
Abstract
The theoretical prediction and characterization of the solid-state structure of organic semiconductors has tremendous potential for the discovery of new high performance materials. To date, the theoretical analysis mostly relied on the availability of crystal structures obtained through X-ray diffraction. However, the theoretical prediction of the crystal structures of organic semiconductor molecules remains a challenge. This review highlights some of the recent advances in the determination of structure–property relationships of the known organic semiconductor single-crystals and summarizes a few available studies on the prediction of the crystal structures of p-type organic semiconductors for transistor applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Charge transfer integral
- Charge transport
- Crystal structure
- Crystal structure prediction
- Mobility
- Organic field-effect transistors
- Organic semiconductors
1 Introduction
Organic compounds with π-electron systems show interesting functionality such as conductivity and semiconductivity [1]. Thanks to their extended π-conjugation, organic semiconductors (OSCs) have delocalized orbitals where charge carriers can move. If the charge carriers are produced extrinsically by injection from electrodes or by the photoelectric effect, electrons or holes can find pathways to go from molecule to molecule or over a conjugated backbone of a polymer (Fig. 1a, b) Thus OSCs have great potential to be used as components in electronics or in solar cells. Especially in inexpensive, flexible, and large area applications such as radio frequency ID tags, chemical/pressure sensors, display drivers, and solar cells [8–11], they have the potential to be an alternative to silicon-based semiconductors.

A notable advantage of OSCs is their design potential. The versatility of carbon provides a vast chemical space which can be explored with in silico strategies. For example, in the Harvard Clean Energy project database, there are 2.6 million theoretical candidate OSC compounds combinatorially designed from 30 molecular fragments as potential semiconductor materials for solar cell applications [12–14]. In this vast compound space, theoretical modeling and prediction have the potential to guide the synthesis of new OSCs thus reducing the high cost of finding materials by trial and error.
In recent years, increasing numbers of new OSCs have been designed and improved through computational modeling. For example, the modification of liquid crystal molecules (Fig. 1c) to enhance columnar organization and optimize intermolecular couplings for enhanced charge transport [4] or the design of polycarbazole derivative donor polymers (Fig. 1d) for organic photovoltaic applications by first-principles screening of the prototypical oligomers [5] proved useful. The synthesis of a new high performance molecular material [6] (Fig. 1e) or diketopyrrolopyrrole (DPP)-based polymers (Fig. 1f) with the help of computational modeling are demonstrated [7]. Despite the common limitations of current theoretical studies based on approximate methods such as density functional theory (DFT), these studies, where theory guided the synthesis of high performance materials, are promising. Now we are just opening a door into a new era where quantum chemistry methods are integrated into material science and engineering problems for more intelligent search of better OSC materials [12, 13, 15–19].
The successful prediction and computational design of high performance OSCs will be possible if three main objectives are achieved. First, a detailed understanding of the structure–property relationship for known OSCs is crucial. The main goal is to know what works, why it works, and how the material properties could be further optimized. Second is the ability to predict the solid-state structure from the molecular structure. This requires the generation of all possible crystal structures and computation of the lattice energies to rank and identify the lowest energy polymorphs. Third is the efficient computational simulation of charge transport to evaluate the crystal structures and identify the best semiconductors for particular applications. In this review we address the first and second objectives and briefly discuss the third.
With an ever-growing number of publications in OSC synthesis, design, and analysis [20, 21], there is now a substantial body of knowledge on the structure–property relationships for the OSCs in transistor applications. Undoubtedly, X-ray diffraction and analysis plays a crucial role in the elucidation of the structures and the derivation of the structure–property relationships experimentally [22]. In addition, theoretical characterization with molecular dynamics, quantum chemistry, especially density functional theory, and charge transport models, helped to derive important structure–property relationships in OSCs [16, 17, 23–25]. We will discuss some of the derived relationships in Sect. 2.
The crystal structure prediction for OSCs, on the other hand, remains mostly unexploited, not only because of the computational challenges but also, we believe, due to the lack of commercial applications that requires immediate attention to possible polymorphs and the structural factors affecting the device performance. One of the most important motivations for the crystal structure prediction for organic molecular solids is the polymorphism in solid forms of drug molecules [26]. For example, an unpredicted lower energy polymorph of the HIV drug Ritonavir [27], which manifested itself during the manufacturing and storage, not only was inactive as a drug but could also act as a seed to convert the active form into the inactive form on contact. In OSCs, interconversion between polymorphs may lead to loss of the desired electrical properties. Thus, we believe in the very near future, as more commercial applications of OSCs appear in the electronics market, the crystal structure prediction of OSCs will gain impetus.
There are various factors that complicate crystal structure prediction for OSCs. As shown in Fig. 1, most OSC molecules are large and flat with quasi-rigid conjugated backbones. Like other organic molecular crystals, they have many polymorphs with similar lattice energies. For example, there is less than 1 kcal/mol difference between C and H polymorphs of pentacene [28]. On one hand, it is advantageous to have a relatively simple molecular structure with higher symmetry that leads to mostly two-dimensional packing. This simplifies the search problem, that is the effective construction of likely packing configurations. On the other hand, phase transitions from one polymorph to another are more likely in these two-dimensional packing patterns. The energy barriers among different polymorphs are not as high as in organic crystals which pack in three-dimensional patterns, where the molecules are locked in place due to a lock and key kind of close-contacts. Therefore, in OSCs, as temperature fluctuates, transitions from one polymorph to another are more likely, sometimes yielding mobility differences as high as an order of magnitude as temperature increases [29]. In additon, solution processible materials usually have long alkyl chains as solubilizing groups [30], with many conformers, increasing the size of the search space dramatically.
With the exception of a few hydrogen bonding OSCs [31, 32], most OSC solids are held together by van der Waals forces, mostly London dispersion forces. These forces are quantum mechanical in nature and hard to capture in classical models such as molecular mechanics, which always treat the van der Waals forces as two body interactions. The transferability of the force field parameters is also an issue [33]. Due to the size of the crystalline systems, the conventional high-accuracy correlated wave function methods are too expensive. Therefore, density functional theory (DFT) methods are widely used. However, conventional DFT approximations cannot describe the attractive van der Waals forces (long-range dispersion) accurately [34]. For example, to rank the lattice energies of polymorphs of a relatively small molecular system such as para-diiodobenzene, DFT falls short, and computationally demanding methods such as diffusion quantum Monte Carlo is necessary [35]. Fortunately, the dispersion corrected DFT methods have now developed to such an extent that many body dispersion interactions can be included in the interaction potentials and can be used to calculate the lattice energies with chemical accuracy [36]. For example, many-body dispersion-corrected DFT has been shown to achieve chemical accuracy for the prediction of the sublimation enthalpies for a set of compounds ranging from pure hydrogen bonding molecules to only van der Waals bonded solids as well as compounds that uses both types of bonding in the solid state [36]. This is an exciting development yet it needs to be put to work for OSC crystal prediction. The practical use of DFT with dispersion to predict small molecular crystals has also been vetted in the last crystal structure blind test organized by Cambridge Structural Database [37].
Blind tests organized by Cambridge Structural Database provides an invalueable opportunity for the test of the predictive power of the state-of-the-art methodologies for crystal structure prediction. In the last blind tests, significant progress towards the prediction of crystal structures of molecular crystals was achieved [37, 38]. The most successful approaches included extensive initial structure searches to span all possible space groups, primary refinement of the initial structures by the tailor-made force fields [39], treatment of the internal degrees of freedom for the flexible molecules correctly [40], and refinement of the lattice energies with dispersion corrected DFT methods [41] or distributed multipole analysis approaches [42]. In the last blind test, from the perspective of the organic semiconductor prediction, especially important was the successful prediction of a compound with large internal flexibility. Two participating groups predicted the crystal structure of the largest (33 heavy atoms) and most flexible (8 rotatable bonds) molecule and reported the right polymorph as their top ranking choice. The positive implication of this success for the prediction of the crystal structures of OSCs is obvious as most of them are large and include solubilizing alkyl chains with many rotatable bonds.
Another challenge in the computational OSC design is the difficulty of testing the theories and structure predictions by comparison with experimental data. The main reason is the multitude of experimental factors affecting the solid-state structures and ensuing device performance. The nanoscale order, for example, is highly dependent on processing factors such as temperature, pressure, type of solvent, impurities, or substrate surface [15]. Small changes in these factors have the potential to affect the experimental outcome drastically. The inclusion of all of these factors into the computer simulations is an important unsolved challenge for the theoretical community. We should also note that theoretical studies usually focus on single crystals, leaving out the polycrystalline materials which unfortunately constitute most of the reported compounds in the literature.
The charge transport processes in OSCs is another hard problem to model. To achieve de novo modeling, one needs general models where different regimes of transport are addressed accurately and efficiently so that libraries of molecules and their solid-state structures can be analyzed to identify the most promising ones. However, in principle, the movement of charge carriers in OSCs requires a many-body quantum mechanical treatment [43]. Since an exact solution is not possible, a number of approximate approaches are adapted such as the density matrix propagation, master equation approaches, dynamical mean-field theory, and so forth. The studies showed that the coupling of the motion of the charge carrier to the molecular and crystal degrees of freedom is crucial to describe the problem accurately and a self-consistent solution of the whole Hamiltonian is necessary [25]. The modeling of charge transport in OSCs is still a developing field and the structural parameters obtained from ab initio quantum chemistry are playing an important role in the development of more realistic models [23–25, 44, 45]. In addition, a comprehensive multi-scale approach also needs to address issues emerging from organic field-effect transistor (OFET) device configurations such as the contact resistance, and short and long-range effects of the gate dielectric, among others.
Thus, in this rapidly advancing field of OSC design, the theoretical characterization and prediction has tremendous potential for growth. Successful routine prediction and screening approaches may make themselves more useful in the future. Before moving on to the theoretical characterization and prediction techniques, in the rest of the introduction we briefly summarize the working principles of an OFET and the important structural parameters of charge transport for an ideal OSC for transistor applications.
1.1 The Organic Field-Effect Transistor
The transistor is the most important single element in an electronic circuit. For organic electronics to find viable commercial applications, a fast, reliable, and inexpensive OFET is crucial [46]. We have come a long way since the first organic transistors in the 1980s [3, 47]. Especially in the last 10 years there has been a great progress in the discovery of new OSCs and the optimization of the process conditions to achieve commercial viability [3, 21]. Today, some organic semiconductor materials surpass amorphous silicon in performance [48] and there are commercial applications of organic transistors in printed electronics [49], displays, and microelectronics [9].
An OFET is a voltage-controlled switch where an external voltage applied between the electrodes creates a current through an OSC layer. Then the flow of current is modulated by a gate voltage applied through the dielectric substrate [50, 51]. In a typical bottom-contact p-type OFET, an OSC crystal or a thin film is placed over a dielectric substrate (usually SiO2) in contact with the source and the drain electrodes (Fig. 2). The negative bias gate voltage applied through the dielectric induces charge carriers (holes), creating a channel between the source and the drain electrodes. Current flows through the channel when a drain bias is applied.

Schematic of an OFET structure (left) and example of herringbone-type molecular packing in the OSC (right)
In p-type materials the holes are the charge carriers. Therefore charge injection into the highest occupied molecular orbital (HOMO) of the OSC requires that the work function of the metal electrodes should match the HOMO energy level of the OSC. The source and drain electrodes are usually chosen from low work function noble metals such as gold (Au work function: 5.1 eV). The dielectric layer separating the gate from the semiconductor could be a Si/SiO2 layer preferentially treated with a hyrophobic self-assembled monolayer of alkylsilanes to promote molecular order and enhance transport [52].
The most important parameters defining the transistor performance are the mobility and drain-source current ratio when the gate voltage switches on and off, I on/off [50]. The mobility characterizes the mean drift velocity of the charge carrier when an electric field is applied and it is derived from the current–voltage curves of the transistor. The higher the mobility and I on/off, the better the transistor performance. For example, in a liquid display, I on/off of 106 and a minimum mobility of 0.1 cm2 V−1 s−1 are needed for good performance [50]. Another important factor which affects the performance is the operating voltage of the transistor. Traditionally, the operating voltages in OFETs could be as high as 100 V. Low operating voltages is a requirement for practical applications and has been demonstrated by the use of high capacitance dielectric layers [53, 54].
OFET devices are very complex since many factors affect their performance, such as the grain size and boundaries, substrate, substrate temperature, or contact resistance. In this review we only focus on the molecular and crystal structural factors affecting the charge transport ability of the OSC layer. Specifically, we consider the single-crystal layers because they provide a more consistent platform to study the structural effects [55].
Since charge carrier transport depends strongly on the π-orbital interactions, the molecular level ordering has important consequences for OSCs. Figure 3 schematically illustrates the correlation of the mobility to molecular level ordering for a set of materials from perfect single-crystals to disordered polymers. It is established now that usually the better the molecular organization of an OSC, the higher the material performance. Surpassing amorphous silicon, the single crystalline OSCs have mobilities in the range of 0.1–40 cm2 V−1 s−1 whereas in polycrystalline and disordered films the mobilities fall down to the range of 0.0001–0.1 cm2 V−1 s−1 [46]. In polycrystalline films the grain boundaries become traps for charge carriers, thus reducing the performance considerably. The anisotropy in the mobilities due to the low symmetry of the molecules compared to inorganic semiconductors, for example, also underlines the importance of the packing patterns and microscopic order in OSCs.

OFET mobility correlates with structural order. As the order of the material decreases, in a progression from single molecular crystals to disordered polymers, carrier transport decreases by orders of magnitude
Despite the success of crystalline small molecule OFETs, the commercialization of these devices has been limited due to their low solubility. Usually, to make single-crystal OFETs, micron-to-millimeter-size crystals are obtained by vacuum deposition and later handpicked to be aligned along the appropriate direction between the electrodes. This is not a yet practical option for commercialization. The large-scale semiconductor coating production similar to printing requires solution processable materials. To achieve solubility, molecular units are usually modified through the addition of aliphatic groups such as long alkyl chains. Although these additions/modifications sometimes increase the performance due the close-packing of the conjugated backbones enforced by the lipophilic interactions of the alkyl chains [31], the grain boundaries reduce the mobility to the range of 0.01–1 cm2 V−1 s−1.
Recently, however, there has been important progress regarding the development of solution-processable and high-performance OSCs. For example, with controlled deposition of tips-pentacene films (Fig. 4a) with solution-shearing through nanoarray surfaces, a mobility of 11 cm2 V−1 s−1 has been demonstrated by Diao et al. [59]. Also in microsheets and microribbons of dithieno[3,2-b:2′,3′-d]thiophene derivatives (Fig. 4b) a mobility of 10 cm2 V−1 s−1 and above is observed [57]. Moreover, a record average high mobility of 16.4 cm2 V−1 s−1 was observed in the single crystals of C8-BTBT (Fig. 4c) deposited in an ink-jet printer [156].

Semiconducting polymers, on the other hand, naturally have the advantages of solution processability, thermal stability, and film uniformity [60]. The polymers usually form semi-crystalline films with lower mobilities as seen in Fig. 3. Compared to the first polymer-based OFET (polythiophene, Fig. 1b) fabricated in 1986 [3], the progress has been remarkable. For example, DPP-based polymers (Fig. 1f) with a record high mobility of 10 cm2 V−1 s−1 were reported recently [61]. Such high mobility for a semicrystalline polymer is very unusual due to the presence of the disorder in these systems. Recent investigations revealed that a certain degree of polymerization is necessary for the high mobility since the polymeric chains connecting crystalline domains can act as bridges for charge transport [62]. Since the structure prediction for polymers is outside the scope of this review, we will not discuss polymer-based OFETs any further. However, we would like to underline the notion that once a good molecular or polymeric material is discovered, through the optimization of the processing conditions it is possible to engineer high-performance materials. This might entail the enhancement of the intermolecular orbital interactions or the reduction of the impurities or other structural traps.
Despite the remarkable progress made in the synthesis and process engineering in OFETs [15], up until now theoretical characterization and prediction has relied on the availability of crystal structures from X-ray diffraction analysis [22]. Once the structure of an OSC is known, the structural parameters affecting the performance are examined through theoretical characterization techniques. The structure–property relationships learned from the study of known OSCs provide a framework upon which new novel materials discovery routes rely. We dedicate the rest of this section to the important parameters in OFET modeling and Sect. 2 to the structure–property relationships.
1.2 Charge Transport Models and Parameters
In contrast to the success of charge transport models for inorganic semiconductors (ISC), models for OSCs are still under development. The major reason for this lack of a comprehensive model is the difficulty of dealing with molecular behavior in the solid state. Unlike in an ISC, the OSC building blocks have high polarizability, low internal symmetry, and weak intermolecular interactions. In addition, the diversity of materials from molecules to polymers and to liquid crystals poses challenges for the development of generalized and consistent models.
To summarize the basic understanding of the charge transport mechanisms and underlying structural factors, it is useful to start with a Hamiltonian for a single particle moving on a periodic lattice. The following Hamiltonian [63] includes all the important energy contributions describing the motion of a charge carrier over regular lattice sites, i and j:
Here, H 0el is the electronic Hamiltonian, a + and a are the creation and annihilation operators, i and j label the molecular sites with energy ϵ, and J is the charge transfer integral which represents the strength of electronic coupling among neighboring sites. H 0ph represents the phonon contributions with frequency ω where q and is the wavevector. The operators b + q and b q denote the creation and annihilation operators for the phonons with energy ℏω q . As can be seen in (1), the electron–phonon coupling term has local (g ii ) and non-local (g ij ) contributions. Within this formulation, the local electron–phonon coupling term modulates the site energies whereas the non-local coupling modulates the site-to-site interaction terms J ij . As a first-order approximation, the local and the non-local electron–phonon coupling terms could be obtained by Taylor expansion of the electronic Hamiltonian along the phonon modes [64].
Depending on the relative strength of the terms of this Hamiltonian, the models could be roughly categorized into band, polaron, and disorder models [63]. In the band transport models, J is the most important term. Due to strong coupling among the sites, the bandwidth is relatively large, and thus a mobility value above 10 cm2 V−1 s−1 is possible. However, in OSCs, since the molecular interactions are weak, injection of a charge polarizes the molecule and its surroundings significantly, leading to formation of a polaron. The polaron is a quasi-particle representing the charge and the polarized lattice around it [43]. Thus in these models the electron–phonon coupling terms play a crucial role and determine the transport characteristics and the temperature dependence in the mobility. In the case of disorder-based models, the fluctuations of the site energies and the modulation of the coupling terms are so large that the charge transfer can be described as a series of uncorrelated hops in a broad density of states [63].
Temperature plays a crucial role in the charge transport in OSCs. It has been demonstrated for pentacene (Fig. 1a) [65] and rubrene (Fig. 4d) [58], for example, that, at low temperatures up to 280 K, the mobility shows an inverse dependence on temperature (T −n) which is the evidence of band transport. The mobility decreases with increasing temperature as more and more scattering of the charge carriers happens due to the phonon modes of the medium. Somewhere around room temperature (280–300 K), the charges carriers localize to the molecular sites due to thermal fluctuations and the transition to activated hopping occurs so that the mobility increases as a function of increasing temperature [66].
Thus, naturally, the choice of the most appropriate model depends on the knowledge of the molecular and solid-state structure. Once the structure is known, ab initio molecular [25, 67] or solid-state electronic structure parameters [68] can be used to make more realistic models based on multi-scale QM/MM or semiclassical schemes [23, 24, 44, 69]. In the following sections we briefly discuss these important parameters for the charge transport models.
1.2.1 Electronic Coupling, J
The electronic coupling term, J, is one of the important structural parameters determining the charge transport properties of an OCS. In the literature, another more common name for the electronic coupling term is the charge transfer integral, or transfer integral.
For p-type OSCs, in a simple dimer approach, the transfer integral can be calculated as the electronic coupling of the HOMO orbitals of adjacent molecules as J = 〈ψ HOMO A |Hel|ψ HOMO B 〉, where the A and B indexes represent the adjacent molecules in a relative geometry extracted from the crystal structure. For symmetrically equivalent molecules, it is simply the splitting of the HOMO molecular orbitals in the dimer configuration. In the case of geometrically non-equivalent molecules, however, the effect of the site energy differences should be taken into account [70–72]. This dimer-based approximation neglects the effect of the crystal environment on the transfer integrals. It is also possible to calculate the transfer integrals from fitting the dispersion of the bands into a tight-binding Hamiltonian [44, 73, 74]. This approach includes the crystal environment effects within the level of the theory used. Experimentally, the transfer integral values can be obtained from the dispersion of the HOMO bands determined by photoemission spectroscopy [68].
In the calculation of transfer integrals, DFT-based methods are usually employed as they provide a good enough accuracy with a reasonable cost. For a medium size molecule such as pentacene, hybrid functionals with double-zeta basis with polarization functions are usually used and shown to provide qualitatively accurate results [75]. For larger molecules, long-range corrected DFT methods provide better accuracy [76]. It has also been shown within the dimer-based approach that the change in the transfer integrals associated with the applied electric field is negligible [77].
The electronic coupling terms show the following characteristic features:
-
1.
Exponentially decreasing as a function of increasing intermolecular distance. The smaller the intermolecular stacking distances, the stronger the molecular orbital couplings for configurations for which the transfer integral is not zero.
-
2.
Oscillatory behavior as a function of molecular displacements in the direction perpendicular to the interaction of π-orbitals. The oscillation as a function of the slip along the long axis is called the D-modulation.
-
3.
Dihedral angle dependence [78].
-
4.
Anisotropy [79].
As can be seen in Fig. 5a–c, the transfer integral strength strongly depends on the dimer geometry of the interacting molecules [25, 64]. The dependence on the π-stacking distance is exponential (Fig. 5a) [25], and thus it is crucial to be able to control it. Slip-stacked geometries as a function of short and long axes show oscillations. Kojima and Mori analyzed the dihedral angle dependence of transfer integrals for a group of molecular OSCs with typical herringbone packing [78]. As expected, all these molecules show the characteristic oscillation behavior as a function of dihedral angle and D-modulation. They observed that the transfer integral strength is strongly dependent on the molecular orbital symmetry as well as the crystal structure packing. Due to the oscillatory nature of the transfer integrals as a function of geometrical orientation of the molecules, achieving charge transport through the optimization of the couplings is challenging.

(a, b) The dependence of the transfer integral on the intermolecular π-stacking distance in tetracene [25], D-modulation in tetracene [25]. (Reprinted with permission from [25]. Copyright (2007) American Chemical Society). (c) Dihedral angle dependence in pentacene [78]. (Reprinted with permission from [78]. Copyright (2011) The Chemical Society of Japan). (d) Anisotropy of the mobility of pentacene in the transport plane (filled squares: maximum mobility, empty squares: −10 V gate voltage). (Reprinted with permission from [79]. Copyright (2006) AIP Publishing LLC)
The strength and the extent of J determine (an)isotropy of the mobility and the bandwidth. As shown in Fig. 5d for pentacene, moderate to strong anisotropy of mobility tensor is a common feature of the transport plane. The bandwidth is also strongly dependent on the transfer integral values. For example, within the tight-binding approximation, the bandwidth for a one-dimensional network of coupled sites is defined as 4J [80, 81]. For a two-dimensional OSC, direct relationships between the transfer integrals and the bandwidth are established [82, 83]. The percolation network’s robustness, fragility, and dimensionality depend on the strength of the transfer integrals between the molecules in each direction. Depending on the packing patterns and the ensuing π-orbital overlap, one-, two-, or sometimes even three-dimensional conductance is possible. For example, Vehoff et al. analyzed the topological connectivity of four different single crystals, namely rubrene, benzo[1,2-b:4,5-b′]bis[b]benzothiophene derivatives with and without C4H9 side chains and indolo[2,3-b]carbazole with CH3 side chains [84] (see Fig. 6). The transfer integrals for the adjacent molecules revealed the dimensionality of the percolation networks as illustrated in Fig. 6.

The percolation networks and the dimensionality of the charge transport determined from the transfer integral calculations for the molecules shown. The centers are the center of mass of the each molecular site. The connectivity and colors refer to the strength and type of the transfer integrals, as indicated by the colored arrows. (Reprinted (adapted) with permission from [84]. Copyright (2010) American Chemical Society)
These observations pertaining to the nature of the transfer integral only include the geometries of the equilibrium structures. At finite temperature, since these materials are weakly bound van der Waals solids, the thermal motion of the molecules significantly affect (modulate) the transfer integrals. Instead of a single equilibrium value, a probability distribution of transfer integral values appears. The shape of the distribution strongly depends on the temperature. This modulation could naturally be described as the coupling of phonon modes of the crystal to the electronic degrees of freedom. There is indeed a great deal of work in the literature dedicated to this subject and we briefly discuss some important aspects of it in the next subsection.
1.2.2 Electron–Phonon Coupling
Following (1), the electron–phonon coupling can be described as a sum of local and non-local electron–phonon coupling terms. This division comes naturally in OSCs as the intermolecular vibrational forces are much weaker compared to the intramolecular ones.
The electron–phonon coupling is usually calculated within the harmonic approximation. A charge carrier over a site changes the potential energy surface for the site such that rearrangement of the nuclei now have a different energy shifted from the original point of the potential well by an amount of g 2 jj ℏω q [23]. This shift in energy is called the polaron binding energy in polaron transport models [25] and the reorganization energy in the terminology of the Marcus electron-transfer theory [85].
The intramolecular portion of the electron–phonon coupling is calculated approximately from the gas-phase geometries of the neutral and charged molecules by measuring the change in the total energy upon charging [25, 86]. This is commonly called the internal reorganization energy. This approach neglects the coupling of the intramolecular modes to the polarization of the neighbour molecules in the crystal environment. The aromatic conjugated molecules are highly polarizable; hence the lattice will distort to accommodate the charges [43]. Nevertheless, the external reorganization energy contributions are found to be small [87, 88], providing some justification for their omission. In addition, the experimentally determined reorganization energy values for rubrene, pentacene, and perfluropentacene are comparable with this local coupling picture [89, 90].
The calculation of the reorganization energy for molecules with rotatable bonds is more complicated than the fused aromatic ones. In molecules with rotatable bonds, geometry of a charged state could be very different from the neutral one. Then the simple harmonic approximation fails to capture the dependency of the total reorganization energy on the anharmonic vibrational modes that are related to the torsional motion of the backbone [91]. Moreover, there is a chance that the gas phase calculations overestimate the reorganization energy of OSCs with rotatable bonds. Although in gas phase calculations the torsional degree of freedom is controlled only by the intramolecular interactions, in the solid state the torsional motion can be hindered due to intermolecular van der Waals interactions [25]. To compensate, sometimes the symmetry constraints of the molecule in the crystal environment are imposed for the gas phase reorganization energy calculations. However, there is no guarantee that this approach will provide an accurate picture in all cases.
The non-local electron–phonon coupling constant, g ij , is more challenging to calculate as it involves the modification of the electronic couplings due to phonon modes of the crystal. Usually, finite temperature dynamics of the lattice is obtained from classical trajectories and the effect of phonon modes of the crystal on the electronic couplings is observed a posteriori by extracting unique dimers from an MD trajectory. Subsequently, the transfer integral calculations based on the dimer approach mentioned earlier are performed along the trajectory [92–95]. This approach provides insight into the thermal modulation of the transfer integrals by providing a probability distribution. How large is the standard derivation compared to the mean value and how strongly does it depends on the temperature? These questions can be answered by the use of this semiclassical approach. For example, for pentacene single-crystals at 300 K, the transfer integrals for three unique dimers have been studied with data from MD trajectories. The standard deviations were found to be of the same order of magnitude as the average transfer integrals [94].
The nonlocal couplings can also be studied by the calculation of the numerical derivatives of the transfer integrals with respect to the distortions of the crystal lattice [96, 97]. These couplings represent the zero Kelvin behaviour of the system. Subsequently, they can be extrapolated to higher temperatures with the use of classical or quantum statistics. With this approach, for pentacene slightly smaller standard deviations of the couplings were observed. However, they were still of the same order of magnitude as that of the mean values. Thus in the larger oligoacenes such as pentacene, it is safe to assume that the non-local electron–phonon coupling are significant and semiclassical models of charge transfer could be of use. We should note, however, that this type of analysis should be extended to other molecular systems, as different behaviour could be observed for systems with comparatively stronger or weaker intermolecular interactions.
2 Structure–Property Relationships
The structure–property relationships in OSC design can be characterized at the molecular and crystal scales. As mentioned previously, a molecular OSC is usually made of molecules which are mostly interacting by van der Waals forces [1]. Thus the formation of an organic solid preserves the molecular properties to a large extent with some perturbation due to the intermolecular interactions. Since the molecule in the solid state has many important characteristics resembling its gas phase properties, the choice of the right molecule is crucial. The molecular properties such as planarity, rigidity, conjugation length, size, symmetry, side groups, and their positions, together with chemical structure determine the electronic structure parameters as well as the packing in the solid state.
On the other hand, although the molecular properties are preserved to a certain degree in a molecular crystal, many new properties emerge. The experimental evidence shows that electronic, optical, or transport characteristics depend highly on the molecular packing [43], such as Davydov splitting [98], line broadening, and bathochromic shifts [99]. From the perspective of the OFET application, the most important feature is the semiconductivity. Since the interactions among the molecules are weak and low internal symmetry of the molecules dictates that only certain packing patterns can have π-orbital interactions, subtle changes in the packing patterns results in big differences in the semiconductance of a material.
2.1 Molecular Structure–Property Relationships
For a successful p-type OSC, the energy and shape of the HOMO is important. First of all, the ionization energy (IE) determines ambient stability and charge carrier polarity of the material, whether it be p-type or n-type.
Before we move on to the discussion of the structure–property relations at molecular scale, we should note here one technical issue, which is how to compare experimental and computational values of the electronic structure of the materials. Although the important quantity for material characteristics is the IE, usually the HOMO energy levels from the quantum chemical calculations are compared to the experimental IEs for practical reasons. As can be seen in Fig. 7, the HOMO energy levels closely follow the IEs from the photoelectron spectrum. In reality the difference between the gas and solid-state IEs is large and depends on the packing and, more precisely, on the nature of the π-orbital interactions. For example, in pentacene the gas phase IE is 6.6 eV whereas in thin films it is around 5 eV depending on the packing patterns; 4.8 eV if the molecules are standing up and 5.35 eV if they are lying over a substrate [101]. From the computational cost perspective, gas phase IE calculations are now routine, but IE calculations for the solid state are much more complicated. Therefore, the HOMO energy levels are usually used instead of the IE values for practical reasons. Our discussion will also follow the same approach.

The frontier orbitals of BTBT, DNTT, and DATT (a) and their photoelectron spectrum in air (b). (Reprinted with permission from [100]. Copyright (2011) Wiley-VCH, Weinheim)
For p-type OSCs, the HOMO level is usually around 5 eV, and thus very close to the oxidation threshold of 5.1 eV. The more extended the conjugation, the higher the HOMO level (see Fig. 7). Thus extending the conjugation without making the material unstable in ambient conditions is a challenge and an active area of research. For example, the introduction of a thienothiophene group in the middle of an acene (Fig. 5) has been shown to help stabilize a longer molecule [102], or the substitutions with electron-withdrawing groups usually leads to deeper HOMO levels [103].
The shape of the HOMO is another important electronic property that affects the performance [100]. If the nodal planes of the HOMO lie on the atoms such as sulfur atoms, which are important for intermolecular interactions, the strength of the intermolecular interactions and thus the electronic coupling are reduced. For example, in molecules with thiophenes, sulfur–sulfur interactions are very important in the crystal and maintaining the short contacts among these atoms is important for stronger couplings. Figure 8 shows the shape of the HOMO level of molecules labeled as DBTDT and BTBT. DBTDT has nodal planes over the sulfur atoms whereas BTBT does not. As suggested by Takimiya and coworkers, DBTDT has larger S⋯S distances as well as much weaker transfer integral values: 11 and 17 meV [100] compared to transfer integral values of 67 and 26 meV in BTBT-C8 [104].

The HOMO orbitals of DBTDT (left) and DNTT (right). (Reprinted with permission from [100]. Copyright (2011) Wiley-VCH, Weinheim)
Together with the chemical structure and availability of the sp 2 hybridized atoms, the size and rigidity of the molecule determines the extent of the conjugation. This, in turn, affects several parameters related to charge transport. One important consequence of the size of the molecule is the strength of the local electron–phonon coupling. Let us assume that a single charge carrier residing on a molecular site with a rigid conjugated backbone can simply be modeled as a particle in a box. Then we can consider that the reorganization energy required to accommodate the charge carrier scales with the length of the box. Usually, the more extended the conjugation, the smaller the reorganization energy and the better the charge transport properties. However, this is only true for a set of similar compounds. In the following examples, we explain what we mean by similar.
Since hexacene has recently been synthesized and its OFET mobility has been measured [105], the oligoacene family of compounds provide a good set to illustrate some of the structure–property relationships discussed above. The charge transport parameters of the members of the oligoacene family with n = 2–6 phenyl rings are presented in Table 1. The transfer integral values listed correspond to the unique dimers shown in Fig. 9b–c.

(a) ORTEP drawing of two adjacent hexacene molecules. (b) Arrangement of hexacene molecules in the ab-plane. (c) Arrays of hexacene along a-axis. The labels T, P, L denote the pairs of molecules used in the transfer integral calculations. (Reprinted (adapted) with permission from [105]. Copyright (2012) Nature Publishing Group)
The trends observed for the reorganization energy, transfer integrals, and ensuing mobilities in oligoacenes confirm the discussion above. First, the more extended the conjugation, the higher the HOMO energy level and the smaller the reorganization energy. Second, although the transfer integral for the parallel arrangement, P, does not improve as the intermolecular distance also gets larger, the edge to face interaction terms, T 1 and T 2, improve going from n = 2 to n = 6. Together with the smaller reorganization energies for the larger oligoacenes, the estimated mobility values improve as well. Similar behavior was also observed for oligothiophenes: the longer the oligomers, the smaller the calculated reorganization energy [106]. Experimental evidence also shows that the longer oligothiophenes, four to six rings, have higher mobilities compared to shorter ones [107]. Usually four or more rings are required in an OSC molecule for good performance.
For structurally different families of compounds, such as heteroacenes, the reorganization energy trends need to be carefully analyzed and molecular similarity needs to be taken into account. For example, the thienoacenes usually have higher reorganization energies compared to oligoacenes (see Fig. 10). However, the polyphenyls have quite large reorganization energy values compared to acenes, even higher than polythiophenes. The trends can depend on structural factors such as the type of bonds, the availability of the torsional angles, or the spatial arrangement of the aromatic rings. Following the classification suggested by Takimiya and coworkers [100], in Figs. 10 and 11 we show that if the structural similarities are taken into account and the acene- and thiophene-based OSC molecules are grouped accordingly, the extended conjugation lowers the reorganization energy. However, the slopes of the curves representing the change in the reorganization energy as a function of the number of rings could be very different for each group of compounds.

The reorganization energies of oligoacene and thienoacene families of homocycles. References for the reorganization energy values: oligoacenes (diamonds) [105], polythiophenes (circles) [106], fused-thiophenes (triangles) [25], polyphenyls (squares). (Atahan-Evrenk and Aspuru-Guzik (2012), unpublished results)

The reorganization energies of thienoacene heterocycles: phenyl substituted dithienothiophenes (diamonds) (Atahan-Evrenk and Aspuru-Guzik (2012). Unpublished results), phenyl substituted dithiophenes (down triangles) (Atahan-Evrenk and Aspuru-Guzik (2012), unpublished results), diacene-fused thienothiophenes (circles) ([6], Atahan-Evrenk and Aspuru-Guzik (2012), unpublished results), benzene-thiophene alternating molecules (pentagons) ([25], Atahan-Evrenk and Aspuru-Guzik (2012), unpublished results), acene-anti-dithiophenes (squares) ([25], Atahan-Evrenk and Aspuru-Guzik (2012), unpublished results), acene-syn-dithiophenes (triangles) ([25], Atahan-Evrenk and Aspuru-Guzik (2012), unpublished results)
One avenue through which to pursue a priori prediction of the high performance OSC molecules is the use of quantum chemical descriptors in quantitative structure–property (QSPR) studies. By combining molecular descriptors from cheminformatics with ab initio quantum chemistry, Misra et al. showed that it is possible to accelerate the search for smaller reorganization energy materials [108]. In a study of a family of 200 polycyclic aromatic hydrocarbons, by assuming that all of the molecules form similar liquid crystalline stacks, the reorganization energy was identified as the dominant factor influencing the charge transfer rates. Based on a QSPR study of various molecular descriptors, a weak correlation of the reorganization energy with the molecular signature (a canonical representation of the atom’s environment up to a preferred height [109]) and another weak correlation with the electronic eigenvalue descriptors were identified. Then these two weakly correlated descriptors were combined into a QSPR model that estimated 80% of the reorganization energies within an error margin of 20 meV. This success rate was quite good as the only quantum chemistry calculation involved was the ground state geometry optimization of the neutral molecular structure.
Due to the benefits of low reorganization energy in the charge transport, a few strategies based on structural modifications are developed. For example, the presence of the nonbonding character in the HOMO level is identified as a reorganization energy lowering strategy. A comparison of tetraceno[2,3-c]thiophene (TcTH) and tetraceno[2,3-b]thiophene (TbTH) showed that the fusing of thiophene in a symmetric manner results in 30 meV smaller reorganization energy in TcTH (Fig. 12a) compared to TbTH (Fig. 12b) [110, 111]. Based on the same principle, the cyanide substitutions, and substitutions by weaker electron-withdrawing groups (for example, chlorination instead of fluorination) has been identified as a reorganization energy lowering strategy [112–115]. In addition, intra-ring substitutions such as the replacement of a carbon atom with nitrogen were also shown to lower the reorganization energy.

Symmetric substitution leads to lower reorganization energy, 97 meV of TbTH compared to 66 meV in TcTH [110]
Last but not the least, choosing the right molecular structure has significant consequences for solid-state packing. The molecular group symmetry, polarizability, side group positions, the intermolecular interactions, etc., play a role in determining the stable lattice conformations, thus leading to enhanced or reduced mobility. For example, relative abundance of C–H (through the peripheral H-atoms) vs C–C interactions affect the tendency to pack in herringbone or π-stacked packing forms [116]. The molecules with H-bonding groups such as quinacridones can form supramolecular synthons [32, 117]. The symmetry of the molecular structures also affects the packing in the solid state. For example, molecules with centrosymmetry can form better short-contact networks [100]. In the following section we further discuss the effect of the modification of the molecular units to achieve high performance and the crystal structure–property relationships.
2.2 Crystal Structure–Property Relationships
Among the crystal structure–property relationship studies in OSCs, perhaps the most striking example is the case of rubrene (Fig. 4d). With a record single crystal OFET mobility of 20 cm2 V−1 s−1 [58], rubrene was a remarkable improvement from tetracene. Although the phenyl groups added to tetracene backbone do not participate in the π-conjugation, they help the tetracene backbone to π-stack right very close to one of the maxima of the transfer integral surface [118]. The phenyl groups also provide a lock-in for the backbone so that displacement along the short axis, which usually yields lower transfer integrals in oligoacenes, is prevented. Understanding of the rubrene crystal structure provided motivation for further studies to control the solid-state packing to enhance performance.
To study further the structure–property relationships in rubrene and its derivatives, the effect of the substitutions of the external phenyl rings on the crystal structure was examined [119, 120]. Haas et al. showed that, depending on the positions of the substitutions, the π-stacking geometries and the interlayer distances between the backbones could be controlled. In particular, they have found that the substitutions on the 5,11 phenyls cause a large twist in the backbone leading to a polymorph with no charge carrier mobility, whereas the tert-butyl substitutions on the 5,12 phenyls showed a similar packing pattern to rubrene with similar OFET characteristics despite a 31% increase in the interlayer spacing. In a recent study, McGarry et al. [120] showed that the interlayer distance could also be controlled by the methyl and trifluoromethyl substitutions of the external phenyl rings. By means of this approach, rubrene derivatives with OFET mobility comparable to rubrene were synthesized. In particular, they had a success with dual substitutions by trifluoromethyls that resulted in ambipolar function as well as a high hole mobility.
Another successful example of charge transport tuning with structural modification is the family of substituted pentacenes [121]. Among the peri-functionalized pentacenes, tips-pentacene (Fig. 4a) with triisopropylsilylethynyl groups at the 6,13 positions emerged as one of the most successful engineering examples [122]. Along with solution processability and ambient stability, the substitutions changed the herringbone type of packing into π-stacking conformation. Subsequently by the solution shearing method a metastable state of tips-pentacene with better performance has been achieved with an order of magnitude increase in the mobility [123]. The theoretical investigation of the transfer integrals corroborated the experimental findings and showed that the solution-sheared films indeed have three times stronger transfer integrals [123]. Recently Bao and co-workers showed even higher mobility of 11 cm2 V−1 s−1 for a solution processed film of tips-pentacene, demonstrating the fact that the control of the solution deposition is crucial for high performance materials [59]. Tips-pentacene and rubrene are among the few OSCs which show band-like transport in an OFET setup.
Another crystal packing control strategy is the engineering of sulfur–sulfur interactions. For example, the chalcogen substitutions at the peri-positions of pentacene promotes the π-stacking interactions [124]. In comparison to the herringbone packing in pentacene, hexathiapentacene has strong π-stacking and unusually short intermolecular sulfur–sulfur distances. These substitutions usually lead to one-dimensional conduction and thus this strategy has the potential to develop semiconducting nanowires. Similarly for benzothiadiazole-tetrathiafulvalene single-crystal nanowires (see Fig. 13), the use of tetrathiafulvalene groups with strong S⋯S interactions and thiadiazole groups promotes the π-stacking interactions, and the intermolecular stacking distances could be controlled and short-contacts such as S⋯S (3.41 Å), S⋯C (3.49 Å), and S⋯N (3.05 Å) could be achieved [125]. Further discussion about additional strategies for inducing π-stacking could be found in [126].

Benzothiadiazole-tetrathiafulvalene with π-stacking interactions for single-crystal nanowire applications (a), short contacts (b). (Reprinted (adapted) with permission from [125]. Copyright (2013) American Chemical Society)
Although one-dimensional transport could be advantageous for nanowire applications, reducing the dimensionality of the mobility has the potential to make the charge transport very susceptible to the presence of defects [127]. A higher dimensional transport network is desirable for OFET applications as it provides alternative pathways for transport. In recent work, for example, the methylated DNTT derivatives are synthesized and significant transfer integrals in all three dimensions is demonstrated [128].
The engineering of the crystal structures through bulky substitutions has also been studied in the case of oligothiophenes with trimethylsilane end groups [129]. It has been shown that through end-substitutions the in plane tilt and the offset of the oligothiophene backbones can be controlled and better or worse electronic couplings among the molecules can be enforced.
Another example of the substitutional engineering of the crystal structure is the end-substituted 5,5′bis(4-alkylphenyl)-2,2′-bithiophenes (P2TPs) [130]. Depending on the even and odd alkyl chain substitutions, the tilt angle and electronic couplings in the two dimensional deposition layers of the SCs can be controlled. The solid-state packing of P2TPs molecules with different length alkyl side chains (n = 3,8) were analyzed with grazing incidence X-ray diffraction. The data showed that having an even or an odd length chain controls the tilt angles of P2TP molecules on the self-assembled monolayers of ODTS. The even length alkyl chains yielded larger tilt angles and higher mobility compared to those with an odd number of carbon atoms. The transfer integrals for the unique dimers extracted from the corresponding crystal structures also corroborated the odd-even trend. In addition, MD simulation of the surfaces showed that the tilt angle is indeed controlled by the parity of the side chains.
In addition to the position and the length of the side groups, stereoisomerism affects the FET mobility [131]. For example, due to RS arrangement of the chiral carbons in DPP(TBFu)2 (labeled 1 and 2 in Fig. 14), the mesomer has been shown to have better π-orbital stacking interaction as well as higher mobility compared to the case of SS or RR isomers. The mesomeric form had a packing distance of 3.38 Å with a flat conjugated backbone compared to 3.47 Å in the case of SS or RR isomers. The mesomeric form also performed better than the case where no stereoisomer was selected (as-synthesized). Therefore, this study highlighted that chirality of the substituted groups also needs to taken into consideration for tuning of properties with structural modifications.

The chemical structure of DPP(TBFu)2 with two chiral carbons highlighted (1 and 2) and the resulting stereoisomers (a), elution profiles for the as-synthesized and isolated stereoisomers (b). (Copyright reference [131])
Another important concept in understanding the crystal structure–property relationships is the polymorphism in OSCs. Like in drug molecules, where polymorphism has direct consequences for the solubility and thus bioavailability of a drug, in OSCs polymorphism has important consequences for the charge transport properties. The polymorphs of an OSC can have totally different mobility tensors. For example, Fig. 15 illustrates the possible variations in the (an)isotropy in the mobility tensor for four polymorphs of pentacene [132]. Among four different polymorphs investigated, apart from one (polymorph IV), all of them showed remarkable anisotropy in the transport plane. This might have important implications for the device configurations.

The mobility tensors in the ab-plane for four polymorphs of pentacene. (Reprinted (adapted) with permission from [132]. Copyright (2005) American Chemical Society)
It is also possible that several polymorphs of an OSC with different charge transport characteristics could be accessible in operating temperatures. It has been shown, for example, that, for a fluorinated derivative of an anthradithiophene, a phase transition between two polymorphs occurs as the temperature rises from 260 K to 300 K [29]. For this system, the field-effect mobility of the films increases as a function of increasing temperature with particular slopes for each polymorph. This finding has immense significance for the consequences of polymorphism in OSCs in commercial applications.
Lastly, we would like to discuss the role of polarizability in the electronic structure and charge transport mechanisms [133]. The electronic structure is highly influenced by the molecular polarizability and the geometrical arrangement of the molecules. In OSCs, there is, for example, about 1 eV difference between the IEs in the gas phase and the solid state. This difference is due the polarization energy [134]. For example, the IE of pentacene in the gas phase is about 6.6 eV compared to about 5 eV in thin films. Moreover, it is 5.35 eV when the molecules lies flat over a substrate, and 4.8 eV when they are standing [101].
An important observation related to the correlation of the molecular polarizability to the band-like transport was discussed recently by Minder et al. [135]. In particular, they correlated the observation of the band-like charge transport in OFETs with the polarizability of the conjugated backbones as well as the collective dielectric response of the OSC crystal. In particular, for a group of compounds shown in Fig. 16, they identified two important factors potentially leading to band-like transport in OFETs: (1) the presence of substituents to influence the coupling among the adjacent OSC layers and (2) the orientation of the molecules and their polarizability tensors with respect to the charge transport direction. They argued that PDIF-CN2 and BTBT-C8 show band-like transport because the dielectric coupling of the adjacent layer as well as the gate is screened by the presence of the side groups. This notion is based on the understanding that the conductance channel is the first layer of the OSC film over the gate dielectric and charge transport mostly happens in this first layer. In addition, the alignment of the molecules with respect to the gate is crucial: the dielectric coupling is smaller if they are parallel to the gate than when they are perpendicular to it. Therefore, band-like transport is also observed in TMTSF, rubrene, and tips-pentacene since the conjugated molecular backbones in these crystals lie parallel to the gate. They also argued that pentacene and oligothiophene have never shown band-like transport in an OFET setting because they are arranged perpendicular to the dielectric surface and have no terminal alkyl chains to reduce the coupling among the adjacent layers. Further work to quantify and measure these observations would be of great value to the design and engineering of high-performance OSC materials.

Analysis of the molecular structure and packing for different organic molecules (from left to right: PDIF-CN2, BTBT-C8, TMTSF, rubrene, TIPS-pentacene, sexithiophene, and pentacene). To date, PDIF-CN2, BTBT-C8, TMTSF, rubrene, and TIPS-pentacene are the only organic semiconductors exhibiting band-like transport in an OFET configuration. The values of mobility are either at room temperature (HT) or low temperature (LT–for the molecules in which band-like transport has been observed). (Reprinted (adapted) with permission from [135]. Copyright (2012) Wiley-VCH, Weinheim)
3 Crystal Structure Prediction for Organic Semiconductors
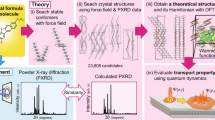
Arguably, the most important piece of information needed for the theoretical characterization of an OSC crystal is the availability of the crystal structure. As illustrated in the previous sections, the literature is full of analyses of the crystal structures of experimentally known materials. The crystal structure prediction for OSCs a priori to synthesis, however, is scarce. Except for a few cases of limited solid-state structure prediction [4, 6, 136], it remains virgin territory. We associate the sporadic interest with lack of commercial applications. In the near future we expect that OSCs will be used in commercial applications ubiquitously and hence a more detailed understanding of the polymorphism or structural stability will be demanded by the market.
From the crystal structure prediction point of view, one advantage of the molecular OSCs with conjugated backbones is that they have mostly two-dimensional geometries and pack in a few specific patterns. For example, many years ago Desiraju and Gavezzotti [116] studied the crystal structures of polynuclear aromatic hydrocarbons. They showed that all of the aromatic hydrocarbons could be grouped into four distinct packing patterns: (1) herringbone pattern (polythiophene, pentacene), (2) pair-wise (sandwich) herringbone pattern (perylene (α phase), pyrene), (3) flattened-herringbone pattern (coronene), and (4) graphitic pattern (tribenzopyrene). Based on their analysis of the structures in terms of the presence of the type of intermolecular interaction, i.e., C⋯H, C⋯C, or H⋯H, they successfully predicted the packing type of some of the unknown structures at the time such as the sandwich structure for the benzo(e)pyrene [137]. Nevertheless, the most practical applications of OSCs involve intra-ring substitutions, heterocycles, or side group additions to the backbone which complicate the crystal structure prediction.
Most crystal structure prediction studies for OSCs reported to date involve the structures of the known crystals of well-studied molecules such as oligoacenes or oligothiophenes. Usually a conventional molecular force field is used in conjunction with electrostatic potential (ESP) fitted point charges to describe the interactions in an experimentally known crystal structure. These studies provide a good basis for understanding the performance of the force fields and the type of improvements required in the force field parameters for particular systems. For example, Marcon and Raos [138] studied the crystalline oligothiophenes with improved MM3 force fields. In particular, they optimized the inter-ring torsion potentials and calculated the atomic charges as well as higher terms of the electrostatic interactions derived from distributed multipole analysis (DMA) with quantum chemistry methods. The systems studied ranged from herringbone packing structures (α-tetrathiophene, α-sexithiophene) to π-stacked configurations as well as an alkyl-chain substituted sexithiophene. Interestingly, they concluded that the MM3 force field with point atomic charges gave satisfactory results for all the p-type OSCs they have studied. Only the n-type OSC, perfluorosexithiophene, required the more accurate electrostatic modeling through the DMA. Therefore, they concluded that the more costly DMA approach is not justified for the p-type OSCs studied. MD simulations at the same level of theory are performed to investigate the effect of the temperature, and to ensure a better comparison of the predicted structures with the structures from room temperature X-ray diffraction. They found that MD simulations at room temperature systematically resulted in approximately 10% lower crystal densities. Among several variations of MM3 force field adapted for the oligothiophenes, MM3 with the ab initio corrected torsion potentials, ESP charges, and adjustment of the dispersion terms with scaling of the dispersion parameters showed the best performance.
The known polymorphs of oligoacenes and oligothiophenes also provide a basis for the test of effective strategies for initial structure search and polymorph prediction. Della Valle and coworkers studied the polymorphs of tetracene [139], pentacene [140, 141], and sexithiophene [142], with the assumption of rigid bodies for the molecular structures. They employed a hybrid approach involving uniform sampling (low-discrepancy Sobol’ sequence) of the energy landscape by including the crystallographic symmetry constraints. The completeness of the search space is maintained by adapting the capture-recapture method from wildlife ecology. The intermolecular potentials are described by atom–atom interactions where electrostatic interactions are defined in terms of the point charges derived from quantum mechanical calculations. In particular, for sexithiophene they employed an AMBER force field with restricted ESP fitted charges, and for pentacene and tetracene an atom–atom Buckingham model with Williams parameter set IV [143] (with ESP charges for the tetracene) is used. For tetracene and pentacene the initial search was constrained to triclinic structures with two independent molecules per unit cell. After the lattice energy minimizations, the crystal space groups are assigned to the optimized lattices by the use of PLATON program [144]. For sexithiophene the initial search was constrained to 16 structural classes with triclinic, monoclinic, and orthorhombic lattice types. In the case of tetracene and pentacene, most of the minima belonged to the P1 and P21/c groups and have the layered herringbone-type packing. The deepest minima for tetracene corresponded to the high temperature-low pressure polymorph known by X-ray diffraction. From the global search for the low energy polymorphs of pentacene, they successfully found the C and H polymorphs as their rank 1 and rank 2 structures. In the case of sexithiophene among nine deepest minima structures, six different space groups are observed, lowest energy polymorphs also having herringbone-type packing (see Fig. 17). Again, the two known polymorphs of sexithiophene have been identified correctly with this methodology. Unfortunately, the same approach failed in the blind tests and Della Valle et al. concluded that the success in the case of tetracene, pentacene, and sexithiophene could be attributed to the symmetry, rigidity, and planarity of these molecules and the presence of the short-range isotropic interactions [142].

Structure of the nine deepest minima, shown with an orientation in which the shortest cell axis (either a or b) is approximately perpendicular to the plane of the page. Minima are labeled by their energy rank N (also indicated in Table 2) and structural class (space group, Z, and site symmetry). (Reprinted (adapted) with permission from [142]. Copyright (2008) American Chemical Society)
Another approach to speed up the optimization of the solid form of materials is based on genetic algorithms (GA), which are widely used in the crystal structure prediction of inorganic materials [145]. Facelli and co-workers [146] adapted the GA for the prediction of the crystal structures of benzene, naphthalene, and anthracene. In the GA approach, genes are a set of geometric parameters defining the spatial arrangements of the rigid bodies in three-dimensional space. Then the genomes are constructed as the collection of parameters defining the crystallographic axes, molecular positions, orientations, and number of molecules in the unit cell. Although no assumptions are made for the crystallographic axes, the unit cell parameters are limited to a certain region of the space to make the problem more tractable. One advantage of this approach is that there are no assumptions on the Z values, i.e., the number of molecules in the unit cell. At every step of the optimization, the lattice energies are calculated with an empirical force field and the lattices leading to higher energies are discarded. Then through the mutations and crossovers performed on the genomes, new structures were generated and minimized until a certain convergence criteria for the lattice energy was reached. Again, here the empirical force fields used were based on van der Waals and electrostatic terms with atomic point charges [143]. Despite the many assumptions involved, the resulting structures were surprisingly successful with lattice energy differences of 1% from the experimental crystals. In addition to the experimentally known structures, a new set of polymorphs was determined by the GA approach. Although this work assumed rigid-bodies, an adaptation of the same algorithm to flexible molecules was also demonstrated [147]. Despite the use of a more advanced force field, the Amber force field used in CHARMM optimizations, the same success for the structure prediction was not observed for a set of flexible molecules. It is perhaps indicated that the conjugated backbone, and hence relative rigidity and flatness of the OSCs, significantly simplify the search problem, and therefore relatively simpler force field parameter sets can provide enough accuracy.
One of the strategies to limit the search space is to take advantage of the fact that 93% of all the space groups of organic molecular solids reported in the Cambridge Structural Database belongs to the 18 most frequent space groups among a total number of 230 [148]. Moreover, about 92% of all organic molecular solids have only one molecule in the asymmetric unit cell [149]. Therefore the initial search space with different packing forms are usually limited to a subset of the most prevalent space groups. We should note however that reported structures in the database does not include all the polymorphs for various reasons [150]. Although the omission of the less likely space groups could provide enough statistics, it does not guarantee that some of the polymorphs of a material might not have been left out. Indeed, in the last blind test, the most successful searches were those that spanned all possible space groups.
For screening many molecules with the potential for high performance, the speed at which the lattice configurations can be ranked is crucial. In the following we summarize an even simpler approach for the structure prediction based on the structural and molecular similarities among molecules. This practical approach is analogous to homology modeling in drug design. Taking advantage of the molecular structural similarities, a known crystal structures is adapted for a new molecule and subsequently optimized with electronic structure methods. A similar strategy, ligand replacement, is also adapted for the prediction of hybrid frameworks [151].
In 2007 Takimiya and coworkers reported two important compounds: BTBT and DNTT (Fig. 7a) with high hole mobility and extraordinary shelf-life. DNTT showed characteristics comparable to pentacene but better ambient stability. Our analysis [152] of the microscopic charge transport parameters of DNTT confirmed that extended aromatic structures have small reorganization energy and mildly anisotropic electronic couplings in the herringbone packing plane. Moreover, the non-local electron–phonon couplings investigated in terms of the thermal modulation of the transfer integrals were found to be weaker, for example, compared to pentacene. The transfer integrals were calculated (at the level of B3LYP/6-31G*) for the unique dimers extracted from an MD trajectory with an MM3 force field with ESP fitted charges. Figure 18 shows the moderate anisotropy of the mobility (Fig. 18b, c) and the weak thermal modulation of the transfer integrals (Fig. 18d).

(a) The packing in the ab-plane in DNTT crystals and molecular dimer pair investigated for transfer integral calculations. (b) The experimental moderate anisotropy in the ab-plane. (Reprinted with permission from [153]. Copyright (2009) AIP Publishing LLC). (c) Effective charge carrier masses obtained from the dispersion of the valence bands [95]. (d) The probability distributions for the transfer integrals extracted from an MD trajectory; the vertical lines correspond to the transfer integrals from the optimized equilibrium structures with the same level of theory. (Reprinted with permission from [95]. Copyright (2010) American Chemical Society)
This analysis motivated us to study a small library of thienoacene derivatives shown in Fig. 19 [6].

DNTT (1) derivatives studied for charge transport capacity [6]
As a first strategy of ranking of the molecules in this library, the gas phase geometries were optimized and internal reorganization energies calculated. The two compounds, 2 and 7, with the smaller reorganization energies (smaller than pentacene reorganization energy of 95 meV) of 85 and 77 meV respectively, were identified as potential high performance materials. Subsequently, the crystal structure minimizations for these two molecules were performed. This involved the optimization of the unit cells derived from the parent compound (DNTT) with Dreiding force field and ESP fitted charges as implemented in the Forcite module [154]. The symmetry imposed (P21) and the symmetry relaxed (P1) optimizations lead to similar packing. Finally, the transfer integrals for the dimers from the optimized structures were calculated and, based on approximate Marcus electron transfer rates, the more promising material was identified as molecule 2. This material, for which a performance better than pentacene had been anticipated, was eventually synthesized and OFET measurements of the single crystals revealed a record high mobility of 12 cm2 V−1 s−1 [6].
The crystal structure was confirmed with powder X-ray with remarkably good agreement. We further adjusted the unit cell parameters by matching the experimental and predicted patterns (see Fig. 20). The lower density predicted by the MM3 with ESP charges is a known property of this approximation unless the dispersion coefficients of the R −6 terms are optimized for the particular system [138]. Although the cell volume error in Table 2 is 6%, in reality it is larger as there is usually an expansion of 6% of the volume as the temperature rises from 0 to 300 K. Since the density of the experimental structure is larger, the ensuing transfer integral values improved. Despite its simplicity, this approach has led to the discovery of a new high-performance materials as well as helped the elucidation of the crystal structures from powder patterns. Again, for molecule 2 the rigid and planar molecular structure simplified the crystal prediction process tremendously. Similarly, Chang et al. predicted the polymorph structures for the substituted tetracenes with Dreiding forcefield with ESP fitted charges [136]. Subsequently, based on the semiclassical charge transfer rate from Marcus theory, they have identified two π-stacking high-performance compounds. These compounds as yet to be synthesized.

The powder X-ray diffraction pattern of molecule 2 from experimental powder (bottom), the re-calculated structure by matching the simulated structure to the experimental powder pattern (middle), and the original predicted structure (top). (Powder simulation wavelength = 0.9758)
Although we have focused on the single-crystals of p-type OSCs, we would like to point out that the prediction of thin-film structures and the effect of various substrates on the film growth have drawn considerable interest over the years and a good summary discussing the recent developments can be found in [155].
4 Conclusions and Outlook
In this chapter we have discussed the molecular and crystal structure–property relationships as well as the state-of-the-art crystal structure prediction for OSCs. We focused on the p-type OSCs for OFET applications. Although we have a good understanding of structure–property relationships through the theoretical and experimental characterization of OSC crystal structures obtained from X-ray diffraction, the prediction of new OSCs from molecular structures is still in its infancy. There have been successful predictions of solid forms of rigid and planar OSC molecules such as pentacene; nevertheless, the more general CSP methodologies to predict crystals of OSCs with functional groups or long alkyl chains remain to be carried out.
The recent blind tests organized by the Cambridge Structural Database showed significant progress towards the prediction of the crystal structures of organic molecular solids [37, 38]. To date, studies on the crystal structure prediction of OSCs have been limited to classical force fields with mostly ESP fitted charges. We believe that there is room for a great deal of improvement by adaptation of the state-of-the-art methodologies that were successful in the blind tests.
If crystal structure prediction for OSCs can be realized, together with theoretical characterization techniques, it can provide valuable guidance for better OSC design. For example, one important area where crystal structure prediction has tremendous potential to facilitate OSC engineering is the optimization of side groups added for solubility [30]. If possible, these studies would tell the synthesis experts what substitutions would lead to what type of crystal structures and whether the substitutions enhance or degrade charge transport. This would facilitate the design process by rational design of the length, type, and positions of the side groups. Moreover, theoretical prediction and characterization can provide insights into the intrinsic limit of a material. The knowledge of whether the intrinsic limit of a semiconductor is reached and what type of changes in the structure can lead to improvement has the potential to facilitate the OSC design tremendously. In its most useful form, the crystal structure prediction can provide the thermodynamically stable structures a priori to synthesis, calculate the energy barriers between low lattice energy configurations, tell us what kind of laboratory conditions would lead to the better performing polymorph, or how to avoid the worse polymorphs with the manipulation of process conditions.
To conclude, we believe that we now have a good understanding of the structure–property relationships in OSCs and, by the adaptation of the state-of-the-art crystal structure prediction methods, there is a great potential to help explore new high performance OSCs for OFET applications. If crystal structure prediction for OSCs can be realized, the same tools can also promote discoveries of new organic photovoltaics, ferrroelectrics, or organic electronics in general.
Abbreviations
- BTBT:
-
[1]Benzothieno[3,2-b][1]benzothiophene
- BTBT-C8 :
-
2,7-Dioctyl[1]benzothieno[3,2-b][1]benzothiophene
- DATT:
-
Dianthra[2,3-b:2′,3′-f]thieno[3,2-b]thiophene
- DMA:
-
Distributed multipole analysis
- DNTT:
-
Dinaphtha[2,3-b:2′,3′-f]thieno[3,2-b]thiophene
- DPP:
-
Diketo-pyrrolo-pyrrole
- DPP(TBFu)2 :
-
3,6-Bis(5-(benzofuran-2-yl)thiophen-2-yl)-2,5-bis(2- ethylhexyl)pyrrolo[3,4-c]pyrrole-1,4-dione
- DTT-Ph-C(8,12):
-
2,6-Bis(4-{octyl,dodecyl}phenyl)-dithieno[3,2-b′:2′,3′-d]thiophene
- GA:
-
Genetic algorithms
- ISC:
-
Inorganic semiconductor
- OSC:
-
Organic semiconductor
- PDIF-CN2 :
-
N,N′-1H,1H-Perfluorobutyldicyanoperylene-carboxydi-imide
- Rubrene:
-
5,6,11,12-Tetraphenyltetracene
- TbTH:
-
Tetraceno[2,3-b]thiophene
- TcTH:
-
Tetraceno[2,3-c]thiophene
- Tips-pentacene:
-
6,13-Bis(triisopropylsilylethynyl)pentacene
- TMTSF:
-
Tetramethyltetraselenafulvalene
References
Schwoerer M, Wolf HC (2007) Organic molecular solids. Wiley-VCH, Weinheim
Katz HE (2004) Recent advances in semiconductor performance and printing processes for organic transistor-based electronics. Chem Mater 16(23):4748–4756. doi:10.1021/cm049781j
Tsumura A, Koezuka H, Ando T (1986) Macromolecular electronic device: field-effect transistor with a polythiophene thin film. Appl Phys Lett 49(18):1210–1212
Feng X, Marcon V, Pisula W, Hansen MR, Kirkpatrick J, Grozema F, Andrienko D, Kremer K, Müllen K (2009) Towards high charge-carrier mobilities by rational design of the shape and periphery of discotics. Nat Mater 8(5):421–426
Blouin N, Michaud A, Gendron D, Wakim S, Blair E, Neagu-Plesu R, Belletête M, Durocher G, Tao Y, Leclerc M (2007) Toward a rational design of poly(2,7-carbazole) derivatives for solar cells. J Am Chem Soc 130(2):732–742. doi:10.1021/ja0771989
Sokolov A, Atahan-Evrenk S, Mondal R, Akkerman HB, Sanchez-Carrera RS, Granados-Focil S, Schrier J, Mannsfeld SCB, Zoombelt AP, Bao Z, Aspuru-Guzik A (2011) From computational discovery to experimental characterization of a high hole mobility organic crystal. Nat Commun 2:437
Jun L, Yan Z, Huei Shuan T, Yunlong G, Chong-An D, Gui Y, Yunqi L, Ming L, Suo Hon L, Yuhua Z, Haibin S, Beng SO (2012) A stable solution-processed polymer semiconductor with record high-mobility for printed transistors. Sci Rep 2:754. doi:10.1038/srep00754
Marien H, Steyeart M, Heremans P (2013) Analog organic electronics, building blocks for organic smart sensor systems on foil. Analog circuits and signal processing. Springer, New York
Gelinck G, Heremans P, Nomoto K, Anthopoulos TD (2010) Organic transistors in optical displays and microelectronic applications. Adv Mater 22(34):3778–3798. doi:10.1002/adma.200903559
Heeger AJ (2010) Semiconducting polymers: the third generation. Chem Soc Rev 39(7):2354–2371. doi:10.1039/b914956m
Zschieschang U, Yamamoto T, Takimiya K, Kuwabara H, Ikeda M, Sekitani T, Someya T, Klauk H (2011) Organic electronics on banknotes. Adv Mater 23(5):654–658. doi:10.1002/adma.201003374
Olivares-Amaya R, Amador-Bedolla C, Hachmann J, Atahan-Evrenk S, Sanchez-Carrera RS, Vogt L, Aspuru-Guzik A (2011) Accelerated computational discovery of high-performance materials for organic photovoltaics by means of cheminformatics. Energy Environ Sci 4:4849–4861
Hachmann J, Olivares-Amaya R, Atahan-Evrenk S, Amador-Bedolla C, Sanchez-Carrera RS, Gold-Parker A, Vogt L, Brockway AM, Aspuru-Guzik A (2011) The Harvard clean energy project: large-scale computational screening and design of organic photovoltaics on the world community grid. J Phys Chem Lett 2(17):2241–2251
Hachmann J, Olivares-Amaya R, Jinich A, Appleton AL, Blood-Forsythe MA, Seress LR, Roman-Salgado C, Trepte K, Atahan-Evrenk S, Er S, Shrestha S, Mondal R, Sokolov A, Bao Z, Aspuru-Guzik A (2013) Lead candidates for high-performance organic photovoltaics from high-throughput quantum chemistry – the Harvard clean energy project. Energy Environ Sci 7:698–704
Mei J, Diao Y, Appleton AL, Fang L, Bao Z (2013) Integrated materials design of organic semiconductors for field-effect transistors. J Am Chem Soc 135(18):6724–6746. doi:10.1021/ja400881n
Kanal IY, Owens SG, Bechtel JS, Hutchison GR (2013) Efficient computational screening of organic polymer photovoltaics. J Phys Chem Lett 4(10):1613–1623. doi:10.1021/jz400215j
O’Boyle NM, Campbell CM, Hutchison GR (2011) Computational design and selection of optimal organic photovoltaic materials. J Phys Chem C 115(32):16200–16210. doi:10.1021/jp202765c
Curtarolo S, Hart GLW, Nardelli MB, Mingo N, Sanvito S, Levy O (2013) The high-throughput highway to computational materials design. Nat Mater 12(3):191–201
Clancy P (2012) Chemical engineering in the electronics industry: progress towards the rational design of organic semiconductor heterojunctions. Curr Opin Chem Eng 1(2):117–122, 10.1016/j.coche.2012.01.001
Holliday S, Donaghey JE, McCulloch I (2013) Advances in charge carrier mobilities of semiconducting polymers used in organic transistors. Chem Mater 26:647–663. doi:10.1021/cm402421p
Wang C, Dong H, Hu W, Liu Y, Zhu D (2011) Semiconducting π-conjugated systems in field-effect transistors: a material odyssey of organic electronics. Chem Rev 112(4):2208–2267. doi:10.1021/cr100380z
Rivnay J, Mannsfeld SCB, Miller CE, Salleo A, Toney MF (2012) Quantitative determination of organic semiconductor microstructure from the molecular to device scale. Chem Rev 112(10):5488–5519. doi:10.1021/cr3001109
Troisi A (2011) Charge transport in high mobility molecular semiconductors: classical models and new theories. Chem Soc Rev 40(5):2347–2358. doi:10.1039/c0cs00198h
Wang L, Nan G, Yang X, Peng Q, Li Q, Shuai Z (2010) Computational methods for design of organic materials with high charge mobility. Chem Soc Rev 39(2):423–434. doi:10.1039/b816406c
Coropceanu V, Cornil J, Da Silva Filho DA, Olivier Y, Silbey R, Bredas J-L (2007) Charge transport in organic semiconductors. Chem Rev 107:926–952
Datta S, Grant DJW (2004) Crystal structures of drugs: advances in determination, prediction and engineering. Nat Rev Drug Discov 3(1):42–57
Bauer J, Spanton S, Henry R, Quick J, Dziki W, Porter W, Morris J (2001) Ritonavir: an extraordinary example of conformational polymorphism. Pharm Res 18(6):859–866. doi:10.1023/a:1011052932607
Della Valle RG, Brillante A, Venuti E, Farina L, Girlando A, Masino M (2004) Exploring the polymorphism of crystalline pentacene. Org Electron 5(1–3):1–6, 10.1016/j.orgel.2003.08.017
Jurchescu OD, Mourey DA, Subramanian S, Parkin SR, Vogel BM, Anthony JE, Jackson TN, Gundlach DJ (2009) Effects of polymorphism on charge transport in organic semiconductors. Phys Rev B 80(8):085201
Mei J, Bao Z (2014) Side chain engineering in solution-processible conjugated polymers for organic solar cells and field-effect transistors. Chem Mater 26(1):604–615. doi:10.1021/cm4020805
Sumrak JC, Sokolov AN, Macgillivray LR (2011) Crystal engineering organic semiconductors. In: Self-organized organic semiconductors. Wiley, New York, pp 1–19. doi:10.1002/9780470949122.ch1
Głowacki ED, Irimia-Vladu M, Kaltenbrunner M, Gsiorowski J, White MS, Monkowius U, Romanazzi G, Suranna GP, Mastrorilli P, Sekitani T, Bauer S, Someya T, Torsi L, Sariciftci NS (2013) Hydrogen-bonded semiconducting pigments for air-stable field-effect transistors. Adv Mater 25(11):1563–1569. doi:10.1002/adma.201204039
Stone AJ (2008) Intermolecular potentials. Science 321(5890):787–789. doi:10.1126/science.1158006
Klimes J, Michaelides A (2012) Perspective: advances and challenges in treating van der Waals dispersion forces in density functional theory. J Chem Phys 137(12):120901
Hongo K, Watson MA, Sánchez-Carrera RS, Iitaka T, Aspuru-Guzik A (2010) Failure of conventional density functionals for the prediction of molecular crystal polymorphism: a quantum Monte Carlo study. J Phys Chem Lett 1(12):1789–1794. doi:10.1021/jz100418p
Reilly AM, Tkatchenko A (2013) Seamless and accurate modeling of organic molecular materials. J Phys Chem Lett 4(6):1028–1033. doi:10.1021/jz400226x
Bardwell DA, Adjiman CS, Arnautova YA, Bartashevich E, Boerrigter SXM, Braun DE, Cruz-Cabeza AJ, Day GM, Della Valle RG, Desiraju GR, van Eijck BP, Facelli JC, Ferraro MB, Grillo D, Habgood M, Hofmann DWM, Hofmann F, Jose KVJ, Karamertzanis PG, Kazantsev AV, Kendrick J, Kuleshova LN, Leusen FJJ, Maleev AV, Misquitta AJ, Mohamed S, Needs RJ, Neumann MA, Nikylov D, Orendt AM, Pal R, Pantelides CC, Pickard CJ, Price LS, Price SL, Scheraga HA, van de Streek J, Thakur TS, Tiwari S, Venuti E, Zhitkov IK (2011) Towards crystal structure prediction of complex organic compounds – a report on the fifth blind test. Acta Crystallogr B 67(6):535–551. doi:10.1107/S0108768111042868
Day GM, Cooper TG, Cruz-Cabeza AJ, Hejczyk KE, Ammon HL, Boerrigter SXM, Tan JS, Della Valle RG, Venuti E, Jose J, Gadre SR, Desiraju GR, Thakur TS, van Eijck BP, Facelli JC, Bazterra VE, Ferraro MB, Hofmann DWM, Neumann MA, Leusen FJJ, Kendrick J, Price SL, Misquitta AJ, Karamertzanis PG, Welch GWA, Scheraga HA, Arnautova YA, Schmidt MU, van de Streek J, Wolf AK, Schweizer B (2009) Significant progress in predicting the crystal structures of small organic molecules - a report on the fourth blind test. Acta Crystallogr B 65(2):107–125. doi:10.1107/S0108768109004066
Neumann MA (2008) Tailor-made force fields for crystal-structure prediction. J Phys Chem B 112(32):9810–9829. doi:10.1021/jp710575h
Kazantsev AV, Karamertzanis PG, Adjiman CS, Pantelides CC (2011) Efficient handling of molecular flexibility in lattice energy minimization of organic crystals. J Chem Theory Comput 7(6):1998–2016. doi:10.1021/ct100597e
Neumann MA, Perrin M-A (2005) Energy ranking of molecular crystals using density functional theory calculations and an empirical van der Waals correction. J Phys Chem B 109(32):15531–15541. doi:10.1021/jp050121r
Price SL, Leslie M, Welch GWA, Habgood M, Price LS, Karamertzanis PG, Day GM (2010) Modelling organic crystal structures using distributed multipole and polarizability-based model intermolecular potentials. Phys Chem Chem Phys 12(30):8478–8490. doi:10.1039/c004164e
Silinsh EA, Capek V (1994) Organic molecular crystals: interaction, localization, and transport phenomena. American Institute of Physics, New York
Ortmann F, Bechstedt F, Hannewald K (2011) Charge transport in organic crystals: theory and modelling. Phys Status Solidi B 248(3):511–525. doi:10.1002/pssb.201046278
Cheng YC, Silbey RJ (2008) A unified theory for charge-carrier transport in organic crystals. J Chem Phys 128(11):114713. doi:10.1063.1.28948.0
Bao Z, Locklin JJ (2007) Organic field-effect transistors. CRC, Boca Raton
Madru M, Guillaud G, Sadoun MA, Maitrot M, Clarisse C, Contellec ML, André JJ, Simon J (1987) The first field effect transistor based on an intrinsic molecular semiconductor. Chem Phys Lett 142(1–2):103–105, 10.1016/0009-2614(87)87259-7
Sakanoue T, Sirringhaus H (2010) Band-like temperature dependence of mobility in a solution-processed organic semiconductor. Nat Mater 9(9):736–740. doi:10.1038/nmat2825
Takamiya M, Sekitani T, Ishida K, Someya T, Sakurai T (2013) Large area electronics with organic transistors. In: Cantatore E (ed) Applications of organic and printed electronics. Integrated circuits and systems. Springer, New York, pp 101–113. doi:10.1007/978-1-4614-3160-2_5
Katz HE, Bao Z (1999) The physical chemistry of organic field-effect transistors. J Phys Chem B 104(4):671–678. doi:10.1021/jp992853n
Horowitz G (2006) Organic transistors. In: Klauk H (ed) Organic electronics, materials, manufacturing and applications. Wiley-VCH, Weinheim, pp 3–32
Ito Y, Virkar AA, Mannsfeld S, Oh JH, Toney M, Locklin J, Bao Z (2009) Crystalline ultrasmooth self-assembled monolayers of alkylsilanes for organic field-effect transistors. J Am Chem Soc 131(26):9396–9404. doi:10.1021/ja9029957
Ukah NB, Granstrom J, Gari RRS, King GM, Guha S (2011) Low-operating voltage and stable organic field-effect transistors with poly (methyl methacrylate) gate dielectric solution deposited from a high dipole moment solvent. Appl Phys Lett 99(24):243302–243303
Zschieschang U, Kang MJ, Takimiya K, Sekitani T, Someya T, Canzler TW, Werner A, Blochwitz-Nimoth J, Klauk H (2012) Flexible low-voltage organic thin-film transistors and circuits based on C10-DNTT. J Mater Chem 22(10):4273–4277. doi:10.1039/c1jm14917b
Horowitz G (1998) Organic field-effect transistors. Adv Mater 10(5):365–377
Anthony JE, Brooks JS, Eaton DL, Parkin SR (2001) Functionalized pentacene: improved electronic properties from control of solid-state order. J Am Chem Soc 123:9482–9483. doi:10.1021/ja0162459
Yang YS, Yasuda T, Kakizoe H, Mieno H, Kino H, Tateyama Y, Adachi C (2013) High performance organic field-effect transistors based on single-crystal microribbons and microsheets of solution-processed dithieno[3,2-b:2′,3′-d]thiophene derivatives. Chem Commun 49(58):6483–6485. doi:10.1039/c3cc42114g
Podzorov V, Menard E, Borissov A, Kiryukhin V, Rogers JA, Gershenson ME (2004) Intrinsic charge transport on the surface of organic semiconductors. Phys Rev Lett 93(8):086602
Diao Y, Tee BCK, Giri G, Xu J, Kim DH, Becerril HA, Stoltenberg RM, Lee TH, Xue G, Mannsfeld SCB, Bao Z (2013) Solution coating of large-area organic semiconductor thin films with aligned single-crystalline domains. Nat Mater 12(7):665–671. doi:10.1038/nmat3650, http://www.nature.com/nmat/journal/v12/n7/abs/nmat3650.html - supplementary-information
Leclerc M, Morin J-F (eds) (2010) Design and synthesis of conjugated polymers. WILEY-VCH Verlag GmbH & Co.KGaA, Weinheim
Li J, Zhao Y, Tan HS, Guo Y, Di C-A, Yu G, Liu Y, Lin M, Lim SH, Zhou Y, Su H, Ong BS (2012) A stable solution-processed polymer semiconductor with record high-mobility for printed transistors. Sci Rep 2:754, http://www.nature.com/srep/2012/121018/srep00754/abs/srep00754.html - supplementary-information
Noriega R, Rivnay J, Vandewal K, Koch FPV, Stingelin N, Smith P, Toney MF, Salleo A (2013) A general relationship between disorder, aggregation and charge transport in conjugated polymers. Nat Mater 12(11):1038–1044
Bassler H, Kohler A (eds) (2012) Charge transport in organic semiconductors, vol 312. Top Curr Chem. Springer, Berlin
Brédas JL, Beljonne D, Coropceanu V, Cornil J (2004) Charge-transfer and energy-transfer processes in pi-conjugated oligomers and polymers: a molecular picture. Chem Rev 104(11):4971–5003. doi:10.1021/Cr040084k
Ostroverkhova O, Cooke DG, Shcherbyna S, Egerton RF, Hegmann FA, Tykwinski RR, Anthony JE (2005) Bandlike transport in pentacene and functionalized pentacene thin films revealed by subpicosecond transient photoconductivity measurements. Phys Rev B 71(3):035204
Karl N (2003) Charge carrier transport in organic semiconductors. Synth Met 133–134:649–657, 10.1016/S0379-6779(02)00398-3
Brédas JL, Calbert JP, da Silva Filho DA, Cornil J (2002) Organic semiconductors: a theoretical characterization of the basic parameters governing charge transport. Proc Natl Acad Sci U S A 99(9):5804–5809. doi:10.1073/pnas.092143399
Hatch RC, Huber DL, Höchst H (2009) HOMO band structure and anisotropic effective hole mass in thin crystalline pentacene films. Phys Rev B 80(8):081411
Ruhle V, Kirkpatrick J, Andrienko D (2010) A multiscale description of charge transport in conjugated oligomers. J Chem Phys 132(13):134103
Norton JE, Brédas JL (2008) Theoretical characterization of titanyl phthalocyanine as a p-type organic semiconductor: short intermolecular pi–pi interactions yield large electronic couplings and hole transport bandwidths. J Chem Phys 128(3):034701. doi:10.1063.1.28068.3
Senthilkumar K, Grozema FC, Bickelhaupt FM, Siebbeles LDA (2003) Charge transport in columnar stacked triphenylenes: effects of conformational fluctuations on charge transfer integrals and site energies. J Chem Phys 119(18):9809–9817
Kirkpatrick J (2008) An approximate method for calculating transfer integrals based on the ZINDO Hamiltonian. Int J Quantum Chem 108(1):51–56. doi:10.1002/qua.21378
Hannewald K, Stojanovic VM, Schellekens JMT, Bobbert PA, Kresse G, Hafner J (2004) Theory of polaron bandwidth narrowing in organic molecular crystals. Phys Rev B 69(7):075211
Ferretti A, Ruini A, Molinari E, Caldas MJ (2003) Electronic properties of polymer crystals: the effect of interchain interactions. Phys Rev Lett 90(8):086401
Huang JS, Kertesz M (2004) Intermolecular transfer integrals for organic molecular materials: can basis set convergence be achieved? Chem Phys Lett 390(1–3):110–115. doi:10.1016/j.cplett.2004.03.141
Mikołajczyk M, Zaleśny R, Czyżnikowska Ż, Toman P, Leszczynski J, Bartkowiak W (2011) Long-range corrected DFT calculations of charge-transfer integrals in model metal-free phthalocyanine complexes. J Mol Model 17(9):2143–2149. doi:10.1007/s00894-010-0865-7
Sancho-Garcia JC, Horowitz G, Bredas JL, Cornil J (2003) Effect of an external electric field on the charge transport parameters in organic molecular semiconductors. J Chem Phys 119(23):12563–12568
Kojima H, Mori T (2011) Dihedral angle dependence of transfer integrals in organic semiconductors with herringbone structures. Bull Chem Soc Jpn 84(10):1049–1056
Lee JY, Roth S, Park YW (2006) Anisotropic field effect mobility in single crystal pentacene. Appl Phys Lett 88(25):252106
Haddon RC, Siegrist T, Fleming RM, Bridenbaugh PM, Laudise RA (1995) Band structures of organic thin-film-transistor materials. J Mater Chem 5(10):1719–1724
Huang JS, Kertesz M (2005) Validation of intermolecular transfer integral and bandwidth calculations for organic molecular materials. J Chem Phys 122(23):234707. doi:10.1063.1.19256.1
Hotta C (2003) Classification of quasi-two dimensional organic conductors based on a new minimal model. J Phys Soc Jpn 72:840
Mori T, Mori H, Tanaka S (1999) Structural genealogy of BEDT-TTF-based organic conductors II. Inclined molecules: theta, alpha, and kappa phases. Bull Chem Soc Jpn 72(2):179–197
Vehoff T, Baumeier B, Troisi A, Andrienko D (2010) Charge transport in organic crystals: role of disorder and topological connectivity. J Am Chem Soc 132(33):11702–11708. doi:10.1021/ja104380c
Marcus RA (1993) Electron transfer reactions in chemistry. Theory and experiment. Rev Mod Phys 65(3):599–610
Reimers JR (2001) A practical method for the use of curvilinear coordinates in calculations of normal-mode-projected displacements and Duschinsky rotation matrices for large molecules. J Chem Phys 115(20):9103–9109
McMahon DP, Troisi A (2010) Evaluation of the external reorganization energy of polyacenes. J Phys Chem Lett 1(6):941–946. doi:10.1021/jz1001049
Norton JE, Brédas JL (2008) Polarization energies in oligoacene semiconductor crystals. J Am Chem Soc 130(37):12377–12384. doi:10.1021/Ja8017797
Duhm S, Xin Q, Hosoumi S, Fukagawa H, Sato K, Ueno N, Kera S (2012) Charge reorganization energy and small polaron binding energy of rubrene thin films by ultraviolet photoelectron spectroscopy. Adv Mater 24(7):901–905. doi:10.1002/adma.201103262
Kera S, Hosoumi S, Sato K, Fukagawa H, Nagamatsu S-I, Sakamoto Y, Suzuki T, Huang H, Chen W, Wee ATS, Coropceanu V, Ueno N (2013) Experimental reorganization energies of pentacene and perfluoropentacene: effects of perfluorination. J Phys Chem C 117(43):22428–22437. doi:10.1021/jp4032089
da Silva Filho DA, Coropceanu V, Fichou D, Gruhn NE, Bill TG, Gierschner J, Cornil J, Brédas JL (2007) Hole-vibronic coupling in oligothiophenes: impact of backbone torsional flexibility on relaxation energies. Philos Trans R Soc A 365(1855):1435–1452. doi:10.1098/rsta.2007.2025
Martinelli NG, Olivier Y, Athanasopoulos S, Ruiz-Delgado MC, Pigg KR, da Silva DA, Sánchez-Carrera RS, Venuti E, Della Valle RG, Brédas JL, Beljonne D, Cornil J (2009) Influence of intermolecular vibrations on the electronic coupling in organic semiconductors: the case of anthracene and perfluoropentacene. ChemPhysChem 10(13):2265–2273. doi:10.1002/cphc.200900298
Nan G, Li Z (2012) Influence of lattice dynamics on charge transport in the dianthra[2,3-b:2′,3′-f]-thieno[3,2-b]thiophene organic crystals from a theoretical study. Phys Chem Chem Phys 14(26):9451–9459. doi:10.1039/c2cp40857k
Troisi A, Orlandi G (2006) Dynamics of the intermolecular transfer integral in crystalline organic semiconductors. J Phys Chem A 110(11):4065–4070. doi:10.1021/Jp055432g
Sánchez-Carrera RS, Atahan S, Schrier J, Aspuru-Guzik A (2010) Theoretical characterization of the air-stable, high-mobility dinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]-thiophene organic semiconductor. J Phys Chem C 114(5):2334–2340. doi:10.1021/jp910102f
Sánchez-Carrera RS, Paramonov P, Day GM, Coropceanu V, Brédas J-L (2010) Interaction of charge carriers with lattice vibrations in oligoacene crystals from naphthalene to pentacene. J Am Chem Soc 132(41):14437–14446. doi:10.1021/ja1040732
Coropceanu V, Sánchez-Carrera RS, Paramonov P, Day GM, Brédas JL (2009) Interaction of charge carriers with lattice vibrations in organic molecular semiconductors: naphthalene as a case study. J Phys Chem C 113(11):4679–4686. doi:10.1021/Jp900157p
Davydov SA (1962) Theory of molecular excitons (trans: M. K, M. O). McGraw-Hill, New York
Pope M, Swenberg CE (1999) Electronic processes in organic crystals and polymers, 2nd edn. Oxford University Press, New York
Takimiya K, Shinamura S, Osaka I, Miyazaki E (2011) Thienoacene-based organic semiconductors. Adv Mater 23(38):4347–4370. doi:10.1002/adma.201102007
Salzmann I, Duhm S, Heimel G, Oehzelt M, Kniprath R, Johnson RL, JrP R, Koch N (2008) Tuning the ionization energy of organic semiconductor films: the role of intramolecular polar bonds. J Am Chem Soc 130(39):12870–12871. doi:10.1021/ja804793a
Kazuo T, Tatsuya Y, Hideaki E, Takafumi I (2007) Design strategy for air-stable organic semiconductors applicable to high-performance field-effect transistors. Sci Technol Adv Mater 8(4):273
Tang ML, Reichardt AD, Wei P, Bao Z (2009) Correlating carrier type with frontier molecular orbital energy levels in organic thin film transistors of functionalized acene derivatives. J Am Chem Soc 131:5264–5273. doi:10.1021/ja809659b
Kobayashi H, Kobayashi N, Hosoi S, Koshitani N, Murakami D, Shirasawa R, Kudo Y, Hobara D, Tokita Y, Itabashi M (2013) Hopping and band mobilities of pentacene, rubrene, and 2,7-dioctyl[1]benzothieno[3,2-b][1]benzothiophene (C8-BTBT) from first principle calculations. J Chem Phys 139:014707
Watanabe M, Chang YJ, Liu S-W, Chao T-H, Goto K, IslamMd M, Yuan C-H, Tao Y-T, Shinmyozu T, Chow TJ (2012) The synthesis, crystal structure and charge-transport properties of hexacene. Nat Chem 4(7):574–578, http://www.nature.com/nchem/journal/v4/n7/abs/nchem.1381.html - supplementary-information
Hutchison GR, Ratner MA, Marks TJ (2005) Hopping transport in conductive heterocyclic oligomers: reorganization energies and substituent effects. J Am Chem Soc 127(7):2339–2350. doi:10.1021/ja0461421
Halik M, Klauk H, Zschieschang U, Schmid G, Ponomarenko S, Kirchmeyer S, Weber W (2003) Relationship between molecular structure and electrical performance of oligothiophene organic thin film transistors. Adv Mater 15(11):917–922. doi:10.1002/adma.200304654
Misra M, Andrienko D, Br B, Faulon J-L, von Lilienfeld OA (2011) Toward quantitative structure–property relationships for charge transfer rates of polycyclic aromatic hydrocarbons. J Chem Theory Comput 7(8):2549–2555. doi:10.1021/ct200231z
Faulon J-L (1994) Stochastic generator of chemical structure. 1. Application to the structure elucidation of large molecules. J Chem Inf Comput Sci 34(5):1204–1218. doi:10.1021/ci00021a031
Kuo M-Y, Liu C-C (2009) Molecular design toward high hole mobility organic semiconductors: tetraceno[2,3-c]thiophene derivatives of ultrasmall reorganization energies. J Phys Chem C 113(37):16303–16306. doi:10.1021/jp9065423
Kwon O, Coropceanu V, Gruhn NE, Durivage JC, Laquindanum JG, Katz HE, Cornil J, Bredas JL (2004) Characterization of the molecular parameters determining charge transport in anthradithiophene. J Chem Phys 120:8186–8194. doi:10.1063.1.16896.6
Kuo M-Y, Chen H-Y, Chao I (2007) Cyanation: providing a three-in-one advantage for the design of n-type organic field-effect transistors. Chemistry 13(17):4750–4758. doi:10.1002/chem.200601803
Chen H-Y, Chao I (2006) Toward the rational design of functionalized pentacenes: reduction of the impact of functionalization on the reorganization energy. ChemPhysChem 7(9):2003–2007. doi:10.1002/cphc.200600266
Sancho-Garcia JC, Perez-Jimenez AJ, Olivier Y, Cornil J (2010) Molecular packing and charge transport parameters in crystalline organic semiconductors from first-principles calculations. Phys Chem Chem Phys 12(32):9381–9388. doi:10.1039/b925652k
Pola S, Kuo C-H, Peng W-T, Islam MM, Chao I, Tao Y-T (2012) Contorted tetrabenzocoronene derivatives for single crystal field effect transistors: correlation between packing and mobility. Chem Mater 24(13):2566–2571. doi:10.1021/cm301190c
Desiraju GR, Gavezzotti A (1989) Crystal structures of polynuclear aromatic hydrocarbons. Classification, rationalization and prediction from molecular structure. Acta Crystallogr B 45(5):473–482. doi:10.1107/S0108768189003794
Glowacki ED, Leonat L, Irimia-Vladu M, Schwodiauer R, Ullah M, Sitter H, Bauer S, Sariciftci NS (2012) Intermolecular hydrogen-bonded organic semiconductors–Quinacridone versus pentacene. Appl Phys Lett 101(2):023304–023305
da Silva Filho DA, Kim EG, Brédas JL (2005) Transport properties in the rubrene crystal: electronic coupling and vibrational reorganization energy. Adv Mater 17(8):1072–1076. doi:10.1002/adma.200401866
Haas S, Stassen AF, Schuck G, Pernstich KP, Gundlach DJ, Batlogg B, Berens U, Kirner HJ (2007) High charge-carrier mobility and low trap density in a rubrene derivative. Phys Rev B 76(11):115203
McGarry KA, Xie W, Sutton C, Risko C, Wu Y, Young VG, Brédas J-L, Frisbie CD, Douglas CJ (2013) Rubrene-based single-crystal organic semiconductors: synthesis, electronic structure, and charge-transport properties. Chem Mater 25(11):2254–2263. doi:10.1021/cm400736s
Anthony JE (2006) Engineered pentacenes. In: Klauk H (ed) Organic electronics: materials, manufacturing and applications. Wiley-VCH, Weinheim, FRG
Anthony JE (2008) The larger acenes: versatile organic semiconductors. Angew Chem Int Ed 47(3):452–483
Giri G, Verploegen E, Mannsfeld SCB, Atahan-Evrenk S, Kim DH, Lee SY, Becerril HA, Aspuru-Guzik A, Toney MF, Bao Z (2011) Tuning charge transport in solution-sheared organic semiconductors using lattice strain. Nature 480:504–508
Zhang L, Fakhouri SM, Liu F, Timmons JC, Ran NA, Briseno AL (2011) Chalcogenoarene semiconductors: new ideas from old materials. J Mater Chem 21(5):1329–1337. doi:10.1039/c0jm02522d
Tucker NM, Briseno AL, Acton O, Yip H-L, Ma H, Jenekhe SA, Xia Y, Jen AKY (2013) Solvent-dispersed benzothiadiazole-tetrathiafulvalene single-crystal nanowires and their application in field-effect transistors. ACS Appl Mater Interfaces 5(7):2320–2324. doi:10.1021/am3025036
Anthony JE (2007) Induced pi-stacking in acenes. In: Muller TJJ, Bunz UHF (eds) Functional organic materials. Wiley-VCH, Weinheim, p 511
Curtis MD, Cao J, Kampf JW (2004) Solid-state packing of conjugated oligomers: from π-stacks to the Herringbone structure. J Am Chem Soc 126(13):4318–4328. doi:10.1021/ja0397916
Kang MJ, Yamamoto T, Shinamura S, Miyazaki E, Takimiya K (2010) Unique three-dimensional (3D) molecular array in dimethyl-DNTT crystals: a new approach to 3D organic semiconductors. Chem Sci 1(2):179–183. doi:10.1039/c0sc00156b
Reese C, Roberts ME, Parkin SR, Bao Z (2009) Tuning crystalline solid-state order and charge transport via building-block modification of oligothiophenes. Adv Mater 21(36):3678–3681. doi:10.1002/adma.200900836
Akkerman HB, Mannsfeld S, Kaushik A, Verploegen E, Burnier L, Zoombelt A, Saathoff J, Hong S, Atahan-Evrenk S, Liu X, Aspuru-Guzik A, Toney M, Clancy P, Bao Z (2013) Effects of odd-even side chain length of alkyl-substituted diphenyl-bithiophenes on first monolayer thin film packing structure. J Am Chem Soc 135(30):11006–11014
Liu J, Zhang Y, Phan H, Sharenko A, Moonsin P, Walker B, Promarak V, Nguyen T-Q (2013) Effects of stereoisomerism on the crystallization behavior and optoelectrical properties of conjugated molecules. Adv Mater 25(27):3645–3650. doi:10.1002/adma.201300255
Troisi A, Orlandi G (2005) Band structure of the four pentacene polymorphs and effect on the hole mobility at low temperature. J Phys Chem B 109(5):1849–1856. doi:10.1021/jp0457489
Bussac MN, Picon JD, Zuppiroli L (2004) The impact of molecular polarization on the electronic properties of molecular semiconductors. Europhys Lett 66(3):392
Topham BJ, Soos ZG (2011) Ionization in organic thin films: electrostatic potential, electronic polarization, and dopants in pentacene films. Phys Rev B 84(16):165405
Minder NA, Ono S, Chen Z, Facchetti A, Morpurgo AF (2012) Band-like electron transport in organic transistors and implication of the molecular structure for performance optimization. Adv Mater 24(4):503–508. doi:10.1002/adma.201103960
Chang Y-f L, Z-y AL-j, J-p Z (2011) From molecules to materials: molecular and crystal engineering design of organic optoelectronic functional materials for high carrier mobility. J Phys Chem C 116(1):1195–1199. doi:10.1021/jp208063h
Reck G, Schulz BW (2006) Benzo(e)pyrene (CSD-CEQGEL)
Marcon V, Raos G (2004) Molecular modeling of crystalline oligothiophenes: testing and development of improved force fields. J Phys Chem B 108(46):18053–18064. doi:10.1021/Jp047128d
Della Valle RG, Venuti E, Brillante A, Girlando A (2006) Inherent structures of crystalline tetracene. J Phys Chem A 110(37):10858–10862. doi:10.1021/jp0611020
Venuti E, Della Valle RG, Brillante A, Masino M, Girlando A (2002) Probing pentacene polymorphs by lattice dynamics calculations. J Am Chem Soc 124(10):2128–2129. doi:10.1021/ja0166949
Della Valle RG, Venuti E, Brillante A, Girlando A (2003) Inherent structures of crystalline pentacene. J Chem Phys 118(2):807–815
Della Valle RG, Venuti E, Brillante A, Girlando A (2008) Are crystal polymorphs predictable? The case of sexithiophene. J Phys Chem A 112(29):6715–6722. doi:10.1021/jp801749n
Williams DE, Starr TL (1977) J Comput Chem 1:13
Spek AL (2003) Platon. J Appl Cryst 36:7
Woodley SM, Catlow R (2008) Crystal structure prediction from first principles. Nat Mater 7(12):937–946
Bazterra VE, Ferraro MB, Facelli JC (2002) Modified genetic algorithm to model crystal structures. I. Benzene, naphthalene and anthracene. J Chem Phys 116(14):5984–5991
Kim S, Orendt AM, Ferraro MB, Facelli JC (2009) Crystal structure prediction of flexible molecules using parallel genetic algorithms with a standard force field. J Comput Chem 30(13):1973–1985. doi:10.1002/jcc.21189
Baur WH, Kassner D (1992) The perils of Cc: comparing the frequencies of falsely assigned space groups with their general population. Acta Crystallogr B 48(4):356–369. doi:10.1107/S0108768191014726
Padmaja N, Ramakumar S, Viswamitra MA (1990) Space-group frequencies of proteins and of organic compounds with more than one formula unit in the asymmetric unit. Acta Crystallogr A 46(9):725–730. doi:10.1107/S0108767390004512
Price S (2013) Why don’t we find more polymorphs? Acta Crystallogr B 69(4):313–328. doi:10.1107/S2052519213018861
Mellot-Draznieks C (2007) Role of computer simulations in structure prediction and structure determination: from molecular compounds to hybrid frameworks. J Mater Chem 17(41):4348–4358. doi:10.1039/b702516p
Sanchez-Carrera RS, Atahan-Evrenk S, Schrier J, Aspuru-Guzik A (2010) Theoretical characterization of the air-stable, high-mobility dinaphtho[2,3-b:2′3′-f]thieno[3,2-b]-thiophene organic semiconductor. J Phys Chem C 114(5):2334–2340
Uno M, Tominari Y, Yamagishi M, Doi I, Miyazaki E, Takimiya K, Takeya J (2009) Moderately anisotropic field-effect mobility in dinaphtho[2,3-b:2(′),3(′)-f]thiopheno[3,2-b]thiophenes single-crystal transistors. Appl Phys Lett 94(22):223308. doi:10.1063.1.31531.9
Accelrys (2006) Materials studio
Clancy P (2011) Application of molecular simulation techniques to the study of factors affecting the thin-film morphology of small-molecule organic semiconductors. Chem Mater 23(3):522–543. doi:10.1021/cm102231b
Minemawari H, Yamada T, Matsui H, Tsutsumi JY, Haas S, Chiba R, Kumai R, Hasegawa T (2011) Inkjet printing of single-crystal films. Nature 475(7356):364–367, http://www.nature.com/nature/journal/v475/n7356/abs/nature10313.html - supplementary-information
Ebata H, Izawa T, Miyazaki E, Takimiya K, Ikeda M, Kuwabara H, Yui T (2007) Highly soluble [1]Benzothieno[3,2-b]benzothiophene (BTBT) derivatives for high-performance, solution-processed organic field-effect transistors. J Am Chem Soc 129(51):15732–15733
Acknowledgements
We thank Semion Saikin and Stéphanie Valleau for stimulating discussions and reading the manuscript. We acknowledge computing facilities at the High Performance Technical Center at the Faculty of Art and Science of Harvard University, XSEDE/Teragrid resources supported by National Science Foundation award number OCI-1053575, and software support from ChemAxon Ltd.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Atahan-Evrenk, Ş., Aspuru-Guzik, A. (2014). Prediction and Theoretical Characterization of p-Type Organic Semiconductor Crystals for Field-Effect Transistor Applications. In: Atahan-Evrenk, S., Aspuru-Guzik, A. (eds) Prediction and Calculation of Crystal Structures. Topics in Current Chemistry, vol 345. Springer, Cham. https://doi.org/10.1007/128_2013_526
Download citation
DOI: https://doi.org/10.1007/128_2013_526
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-05773-6
Online ISBN: 978-3-319-05774-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)