
Abstract
Colorectal cancer (CRC) is one of the most prevalent cancers and the second leading cause of cancer mortality worldwide. Survival in the metastatic setting has been gradually improved by the addition to cytotoxic chemotherapy of agents targeting the vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR). Considerable heterogeneity exists within CRC due to the varied genetic and epigenetic mechanisms involved in differing pathways of carcinogenesis. The knowledge of molecular abnormalities underlying colorectal tumourigenesis and the progression of dysplastic precursors to invasive and ultimately metastatic lesions has advanced in recent years by comprehensive sequencing studies. From these genome-scale analyses, we know that a handful of genes are commonly affected by somatic mutations, whereas recurrent copy-number alterations and chromosomal translocations are rarer in this disease. Even though some of these molecular abnormalities make genes acting as drivers of cancer progression, translation of this recognition for therapeutic purposes is still limited, encompassing only as standard of care the exclusion of RAS-mutated cancers for better selecting patients to candidate to EGFR-targeted therapy with monoclonal antibodies. However, the effort of ameliorating molecular selection should not be considered exhausted by demonstration of RAS and BRAF-induced resistance, as the genomic landscape of response to EGFR blockade has been demonstrated to be wider and dynamically multifaceted. In this chapter we will review main molecular biomarkers of de novo (primary) and acquired (secondary) resistance to EGFR-targeted monoclonal antibodies in metastatic CRC and discuss therapeutic implications.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
1 Introduction
Colorectal cancer (CRC) is one of the most prevalent cancers and the second leading cause of cancer mortality worldwide (Siegel et al. 2017). Survival in the metastatic setting has been gradually improved by the use of fluorouracil/leucovorin in doublet or triplet combinations with oxaliplatin (FOLFOX) and/or irinotecan (FOLFIRI) together with agents targeting the vascular endothelial growth factor (VEGF) and epidermal growth factor receptor (EGFR) (Ciombor et al. 2015). Nowadays, precision oncology significantly influences current and emerging therapies for metastatic CRC patients by the demonstration that the molecular refinement has led to substantial improvements in clinical outcomes (overall survival, OS and progression-free survival, PFS) (Douillard et al. 2013). This approach, together with advancement in surgical resection for selected patients with limited liver and/or lung involvement, has indeed significantly improved median overall survival to over 40 months from diagnosis (Heinemann et al. 2014; Schwartzberg et al. 2014; Van Cutsem et al. 2011; Venook et al. 2014).
Considerable heterogeneity exists within colorectal tumours due to the varied genetic and epigenetic mechanisms involved in differing pathways of carcinogenesis. The knowledge of molecular abnormalities underlying colorectal tumourigenesis and the progression of dysplastic precursors to invasive and ultimately metastatic lesions has advanced in recent years by comprehensive sequencing studies (Cancer Genome Atlas Network 2012). From these genome-scale analyses, we know that a handful of genes are commonly affected by somatic mutations, whereas recurrent copy-number alterations and chromosomal translocations are rarer in this disease (Vogelstein et al. 2013). Even though some of these molecular abnormalities make genes acting as drivers of cancer progression, translation of this recognition for therapeutic purposes is still limited, encompassing only as standard of care the exclusion of RAS-mutated cancers for better selecting patients to candidate to EGFR-targeted therapy with monoclonal antibodies. It should be acknowledged that the process of refining molecular selection for these therapeutics has paralleled, and in some instances enhanced the quest for targets actionable at the clinical level. In particular, well-known oncogenes such as BRAF and ERBB2, that are now among most promising targets in this tumour (Corcoran et al. 2014; Sartore-Bianchi et al. 2016b), have been studied as biomarkers of resistance to anti-EGFR therapies. On the other hand, the effort of ameliorating molecular selection should not be considered exhausted by demonstration of RAS and BRAF-induced resistance, as the genomic landscape of response to EGFR blockade has been demonstrated to be wider (Bertotti et al. 2015) and dynamically multifaceted (Siravegna et al. 2015). In this chapter we will review main molecular biomarkers of de novo (primary) and acquired (secondary) resistance to EGFR-targeted monoclonal antibodies in metastatic CRC and discuss therapeutic implications.
2 Primary Resistance
Primary resistance is defined as ab initio refractoriness to anticancer treatment. It could be explained by resistance-conferring factors pre-existing in the bulk of tumour cells (Leto and Trusolino 2014) that we may not recognize due to tumour heterogeneity (Tannock and Hickman 2016). Intratumour heterogeneity is present early in cancer development and cancer treatment selects for resistant subclones. The main therapeutic implication is that a single drug may not be adequate enough to treat a genetically heterogeneous tumour, since a pre-treatment cancer cell population harbouring resistance genetic alteration, even if present at a low frequency, can contribute to therapeutic failure and poor outcome in a Darwinian fashion (Fisher et al. 2013; Tannock and Hickman 2016; Misale et al. 2014; Sartore-Bianchi et al. 2016a). Among molecular biomarkers of resistance to EGFR-targeted therapies in CRC, alterations in the RAS/RAF/MAPK pathway have been the most consistently shown to predict resistance, and RAS mutations have been the only reaching clinical grade. The landscape of molecular alterations impacting on sensitivity or resistance to these therapeutics is still interspersed with many other biomarkers, however they should be considered only in the context of translational research.
RAS
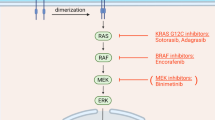
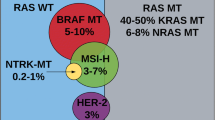
In the EGFR signalling pathway a dominant downstream direction involves the activation of the G-protein intermediate RAS, and subsequent signalling through BRAF, MEK, and ERK (the MAP kinase pathway). Mutations in the RAS family of proto-oncogenes (KRAS, NRAS, HRAS) result in constitutive activation of MAP kinase pathway signalling that is independent of activation of receptor tyrosine kinases such as EGFR. In CRC KRAS is the predominantly mutated isoform, whereas NRAS mutations are found more rarely and the HRAS mutated isoform is extremely uncommon and therefore not tested by routine (Prior et al. 2012). As a consequence, the upstream pharmacological blockade of the receptor can be circumvented for cancer progression by constitutive signalling of activated GTP-bound RAS forms (Bardelli and Siena 2010). This discovery by our group and others has been acknowledged as a landmark step for the evolution of precision medicine in the field of CRC (Benvenuti et al. 2007; Lièvre et al. 2006; Ushijima and Yoshino 2016). The most common RAS mutations in colon cancer occur at exon-2 (codons 12 and 13) of KRAS, and are present in about 42% of cases (Peeters et al. 2015). It is estimated that among tumours classified as KRAS exon 2 wild type, about one out of five carries other mutations in KRAS exon 3 (3.8–4.3%), KRAS exon 4 (6.2–6.7%), NRAS exon 2 (2.9–3.8%), NRAS exon 3 (4.2–4.8%) or NRAS exon 4 (0.3–0.5%), overall accounting for an additional total 11% of the so-called extended RAS or pan-RAS mutated CRCs (Peeters et al. 2015; Sorich et al. 2015). Initially, resistance to anti-EGFR mAbs was reported as associated with mutations confined to those occurring in codons 12 and 13 of exon 2 of the KRAS gene (Benvenuti et al. 2007; Lièvre et al. 2006). These findings were subsequently confirmed in retrospective analyses of large clinical trials (Amado et al. 2008; Van Cutsem et al. 2011) and reached clinical grade (Schmoll et al. 2012). However, retrospective analyses of multiple trials demonstrated that also additional mutations in exons 2, 3 and 4 of KRAS/NRAS exerted a similar predictive negative effect (De Roock et al. 2010), and this has been confirmed by analyses of phase III pivotal studies for development of anti-EGFR moAbs (Douillard et al. 2013; Van Cutsem et al. 2015). A meta-analysis of nine randomized controlled trials also confirmed that the treatment with both cetuximab and panitumumab had superior efficacy in terms of PFS and OS for extended RAS WT (i.e. KRAS exons 3 and 4 and NRAS exons 2, 3 and 4) compared with the expanded RAS mutant subgroup, and the efficacy was not significantly different between the expanded RAS mutant and KRAS exon 2 mutant subgroups (Sorich et al. 2015). Based on these studies, the screening of expanded RAS mutations for patients with metastatic CRC is currently recommended by principal treatment guidelines and included in the license for panitumumab and cetuximab for metastatic CRC (Van Cutsem et al. 2016) (Fig. 1). However, even though the presence of RAS mutations is a prerequisite for anti-EGFR moAbs, efficacy, its absence does not warrant response, as only 40–50% of patients with RAS WT tumours achieve objective response to treatment (Misale et al. 2014). Finally, the KRAS gene has been found not only to be mutated but also amplified, although in a very small percentage of CRC patients (0.7%), and this amplification has been observed as a mechanism in both primary and acquired resistance to EGFR inhibitors (Valtorta et al. 2013).

Parallel development of approval processes and evolution of the knowledge of molecular mechanisms of resistance to EGFR-targeted monoclonal antibodies in metastatic colorectal cancer
BRAF
BRAF is an oncogene that encodes a downstream effector of KRAS in the MAPK pathway. In CRC tumours, mutations leading to constitutive BRAF activation have been reported in 47% of hyper-mutated tumours and 3% of non-hypermutated tumours (Cancer Genome Atlas Network 2012), and approximately 5–10% of CRC tumours overall (De Roock et al. 2010). Of note, KRAS and BRAF mutations are mainly mutually exclusive in CRC (Richman et al. 2009). The presence of an activating mutation in BRAF conveys a strong prognostic significance, with mutated tumours conferring a poor prognosis with aggressive tumour biology and shorter OS, regardless of the treatment regimen (Safaee Ardekani et al. 2012). It should be noted that this prognostic impact should be regarded as confined to the BRAF V600E mutation, whereas BRAF mutations affecting codons 594 and 596, occurring in <1% of CRCs, display differences in terms of clinicopathologic features and less adverse clinical outcome (Cremolini et al. 2015).
Many different retrospective studies and meta-analyses by our group and others have suggested that the presence of a BRAF mutation also confers a weaker benefit from anti-EGFR monoclonal antibodies, negatively interfering with EGFR blockade (Benvenuti et al. 2007; Bertotti et al. 2015; Di Nicolantonio et al. 2008; Yuan et al. 2013). For these patients, even though there is not a formal demonstration by first-line combination trials that cetuximab or panitumumab are not effective (Douillard et al. 2013; Van Cutsem et al. 2015; Rowland et al. 2015), a general consensus exists for using an ab initio more intense chemotherapy regimens or enrolling in clinical studies with BRAF-directed strategies (Sartore-Bianchi et al. 2016a) to counteract the poor prognosis, making anti-EGFR treatment a less preferable option.
Family of HER
HER2 – the human epidermal growth factor receptor 2 (HER2/neu) is a well-established oncogenic driver in breast and gastric cancer, and recent data are highlighting a renewed role for this molecular target in CRC (Sartore-Bianchi et al. 2016a, c). Expression rates in historical series for CRC range widely from 1.6% (Ingold Heppner et al. 2014) to 47.4% (Park et al. 2007), but the sample size inclusion of distinct subgroups and the use of different diagnostic methods and scoring systems may account for this variability. In three of the most recent series, the rate of HER2 positivity (immunohistochemistry [IHC] score of 2+/3+, or HER2 gene amplification by in-situ hybridization) ranged from 1.6 to 6.3% (Ingold Heppner et al. 2014; Seo et al. 2014; Richman et al. 2016). In a consensus study aimed at defining CRC-specific criteria for HER2 positivity (Valtorta et al. 2015), we demonstrated that there is a clinically sizeable 5% fraction of KRAS wild-type CRC patients harbouring HER2-positive tumours, and this knowledge paralleled translational research with demonstration of HER2 as a therapeutic target (Sartore-Bianchi et al. 2016c).
On the other hand, HER2 amplification has been proposed as a biomarker of resistance to anti-EGFR antibodies. In 2011, we recognized HER2 amplification as a potential mechanism of primary resistance to cetuximab within a “quadruple wild type” population (KRAS, NRAS, BRAF and PIK3CA wild type) of immune-compromised mice harbouring CRC xenograft (patient-derived xenografts, PDX, a.k.a. “xenopatients”) (Bertotti et al. 2011). The same adverse effect was shown by retrospective analyses of patients treated with cetuximab (Yonesaka et al. 2011) or panitumumab (Martin et al. 2013; Sartore-Bianchi et al. 2016c). Interestingly, this effect has been demonstrated to be dependent on the level of amplification (Martin et al. 2013) and to contribute to both de novo and acquired drug resistance (Yonesaka et al. 2011). These results altogether suggest that patients with HER2-amplified CRC should be enrolled in clinical trials with HER2-targeted therapies regardless of having received a previous anti-EGFR inhibitor (Sartore-Bianchi et al. 2016c).
Together with amplification, also HER2 somatic mutations have been reported to occur in CRC at a frequency of about 3%, independently or concomitantly with amplification (Cancer Genome Atlas Network 2012). HER2 activating mutations tend to fall in several hotspots (residues 309–310 in the extracellular domain and residues 755–781 and 842 in the kinase domain), and cause oncogenic transformation of colon epithelial cells (Kavuri et al. 2015). It has been shown that these mutations produce resistance to cetuximab and panitumumab in preclinical models of CRC including PDXs and that dual HER2-targeted therapy with either trastuzumab plus neratinib or trastuzumab plus lapatinib can induce tumour regression (Kavuri et al. 2015).
HER3 also has been described to have a role as a potential biomarker of resistance to anti-EGFR treatments. In a cohort of metastatic CRC patients treated in second- or third-line therapy with irinotecan and cetuximab, HER3 overexpression was associated with shorter PFS and OS (Scartozzi et al. 2011). HER3 has been found to be also mutated in approximately 11% of CRC patients (Jaiswal et al. 2013), even though further studies are needed to elucidate the predictive role in conferring primary or acquired resistance to EGFR inhibitors.
cMET
The mesenchymal-epithelial transition (MET) protooncogene encodes for c-MET, a receptor with tyrosine kinase activity targeting hepatocyte growth factor (HGF) (Trusolino et al. 2010). Activation of this pathway by gene amplification has been implicated in metastatic progression of CRC, since MET has been historically reported to be overexpressed in 50% and amplified in 2–10% of primary CRCs, with the rate of amplification increasing up to 18–89% in metastases (Di Renzo et al. 1995; Zeng et al. 2008). However, more recent studies adopting ISH technologies together with newer NGS approaches clearly indicate that MET amplification ranges between 0.4 and 2.2% of cases (Cancer Genome Atlas Network 2012; Raghav et al. 2016), in both primary tumours and metastatic deposits (Raghav et al. 2016). As far as resistance to anti-EGFR therapies is concerned, in a recent complete exome sequence and copy number analyses of 129 PDX and targeted genomic analyses of 55 patient tumours, MET amplification was identified as a cause of primary resistance to cetuximab in 2.3% of cases (Bertotti et al. 2015). All in all, MET amplification rarely occurs and accounts for primary resistance in CRC, even though it is among alterations predominantly involved in acquired resistance (see below section on acquired resistance).
3 Biomarkers of Sensitivity
EGFR Gene Copy Number Gain
During the initial development of EGFR antibodies in metastatic CRC, it was predicted that EGFR protein expression would be required for therapeutic efficacy. Therefore, initially only EGFR-expressing CRCs were allowed into clinical trials (Saltz et al. 2004; Cunningham et al. 2004). However, subsequent analyses demonstrated a lack of association between EGFR expression and response (Chung et al. 2005; Moroni et al. 2008), eventually leading to decline of this restriction in label. On the other hand, an association was found with an EGFR gene copy number increase (Moroni et al. 2005) and this was originally postulated by our group as a biomarker of sensitivity to anti-EGFR therapy (Moroni et al. 2005). This finding was confirmed in preclinical experiments (Bertotti et al. 2015) as well as clinical cohorts (Personeni et al. 2008; Sartore-Bianchi et al. 2007; Scartozzi et al. 2009), however overt EGFR gene amplification is rarely observed in CRC, and correlation with response has been mainly based on balanced chromosome 7 polysomy rather than amplification, even though it is unknown whether the former could have an equivalent biologic effect in driving cancer progression and predicting response to EGFR-targeted agents. Also, detection by ISH methods of copy number gain suffers from issues of standardization, as it was shown by an interlaboratory reproducibility ring study conducted by our group (Sartore-Bianchi et al. 2012). In the end, even though an association between an increase in EGFR gene dosage and response has been established and confirmed especially in case of amplification (Bertotti et al. 2015), still this is not a validated biomarker for EGFR-targeted agents.
IRS2
The insulin receptor substrate (IRS) family of proteins are cytoplasmic adaptor that mediate through phosphorylation signalling between receptor tyrosine kinases of IGF1R and downstream effectors with roles in normal growth, metabolism and differentiation such as PI3K activation. IRS2 over-expression has been reported in 6–7% of MSS CRCs (Cancer Genome Atlas Network 2012; Nunes et al. 2015). A preclinical study has shown that IRS2 over-expression in the absence of an upstream activator leads to AKT phosphorylation and also increases CRC cell adhesion (Day et al. 2013). Amplifications and sequence alterations in the tyrosine kinase receptor adaptor gene IRS2 have been identified also in tumours with increased sensitivity to anti-EGFR therapy. Expression analyses of 100 CRC PDXs with wild-type KRAS, NRAS, BRAF and PIK3CA showed increased IRS2 levels as a significant predictor of cetuximab sensitivity in cases without other mechanisms of resistance to EGFR therapy (Bertotti et al. 2015).
4 Acquired (Secondary) Resistance
Acquired (or secondary) resistance refers to disease progression during an ongoing treatment that was initially effective. This occurs eventually in all metastatic CRCs and can be caused by gene mutations of the molecular target arising during treatment, expansion of resistant subclones in the context of intratumour heterogeneity selected under the pressure of cancer treatment, upregulation of a partially inhibited pathway or activation of alternative pathways. Even in patients with a refined RAS extended wild-type status, the tumour becomes refractory after a median of 5.2 months by developing secondary resistance (Kim et al. 2016). Liquid biopsy for monitoring of circulating tumour DNA (ctDNA) has been demonstrated by our group and others to be a powerful diagnostic tool for understanding dynamic mechanisms of tumour evolution in CRC (Misale et al. 2014; Van Emburgh et al. 2014). Molecular analysis performed on a tissue biopsy from a tumour is indeed a single snapshot in time subjected to selection bias due to spatial tumour heterogeneity, whereas analyses performed by liquid biopsy overcome this limitation and unveiled main mechanisms of acquired resistance to EGFR inhibitors.
RAS and BRAF
Various studies from our group and others have already demonstrated concordance between liquid biopsy and tumour-tissue biopsy for molecular characterization of clinically validated biomarkers for CRC such as KRAS and BRAF mutations (Thierry et al. 2014; Siravegna et al. 2015). At the same time, experiments in preclinical models (Misale et al. 2012) and translational studies with longitudinal monitoring by liquid biopsy have revealed clonal evolution during therapies with anti-EGFR antibodies showing that mutant RAS clones rise in blood during EGFR blockade (Misale et al. 2012) and decline upon withdrawal of treatment (Siravegna et al. 2015). It is conceivable that these subclones are less fit in the untreated tumour and acquire fitness as a consequence of adaptation to the perturbation induced by the treatment itself. Further, anti-EGFR pressure gives rise to multiple emergent circulating mutations of MAPK pathway within the same patient, with and individual average of almost three mutations (Bettegowda et al. 2014), in what has been called a “war of clones”. Interestingly, the relative frequency of individual KRAS alleles is similar but not identical in primary and acquired resistance: we firstly reported the secondary occurrence of codon 61 mutations (Misale et al. 2012) that rarely occur in anti-EGFR naïve patients, and now it is established that these mutations in either the KRAS or NRAS genes are more prevalent in the acquired than in the primary resistance setting (Bettegowda et al. 2014).
Family of HER
HER2 amplification has been associated also to acquired resistance to anti-EGFR moAbs. Yonesaka et al. showed that patients with acquired resistance to cetuximab had an increased percentage of HER2 amplification in post-treatment samples compared to the proportion present in pretreatment tumour cells (Yonesaka et al. 2011). These authors reported that in this context hyper-activation of HER2 signalling is triggered not only by HER2 amplification but also by overproduction of heregulin, a HER3 ligand.
MET
HGF-induced MET activation has been proposed as a mechanism of cetuximab resistance in CRC (Liska et al. 2011). We firstly reported by tissue analysis and longitudinal ctDNA monitoring that MET amplification is also associated with secondary resistance to anti-EGFR monoclonal antibodies (Bardelli et al. 2013). This observation was paralleled by functional analysis in CRC preclinical models indicating that HGF plays an important role in driving MET-mediated resistance to anti-EGFR monoclonal antibodies. HGF stimulation was demonstrated to be sufficient to confer cetuximab and panitumumab resistance both in vitro and in vivo, thus supporting the possibility that HGF overexpression by cancer cells or the surrounding stroma might be an independent mechanism of acquired (or primary) resistance to cetuximab (Bardelli et al. 2013). Further, we recently showed that MET amplification can simultaneously arise together with KRAS amplification within the same patient after initial response to EGFR inhibition in a context of substantial intrapatient heterogeneity (Sartore-Bianchi et al. 2016d).
EGFR External Domain Mutations
Mutations affecting the extracellular domain of the EGFR have not been reported in the absence of treatment with EGFR inhibitors in metastatic CRC (Esposito et al. 2013; Montagut et al. 2012). In 2012 it was firstly demonstrated that that cell lines with acquired resistance to cetuximab showed a mutation of the extracellular domain of the EGFR, 1476C>A, leading to a substitution of serine to arginine at amino acid 492 (S492R) (Montagut et al. 2012). This mutation interferes with binding to cetuximab but not to panitumumab in preclinical models and parallel observations in patients with acquired resistance to cetuximab. These findings have been confirmed subsequently in retrospective cohorts (Arena et al. 2015) and at the clinical level in the ASPECCT trial of cetuximab versus panitumumab for chemorefractory metastatic CRC, where the EGFR S492R was detected in 1% of patients in the panitumumab arm and 16% in the cetuximab arm in post-treatment plasma ctDNA samples (Newhall et al. 2014). It has been subsequently discovered that several other mutations in the EGFR extracellular domain can occur in preclinical models of cetuximab resistance (S464L, G465R and I491M) and in patients (R451C and K467T), mainly located in the cetuximab-binding region, except for the R451C mutant (Arena et al. 2015). From a clinical standpoint there are important therapeutic implications, since EGFR ectodomain mutations prevent binding to cetuximab but a subset is permissive for interaction with panitumumab (Arena et al. 2015), and it has been shown that new generation EGFR inhibitors such as the anti-EGFR antibody mixture Sym004 that bound and abrogated ligand-induced phosphorylation of EGFR mutants can overcome cetuximab/panitumumab resistance mediated by EGFR mutations (Sánchez-Martín et al. 2016). Interestingly, emerging knowledge is also indicating that there is a differential kinetic in the appearance of EGFR versus RAS mutated alleles during EGFR-targeted treatment, since RAS mutations emerge earlier than EGFR ECD variants (Van Emburgh et al. 2014). Subclonal RAS, but not EGFR extracellular domain mutations, have been shown indeed to be present in CRC samples obtained before exposure to EGFR blockade. Finally, retrospective analysis in a clinical cohort indicates that patients who experience greater and longer responses to EGFR blockade preferentially develop EGFR extracellular domain mutations, while RAS mutations emerge more frequently in patients with smaller tumour shrinkage and shorter progression-free survival (Van Emburgh et al. 2016).
5 Conclusions
Approved anti-EGFR antibodies cetuximab and panitumumab provide significant clinical benefit for the treatment of CRC, with patients in the metastatic setting now reaching an OS of more than 30 months. These advances have been made thanks to evolution of surgical techniques for metastasectomy, introduction of new agents, but to an important extent also to a refinement of molecular selection based on newer pharmacogenomics strategies. In this regard, the discovery of mechanisms of resistance to anti-EGFR monoclonal antibodies has enhanced clinical results, paving the way for a precision medicine approach in this disease. This effort should not be considered exhausted, as the genomic landscape of response to EGFR blockade has been demonstrated to be starred by a myriad of molecular abnormalities and, thanks to application of liquid biopsy for longitudinal analysis of the instable tumour genome, dynamically multifaceted (Fig. 2). Next challenges in this field will include the translation of current knowledge of tumour evolution mechanisms in the clinic for preventing or overcoming resistance at the individual patient level.

Biomarkers of resistance to EGFR-targeted monoclonal antibodies and known actionable targets for therapy in metastatic colorectal cancer. Evolution from 2006 until today
References
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) J Clin Oncol 26:1626–1634
Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S, González I, Iglesias M, Gavilan E, Corti G, Hobor S, Crisafulli G, Salido M, Sánchez J, Dalmases A, Bellmunt J, De Fabritiis G, Rovira A, Di Nicolantonio F, Albanell J, Bardelli A, Montagut C (2015) Clin Cancer Res 21:2157–2166
Bardelli A, Siena S (2010) J Clin Oncol 28:1254–1261
Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, Apicella M, Migliardi G, Galimi F, Lauricella C, Zanon C, Perera T, Veronese S, Corti G, Amatu A, Gambacorta M, Diaz LA, Sausen M, Velculescu VE, Comoglio P, Trusolino L, Di Nicolantonio F, Giordano S, Siena S (2013) Cancer Discov 3:658–673
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A (2007) Cancer Res 67:2643–2648
Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C, Ribero D, Russolillo N, Muratore A, Massucco P, Pisacane A, Molinaro L, Valtorta E, Sartore-Bianchi A, Risio M, Capussotti L, Gambacorta M, Siena S, Medico E, Sapino A, Marsoni S, Comoglio PM, Bardelli A, Trusolino L (2011) Cancer Discov 1:508–523
Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, Sassi F, Cottino F, Migliardi G, Zanella ER, Ribero D, Russolillo N, Mellano A, Muratore A, Paraluppi G, Salizzoni M, Marsoni S, Kragh M, Lantto J, Cassingena A, Li QK, Karchin R, Scharpf R, Sartore-Bianchi A, Siena S, Diaz LA, Trusolino L, Velculescu VE (2015) Nature 526:263–267
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, Bartlett BR, Wang H, Luber B, Alani RM, Antonarakis ES, Azad NS, Bardelli A, Brem H, Cameron JL, Lee CC, Fecher LA, Gallia GL, Gibbs P, Le D, Giuntoli RL, Goggins M, Hogarty MD, Holdhoff M, Hong S-M, Jiao Y, Juhl HH, Kim JJ, Siravegna G, Laheru DA, Lauricella C, Lim M, Lipson EJ, Marie SKN, Netto GJ, Oliner KS, Olivi A, Olsson L, Riggins GJ, Sartore-Bianchi A, Schmidt K, Shih L-M, Oba-Shinjo SM, Siena S, Theodorescu D, Tie J, Harkins TT, Veronese S, Wang T-L, Weingart JD, Wolfgang CL, Wood LD, Xing D, Hruban RH, Wu J, Allen PJ, Schmidt CM, Choti MA, Velculescu VE, Kinzler KW, Vogelstein B, Papadopoulos N, Diaz LA (2014) Sci Transl Med 6:224ra24
Cancer Genome Atlas Network (2012) Nature 487:330–337
Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, Klimstra DS, Fridman D, Kelsen DP, Saltz LB (2005) J Clin Oncol 23:1803–1810
Ciombor KK, Wu C, Goldberg RM (2015) Annu Rev Med 66:83–95
Corcoran RB, Atreya CE, Falchook GS, Infante JR, Hamid O, Messersmith WA, Daud A, Kwak EL, Ryan D, Kurzrock R, Sun P, Cunningham EA, Orford KW, Motwani M, Bai Y, Patel K, Venook AP, Kopetz S (2014) J Clin Oncol 32:5s
Cremolini C, Di Bartolomeo M, Amatu A, Antoniotti C, Moretto R, Berenato R, Perrone F, Tamborini E, Aprile G, Lonardi S, Sartore-Bianchi A, Fontanini G, Milione M, Lauricella C, Siena S, Falcone A, de Braud F, Loupakis F, Pietrantonio F (2015) Ann Oncol Off J Eur Soc Med Oncol 26:2092–2097
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E (2004) N Engl J Med 351:337–345
Day E, Poulogiannis G, McCaughan F, Mulholland S, Arends MJ, Ibrahim AEK, Dear PH (2013) Int J Exp Pathol 94:203–211
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S (2010) Lancet Oncol. 11:753–762
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2008) J Clin Oncol 26:5705–5712
Di Renzo MF, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, Giordano S (1995) Clin Cancer Res 1:147–154
Douillard J-Y, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD (2013) N Engl J Med 369:1023–1034
Esposito C, Rachiglio AM, La Porta ML, Sacco A, Roma C, Iannaccone A, Tatangelo F, Forgione L, Pasquale R, Barbaro A, Botti G, Ciardiello F, Normanno N (2013) Cancer Biol Ther 14:1143–1146
Fisher R, Pusztai L, Swanton C (2013) Br J Cancer 108:479–485
Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran S-E, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, Hielscher J, Scholz M, Müller S, Link H, Niederle N, Rost A, Höffkes H-G, Moehler M, Lindig RU, Modest DP, Rossius L, Kirchner T, Jung A, Stintzing S (2014) Lancet Oncol 15:1065–1075
Ingold Heppner B, Behrens H-M, Balschun K, Haag J, Krüger S, Becker T, Röcken C (2014) Br J Cancer 111:1977–1984
Jaiswal BS, Kljavin NM, Stawiski EW, Chan E, Parikh C, Durinck S, Chaudhuri S, Pujara K, Guillory J, Edgar KA, Janakiraman V, Scholz R-P, Bowman KK, Lorenzo M, Li H, Wu J, Yuan W, Peters BA, Kan Z, Stinson J, Mak M, Modrusan Z, Eigenbrot C, Firestein R, Stern HM, Rajalingam K, Schaefer G, Merchant MA, Sliwkowski MX, de Sauvage FJ, Seshagiri S (2013) Cancer Cell 23:603–617
Kavuri SM, Jain N, Galimi F, Cottino F, Leto SM, Migliardi G, Searleman AC, Shen W, Monsey J, Trusolino L, Jacobs SA, Bertotti A, Bose R (2015) Cancer Discov 5:832–841
Kim TW, Elme A, Kusic Z, Park JO, Udrea AA, Kim SY, Ahn JB, Villalobos Valencia R, Srinivasan K, Bilic A, Manojlovic N, Dong J, Guan X, Lofton-Day C, Jung AS, Vrdoljak E (2016) J Clin Oncol 34:642–642
Leto SM, Trusolino L (2014) J Mol Med Berl Ger 92:709–722
Lièvre A, Bachet J-B, Le Corre D, Boige V, Landi B, Emile J-F, Côté J-F, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P (2006) Cancer Res 66:3992–3995
Liska D, Chen C-T, Bachleitner-Hofmann T, Christensen JG, Weiser MR (2011) Clin Cancer Res 17:472–482
Martin V, Landi L, Molinari F, Fountzilas G, Geva R, Riva A, Saletti P, De Dosso S, Spitale A, Tejpar S, Kalogeras KT, Mazzucchelli L, Frattini M, Cappuzzo F (2013) Br J Cancer 108:668–675
Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen C-T, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A (2012) Nature 486:532–536
Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A (2014) Cancer Discov 4:1269–1280
Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, Salido M, Gallen M, Marsters S, Tsai SP, Minoche A, Seshagiri S, Somasekar S, Serrano S, Himmelbauer H, Bellmunt J, Rovira A, Settleman J, Bosch F, Albanell J (2012) Nat Med 18:221–223
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A (2005) Lancet Oncol 6:279–286
Moroni M, Veronese S, Sartore-Bianchi A, Artale S, Siena S (2008) Target Oncol 3:127–130
Newhall K, Price T, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, Suresh AS, Thomas A, Tjulandin S, Ogbagabriel S, Boedigheimer M, Sexson S, Zhang K, Murugappan S, Sidhu R (2014) Ann Oncol 25:ii109
Nunes M, Vrignaud P, Vacher S, Richon S, Lièvre A, Cacheux W, Weiswald L-B, Massonnet G, Chateau-Joubert S, Nicolas A, Dib C, Zhang W, Watters J, Bergstrom D, Roman-Roman S, Bièche I, Dangles-Marie V (2015) Cancer Res 75:1560–1566
Park DI, Kang MS, Oh SJ, Kim HJ, Cho YK, Sohn CI, Jeon WK, Kim BI, Han WK, Kim H, Ryu SH, Sepulveda AR (2007) Int J Color Dis 22:491–497
Peeters M, Kafatos G, Taylor A, Gastanaga VM, Oliner KS, Hechmati G, Terwey J-H, van Krieken JH (2015) Eur J Cancer Oxford Engl 51:1704–1713
Personeni N, Fieuws S, Piessevaux H, De Hertogh G, De Schutter J, Biesmans B, De Roock W, Capoen A, Debiec-Rychter M, Van Laethem J-L, Peeters M, Humblet Y, Van Cutsem E, Tejpar S (2008) Clin Cancer Res 14:5869–5876
Prior IA, Lewis PD, Mattos C (2012) Cancer Res 72:2457–2467
Raghav K, Morris V, Tang C, Morelli P, Amin HM, Chen K, Manyam GC, Broom B, Overman MJ, Shaw K, Meric-Bernstam F, Maru D, Menter D, Ellis LM, Eng C, Hong D, Kopetz S (2016) Oncotarget 7:54627–54631
Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, Taylor G, Barrett JH, Quirke P (2009) J Clin Oncol 27:5931–5937
Richman SD, Southward K, Chambers P, Cross D, Barrett J, Hemmings G, Taylor M, Wood H, Hutchins G, Foster JM, Oumie A, Spink KG, Brown SR, Jones M, Kerr D, Handley K, Gray R, Seymour M, Quirke P (2016) J Pathol 238:562–570
Rowland A, Dias MM, Wiese MD, Kichenadasse G, McKinnon RA, Karapetis CS, Sorich MJ (2015) Br J Cancer 112:1888–1894
Safaee Ardekani G, Jafarnejad SM, Tan L, Saeedi A, Li G (2012) PLoS One 7:e47054
Saltz LB, Meropol NJ, Loehrer PJ, Needle MN, Kopit J, Mayer RJ (2004) J Clin Oncol 22:1201–1208
Sánchez-Martín FJ, Bellosillo B, Gelabert-Baldrich M, Dalmases A, Cañadas I, Vidal J, Martinez A, Argilés G, Siravegna G, Arena S, Koefoed K, Visa L, Arpí O, Horak ID, Iglesias M, Stroh C, Kragh M, Rovira A, Albanell J, Tabernero J, Bardelli A, Montagut C (2016) Clin Cancer Res 22:3260–3267
Sartore-Bianchi A, Moroni M, Veronese S, Carnaghi C, Bajetta E, Luppi G, Sobrero A, Barone C, Cascinu S, Colucci G, Cortesi E, Nichelatti M, Gambacorta M, Siena S (2007) J Clin Oncol 25:3238–3245
Sartore-Bianchi A, Fieuws S, Veronese S, Moroni M, Personeni N, Frattini M, Torri V, Cappuzzo F, Vander Borght S, Martin V, Skokan M, Santoro A, Gambacorta M, Tejpar S, Varella-Garcia M, Siena S (2012) J Clin Pathol 65:218–223
Sartore-Bianchi A, Loupakis F, Argilés G, Prager GW (2016a) Ann. Oncol 27:1456–1466
Sartore-Bianchi A, Siena S, Tonini G, Bardelli A, Santini D (2016b) Cancer Treat Rev 51:54–62
Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, Bertotti A, Siravegna G, Torri V, Amatu A, Ghezzi S, Marrapese G, Palmeri L, Valtorta E, Cassingena A, Lauricella C, Vanzulli A, Regge D, Veronese S, Comoglio PM, Bardelli A, Marsoni S, Siena S (2016c) Lancet Oncol 17:738–746
Sartore-Bianchi A, Valtorta E, Amatu A, Veronese S, Lauricella C, Bonazzina E, Siravegna G, Truini M, Bardelli A, Siena S (2016d) ESMO Open 1:e000079
Scartozzi M, Bearzi I, Mandolesi A, Pierantoni C, Loupakis F, Zaniboni A, Negri F, Quadri A, Zorzi F, Galizia E, Berardi R, Biscotti T, Labianca R, Masi G, Falcone A, Cascinu S (2009) BMC Cancer 9:303
Scartozzi M, Mandolesi A, Giampieri R, Bittoni A, Pierantoni C, Zaniboni A, Galizia E, Giustini L, Silva RR, Bisonni R, Berardi R, Biscotti T, Biagetti S, Bearzi I, Cascinu S (2011) Oncologist 16:53–60
Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, Nagtegaal ID, Beets-Tan RG, Arnold D, Ciardiello F, Hoff P, Kerr D, Köhne CH, Labianca R, Price T, Scheithauer W, Sobrero A, Tabernero J, Aderka D, Barroso S, Bodoky G, Douillard JY, El Ghazaly H, Gallardo J, Garin A, Glynne-Jones R, Jordan K, Meshcheryakov A, Papamichail D, Pfeiffer P, Souglakos I, Turhal S, Cervantes A (2012) Ann Oncol 23:2479–2516
Schwartzberg LS, Rivera F, Karthaus M, Fasola G, Canon J-L, Hecht JR, Yu H, Oliner KS, Go WY (2014) J Clin Oncol 32:2240–2247
Seo AN, Kwak Y, Kim D-W, Kang S-B, Choe G, Kim WH, Lee HS (2014) PLoS One 9:e98528
Siegel RL, Miller KD, Jemal A (2017) CA Cancer J Clin 67:7–30
Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu A, Lauricella C, Lamba S, Hobor S, Avallone A, Valtorta E, Rospo G, Medico E, Motta V, Antoniotti C, Tatangelo F, Bellosillo B, Veronese S, Budillon A, Montagut C, Racca P, Marsoni S, Falcone A, Corcoran RB, Di Nicolantonio F, Loupakis F, Siena S, Sartore-Bianchi A, Bardelli A (2015) Nat Med 21:795–801
Sorich MJ, Wiese MD, Rowland A, Kichenadasse G, McKinnon RA, Karapetis CS (2015) Ann Oncol 26:13–21
Tannock IF, Hickman JA (2016) N Engl J Med 375:1289–1294
Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, Gillet B, Gongora C, Dechelotte P, Robert B, Del Rio M, Lamy P-J, Bibeau F, Nouaille M, Loriot V, Jarrousse A-S, Molina F, Mathonnet M, Pezet D, Ychou M (2014) Nat Med 20:430–435
Trusolino L, Bertotti A, Comoglio PM (2010) Nat Rev Mol Cell Biol 11:834–848
Ushijima T, Yoshino T (2016) Cancer Res 76:6443–6444
Valtorta E, Misale S, Sartore-Bianchi A, Nagtegaal ID, Paraf F, Lauricella C, Dimartino V, Hobor S, Jacobs B, Ercolani C, Lamba S, Scala E, Veronese S, Laurent-Puig P, Siena S, Tejpar S, Mottolese M, Punt CJA, Gambacorta M, Bardelli A, Di Nicolantonio F (2013) Int J Cancer 133:1259–1265
Valtorta E, Martino C, Sartore-Bianchi A, Penaullt-Llorca F, Viale G, Risio M, Rugge M, Grigioni W, Bencardino K, Lonardi S, Zagonel V, Leone F, Noe J, Ciardiello F, Pinto C, Labianca R, Mosconi S, Graiff C, Aprile G, Frau B, Garufi C, Loupakis F, Racca P, Tonini G, Lauricella C, Veronese S, Truini M, Siena S, Marsoni S, Gambacorta M (2015) Mod Pathol 28:1481–1491
Van Cutsem E, Köhne C-H, Láng I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F (2011) J Clin Oncol 29:2011–2019
Van Cutsem E, Lenz H-J, Köhne C-H, Heinemann V, Tejpar S, Melezínek I, Beier F, Stroh C, Rougier P, van Krieken JH, Ciardiello F (2015) J Clin Oncol 33:692–700
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson A, Bodoky G, Ciardiello F, D’Hoore A, Diaz-Rubio E, Douillard J-Y, Ducreux M, Falcone A, Grothey A, Gruenberger T, Haustermans K, Heinemann V, Hoff P, Köhne C-H, Labianca R, Laurent-Puig P, Ma B, Maughan T, Muro K, Normanno N, Österlund P, Oyen WJG, Papamichael D, Pentheroudakis G, Pfeiffer P, Price TJ, Punt C, Ricke J, Roth A, Salazar R, Scheithauer W, Schmoll HJ, Tabernero J, Taïeb J, Tejpar S, Wasan H, Yoshino T, Zaanan A, Arnold D (2016) Ann Oncol 27:1386–1422
Van Emburgh BO, Sartore-Bianchi A, Di Nicolantonio F, Siena S, Bardelli A (2014) Mol Oncol 8:1084–1094
Van Emburgh BO, Arena S, Siravegna G, Lazzari L, Crisafulli G, Corti G, Mussolin B, Baldi F, Buscarino M, Bartolini A, Valtorta E, Vidal J, Bellosillo B, Germano G, Pietrantonio F, Ponzetti A, Albanell J, Siena S, Sartore-Bianchi A, Di Nicolantonio F, Montagut C, Bardelli A (2016) Nat Commun 7:13665
Venook AP, Niedzwiecki D, Lenz H-J, Innocenti F, Mahoney MR, O’Neil BH, Shaw JE, Polite BN, Hochster HS, Atkins JN, Goldberg RM, Mayer RJ, Schilsky RL, Bertagnolli MM, Blanke CD, Alliance CLGB, SWOG, ECOG (2014) J Clin Oncol 32:5s
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW (2013) Science 339:1546–1558
Yonesaka K, Zejnullahu K, Okamoto I, Satoh T, Cappuzzo F, Souglakos J, Ercan D, Rogers A, Roncalli M, Takeda M, Fujisaka Y, Philips J, Shimizu T, Maenishi O, Cho Y, Sun J, Destro A, Taira K, Takeda K, Okabe T, Swanson J, Itoh H, Takada M, Lifshits E, Okuno K, Engelman JA, Shivdasani RA, Nishio K, Fukuoka M, Varella-Garcia M, Nakagawa K, Jänne PA (2011) Sci Transl Med 3:99ra86
Yuan Z-X, Wang X-Y, Qin Q-Y, Chen D-F, Zhong Q-H, Wang L, Wang J-P (2013) PLoS One 8:e65995
Zeng Z-S, Weiser MR, Kuntz E, Chen C-T, Khan SA, Forslund A, Nash GM, Gimbel M, Yamaguchi Y, Culliford AT, D’Alessio M, Barany F, Paty PB (2008) Cancer Lett 265:258–269
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Sartore-Bianchi, A., Siena, S. (2017). Plasticity of Resistance and Sensitivity to Anti-Epidermal Growth Factor Receptor Inhibitors in Metastatic Colorectal Cancer. In: Mandalà, M., Romano, E. (eds) Mechanisms of Drug Resistance in Cancer Therapy. Handbook of Experimental Pharmacology, vol 249. Springer, Cham. https://doi.org/10.1007/164_2017_19
Download citation
DOI: https://doi.org/10.1007/164_2017_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-10506-8
Online ISBN: 978-3-030-10507-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)