB, Unutmaz D, Torres VJ (2013) Staphylococcus Aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 14:453–459
Rigby KM, DeLeo FR (2012) Neutrophils in innate host defense against Staphylococcus Aureus infections. Semin Immunopathol 34:237–259
Rogers DE (1956) Studies on bacteriemia. I. Mechanisms relating to the persistence of bacteriemia in rabbits following the intravenous injection of staphylococci. J Exp Med 103:713–742
Rooijakkers SH, van Wamel WJ, Ruyken M, van Kessel KP, van Strijp JA (2005a) Anti-opsonic properties of staphylokinase. Microbes Infect 7:476–484
Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA (2005b) Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol 6:920–927
Rooijakkers SH, Ruyken M, van Roon J, van Kessel KP, van Strijp JA, van Wamel WJ (2006) Early expression of SCIN and CHIPS drives instant immune evasion by Staphylococcus Aureus. Cell Microbiol 8:1282–1293
Root RK, Rosenthal AS, Balestra DJ (1972) Abnormal bactericidal, metabolic, and lysosomal functions of Chediak-Higashi syndrome leukocytes. J Clin Invest 51:649–665
Ruiz-Perez F, Wahid R, Faherty CS, Kolappaswamy K, Rodriguez L, Santiago A, Murphy E, Cross A, Sztein MB, Nataro JP (2011) Serine protease autotransporters from Shigella flexneri and pathogenic Escherichia Coli target a broad range of leukocyte glycoproteins. Proc Natl Acad Sci U S A 108:12881–12886
Seilie ES, Bubeck Wardenburg J (2017) Staphylococcus Aureus Pore-forming toxins: the interface of pathogen and host complexity. Semin Cell Dev Biol. https://doi.org/10.1016/j.semcdb.2017.04.003
Senior BW, Dunlop JI, Batten MR, Kilian M, Woof JM (2000) Cleavage of a recombinant human immunoglobulin A2 (IgA2)-IgA1 hybrid antibody by certain bacterial IgA1 proteases. Infect Immun 68:463–469
Serruto D, Rappuoli R, Scarselli M, Gros P, van Strijp JA (2010) Molecular mechanisms of complement evasion: learning from staphylococci and meningococci. Nat Rev Microbiol 8:393–399
Spaan AN, Surewaard BGJ, Nijland R, van Strijp JAG (2013a) Neutrophils versus Staphylococcus Aureus: a biological tug of war. Annu Rev Microbiol 67:629–650
Spaan AN, Henry T, van Rooijen WJ, Perret M, Badiou C, Aerts PC, Kemmink J, de Haas CJ, van Kessel KP, Vandenesch F, Lina G, van Strijp JA (2013b) The staphylococcal toxin Panton-valentine Leukocidin targets human C5a receptors. Cell Host Microbe 13:584–594
Spaan AN, van Strijp JAG, Torres VJ (2017) Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15:435–447
Surewaard BG, Nijland R, Spaan AN, Kruijtzer JA, de Haas CJ, van Strijp JA (2012) Inactivation of staphylococcal phenol soluble modulins by serum lipoprotein particles. PLoS Pathog 8:e1002606
Surewaard BG, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, Omri A, Yates RM, Kubes P (2016) Identification and treatment of the Staphylococcus Aureus reservoir in vivo. J Exp Med 213:1141–1151
Surewaard BGJ, Kubes P (2017) Measurement of bacterial capture and phagosome maturation of Kupffer cells by intravital microscopy. Methods 128:12–19
Surewaard BGJ, de Haas CJC, Vervoort F, Rigby KM, DeLeo FR, Otto M, van Strijp JAG, Nijland R. 2013. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell Microbiol: n/a-n/a
Thammavongsa V, Kim HK, Missiakas D, Schneewind O (2015) Staphylococcal manipulation of host immune responses. Nat Rev Microbiol 13:529–543
Thwaites GE, Gant V (2011) Are bloodstream leukocytes Trojan horses for the metastasis of Staphylococcus Aureus? Nat Rev Microbiol 9:215–222
Tuchscherr L, Medina E, Hussain M, Volker W, Heitmann V, Niemann S, Holzinger D, Roth J, Proctor RA, Becker K, Peters G, Loffler B (2011) Staphylococcus Aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol Med 3:129–141
Uchiyama S, Carlin AF, Khosravi A, Weiman S, Banerjee A, Quach D, Hightower G, Mitchell TJ, Doran KS, Nizet V (2009) The surface-anchored NanA protein promotes pneumococcal brain endothelial cell invasion. J Exp Med 206:1845–1852
Vandenesch F, Naimi T, Enright MC, Lina G, Nimmo GR, Heffernan H, Liassine N, Bes M, Greenland T, Reverdy ME, Etienne J (2003) Community-acquired methicillin-resistant Staphylococcus Aureus carrying Panton-valentine leukocidin genes: worldwide emergence. Emerg Infect Dis 9:978–984
Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Saïd-Salim B, Porcella SF, Long RD, Dorward DW, Gardner DJ, Kreiswirth BN, Musser JM, DeLeo FR (2005) Insights into mechanisms used by Staphylococcus Aureus to avoid destruction by human neutrophils. J Immunol 175:3907–3919
Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M (2007) Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13:1510–1514
Wanner S, Schade J, Keinhorster D, Weller N, George SE, Kull L, Bauer J, Grau T, Winstel V, Stoy H, Kretschmer D, Kolata J, Wolz C, Broker BM, Weidenmaier C (2017) Wall teichoic acids mediate increased virulence in Staphylococcus Aureus. Nat Microbiol 2:16257
Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S, Henriques-Normark B (2007) Capsule and D-alanylated lipoteichoic acids protect Streptococcus Pneumoniae against neutrophil extracellular traps. Cell Microbiol 9:1162–1171
Weidenmaier C, Goerke C, Wolz C (2012) Staphylococcus Aureus determinants for nasal colonization. Trends Microbiol 20:243–250
Weinberger DM, Malley R, Lipsitch M (2011) Serotype replacement in disease after pneumococcal vaccination. Lancet 378:1962–1973
Weiser JN (2010) The pneumococcus: why a commensal misbehaves. J Mol Med 88:97–102
Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL (2005) The role of nasal carriage in Staphylococcus Aureus infections. Lancet Infect Dis 5:751–762
Yesilkaya H, Andisi VF, Andrew PW, Bijlsma JJ (2013) Streptococcus Pneumoniae and reactive oxygen species: an unusual approach to living with radicals. Trends Microbiol 21:187–195
Yipp BG, Petri B, Salina D, Jenne CN, Scott BN, Zbytnuik LD, Pittman K, Asaduzzaman M, Wu K, Meijndert HC, Malawista SE, de Boisfleury Chevance A, Zhang K, Conly J, Kubes P (2012) Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med 18:1386–1393
Zhang J-R, Mostov KE, Lamm ME, Nanno M, Shimida S-I, Ohwaki M, Tuomanen E (2000) The polymeric immunoglobulin receptor Translocates Pneumococci across human nasopharyngeal epithelial cells. Cell 102:827–837
Zhu W, Murray PR, Huskins WC, Jernigan JA, McDonald LC, Clark NC, Anderson KF, McDougal LK, Hageman JC, Olsen-Rasmussen M, Frace M, Alangaden GJ, Chenoweth C, Zervos MJ, Robinson-Dunn B, Schreckenberger PC, Reller LB, Rudrik JT, Patel JB (2010) Dissemination of an Enterococcus Inc18-like vanA plasmid associated with vancomycin-resistant Staphylococcus Aureus. Antimicrob Agents Chemother 54:4314–4320
Zipfel PF, Skerka C (2014) Staphylococcus Aureus: the multi headed hydra resists and controls human complement response in multiple ways. Int J Med Microbiol 304:188–194
Acknowledgements
B.G.J Surewaard is partially funded by Marie Curie Actions (FP7-PEOPLE-2013-IOF; grant no. 627575) and a postgraduate fellowship from the Canadian Institutes of Health Research. M.L. Lewis is supported by the Branch Out Foundation. We thank Justin F. Deniset for carfully proofreading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lewis, M.L., Surewaard, B.G.J. Neutrophil evasion strategies by Streptococcus pneumoniae and Staphylococcus aureus . Cell Tissue Res 371, 489–503 (2018). https://doi.org/10.1007/s00441-017-2737-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2737-2