
Abstract
Aging is a universal phenomenon in metazoans, characterized by a general decline of the organism physiology associated with an increased risk of mortality and morbidity. Aging of an organism correlates with a decline in function of its cells, as shown for muscle, immune, and neuronal cells. As the DNA content of most cells within an organism remains largely identical throughout the life span, age-associated transcriptional changes must be achieved by epigenetic mechanisms. However, how aging may impact on the epigenetic state of cells is only beginning to be understood. In light of a growing number of studies demonstrating that noncoding RNAs can provide molecular signals that regulate expression of protein-coding genes and define epigenetic states of cells, we hypothesize that noncoding RNAs could play a direct role in inducing age-associated profiles of gene expression. In this context, the role of long noncoding RNAs (lncRNAs) as regulators of gene expression might be important for the overall transcriptional landscape observed in aged human cells. The possible functions of lncRNAs and other noncoding RNAs, and their roles in the regulation of aging-related cellular pathways will be analyzed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Aging is a universal and multifactorial process in complex living systems, characterized by a general decline of the organism physiology associated with an increased risk of mortality and morbidity. Due to its intrinsic complexity, models for studying organism aging are often inadequate and partial, being difficult to distinguish between causes and consequences of the aging phenomenon. At the phenotype level, aged organisms show a characteristic panoply of features always related to their physiological deterioration (Madrigano et al. 2012). However, the molecular mechanisms underlying this phenotype are far to be globally understood.
In complex organisms, aging appeared to be caused by the individual cell aging. In several types of tissues, the function of somatic cells declines with age. The term senescence was applied to these cells that ceased to divide in culture, based on the speculation that their behavior recapitulated organism aging. Consequently, cellular senescence is sometimes termed cellular aging or replicative senescence. Global aging of an organism is directly related to the individual cell aging. Increasing data suggest that cell aging is not merely an accumulation of damage, but an accumulation of damage associated with an altered transcriptional profile (Kato et al. 2011). There is not likely to be a single gene responsible for aging. Rather, a complex network of genomic interactions probably exists, which currently remains unknown. In order to support this idea, a coherent and integrative view has recently emerged with the major goal of studying the genetic mechanism subjacent to cell aging by combining systems biology with Genomics and Proteomics (Madrigano et al. 2012).
Spatial genome organization can critically affect gene expression in aging. While it is well known that chromatin composition can directly shape gene activity, three-dimensional chromatin organization is also emerging as an important gene regulation mechanism in aging (Collado et al. 2007). There are many ways by which chromatin interactions could be regulated: first, by modifying the DNA itself with cytosine methylation and consequently altering protein association (Fraga et al. 2007). Chromatin contacts could also be regulated by controlling access to DNA sequences with post-translational histone modifications (PTMs), the use of histone variants or by altering nucleosome positioning. Similarly, post-translational modification or changes in expression level of non-histone chromatin-binding proteins could represent important mechanisms to regulate chromatin contacts. Additionally, noncoding RNAs (ncRNAs) and their protein complexes could regulate the three-dimensional architecture of our genome. ncRNAs are a broad class of RNAs consisting of structural (rRNAs, tRNAs, snRNAs, snoRNAs, etc.), regulatory (miRNAs, piRNAs, etc.), and of sense/antisense transcripts, whose functions remain mostly uncharacterized (Mattick 2009). RNA is an ideal molecule to regulate biological networks, since it encodes sequence information and possess a great structural plasticity. The intrinsic relevance of ncRNAs in the regulation of genomic output has been rapidly unveiled during the last decade (Kato et al. 2011; Liao et al. 2011). However, the functional elements in the primary sequence of the majority of ncRNAs that determine their regulatory role remain unknown.
This chapter will analyze the molecular aging events and discuss the possible role of small and long ncRNAs in the regulation of pathways and processes related to aging at the cellular level, emphasizing their importance as modulators of the aging-mediated deterioration of cell physiology.
2 Pathways and Key Topics of Human Cellular Aging
2.1 Molecular Damage as a Driving Factor for Cell Aging
In humans, aging is thought to correlate with a recession in function of its cells and tissues, namely immune and neuronal cells (Grolleau-Julius et al. 2010; Lu et al. 2004). At the tissue and organ level, aging can be also characterized by the accumulation of senescent cells. Senescence is a physiological process in which normal cells cease to divide and can be induced by nutrient starvation (replicative senescence), DNA damage, telomere shortening, or by the expression of some genes (oncogene-induced senescence) (Lopez-Otin et al. 2013). In normal tissues, senescent cells are part of a mechanism devoted to the tissue regeneration which selectively eliminates damaged and dysfunctional cells. Recently, Muñoz-Espín and Serrano (2014) proposed that the accumulation of senescent cells in aged tissues could be the result of the lack of proper clearance of damaged cells by the immune system.
At the cellular level, aging is characterized by the presence of increasing amounts of molecular damage, which leads to a physiological imbalance and decline of cell metabolic functions. How and when cellular functions begin to decline due to aging is unknown; however, this decline is founded within a molecular basis. It is difficult to determine whether this molecular damage is the main cause of aging, but its presence can be related to the impairment of the control mechanisms that happens during organism aging (Rattan 2008). Seminal research by Wulf and coworkers showed early in the 1960s that the aging tissues and cells are unbalanced for the production of RNA molecules (Wulff et al. 1962). The authors postulated that the accumulation of mutations at the DNA level during aging would lead to the production of faulty RNAs, responsible at least in part for the aged phenotype. DNA damage leads to a misreading of the genetic information, and consequently to the possibility of faulty transmission of its message (Fukada et al. 2014). The correct functionality of the repair systems that correct DNA lesions has been related to an increased life span in mouse models (Brenerman et al. 2014).
The main sources of molecular damage during aging in DNA, RNA, proteins, and other biomolecules such as lipids come from free radicals or oxidative chemicals that can be originated either internally or externally to the cell (Fig. 1). In this context, the mitochondrial respiratory chain is responsible for the generation of reactive oxygen species (ROS), which include some free radicals, hydrogen peroxide, and the very reactive superoxide anion (Poyton et al. 2009). Extrinsic factors such as radiation and UV light can also trigger the production of ROS and free radicals. ROS are extremely reactive species, able to covalently modify many macromolecules, altering their functional and structural properties, and being responsible by the so-called oxidative stress (Nunomura et al. 2012; Di Domenico et al. 2010). Healthy cells harbor different mechanisms to destroy the free radicals and ROS in order to avoid their oxidative action over biomolecules, mainly based on the antioxidant molecules.

Molecular damage induced by internal or external factors contribute to the cellular aging process, founded in four pillars: DNA damage, transcriptional imbalance, accumulation of unfolded proteins and mitochondrial dysfunction. The molecular imbalance observed in aged cells is mainly triggered by chemical or physical stress, produced from either internal or external sources. Chemical stress can be originated by external chemicals or by the reactive oxygen species (ROS) formed as a consequence of the cellular oxidative metabolism
Enzymes such as superoxide-dismutase, catalase and thioredoxin and small organic molecules such as glutathione, are defense systems against the oxidative action of reactive chemical species (Mari and Cederbaum 2001; Sims-Robinson et al. 2013; Fukui and Zhu 2010). When the molecular damage is already caused, the disturbed biomolecule must be either repaired or destroyed. DNA molecules are typically repaired by several complex mechanisms involving macromolecular complexes assembled at the damaged loci, which are globally triggered in the presence of specific DNA lesions (Huen and Chen 2010; Lord and Ashworth 2012). In the unlikely event of an unrepaired DNA lesion, a global DNA damage response is activated and the cell will enter a cell-cycle arrest phase or become senescent in order to ensure genome maintenance and stability (Tian et al. 2014). DNA damage-induced senescence is also a natural mechanism to protect cells against cancer, but its relationship with the aging process is still not clear (Lieberman 2008; Tian et al. 2014). In mouse models during aging, some tissues appeared to be more prone to be enriched in senescent cells induced by DNA damage or telomere shortening (Wang et al. 2009). The same phenomenon is observed in human progeroid syndromes of accelerated aging like Werner’s syndrome, where genetic mutations disrupt totally or partially the molecular machinery responsible for the genomic integrity (Pichierri et al. 2001). RNA molecules can also be targets of oxidative damage during aging as described previously in neural cells (Nunomura et al. 2012). These alterations would lead to faulty transcription and an imbalance in the cellular RNA content (Fig. 1). When the affected molecules are ncRNA transcripts, defects in their regulatory activities are also expected.
Proteostasis, understood as the maintenance of a functional proteome, also declines with aging (Perez et al. 2009). A functional and healthy proteome is related to the chemical integrity of its components and their proper folding into a 3D space. Many cellular and external factors can challenge the proteome to cause protein instability or misfolding. Among them, the stress that lead to covalent modifications such as oxidation, the translational errors, and the presence of genome mutations are the most frequent. Misfolding can affect globular proteins or their domains when those have a consistent three-dimensional structure. In consequence, proteins lacking stable structure often denominated as intrinsically disordered proteins or IDPs are less sensitive to cellular stress and mutations (Light et al. 2013). Accumulation of misfolded proteins have negative consequences to the cell, since mutated and destabilized proteins often expose hydrophobic regions that tend to aggregate or to interact with cellular structures (Chiti et al. 2003; Stefani and Dobson 2003).
Time-dependent decline in protein functions during aging induces a stress over the physiological mechanisms devoted to the clearance of faulty protein molecules, mainly the proteasome and the lysosomes (Miller et al. 2014; Taylor et al. 2011). Intermediate quality control sensors and effectors, also known as protein chaperones, are also submitted to pressure during aging due to the accumulation of unfolded proteins (Brehme et al. 2014; Taylor et al. 2011). In model systems such as Caenorhabditis elegans, recent work demonstrated that the levels of ribosomal and mitochondrial proteins were decreased in aged worms, supporting the notion that proteostasis is altered during organism aging (Liang et al. 2014). Moreover, mitochondrial enzymes of the Kreb’s cycle and electron transport chain were diminished in aged animals, being consistent with the observed age-associated energy impairment (Ben-Zvi et al. 2009). Also in Drosophila, impaired proteasome function promoted aging phenotypes and reduced life span among individuals (Tsakiri et al. 2013). In humans, proteostasis networks centered in the protein chaperones have been characterized in relationship with neurodegenerative and aging-related diseases (Brehme et al. 2014). Interestingly, the mass spectrometry characterization of the proteome of human cells during aging also showed a consistent picture of decreased levels of proteins involved in cell death, cell differentiation and organization, response to stress, translation, RNA metabolism, and proteostasis control during aging (Waldera-Lupa et al. 2014).
2.2 Aging-Related Metabolic Pathways
Despite its multifactorial nature, aging is regulated by specific metabolic pathways including hormone-regulated signaling cascades and environmental nutrient sensing systems (Barzilai et al. 2012). The main cellular pathways involved in the control of life span in complex organisms are summarized in Fig. 2. All these pathways together form an entangled and interconnected regulatory framework which is part of the aging hallmarks (Lopez-Otin et al. 2013).

Metabolic pathways involved in the control of life span in humans. Nutrients and different external signals can act as triggers for pathways that ultimately control the cell fate under diverse biological circumstances
Insulin, insulin-like growth factor, and mTOR pathways showed crucial roles over organism life span, being highly conserved among species (Greer and Brunet 2008). IGF and insulin pathways are activated via their cognate membrane receptors inducing a signaling cascade centered in the AKT family of protein kinases that is related to a reduction in life span in model organisms (Miyauchi et al. 2004). On the other hand, the nutrient-dependent activation of mTOR pathway induces a metabolic alteration toward cell growth upon regulation of catabolism mediated by autophagy (Kapahi et al. 2010). Inhibition of this pathway extends life span in model organisms and confers protection against a wide range of age-related pathologies (Johnson et al. 2013). Autophagy, a well-characterized process that protects cell integrity by removing the damaged cell components is impaired during aging leading to the accumulation of molecular damage. This phenomenon has been observed in model organisms and in some human tissues (Carnio et al. 2014; Zou et al. 2014). Moreover, aging can be also considered as a chronic low-intensity inflammation state, where cytokine activation of the NF-kB pathway plays an important role. This cytokine-mediated activation is extremely relevant in the global aging process of a particular organism since it can be related to the accumulation of senescent cells and their secretory phenotype which can collaborate to the tissue function impairment (Coleman et al. 2013). Additional modulators of cell survival like sirtuins which are responsible for an extended life span in complex organisms, as well as for the introduction of more complexity into the aging-related pathways (Michan 2014).
Within this context, the changes in the expression of genes encoding proteins involved in aging-related metabolic pathways have been used as quantifiable biomarkers and possible causes of aging. Next-generation sequencing technologies have improved the resolution and information obtained from transcriptional data related to aging. To date, multiple studies profiled age-related transcriptional changes in mouse and human cells revealed important insight into the molecular mechanisms of aging. Namely, a set of age-regulated genes were identified, including genes associated with immunity and the inflammatory response, metabolic energy and degradation pathways, and extracellular matrix components (de Magalhaes et al. 2009). A parallel approach relied on genetics to search for single-gene mutations that extend life span in model organisms. These studies found that mutations affecting genes of the insulin signaling pathway increase the life span of C. elegans (Kenyon et al. 1993), Drosophila (Satomura et al. 2001; Clancy et al. 2001), and mice (Bluher et al. 2003; Holzenberger et al. 2003). Despite such striking evolutionary conservation, the genes that appear differentially expressed in mutant nematodes, flies, and mice tend to be species-specific (McElwee et al. 2007), highlighting the importance of investigating biological processes rather than individual genes to understand the molecular mechanisms underlying aging. More recent work has further contributed to pinpoint an intimate interplay between age-related transcriptional changes including those observed in the noncoding genome, alterations in chromatin structure and epigenetic modifications, and persistence of irreparable DNA lesions in chromosomal and mitochondrial DNA (Burgess et al. 2012).
3 Small Noncoding RNAs in the Aging Context
Noncoding (nc) RNAs represent an additional layer of gene regulation implicated in aging (Jung and Suh 2012). The ncRNAs are a remarkably diverse universe of RNAs that are not templates for protein synthesis but can regulate their expression in the context of human physiology and pathology. Several classes of small (typically 20–30 nucleotides) and long (>200 nucleotides) ncRNAs have been identified and shown to act as key regulators of protein gene expression in several biological processes (Grammatikakis et al. 2014; Di Leva and Croce 2013; Jung and Suh 2012). The medical relevance of ncRNAs is well established, particularly for some of the small members of the group such as the miRNAs (Esteller 2011). The vast majority of miRNAs act as posttranscriptional repressors of protein gene expression by binding the untranslated regions (UTRs) of target mRNAs. The miRNA regulatory effect over a selected transcript is relatively mild and could be described as a “fine-tuning” mechanism of post-transcriptional regulation (Grosshans and Filipowicz 2008). In contrast, a single miRNA could act over hundreds of different mRNAs, constituting an overall control layer that modulated the products of gene expression. Taking into account this fact, it is very tempting to relate miRNAs and their mechanism of action with global cell phenomena such as differentiation, senescence, cancer, or aging (Lafferty-Whyte et al. 2009; Bates et al. 2009b). MiRNAs have been implicated in many biological and pathological processes, ranging from development to cancer and life span (Jung and Suh 2012). Among them, the miRNA Lin-4 was first shown to regulate life span in C. elegans (Boehm and Slack 2005); lin-4 was subsequently found to be part of a group of miRNAs that change in expression as animals grow older (Ibanez-Ventoso et al. 2006). More recently, additional miRNAs were identified that influence life span in C. elegans both positively and negatively (de Lencastre et al. 2010). Age-related changes in miRNA expression were also reported in mouse brain (Inukai et al. 2012) and in human peripheral blood mononuclear cells (Noren Hooten et al. 2010). Also in model systems, a particularly interesting case is the Ames dwarf mouse, a mouse that shows increased delay in the onset of aging: miR-27a has been described as a main regulator of some intermediate metabolic enzymes that are related to the delayed aging of these animals (Bates et al. 2009a).
Moreover, the identification of mRNAs regulated by these miRNAs is further providing clues to understand how alterations in miRNA expression can contribute to the age-associated physiological decline. For instance, miR-146a, which is highly expressed in aged mice, down-regulates the expression of IL-1β and IL-6 leading to a lack of response of macrophages to proinflammatory stimuli (Jiang et al. 2012). Other aging-related pathways such as Wnt-mediated signaling (Vinas et al. 2013) and insuling/IGF-1 regulatory axis (Jordan et al. 2011) are also under the control of miRNAs. Interestingly, miRNAs are also regulatory players that can respond to hormonal stimuli, constituting feedback regulatory loops that ensure the tight control of metabolic signals (Martin et al. 2012).
Recent studies suggested that miRNAs and their biogenesis could control specifically the aging process by targeting several apparently unrelated genes. The nuclear work supporting this evidence has been performed using specific tissue or organs from aging mice. For instance, some murine miRNAs such as miR-93 and miR-214 have been found to be up-regulated in extremely old liver tissues (Li et al. 2009). Defects in the biogenesis of miRNAs have been also related to the induction of a senescence phenotype (Mudhasani et al. 2008), and the regulatory activities of miRNAs over cell aging and senescence-related pathways were proposed to act as pro- and antilongevity factors (Murphy 2010; de Lencastre et al. 2010). The term senescence was coined to describe cells that cease to divide in culture, assuming that this behavior recapitulates organism aging. However, several lines of evidence argue that replicative senescence and cell aging are not overlapping processes (Wennmalm et al. 2005; Bai et al. 2011). MiRNAs are also able to revert some senescence phenotypes induced by oncogenic factors such RAS (Borgdorff et al. 2010) and also to actively induce senescence and aging phenotypes in a variety of cell and organism models (Tazawa et al. 2007; Li et al. 2011; Liu et al. 2012).
It is well known that miRNAs can be actively secreted by cells, being detectable in all biological fluids. The mechanistic reasons for this phenomenon are far to be completely understood, but probably the secreted miRNAs could function as slow-action hormones able to regulate gene expression within cells located in organs or tissues far from they were synthesized (Creemers et al. 2012). Circulating miRNAs have been also considered as powerful biomarkers for the diagnosis and prognosis of several human conditions including aging (Weilner et al. 2013). Several authors proposed an active role for circulating miRNAs during aging, for instance acting as modulators of the chronic inflammatory phenotype observed in aged individuals (Olivieri et al. 2013b). Recently, a group of up-regulated age-related circulating miRNAs has been identified in mouse models. This group of circulating miRNA appeared to be up-regulated in aged animals and this effect can be reverted by caloric restriction. The genes targeted by this cohort of age-modulated circulating miRNAs are predicted to regulate biological processes linked to the phenotypic manifestations of aging, including metabolic changes, demonstrating the growing importance of this circulating regulators and their roles in the global context of organisms aging (Dhahbi et al. 2013).
4 Regulatory Long Noncoding RNAs as Modulators of Aging Metabolic Pathways
4.1 Common Functional Features of lncRNAs Within the Aging Transcriptional Landscape
In addition to miRNA genes, the human genome contains over 15,000 long noncoding RNA genes (lncRNAs) (Volders et al. 2013; Bu et al. 2012). This class of RNAs are by definition >200 bp in length, lacking significant protein-coding capacity. Their synthesis and structure are similar to protein-coding mRNAs, as they contain introns, have their 5′ and 3′ ends capped, and are frequently polyadenylated. These transcripts have initially been suggested to represent only the bystander’s transcription within protein-coding regions. However, histone markers of active transcription have been identified them outside protein-coding regions (Guttman et al. 2010). A subset of lncRNAs can be highly cell- and tissue-specific (Guttman et al. 2011) and show precise temporal specific patterns of expression as well as a certain degree of evolutionary conservation (Cabili et al. 2011). Meanwhile, only a small number of thousands of known noncoding RNAs have been implicated in a specific biological function.
Loss-of-function experiments have provided further evidence of lncRNAs functional importance on the regulation of gene expression patterns that control cell pluripotency, differentiation and survival, as well as epithelial-to-mesenchymal transition (Beltran et al. 2008). They also act as regulators of development and morphogenesis (Ulitsky et al. 2011), chromosomal dosage compensation (Tian et al. 2010), control of imprinting (Sleutels et al. 2002), cell-cycle regulation, and alternative splicing (Tripathi et al. 2010). LncRNAs exert the regulatory function in cis, modulating nearby genes on the same allele, or in trans by affecting genes at long genomic distances (Court et al. 2011). LncRNAs also interact with genomic DNA as well as RNA, and they function as flexible molecular scaffolds for the recruitment of chromatin modifying enzymes and transcription factors (Saxena and Carninci 2011; Gupta et al. 2010), driving their correct localization to genomic DNA targets. LncRNAs have also been shown to regulate the activity of other ncRNAs, specifically miRNAs, by acting as “sponges” that titrate miRNAs away from natural mRNA targets (thereby acting as competing endogenous RNAs; ceRNAs) (Cesana et al. 2011). However, the role of lincRNAs in cell aging needs to be further investigated.
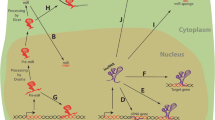
Despite the lack of experimental data, an altered expression pattern of the noncoding transcriptome is also expected in aging. In fact, lncRNAs are known to be involved in the control and regulation of cell fate decisions, including cell lineage commitment (Lin et al. 2014) and stemness (Guttman et al. 2011). Similar regulatory circuits based on the ncRNAs have been proposed to be on the basis of the age-dependent evolution of some human diseases such as cognitive disorders (Qureshi and Mehler 2011). In a small number of cases, the noncoding transcriptome was used to characterize the aging process and their phenotypic consequences (Chang et al. 2013). Also very recently, a specific cohort of lncRNAs has been characterized as implicated in replicative cell senescence (Abdelmohsen et al. 2013). However, in the majority of the studied cases, the relationships between lncRNAs and aging can only be depicted by the particular regulatory action exerted over a gene or an aging-related pathway, and not over the global aging process (Fig. 3). Moreover, these regulatory mechanisms have been frequently characterized outside the aging phenomenon itself and related to other biological problems such as cell differentiation, lineage commitment, or cancer (Table 1).

Potential regulatory role of selected lncRNAs within the aging pathways. Aging-related pathways are schematically represented using only the main key players, connected with lines to pinpoint their regulatory relationships. The lncRNAs are depicted in solid hexagons, connected with their regulated targets by dotted lines
4.2 lncRNAs and DNA Damage
As already discussed, the cell capacity to respond to DNA damage is essential to avoid the deleterious accumulation of functional mutations during aging (Jackson 2009). Several lncRNAs have been recently characterized as regulator of the cellular DNA damage response. One of the initial evidences of the regulatory role of a lncRNA on the DNA damage response was observed for RoR, a strong negative regulator of P53. Interestingly, and unlike other P53 regulators such as MDM2 which causes an ubiquitin-mediated P53 degradation, the lncRNA RoR suppresses the translation of P53 protein by direct interaction with hnRNPI (Zhang et al. 2013).
Other lncRNAs acting as modulators of the DNA damage response (DDR) include the CDKN2B antisense transcript also known as ANRIL (Wan et al. 2013b). Globally, ANRIL contributes to the maintenance of cellular responses triggered by DNA damage, via its regulation of cell-cycle checkpoints, apoptosis, and DNA repair (Wan et al. 2013b). ANRIL is transcriptionally activated by E2F1, and functions as homeostatic regulator by inhibiting P53 protein and thus bringing down the DNA damage response. In the particular case of cancer cells, the aberrant expression of this lncRNA would imbalance the DDR and eventually cause the blockage of this defense mechanism (Wan et al. 2013b).
The role of chromatin structure in DDR has been extensively studied, including chromatin modifications. Recently, the lncRNA-JADE which is induced after DDR has been characterized as an inducer of histone H4 acetylation. The histone acetylation is ensured via activation of the closing coding gene JADE1, a component of the HBO1 histone acetylation complex (Wan et al. 2013a).
More recently, a group of long intergenic radiation-responsive ncRNAs (LIRRs) have been shown to have an important role in the p53-mediated DDR. The expression of these lncRNAs is induced after a radiation-induced cell injury. A member of this family, LIRR1, has been characterized as an important regulator of the DDR. Its overexpression in human cells led to a decreased expression of several DNA repair proteins, an activation of p53, induction of p21 expression, and a cell-cycle G1 phase arrest (Jiao et al. 2015).
Other lncRNAs potentially involved in the mechanisms of DNA damage repair are TARID, which has been characterized as a regulator of DNA demethylation involved in base excision repair (Arab et al. 2014) and PCAT-1, a lncRNA identified in prostate cancers which negatively regulates the homologous recombination mechanism via repression of the tumor suppressor BRCA2 (Prensner et al. 2014).
4.3 lncRNAs and Inflammation
Human aging is characterized by a low-degree chronic inflammatory state, being a significant risk factor for morbidity and mortality in elderly individuals. The etiology of human chronic inflammation during aging remains unknown; however, the identification of pathways and modulators that control this phenotype is important in order to understand whether specific treatments that control inflammation can be beneficial to elderly people (Franceschi and Campisi 2014). In this context, the role of noncoding RNAs, inflammation, and aging has been extensively explored for the case of the miRNAs and reviewed elsewhere (Olivieri et al. 2013a). On the other hand, the evidences of the regulatory role of lncRNAs on the inflammation process are relatively more recent and derived from isolated observations.
Probably, one of the first lncRNAs characterized as a modulator of the inflammatory signals is Lethe. This mouse lncRNA is selectively induced by proinflammatory cytokines via NF-kappaB or glucocorticoid receptor agonists, and functions as a negative regulator in a feedback signaling to NF-kappaB. Lethe is able to interact with the RelA subunit of the NF-kappaB, inhibiting the RelA binding to the DNA targets and their activation (Rapicavoli et al. 2013). Interestingly, Lethe decreases with the organism aging, which is associated with an increase in the proinflammatory signals mediated by NF-kappaB pathway in several human tissues (Maqbool et al. 2013; Sriram et al. 2011).
Another lncRNA, PACER (p50-associated COX-2 extragenic RNA), has been recently characterized as a modulator of the inflammation also within the cancer context; however, its regulatory functions could be extended far from this disease to the overall inflammatory phenotype observed in aging (Krawczyk and Emerson 2014). PACER lncRNA is able to interact with p50, a repressive subunit of the NF-kappaB leading to an activation of competent NF-kappaB p65/p50 dimers. This mechanism will further enable the recruitment of histone acetyltransferases, a genome-wide histone acetylation, and RNApol II initiation complex assembly, constituting a global modulator of the inflammatory process (Krawczyk and Emerson 2014). In the same context, a lncRNA transcript which partially overlaps the gene encoding the interleukin-7 receptor alpha-subunit (IL7R) designated as lnc-IL7R has been characterized as a modulator of the inflammatory response via epigenetic regulation of the promoters of several inflammatory mediators (Cui et al. 2014). Indirect evidences have also linked the role of MALAT1 (Liu et al. 2014) and HOTAIR (Liu et al. 2015) lncRNAs to the regulation of the inflammatory response.
Also in the case of acute inflammatory events, the role of lncRNAs is becoming to be relevant. Recently, Li and coworkers have characterized a group of around 160 lncRNA founded to be differentially expressed upon innate activation of THP1 macrophages (Li et al. 2014a). Among them, a lincRNA called THRIL (TNFalpha and hnRNPL related immunoregulatory lincRNA) was required for expression of many immune response genes including cytokines and transcriptional and posttranscriptional regulators of TNFalpha expression (Li et al. 2014a). The authors were also able to correlate the levels of THRIL lncRNA with the severity of the symptoms of acute inflammatory diseases as Kawasaki syndrome.
4.4 Regulation of Senescence by lncRNAs
Senescence is an essential process to understand organism aging, since aged tissues have the tendency to accumulate senescent cells. The senescent phenotype can be reached by several biological routes involving different external stimuli and signaling cascades (Munoz-Espin and Serrano 2014). Early work by Gorospe’s laboratory showed that human senescent cells are characterized by a specific pattern of differentially expressed lncRNAs when compared to replicative cells. (Abdelmohsen et al. 2013). Further work has described different lncRNAs involved in the modulation of the senescence process.
HOTAIR lncRNA is clearly up-regulated in senescent cells, being associated with ubiquitin ligases to constitute a platform for protein ubiquitination. In senescent cells, HOTAIR helps to ubiquitinate Ataxin-1 and Snurportin-1, accelerating their degradation and preventing premature senescence (Yoon et al. 2013). Another lncRNAs such as UCA1 is involved in a more directed control of the senescence process. In fact, the lncRNA UCA1 is able to bind and sequester hnRNPA1, stabilizing the CDKN2A-p16INK complex and inducing senescence (Kumar et al. 2014). Interestingly, down-regulation of NEAT1, a lncRNA located in nuclear paraspeckles, has been also related to the induction of replicative senescence since it controls the overall nuclear organization (Yoon et al. 2014).
Also recently, Kumar and coworkers characterized a lncRNA called PANDA that is able to differentially interact with polycomb repressive complexes (PRC1 and PRC2) and the transcription factor NF-YA to promote or suppress senescence. In proliferation cells, the scaffold-attachment protein factor SAFA and the PANDA lncRNA recruit polycomb complexes to repress senescence-promoting genes (Puvvula et al. 2014).
In this context, study of several types of tumors and their development allowed the identification of an additional lncRNA denominated as FAL1 (Focally amplified lncRNA on chromosome 1), which was overexpressed in cancers with poor outcome. Molecular characterization of FAL1 transcript determined its ability to interact with the epigenetic repressor BMI1 to modulate the transcription of some genes including CDKN1A. FAL1 overexpression in tumors maintains the cells in the proliferative state. In consequence, FAL1 can be considered as a classical oncogene, mainly because of its ability to repress p21, a CDK inhibitor which is an inductor of senescence (Hu et al. 2014).
4.5 Regulatory LncRNAs and the Insulin Pathway
The insulin/IGF-1 metabolic axis is an essential regulatory pathway that is involved in organism development and aging. In fact, the levels of growing hormone and IGF-1 declined with aging. Low peripheral levels of IGF-1 are associated with increased aging-dependent risk of several conditions such as sarcopenia and osteoporosis (Barzilai et al. 2012). Moreover, in humans the aging process is accompanied by a phenomenon known as “insulin resistance” (IR), characterized by a lack of response of insulin receptors across the body. The IR syndrome is compensated by a hyperinsulinemia which can be considered as a risk factor for age-related diseases (Erol 2007). Epigenetic factors, including the regulatory effects of the noncoding transcriptome, could be potential modulators of this age-dependent decline of the insulin signaling pathway (Koerner et al. 2012).
One of the first lncRNAs characterized as a direct global regulator of the insulin signaling pathway is CRNDE. This lncRNAs has been firstly characterized as an overexpressed noncoding transcript in human colorectal cancer, being able to promote metabolic changes to support the aerobic glycolytic metabolism in cancer cells. Selective knockdown of CRNDE lncRNA by RNAi experiments affected the expression of many genes, which showed correlation with insulin/IGF-1 signaling pathway components and responses, including lipid and sugar metabolism (Ellis et al. 2014).
Other recently characterized lncRNAs included E330013P06, a mouse lncRNA up-regulated in macrophages obtained from diet-induced insulin-resistant type 2 diabetic mice, but not in type 1 diabetic mice. Reddy and coworkers determined that this lncRNA must constitute a link between insulin and inflammation pathways, since its knockdown inhibited the expression of inflammatory genes induced by diabetic stimuli (Reddy et al. 2014).
4.6 Regulatory lncRNAs in the WNT, mTOR, and Sirtuin Pathways
Aging-related metabolic pathways are closely related to those observed as dis-regulated in tumors, empowering the propensity of elderly people to suffer cancer. For instance, WNT and downstream effectors regulate processes that are relevant for cancer progression such as cell senescence and death which are also significant for complex organism aging (Anastas and Moon 2013). Some lncRNAs have been characterized recently as possibly involved in the regulation of WNT signaling pathway. The most relevant is probably lncRNA-p21, a long noncoding RNA which represses the WNT/β-catenin signaling axis (Wang et al. 2014a). Inversely, CCAT2 a lncRNA related to metastases in colon cancer has shown to be an enhancer of WNT signaling activity. Its mechanism of action involves a direct interaction with the TCF7L2 transcription factor, being itself also a downstream target of WNT (Ling et al. 2013).
Additional aging-related pathways such as mTOR signaling are also susceptible to the modulation exerted by lncRNA. In this context, the work by Li and coworkers proposed a new role for the UCA1 lncRNA (Li et al. 2014b). Experimental evidences linked the molecular regulatory mechanism of UCA1 lncRNA to the glucose and energy metabolism. This lncRNA is able to induce the expression of hexokinase 2 (HK2) in tumor cells by a mechanism that involves the activation of mTOR pathway (Li et al. 2014b). Regulation of mTOR signaling in the context of aging is related in part with autophagy. Also very recently, a lncRNA designated as FLJ1181 and derived from the 3′-UTR of the TGFB2 gene was characterized as a complementary endogenous ncRNA (ceRNA) involved in the regulation of autophagy via mTOR (Ge et al. 2014). Complementary endogenous RNAs or ceRNAs are lncRNAs which sense and capture miRNAs, acting as sponges that remove miRNAs from their action places. FLJ1181 binds miR-4459 which is a regulator of the autophagy-related 13 protein (ATG13). In consequence, FLJ1181 is a mTOR activator which acts as a link with the autophagy process (Ge et al. 2014).
Sirtuins are a wide group of enzymes with deacylase or mono-ADP ribosyl-transferase activity, classically related to cell differentiation processes and also with aging and extended life span in complex organisms (Liu and Sun 2011; Mantel and Broxmeyer 2008). Some recent evidences have pointed out the possible role of ncRNAs in the regulation of sirtuin activity. Wang and coworkers identified a natural antisense transcript (NAT) derived from divergent antisense transcription of Sirt1 gene (Wang et al. 2014b). This NAT has been characterized in myogenic differentiation of mouse model cells and showed regulatory activity of the Sirt1 gene. Due to their ubiquity, diversity of functions, and inter-species conservation, NATs are good functional candidates to be studied in within the context of the aging process (Werner 2013).
5 Conclusions and Further Perspectives
Eukaryotic genomes are pervasively transcribed into hundreds of RNA transcripts, many of them without evident capacity for coding proteins. The degree of organism complexity strongly correlates with the relative proportion of noncoding DNA in their genomes. Noncoding RNA transcripts have been pointed out as essential modulators of many biologically relevant processes. The ability of noncoding RNAs to regulate biological processes is mainly related to the intrinsic nature of the RNA molecules, able to carry sequence information as DNA but also to fold into complex structures and to have catalytic activity as proteins.
The specific role of some families of ncRNAs such as miRNAs and lcnRNAs is starting to be unveiled. As described along this review, some of the evidences pointing specific ncRNAs to their regulatory functions within the aging context are still circumstantial and in the majority of the cases extracted in an indirect fashion using aging-related diseases or cancer. Taking into account the complexity of the pathways and regulatory events involved in human aging, a single “master regulator” is not expected. Moreover, the accumulation of data obtained with the study of specific metabolic pathways involved in aging clearly suggests the presence of an important regulatory layer modulating aging-related processes which is ensured by the action of specific ncRNAs. Indeed, the aberrant ncRNA expression could be a new factor contributing to aging and aging-associated conditions in humans. The presence of aberrantly expressed ncRNAs in aging-related diseases opens room for RNA-based therapeutics using oligonucleotide-based drugs.
Our knowledge of the roles and rules of the noncoding transcriptome within the human aging context is still in its infancy, with only a few examples of miRNAs and lncRNAs characterized as regulators of aging-related pathways. One of the main weaknesses to develop functional aging studies is the lack of strong models of the process, which is more relevant in the case of the ncRNAs since they are not conserved across species. Future trends need to be focused in the development of new aging models, but also on the dissection of the molecular mechanism underlying the action of the already characterized ncRNAs and in the discovery of new relevant ones. The use of new techniques to characterize the function and structure of the genome at its output will be essential to understand the particular role of each ncRNA in the complex aging landscape. In this context, the combination of chromosome conformation capture techniques with the determination of structural features of the transcribed RNAs will open a new field of research to understand the wide range of functional genomic changes associated with the aging process and the role of ncRNAs in the regulation of these events.
References
Abdelmohsen K, Panda A, Kang MJ, Xu J, Selimyan R, Yoon JH, Martindale JL, De S, Wood WH 3rd, Becker KG, Gorospe M (2013) Senescence-associated lncRNAs: senescence-associated long noncoding RNAs. Aging Cell 12(5):890–900
Anastas JN, Moon RT (2013) WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer 13(1):11–26
Arab K, Park YJ, Lindroth AM, Schafer A, Oakes C, Weichenhan D, Lukanova A, Lundin E, Risch A, Meister M, Dienemann H, Dyckhoff G, Herold-Mende C, Grummt I, Niehrs C, Plass C (2014) Long noncoding RNA TARID directs demethylation and activation of the tumor suppressor TCF21 via GADD45A. Mol Cell 55(4):604–614
Bai XY, Ma Y, Ding R, Fu B, Shi S, Chen XM (2011) miR-335 and miR-34a promote renal senescence by suppressing mitochondrial antioxidative enzymes. J Am Soc Nephrol 22(7):1252–1261
Barzilai N, Huffman DM, Muzumdar RH, Bartke A (2012) The critical role of metabolic pathways in aging. Diabetes 61(6):1315–1322
Bates DJ, Li N, Liang R, Sarojini H, An J, Masternak MM, Bartke A, Wang E (2009a) MicroRNA regulation in Ames dwarf mouse liver may contribute to delayed aging. Aging Cell
Bates DJ, Liang R, Li N, Wang E (2009b) The impact of noncoding RNA on the biochemical and molecular mechanisms of aging. Biochim Biophys Acta 1790(10):970–979
Beltran M, Puig I, Pena C, Garcia JM, Alvarez AB, Pena R, Bonilla F, de Herreros AG (2008) A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial-mesenchymal transition. Genes Dev 22(6):756–769
Ben-Zvi A, Miller EA, Morimoto RI (2009) Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci USA 106(35):14914–14919
Bluher M, Kahn BB, Kahn CR (2003) Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 299(5606):572–574
Boehm M, Slack F (2005) A developmental timing microRNA and its target regulate life span in C. elegans. Science 310(5756):1954–1957
Borgdorff V, Lleonart ME, Bishop CL, Fessart D, Bergin AH, Overhoff MG, Beach DH (2010) Multiple microRNAs rescue from Ras-induced senescence by inhibiting p21(Waf1/Cip1). Oncogene 29(15):2262–2271
Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9(3):1135–1150
Brenerman BM, Illuzzi JL, Wilson DM 3rd (2014) Base excision repair capacity in informing healthspan. Carcinogenesis 35(12):2643–2652
Bu D, Yu K, Sun S, Xie C, Skogerbo G, Miao R, Xiao H, Liao Q, Luo H, Zhao G, Zhao H, Liu Z, Liu C, Chen R, Zhao Y (2012) NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res 40 (Database issue):D210–D215
Burgess RC, Misteli T, Oberdoerffer P (2012) DNA damage, chromatin, and transcription: the trinity of aging. Curr Opin Cell Biol 24(6):724–730
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25(18):1915–1927
Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R, Sandri M (2014) Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8(5):1509–1521
Carrion K, Dyo J, Patel V, Sasik R, Mohamed SA, Hardiman G, Nigam V (2014) The long non-coding HOTAIR is modulated by cyclic stretch and WNT/beta-CATENIN in human aortic valve cells and is a novel repressor of calcification genes. PLoS ONE 9(5):e96577
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I (2011) A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147(2):358–369
Chang AL, Bitter PH Jr, Qu K, Lin M, Rapicavoli NA, Chang HY (2013) Rejuvenation of gene expression pattern of aged human skin by broadband light treatment: a pilot study. J Invest Dermatol 133(6):1691
Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM (2003) Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature 424(6950):805–808
Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L (2001) Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292(5514):104–106
Coleman PR, Chang G, Hutas G, Grimshaw M, Vadas MA, Gamble JR (2013) Age-associated stresses induce an anti-inflammatory senescent phenotype in endothelial cells. Aging (Albany NY) 5(12):913–924
Collado M, Blasco MA, Serrano M (2007) Cellular senescence in cancer and aging. Cell 130(2):223–233
Court F, Baniol M, Hagege H, Petit JS, Lelay-Taha MN, Carbonell F, Weber M, Cathala G, Forne T (2011) Long-range chromatin interactions at the mouse Igf2/H19 locus reveal a novel paternally expressed long non-coding RNA. Nucleic Acids Res 39(14):5893–5906
Creemers EE, Tijsen AJ, Pinto YM (2012) Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res 110(3):483–495
Cui H, Xie N, Tan Z, Banerjee S, Thannickal VJ, Abraham E, Liu G (2014) The human long noncoding RNA lnc-IL7R regulates the inflammatory response. Eur J Immunol 44(7):2085–2095
de Lencastre A, Pincus Z, Zhou K, Kato M, Lee SS, Slack FJ (2010) MicroRNAs both promote and antagonize longevity in C. elegans. Curr Biol 20(24):2159–2168
de Magalhaes JP, Budovsky A, Lehmann G, Costa J, Li Y, Fraifeld V, Church GM (2009) The human ageing genomic resources: online databases and tools for biogerontologists. Aging Cell 8(1):65–72
Dhahbi JM, Spindler SR, Atamna H, Yamakawa A, Guerrero N, Boffelli D, Mote P, Martin DI (2013) Deep sequencing identifies circulating mouse miRNAs that are functionally implicated in manifestations of aging and responsive to calorie restriction. Aging (Albany NY) 5(2):130–141
Di Domenico F, Perluigi M, Butterfield DA, Cornelius C, Calabrese V (2010) Oxidative damage in rat brain during aging: interplay between energy and metabolic key target proteins. Neurochem Res 35(12):2184–2192
Di Leva G, Croce CM (2013) miRNA profiling of cancer. Curr Opin Genet Dev 23(1):3–11
Ellis BC, Graham LD, Molloy PL (2014) CRNDE, a long non-coding RNA responsive to insulin/IGF signaling, regulates genes involved in central metabolism. Biochim Biophys Acta 1843(2):372–386
Erol A (2007) Insulin resistance is an evolutionarily conserved physiological mechanism at the cellular level for protection against increased oxidative stress. BioEssays 29(8):811–818
Esteller M (2011) Non-coding RNAs in human disease. Nat Rev Genet 12(12):861–874
Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y (2014) Long non-coding RNA UCA1 increases chemoresistance of bladder cancer cells by regulating Wnt signaling. FEBS J 281(7):1750–1758
Feldstein O, Nizri T, Doniger T, Jacob J, Rechavi G, Ginsberg D (2013) The long non-coding RNA ERIC is regulated by E2F and modulates the cellular response to DNA damage. Mol Cancer 12(1):131
Fraga MF, Agrelo R, Esteller M (2007) Cross-talk between aging and cancer: the epigenetic language. Ann N Y Acad Sci 1100:60–74
Franceschi C, Campisi J (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 69(Suppl 1):S4–S9
Fukada S, Ma Y, Uezumi A (2014) Adult stem cell and mesenchymal progenitor theories of aging. Front Cell Dev Biol 2:10
Fukui M, Zhu BT (2010) Mitochondrial superoxide dismutase SOD2, but not cytosolic SOD1, plays a critical role in protection against glutamate-induced oxidative stress and cell death in HT22 neuronal cells. Free Radic Biol Med 48(6):821–830
Ge D, Han L, Huang S, Peng N, Wang P, Jiang Z, Zhao J, Su L, Zhang S, Zhang Y, Kung H, Zhao B, Miao J (2014) Identification of a novel MTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy 10(6):957–971
Grammatikakis I, Panda AC, Abdelmohsen K, Gorospe M (2014) Long noncoding RNAs (LncRNAs) and the molecular hallmarks of aging. Aging (Albany NY)
Greer EL, Brunet A (2008) Signaling networks in aging. J Cell Sci 121(Pt 4):407–412
Grolleau-Julius A, Ray D, Yung RL (2010) The role of epigenetics in aging and autoimmunity. Clin Rev Allergy Immunol 39(1):42–50
Grosshans H, Filipowicz W (2008) Proteomics joins the search for microRNA targets. Cell 134(4):560–562
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464(7291):1071–1076
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, Yang X, Amit I, Meissner A, Regev A, Rinn JL, Root DE, Lander ES (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477(7364):295–300
Guttman M, Garber M, Levin JZ, Donaghey J, Robinson J, Adiconis X, Fan L, Koziol MJ, Gnirke A, Nusbaum C, Rinn JL, Lander ES, Regev A (2010) Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28(5):503–510
Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, Even PC, Cervera P, Le Bouc Y (2003) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421(6919):182–187
Hu X, Feng Y, Zhang D, Zhao SD, Hu Z, Greshock J, Zhang Y, Yang L, Zhong X, Wang LP, Jean S, Li C, Huang Q, Katsaros D, Montone KT, Tanyi JL, Lu Y, Boyd J, Nathanson KL, Li H, Mills GB, Zhang L (2014) A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell 26(3):344–357
Huen MS, Chen J (2010) Assembly of checkpoint and repair machineries at DNA damage sites. Trends Biochem Sci 35(2):101–108
Ibanez-Ventoso C, Yang M, Guo S, Robins H, Padgett RW, Driscoll M (2006) Modulated microRNA expression during adult lifespan in Caenorhabditis elegans. Aging Cell 5(3):235–246
Inukai S, de Lencastre A, Turner M, Slack F (2012) Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE 7(7):e40028
Jackson SP (2009) The DNA-damage response: new molecular insights and new approaches to cancer therapy. Biochem Soc Trans 37(Pt 3):483–494
Jiang M, Xiang Y, Wang D, Gao J, Liu D, Liu Y, Liu S, Zheng D (2012) Dysregulated expression of miR-146a contributes to age-related dysfunction of macrophages. Aging Cell 11(1):29–40
Jiao Y, Liu C, Cui FM, Xu JY, Tong J, Qi XF, Wang LL, Zhu W (2015) Long intergenic non-coding RNA induced by X-ray irradiation regulates DNA damage response signaling in the human bronchial epithelial BEAS-2B cell line. Oncol Lett 9(1):169–176
Johnson SC, Rabinovitch PS, Kaeberlein M (2013) mTOR is a key modulator of ageing and age-related disease. Nature 493(7432):338–345
Jordan SD, Kruger M, Willmes DM, Redemann N, Wunderlich FT, Bronneke HS, Merkwirth C, Kashkar H, Olkkonen VM, Bottger T, Braun T, Seibler J, Bruning JC (2011) Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol 13(4):434–446
Jung HJ, Suh Y (2012) MicroRNA in aging: from discovery to biology. Curr Genomics 13(7):548–557
Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L (2010) With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab 11(6):453–465
Kato M, Chen X, Inukai S, Zhao H, Slack FJ (2011) Age-associated changes in expression of small, noncoding RNAs, including microRNAs in C. elegans. RNA 17(10):1804–1820
Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R (1993) A C. elegans mutant that lives twice as long as wild type. Nature 366(6454):461–464
Koerner MV, Pauler FM, Hudson QJ, Santoro F, Sawicka A, Guenzl PM, Stricker SH, Schichl YM, Latos PA, Klement RM, Warczok KE, Wojciechowski J, Seiser C, Kralovics R, Barlow DP (2012) A downstream CpG island controls transcript initiation and elongation and the methylation state of the imprinted airn macro ncRNA promoter. PLoS Genet 8(3):e1002540
Krawczyk M, Emerson BM (2014) p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-kappaB complexes. Elife 3:e01776
Kumar PP, Emechebe U, Smith R, Franklin S, Moore B, Yandell M, Lessnick SL, Moon AM (2014) Coordinated control of senescence by lncRNA and a novel T-box3 co-repressor complex. Elife 3
Lafferty-Whyte K, Cairney CJ, Jamieson NB, Oien KA, Keith WN (2009) Pathway analysis of senescence-associated miRNA targets reveals common processes to different senescence induction mechanisms. Biochim Biophys Acta 1792(4):341–352
Li N, Bates DJ, An J, Terry DA, Wang E (2009) Up-regulation of key microRNAs, and inverse down-regulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol Aging
Li N, Muthusamy S, Liang R, Sarojini H, Wang E (2011) Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech Ageing Dev 132(3):75–85
Li Z, Chao TC, Chang KY, Lin N, Patil VS, Shimizu C, Head SR, Burns JC, Rana TM (2014a) The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci USA 111(3):1002–1007
Li Z, Li X, Wu S, Xue M, Chen W (2014b) Long non-coding RNA UCA1 promotes glycolysis by upregulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci 105(8):951–955
Liang V, Ullrich M, Lam H, Chew YL, Banister S, Song X, Zaw T, Kassiou M, Gotz J, Nicholas HR (2014) Altered proteostasis in aging and heat shock response in C. elegans revealed by analysis of the global and de novo synthesized proteome. Cell Mol Life Sci 71(17):3339–3361
Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, Zhao G, Luo H, Bu D, Zhao H, Skogerbo G, Wu Z, Zhao Y (2011) Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic Acids Res 39(9):3864–3878
Lieberman HB (2008) DNA damage repair and response proteins as targets for cancer therapy. Curr Med Chem 15(4):360–367
Light S, Sagit R, Sachenkova O, Ekman D, Elofsson A (2013) Protein expansion is primarily due to indels in intrinsically disordered regions. Mol Biol Evol 30(12):2645–2653
Lin ST, Heng MY, Ptacek LJ, Fu YH (2014) Regulation of myelination in the central nervous system by nuclear lamin B1 and non-coding RNAs. Transl Neurodegener 3(1):4
Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS, Nishida N, Gafa R, Song J, Guo Z, Ivan C, Barbarotto E, De Vries I, Zhang X, Ferracin M, Churchman M, van Galen JF, Beverloo BH, Shariati M, Haderk F, Estecio MR, Garcia-Manero G, Patijn GA, Gotley DC, Bhardwaj V, Shureiqi I, Sen S, Multani AS, Welsh J, Yamamoto K, Taniguchi I, Song MA, Gallinger S, Casey G, Thibodeau SN, Le Marchand L, Tiirikainen M, Mani SA, Zhang W, Davuluri RV, Mimori K, Mori M, Sieuwerts AM, Martens JW, Tomlinson I, Negrini M, Berindan-Neagoe I, Foekens JA, Hamilton SR, Lanza G, Kopetz S, Fodde R, Calin GA (2013) CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res 23(9):1446–1461
Liu JY, Yao J, Li XM, Song YC, Wang XQ, Li YJ, Yan B, Jiang Q (2014) Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis 5:e1506
Liu N, Landreh M, Cao K, Abe M, Hendriks GJ, Kennerdell JR, Zhu Y, Wang LS, Bonini NM (2012) The microRNA miR-34 modulates ageing and neurodegeneration in Drosophila. Nature 482(7386):519–523
Liu Y, Luo F, Xu Y, Wang B, Zhao Y, Xu W, Shi L, Lu X, Liu Q (2015) Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol Appl Pharmacol 282(1):9–19
Liu Z, Sun LY (2011) Complex roles of Sirtuin 1 in cancer and aging. Transl Res 157(5):273–275
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153(6):1194–1217
Lord CJ, Ashworth A (2012) The DNA damage response and cancer therapy. Nature 481(7381):287–294
Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA (2004) Gene regulation and DNA damage in the ageing human brain. Nature 429(6994):883–891
Madrigano J, Baccarelli A, Mittleman MA, Sparrow D, Vokonas PS, Tarantini L, Schwartz J (2012) Aging and epigenetics: Longitudinal changes in gene-specific DNA methylation. Epigenetics 7 (1)
Mantel C, Broxmeyer HE (2008) Sirtuin 1, stem cells, aging, and stem cell aging. Curr Opin Hematol 15(4):326–331
Maqbool A, Lattke M, Wirth T, Baumann B (2013) Sustained, neuron-specific IKK/NF-kappaB activation generates a selective neuroinflammatory response promoting local neurodegeneration with aging. Mol Neurodegener 8:40
Mari M, Cederbaum AI (2001) Induction of catalase, alpha, and microsomal glutathione S-transferase in CYP2E1 overexpressing HepG2 cells and protection against short-term oxidative stress. Hepatology 33(3):652–661
Martin EC, Bratton MR, Zhu Y, Rhodes LV, Tilghman SL, Collins-Burow BM, Burow ME (2012) Insulin-like growth factor-1 signaling regulates miRNA expression in MCF-7 breast cancer cell line. PLoS ONE 7(11):e49067
Mattick JS (2009) The genetic signatures of noncoding RNAs. PLoS Genet 5(4):e1000459
McElwee JJ, Schuster E, Blanc E, Piper MD, Thomas JH, Patel DS, Selman C, Withers DJ, Thornton JM, Partridge L, Gems D (2007) Evolutionary conservation of regulated longevity assurance mechanisms. Genome Biol 8(7):R132
Michan S (2014) Calorie restriction and NAD(+)/sirtuin counteract the hallmarks of aging. Front Biosci (Landmark Ed) 19:1300–1319
Miller BF, Drake JC, Naylor B, Price JC, Hamilton KL (2014) The measurement of protein synthesis for assessing proteostasis in studies of slowed aging. Ageing Res Rev 18C:106–111
Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I (2004) Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J 23(1):212–220
Mudhasani R, Zhu Z, Hutvagner G, Eischen CM, Lyle S, Hall LL, Lawrence JB, Imbalzano AN, Jones SN (2008) Loss of miRNA biogenesis induces p19Arf-p53 signaling and senescence in primary cells. J Cell Biol 181(7):1055–1063
Munoz-Espin D, Serrano M (2014) Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol 15(7):482–496
Murphy CT (2010) Aging: miRacles of longevity? Curr Biol 20(24):R1076–R1078
Noren Hooten N, Abdelmohsen K, Gorospe M, Ejiogu N, Zonderman AB, Evans MK (2010) microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 5(5):e10724
Nunomura A, Moreira PI, Castellani RJ, Lee HG, Zhu X, Smith MA, Perry G (2012) Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res 22(3):231–248
Olivieri F, Rippo MR, Monsurro V, Salvioli S, Capri M, Procopio AD, Franceschi C (2013a) MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res Rev 12(4):1056–1068
Olivieri F, Rippo MR, Procopio AD, Fazioli F (2013b) Circulating inflamma-miRs in aging and age-related diseases. Front Genet 4:121
Perez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B, Ward W, Richardson A, Chaudhuri A (2009) Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc Natl Acad Sci USA 106(9):3059–3064
Pichierri P, Franchitto A, Mosesso P, Palitti F (2001) Werner’s syndrome protein is required for correct recovery after replication arrest and DNA damage induced in S-phase of cell cycle. Mol Biol Cell 12(8):2412–2421
Pickard MR, Williams GT (2014) Regulation of apoptosis by long non-coding RNA GAS5 in breast cancer cells: implications for chemotherapy. Breast Cancer Res Treat 145(2):359–370
Poyton RO, Ball KA, Castello PR (2009) Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab 20(7):332–340
Prensner JR, Chen W, Iyer MK, Cao Q, Ma T, Han S, Sahu A, Malik R, Wilder-Romans K, Navone N, Logothetis CJ, Araujo JC, Pisters LL, Tewari AK, Canman CE, Knudsen KE, Kitabayashi N, Rubin MA, Demichelis F, Lawrence TS, Chinnaiyan AM, Feng FY (2014) PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res 74(6):1651–1660
Puvvula PK, Desetty RD, Pineau P, Marchio A, Moon A, Dejean A, Bischof O (2014) Long noncoding RNA PANDA and scaffold-attachment-factor SAFA control senescence entry and exit. Nat Commun 5:5323
Qureshi IA, Mehler MF (2011) Non-coding RNA networks underlying cognitive disorders across the lifespan. Trends Mol Med 17(6):337–346
Rapicavoli NA, Qu K, Zhang J, Mikhail M, Laberge RM, Chang HY (2013) A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife 2:e00762
Rattan SI (2008) Increased molecular damage and heterogeneity as the basis of aging. Biol Chem 389(3):267–272
Reddy MA, Chen Z, Park JT, Wang M, Lanting L, Zhang Q, Bhatt K, Leung A, Wu X, Putta S, Saetrom P, Devaraj S, Natarajan R (2014) Regulation of inflammatory phenotype in macrophages by a diabetes-induced long noncoding RNA. Diabetes 63(12):4249–4261
Satomura S, Yokota I, Tatara K, Naito E, Ito M, Kuroda Y (2001) Paradoxical weight loss with extra energy expenditure at brown adipose tissue in adolescent patients with Duchenne muscular dystrophy. Metabolism 50(10):1181–1185
Saxena A, Carninci P (2011) Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. BioEssays 33(11):830–839
Sims-Robinson C, Hur J, Hayes JM, Dauch JR, Keller PJ, Brooks SV, Feldman EL (2013) The role of oxidative stress in nervous system aging. PLoS ONE 8(7):e68011
Sleutels F, Zwart R, Barlow DP (2002) The non-coding air RNA is required for silencing autosomal imprinted genes. Nature 415(6873):810–813
Sriram S, Subramanian S, Sathiakumar D, Venkatesh R, Salerno MS, McFarlane CD, Kambadur R, Sharma M (2011) Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-kappaB. Aging Cell 10(6):931–948
Stefani M, Dobson CM (2003) Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med (Berl) 81(11):678–699
Taylor JR, Lehmann BD, Chappell WH, Abrams SL, Steelman LS, McCubrey JA (2011) Cooperative effects of Akt-1 and Raf-1 on the induction of cellular senescence in doxorubicin or tamoxifen treated breast cancer cells. Oncotarget 2(8):610–626
Tazawa H, Tsuchiya N, Izumiya M, Nakagama H (2007) Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 104(39):15472–15477
Tian D, Sun S, Lee JT (2010) The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell 143(3):390–403
Tian H, Gao Z, Li H, Zhang B, Wang G, Zhang Q, Pei D, Zheng J (2014) DNA damage response—a double-edged sword in cancer prevention and cancer therapy. Cancer Lett
Tripathi V, Ellis JD, Shen Z, Song DY, Pan Q, Watt AT, Freier SM, Bennett CF, Sharma A, Bubulya PA, Blencowe BJ, Prasanth SG, Prasanth KV (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell 39(6):925–938
Tsakiri EN, Sykiotis GP, Papassideri IS, Terpos E, Dimopoulos MA, Gorgoulis VG, Bohmann D, Trougakos IP (2013) Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 12(5):802–813
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147(7):1537–1550
Vinas JL, Ventayol M, Brune B, Jung M, Sola A, Pi F, Mastora C, Hotter G (2013) miRNA let-7e modulates the Wnt pathway and early nephrogenic markers in mouse embryonic stem cell differentiation. PLoS ONE 8(4):e60937
Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K, Vandesompele J, Mestdagh P (2013) LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res 41(Database issue):D246–D251
Waldera-Lupa DM, Kalfalah F, Florea AM, Sass S, Kruse F, Rieder V, Tigges J, Fritsche E, Krutmann J, Busch H, Boerries M, Meyer HE, Boege F, Theis F, Reifenberger G, Stuhler K (2014) Proteome-wide analysis reveals an age-associated cellular phenotype of in situ aged human fibroblasts. Aging (Albany NY) 6(10):856–878
Wan G, Hu X, Liu Y, Han C, Sood AK, Calin GA, Zhang X, Lu X (2013a) A novel non-coding RNA lncRNA-JADE connects DNA damage signalling to histone H4 acetylation. EMBO J 32(21):2833–2847
Wan G, Mathur R, Hu X, Liu Y, Zhang X, Peng G, Lu X (2013b) Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal 25(5):1086–1095
Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T (2009) DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 8(3):311–323
Wang G, Li Z, Zhao Q, Zhu Y, Zhao C, Li X, Ma Z, Zhang Y (2014a) LincRNA-p21 enhances the sensitivity of radiotherapy for human colorectal cancer by targeting the Wnt/beta-catenin signaling pathway. Oncol Rep 31(4):1839–1845
Wang Y, Pang WJ, Wei N, Xiong Y, Wu WJ, Zhao CZ, Shen QW, Yang GS (2014b) Identification, stability and expression of Sirt1 antisense long non-coding RNA. Gene 539(1):117–124
Weilner S, Schraml E, Redl H, Grillari-Voglauer R, Grillari J (2013) Secretion of microvesicular miRNAs in cellular and organismal aging. Exp Gerontol 48(7):626–633
Wennmalm K, Wahlestedt C, Larsson O (2005) The expression signature of in vitro senescence resembles mouse but not human aging. Genome Biol 6(13):R109
Werner A (2013) Biological functions of natural antisense transcripts. BMC Biol 11:31
Wulff VJ, Quastler H, Sherman FG (1962) An hypothesis concerning RNA metabolism and aging. Proc Natl Acad Sci USA 48:1373–1375
Yoon JH, Abdelmohsen K, Kim J, Yang X, Martindale JL, Tominaga-Yamanaka K, White EJ, Orjalo AV, Rinn JL, Kreft SG, Wilson GM, Gorospe M (2013) Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat Commun 4:2939
Yoon JH, De S, Srikantan S, Abdelmohsen K, Grammatikakis I, Kim J, Kim KM, Noh JH, White EJ, Martindale JL, Yang X, Kang MJ, Wood WH 3rd, Noren Hooten N, Evans MK, Becker KG, Tripathi V, Prasanth KV, Wilson GM, Tuschl T, Ingolia NT, Hafner M, Gorospe M (2014) PAR-CLIP analysis uncovers AUF1 impact on target RNA fate and genome integrity. Nat Commun 5:5248
Zhang A, Zhou N, Huang J, Liu Q, Fukuda K, Ma D, Lu Z, Bai C, Watabe K, Mo YY (2013) The human long non-coding RNA-RoR is a p53 repressor in response to DNA damage. Cell Res 23(3):340–350
Zou CG, Ma YC, Dai LL, Zhang KQ (2014) Autophagy protects C. elegans against necrosis during Pseudomonas aeruginosa infection. Proc Natl Acad Sci USA 111(34):12480–12485
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Costa, M.C., Leitão, A.L., Enguita, F.J. (2015). Noncoding Transcriptional Landscape in Human Aging. In: Morris, K. (eds) Long Non-coding RNAs in Human Disease. Current Topics in Microbiology and Immunology, vol 394. Springer, Cham. https://doi.org/10.1007/82_2015_460
Download citation
DOI: https://doi.org/10.1007/82_2015_460
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-23906-4
Online ISBN: 978-3-319-23907-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)