
Abstract
Metal-organic frameworks (MOFs) are porous crystals that are synthesized in a building-block approach that greatly facilitates rational design. MOFs are promising materials for gas storage and separation applications, but they are also intriguing for their potential use as catalysts, electrodes, and drug delivery vehicles. For these reasons, MOFs have spurred a renewed interest in the concept of “crystal engineering,” where the crystal structure of a material is designed to meet application-specific criteria. This chapter reviews recent work in the computational design of MOFs, with an emphasis on high-throughput methods that generate and screen many thousands of candidates automatically.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
1 Unique Properties of Metal-Organic Frameworks
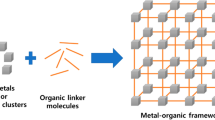
Metal-organic frameworks (MOFs) are crystalline materials that share much in common with (non-crystalline) highly cross-linked polymers [1, 2]. Like conventional polymers, MOFs are synthesized by the self-assembly of molecular “building blocks” that form into an extended structure. Unlike most polymers, however, the extended structure is a rigid, three-dimensional porous crystal whose order can be maintained over millimeter scales. MOFs are so named because the monomers are divided into two distinct groups: metal ions (derived from dissolved metal salts) and the organic ligands that coordinate to them (see Fig. 1). Before the term MOF came into widespread use [3, 4] these materials were (and still are) referred to as coordination polymers [5–7] and, more recently, porous coordination polymers (PCPs) [1, 8, 9]. Note, however, that amorphous materials have been referred to as coordination polymers [7], but MOFs are exclusively crystalline.

Schematic of MOF self-assembly. MOFs are synthesized by the self-assembly of organic and metal-containing (inorganic) building blocks to form extended crystalline frameworks. Note that MOFs can have a wide variety of framework topologies beyond the cubic framework depicted here
A significant milestone in the history of MOFs was reached when it was discovered that the solvent (used in the synthesis procedure) could be removed, leaving behind a freestanding “permanently porous” structure with a very high internal surface area [3, 4, 10, 11].
Two of the earliest permanently porous MOF structures are HKUST-1, reported by Chui et al. [10], and MOF-5 (later named IRMOF-1) by Li et al. [4]. The former is formed from the self-assembly of benzene-1,3,5-tricarboxylic acid and a copper salt and the latter from benzene-1,4-dicarboxylic acid and a zinc salt. In both cases the metal salt and organic ligand were dissolved in dimethylformamide (DMF) solvent at elevated temperatures for 12–24 h. In each case, the synthesis resulted in micrometer scale crystals that could be analyzed by X-ray diffraction to obtain the detailed crystal structures (see Fig. 2). The discovery of these and other permanently porous MOFs [8, 11] over a decade ago catapulted MOFs from being materials of purely scientific interest to materials of potential industrial importance.

Building blocks and crystal structures of (a–c) HKUST-1 and (d–f) IRMOF-1. (a) Benzene-1,3,5-tricarboxylic acid and copper ions, which arrange into octahedral “paddle wheel” clusters in solution, form (b, c) the HKUST-1 structure [10]. (d) Benzene-1,4-dicarboxylic acid and zinc ions, which form Zn4O tetrahedral clusters in solution, form (e, f) the IRMOF-1 structure [4]. Grey, white, red, cyan, and blue spheres represent carbon, hydrogen, oxygen, copper, and zinc atoms, respectively. The purple sphere (f) indicates the size of the pore that is available to guest molecules
1.1 Utility in Gas Storage and Separation Applications
In the past decade a significant impetus for studying MOFs has been their potential use as adsorbents for industrial gas storage and separations applications [12]. MOFs can have surface areas as high as 7,200 m2/g, significantly higher than the best activated carbon or zeolite materials [13]. Because of their high internal surface areas, MOFs have a high density of adsorption sites that can bind gases of interest, which collectively concentrate the gas without increasing the pressure. This phenomenon can have a dramatic effect on gas storage under certain conditions. For example, at 35 bar and 298 K, a vessel filled with MOF-177 would store as much CO2 as nine equally sized vessels without a sorbent material [14]. There has also been marked interest in developing MOFs to store gaseous fuels, such as methane [15, 16] and hydrogen [17, 18], compactly in vehicles.
In addition to storage, MOFs can selectively adsorb certain gases over others, which makes them promising for separating and purifying mixtures of gases [19]. Adsorption-based gas separation is an alternative to distillation, which is a pervasive and energetically costly process in the chemicals industry [20]. In the recent literature, MOFs have been reported as promising adsorbents for separating mixtures of H2/CO2 [21], H2/NH3 [21], CH4/CO2 [22], olefin/paraffin mixtures [23], and p-, o-, m-xylene mixtures [24, 25], among many others [26–30].
It would be particularly beneficial to industry to design a MOF that is optimal for a particular industrial process, as opposed to relying on serendipitous discovery. This is not an unrealistic goal, since both the crystal structure and gas adsorption behavior of a MOF can potentially be computationally predicted a priori.
1.2 Predictable Self-Assembly
Since the molecular building blocks used in their self-assembly only coordinate in very specific orientations and stoichiometries, MOF structures can be relatively straightforward to predict. The creation of new MOFs based on the known shape and connectivity of the building blocks has come to be called “reticular design” [31] and reports of thousands of new structures over the past decade are a testament to the reliability of this approach [32].
However, knowledge of the final structure does not equal knowledge of the detailed synthesis pathway. It is still a significant challenge in the field of MOFs to find the synthesis conditions that lead to the formation of a desired crystal structure. To this end, high throughput robotics have been developed to test rapidly thousands of synthesis conditions given a particular choice of building blocks [33, 34].
Even with predictable self-assembly, the development of MOFs for industrial applications must rely on chemical intuition and trial-and-error testing unless we can predict how the crystal structure determines the gas adsorption behavior. Fortunately, significant advances have been made in accurately modeling gas adsorption in MOFs using molecular simulations.
1.3 Predictable Gas Adsorption Behavior
For gases that are relatively inert, molecules interact weakly with the walls of a MOF and do not appreciably change their electronic structure (i.e., no new bonds are formed or broken). Thus, under these conditions (i.e., physisorption rather than chemisorption) the details of the electronic structure for both the gas molecules and framework structure can be ignored and all atoms can be modeled as classical particles where inter-atomic interactions are governed by Lennard Jones and Coulombic potentials (for more details see Sect. 5.2) [35, 36]. While such models may seem overly simple, they have often predicted gas adsorption behavior in remarkable agreement with experimental measurements for a variety of gases over a wide range of temperatures and pressures (e.g., see Fig. 3) [15, 16, 35, 37, 38]. Such close agreement between experimental measurements and simulation data would not be possible if not for the crystalline nature of MOFs.

Accuracy of molecular simulations for predicting gas adsorption in two different MOFs. (a–c) Simulated (dashed lines) and experimentally measured (solid lines) adsorption isotherms for three different gases over a wide range of temperatures for the MOF NU-125 [16]. (d–f), Simulated (dashed lines) and experimentally measured (solid lines) adsorption isotherms for three different gases over a wide range of temperatures for the MOF NU-111. Note that no fitting was used. Figure parts (a–c) adapted from [16] with permission from The Royal Society of Chemistry. Figure parts ( d–f) adapted from [38] with permission from The Royal Society of Chemistry
Certain gas-temperature combinations are more challenging to model accurately than others. Cryogenic hydrogen adsorption requires taking quantum diffraction effects into account [35, 39–41]. Notably, at low pressures and temperatures, where adsorption is dominated by host–guest interactions (rather than guest–guest) [42], the accuracy of adsorption predictions will depend strongly on how the strongest interaction sites are modeled. For example, simulations of water adsorption [43–45] and CO2 adsorption at sub-atmospheric pressures are often challenging [46, 47] due to strong interactions that occur at the open metal sites [48, 49], which are not well described by Lennard Jones potentials [35, 50].
However, even when molecular simulations are unable to predict gas adsorption accurately, computational analysis of MOFs can precisely calculate pore sizes, surface areas, and other properties of indirect importance to the application of interest. It is also sometimes possible to use molecular simulations to rank correctly MOFs from best to worst, even when the predictions are not quantitatively accurate [46, 47, 51].
1.4 Structure Tunability
Perhaps the most attractive feature of MOFs is the enormous diversity of possible structures, due to the practically unlimited variety of organic ligands that can be used. A small subset of the many organic ligands that have been incorporated in MOF structures is shown in Fig. 4. Therefore, MOFs can have a very wide range of possible pore geometries and surface chemistries [31, 52]. By choosing the building blocks appropriately, one can tune the properties of the resulting MOF to behave in an optimal way for a given context [53]. For example, in the context of membranes for gas separations, one might create a material with pores tuned to be just large enough to let through one gas species (perhaps the desired product), but too small for the other species in the mixture. Recent reports of “post-synthesis” functionalization [54] and “solvent assisted ligand exchange” [55, 56] even allow for nuanced modifications to MOF structures and continue to expand what it is possible to synthesize.

A selection of organic ligand building blocks for MOFs. Figure adapted and reprinted with permission from [58]. Copyright 2013 American Chemical Society
This tunability presents a challenge however: given the ability to create almost any kind of MOF structure, which one should we make? This chapter mainly addresses this challenge.
2 Challenges in MOF Design
2.1 Large Space of Possible MOFs
As alluded to above, the space of possible MOFs is vast. Even when considering only hundreds of molecular building blocks (i.e., organic ligands and metal salts), the combinatorial possibilities allow for millions of hypothetical MOF structures [57]. Given that the universe of organic chemistry from which the ligands are chosen is itself vast (and growing), exploring the space of possible MOFs appears daunting. Although design by intuition continues to yield materials with impressive and successively better properties, maximizing the potential of MOFs will be greatly accelerated by efficient methods to search this enormous space for optimal candidates.
2.2 Difficult to Predict Structural Details
While from the geometry and connectivity of the building blocks it is possible to get an approximate sense of the self-assembled MOF structure, certain structural details can be difficult to estimate without appealing to more sophisticated calculations. For example, even after organic ligands coordinate to the metal ions in the framework, they may retain rotational or other non-translational degrees of freedom [59, 60]. The detailed geometry of the pores of a MOF in such cases may be temperature dependent, independent of expected thermal expansion effects. If the organic ligands exhibit conformational degrees of freedom then even the resulting framework topology can depend on the synthesis conditions, resulting in supramolecular isomerism [53, 61].
MOFs are also able to self-assemble such that one (or more) framework is interpenetrated (also called “catenated”) within another [62, 63] (see Fig. 5). A common rule of thumb is that if there is enough space for another framework, then the MOF will interpenetrate. However, it is possible to control interpenetration via the synthesis procedure [63]. For example, an intermediate MOF can be constructed from an organic ligand with a bulky leaving group that does not leave room for interpenetration. By subsequently removing this bulky leaving group, a non-interpenetrated MOF with “extra” space can be synthesized.

Framework interpenetration can occur in MOFs, where one framework grows inside another. Whether or not this occurs can depend on the synthesis path taken to produce the final structure. Figure adapted and reprinted with permission from [63]. Copyright 2010 American Chemical Society
It is not only challenging to predict whether or not MOFs will (or have the ability to) interpenetrate; it is also difficult to know how the interpenetrated frameworks will pack relative to each other. Whether or not the interpenetrated frameworks are packed tightly (see Fig. 6) can have a significant impact on the gas adsorption properties [64].

Based on the choice of building blocks, it is sometimes easy to predict the structure of the framework (a), but not whether multiple frameworks will interpentrate (b, c). Additionally, the spacing between the interpenetrated frameworks, which can be small (b) or large (c), can be difficult to predict
The inorganic building blocks, referred to sometimes as secondary building units (SBUs) [65, 66], are themselves self-assembled from dissolved metal salts. The structure of these precursor assemblies would be very challenging to predict from ab initio calculations, but MOF design is broadly based on the assumption that, under similar conditions, dissolved metal salts will always form the same SBUs. Thus, the SBUs can be thought of as rigid building blocks in the same way as the rigid organic ligands. Nevertheless, many distinct SBUs are derived from the same metal salts but under different conditions, and so one should be cautious when predicting MOF structures on the assumption that a particular SBU will form.
Finally, it is perhaps fundamentally impossible to predict the location and arrangement of defects (e.g., missing organic ligands) or of interchangeable ligands in so-called multivariate MOFs (MTV-MOFs) [67]. In the latter, ligands that are identical except for having different chemical functional groups are allowed to self-assemble simultaneously, resulting in crystalline materials with a random spatial distribution of functional groups. Creating MOFs with multiple chemical functional groups in a single crystal is attractive for catalytic applications and for gas masks, where each functional group can respond to a different toxic molecule. Unfortunately, designing such an MTV-MOF cannot currently rely on any particular arrangement of functional groups.
2.3 Unclear Structure–Property Relationships
Even if it were possible to know the detailed MOF structure based on the choice of building blocks, that would still not answer the question of which MOF should be designed for a particular engineering problem. This is because, in general, we do not know the relationship of the crystal structure to the gas adsorption property of interest (without performing either experiments or detailed molecular simulations).
For a single MOF, or a small set, it is possible to predict the gas adsorption properties of each structure using molecular simulations. However, as described above, the space of possible MOFs is so vast that even computational trial-and-error is inefficient. The discovery of structure–property relationships, ideally ones that could be expressed as analytical equations, would be the ultimate tool in designing optimal MOF structures. Unfortunately, only a few such relationships have been tentatively proposed in the literature [42, 68, 69], and those only apply to specific gases under a limited range of conditions (see Fig. 7.)

The relationship between CO2 adsorption, N 1 , and the heat of adsorption, Q st, in over 40 different MOFs in 4 cases corresponding to different pressures. The points represent experimental measurements and the solid black lines represent linear fits to the data, which are drawn for the purpose of identifying structure–property relationships. Cases 1–4 correspond to pressures of 0.5, 2.5, 0.5, and 0.1 bar, respectively. Materials labelled a–d were not used in the linear fit. Figure obtained from [68]. Copyright 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim
As will be discussed later in the chapter, the use of high throughput computational screening methods has provided unprecedented clarity in the form of highly resolved structure–property relationships for certain gases. However, this is still an area where much work needs to be done.
3 Strategies for MOF Design
3.1 High Throughput Experimental Synthesis
Although the focus of this chapter is on computational methods, it is worth mentioning that a valid approach to finding useful MOFs from the vast sea of possibilities is high throughput experimental synthesis using sophisticated robotic equipment [33, 34, 70]. While robotic equipment presents a tremendous improvement in speed over traditional synthesis workflows, it is unlikely to become fast enough to cover a significant fraction of the space of possible MOF structures. To truly harness the vast space of MOFs, high speed computational methods are needed.
3.2 Simulating the Self-Assembly of Hypothetical MOF Structures
An intuitive computational approach to exploring hypothetical MOF structures is to model the self-assembly process that takes place during synthesis for many combinations of molecular building blocks. Based on what is known today, molecular simulations that take into account chemical reactions amongst thousands of molecules simultaneously are either prohibitively costly or insufficiently accurate. However, if only the final crystal structure is desired, rather than the intermediate details, then the self-assembly process can be modeled in a simpler way that ignores much of the unimportant physics.
This is the approach taken by Mellot-Draznieks et al. in what is called the “automated assembly of secondary building units” (AASBU) approach [71]. Here, building blocks referred to as SBUs, are treated as rigid bodies that are assigned “sticky sites” that have no physical significance except to cause SBUs to bind to one another via a Lennard–Jones interaction potential. The SBUs are initially distributed randomly in a periodic unit cell and then allowed to settle into an ordered crystal structure by simulated annealing Monte Carlo [72]. This procedure can generate thousands of plausible structures in a short period of time.
The AASBU approach allows one to explore the phase space of MOFs orders of magnitude more quickly than by high throughput experimental methods. However, the iterative nature of energy minimization schemes requires a baseline level of computational expense that can potentially be avoided by other approaches. In the following sections we describe non-iterative methods of generating hypothetical MOF structures.
3.3 Non-iterative Generation of Hypothetical MOF Structures
Rather than arranging building blocks randomly in space and then minimizing the energy of the system, one can potentially choose the initial configuration in such a way that minimization is practically unnecessary. Here, the computational complexity is shifted from iterative energy minimization to searching the space of logical arrangements of chemical building blocks (i.e., candidate “initial configurations”) based on geometrical, topological, and chemical considerations.
While there are potentially many ways to approach non-iterative MOF generation, we have broadly defined two categories: “top-down” and “bottom-up” generation.
3.3.1 “Top-Down” Generation: From Network Topology to Hypothetical MOF
For over a century there has been interest in categorizing crystalline structures by various mathematical descriptions [73–75]. One approach, in which crystal structures are described by periodic graphs called nets, has played a central role in MOF design. Each node and edge in a net represents a group of atoms, with the atoms of the inorganic building blocks often grouped as nodes (see Fig. 8). The assignment of atoms to nodes and edges is subjective, but the study of nets has nevertheless been very significant in predicting how building blocks can connect into periodic structures.

Examples of six different periodic graphs, called nets, that can represent the underlying topology of a MOF. Reprinted with permission from [75]. Copyright 2008 American Chemical Society
Many experimentally synthesized zeolites and MOF crystal structures are described a posteriori as corresponding to a particular net. Therefore, a potential strategy for generating a candidate MOF structure computationally is to begin with a known net, and then substitute chemical building blocks in place of the nodes and edges as appropriate. For this approach, however, a database of nets or a net generation algorithm is needed.
It is possible to use nets from experimentally discovered MOFs, such as those kept in the Reticular Chemistry Structure Resource (RCSR) database [75]. However, the number of experimentally synthesized MOFs thus far is a negligible fraction of the total MOF space (there are 2,031 nets available as of writing in the RCSR database). There may also be specific interest in designing MOFs corresponding to new nets, which is fundamentally not possible if only the known nets are used to generate MOF structures.
There have recently been significant advances in the mathematical understanding of nets, and this has led to the development of algorithms that can systematically enumerate them (see Fig. 9) [73, 76–78]. An exciting property of enumerative algorithms is that there is the possibility of comprehensively generating all possible nets (within certain constraints on complexity). In the context of computational screening in the search of optimal materials, it is reassuring to know that no structure, or subset of structures, was missed that might have had better properties.

Such algorithms are potentially ideal for generating hypothetical MOF structures since they provide a large and potentially comprehensive set of nets onto which chemical building blocks can be added (see Fig. 10.) This strategy has been used to generate zeolite-like microporous solids systematically [79], and recently Bureekaew and Schmid reported a hypothetical covalent organic framework (COF) generation scheme that follows a top-down approach [80]. It is also worth pointing out that non-systematic top-down approaches have been used to design new MOFs one at a time, such as with the design of NU-100 [81].

Schematic illustration of the top-down approach to generating hypothetical MOFs. First a net is chosen (far left), and then chemical building blocks are appended to the net, resulting in a chemically detailed MOF structure (far right)
For a given net, only certain combinations of chemical building blocks will match the topology of the net (e.g., a net may have both vertices with three edges and vertices with two edges, which would require a combination of 2-connected and 3-connected chemical building blocks). Since the number of possible nets is very large, a large fraction of nets may not be compatible with the building blocks in one’s library. Another challenge is that the geometry of the building blocks, which is not explicitly specified by the net, can affect the topology of the crystal structure. In the supramolecular chemistry literature it is known that the assembly of building blocks into larger structures depends on the shape and size of the building blocks, in addition to the degree of connectivity [82]. A 2-connected organic building block self-assembling with a 2-connected metal node may form an infinite chain, square, or rod, depending on the bend angle of the linker, as shown in Fig. 11.

The shape of the building block, in addition to its connectivity, plays a critical role in determining the shape and even dimensionality of the final self-assembled structure. An organic 2-connected building block can self-assemble with a 2-connected inorganic building block to form (a) linear chains, (b) squares, or (c) rods, depending on the organic building block’s shape
Therefore, the top-down approach requires matching a net to a set of compatible building blocks in terms of both connectivity and shape. An interesting strategy may be to generate de novo building blocks that are compatible with a given net and are constrained to be reasonable candidates for chemical synthesis.
3.3.2 “Bottom-Up” Generation: Connecting Building Blocks into Crystals
Another approach, which does not make use of nets explicitly, is to begin with the building blocks and connect them together sequentially until they form a logical periodic structure (i.e., where all connection points are satisfied) (see Fig. 12). Although it is not known a priori whether any particular combination of building blocks is able to form a plausible MOF structure, the scale of the computational search is a function of the size of the building block library (whereas the space of possible nets is independent of the building block library size). It is not clear whether the top-down or bottom-up approach is computationally more efficient. As will be described below, in our first implementation of a bottom-up strategy, over 100,000 hypothetical MOF structures could be generated in less than 24 h on a single CPU [57].

A schematic of bottom-up generation of hypothetical MOF structures. Chemical building blocks are connected together until they form a periodic, chemically detailed structure
4 Bottom-Up MOF Generation Details
4.1 Creating MOF Building Blocks
We created a library of building blocks by extracting fragments from experimentally determined MOF crystal structures. These building blocks were later recombined in various ways to form new hypothetical MOF structures, as shown in Fig. 13. Although it is beyond the scope of this chapter, it is worth noting that the partitioning of a crystal structure into fragments in such a way that they can be recombined into many different structures is a challenging problem; we relied on human intuition and manual inspection, but potentially pattern recognition and other techniques from computer science could have been used. An important aspect to creating these fragments such that they were modular was to partition the MOF at junctures that occurred frequently in MOF materials (e.g., the point where carboxyl-terminated ligands coordinate to a metal).

Visual summary of the bottom-up hypothetical MOF generation strategy. Crystal structures of existing MOFs are obtained from X-ray diffraction data (left), and are subsequently divided into building blocks (middle) that can then be recombined to form new, hypothetical MOFs (right). Figure adapted and reprinted with permission from [57]. Copyright 2012 Nature Publishing Group
Each extracted fragment is assigned a number of connection sites, and each site contains information about the chemical details at the fragment boundary, such as which building blocks can combine with each other. Information about the relative spatial arrangement of two fragments before they were extracted is preserved by a set of vectors associated with each connection site, as shown in Fig. 14.

Encoded in each fragment (i.e., building block) are the (a) atom composition and geometry, (b) topological information via numbered connection sites, and (c) geometrical information in the form of orientation vectors. To allow for rotational degrees of freedom (i.e., for building blocks that can rotate relative to one another), a list of angles for alternative orientations is also included. Reprinted with permission [57]. Copyright 2012 Nature Publishing Group
4.2 Assembling MOFs Block by Block
Generating a MOF can be described as a sequence of decisions, starting with choosing a building block in the library, then choosing a second building block and how it will connect to the first one, and so on until a complete structure is formed. This process is depicted in Fig. 15. Building blocks are combined stepwise, and if an atomic overlap occurs at a particular step, a different building block is chosen or a different connection site, until all possibilities are exhausted. While the total number of steps in each generation process can vary, there are always three steps when, instead of adding a building block, a periodic boundary is imposed by connecting any two building blocks (see steps 2 and 4 in Fig. 15). When no more building blocks can be added, the crystal generation procedure is complete. (Note that no force field or quantum mechanical energy minimizations are involved.)

The assembly process occurs by stepwise addition of building blocks (1), which are attached at their connection sites (purple Xs). Building blocks are also connected across periodic boundaries (2, hashed circles indicate mirror images ). The process repeats (3, 4) until all connection sites are utilized. An interpenetrated MOF may be generated if enough space exists (5, black circles indicate atoms belonging to one of two interpenetrated frameworks ). Gray, red, blue, and turquoise spheres represent carbon, oxygen, nitrogen, and zinc atoms, respectively. Hydrogen atoms have been omitted for clarity. Reprinted with permission from [57]. Copyright 2012 Nature Publishing Group
Since after deciding on the first building block there are many distinct second decisions, the space of all MOF generation attempts can be described by a decision tree. The number of branches that lead to failed attempts (i.e., illogical structures with only partially connected building blocks or building blocks that overlap sterically) is vastly greater than the number of successful structures (i.e., plausible hypothetical MOFs).
Here we attempt to crudely quantify lower bounds on the size of this decision tree. The number of possible hypothetical MOFs (where we consider every decision sequence a “possible” hypothetical MOF) can be estimated based on the size of the library of modular building blocks (from here on assumed to be 100) and a few simplifying assumptions.
Let’s consider the case of MOFs composed of only one type of inorganic building block and one type of organic building block. Let L be the number of organic building blocks (L as in “linkers”) and C be the number of inorganic building blocks to choose from (C as in “corners”). Linkers may only connect with corners, and vice versa. The number of possible MOFs, N, is simply N = L × C, which corresponds, for example, to 900 for L = 90 and C = 10.
Now we can consider the case where a unit-cell of a MOF contains M linkers (not to be confused with L: the number of linker types), which can be either of two types: A or B. Here the diversity of possible structures spans two dimensions: the ratio of A-linkers to B-linkers, and the number of possible arrangements of A and B linkers at a fixed ratio (see Fig. 16).

MOFs that contain two distinct linkers (A-type, blue and B-type, yellow) (a) may vary in the ratio of A to B linkers (b – left vs middle ) or in the arrangement of those linkers at a fixed ratio (b – left vs right ). A larger fragment of the schematic MOF framework is shown in (c) for clarity. Reprinted with permission from [57]. Copyright 2012 Nature Publishing Group
We can estimate a lower bound on the number of unique MOFs by the number of ratios of component types (i.e., a unit-cell with two A-linkers and one B-linker cannot be the same crystal as one with one A-linker and 2 B-linkers). Calculating this lower bound is equivalent to finding the number of unordered sets of M balls of L colors (the answer is: M + L – 1 choose L – 1). However, two crystals, both with two A-linkers and one B-linker but in different positions, can either be physically identical (i.e., related by a symmetry operation) or unique (for example, if the corner is asymmetrical as in Fig. 16). Thus, we can set an upper bound on the number of possible crystals by forming strings such as “BBA,” “BAA,” “BAB,” and so forth. Thus, with a meager library of one corner and two linkers, the number, N, of possible MOFs is
If we allow for more corners and linkers in our library (for example, C = 10, L = 90) but keep the constraint that MOFs may only use two linkers simultaneously, then we arrive at the modified expression
This analysis underestimates the size of the decision tree because, among other things, conformational degrees of freedom and topological variations were neglected (which would have a further multiplying effect on the possibility space). We have also neglected to consider decision sequences that are not equal, but that can lead to the same MOF structure (e.g., by changing the order in which two building blocks were added). Collapsing all such “degenerate” decision sequences into one is itself a computationally expensive procedure. Therefore it is possible that searching a larger decision tree with degeneracies is comparable to the computation cost of searching a reduced non-degenerate decision tree.
Finding plausible hypothetical MOFs in this decision tree can be carried out using a depth-first or breadth-first search along with established optimization techniques, such as various branch pruning methods. In our first implementation of this approach we used a depth-first search and implemented a fail-safe measure that would abort a branch of the tree and jump ahead by a random increment to another branch to prevent getting stuck. One should be cautious about the use of a random increment without precise control of the random number generator because it can hinder reproducibility of generated results.
Using the above approach, we were able to efficiently create a database of hypothetical MOFs that could be screened (or searched through) for applications in natural gas storage [57], xenon/krypton separations [83], and CO2 separation and capture [84]. These investigations have largely served as proof-of-concept demonstrations of how a database of hypothetical MOFs can help find candidate MOFs for synthesis and also help elucidate structure–property relationships.
5 Large-Scale Screening of Hypothetical MOFs for Gas Storage and Separations
5.1 Motivations
Independent of the method, generating and screening hypothetical MOF structures computationally en masse serves two distinct purposes: helping to identify MOFs that can be synthesized and tested experimentally, and identifying structure–property relationships that can also reveal important physical limits on gas adsorption.
5.1.1 Identifying Promising Candidates for Experimental Synthesis
Generating hypothetical MOF structures to find promising candidates for experimental synthesis saves significant time and resources. There are already a few reported cases where a de novo MOF was designed, simulated, and later synthesized and found to have properties in nearly perfect agreement with the simulation predictions. Notably, Farha et al. demonstrated this approach for the MOF NU-100 [81], which had nearly the highest BET surface area for any porous material at the time of publication (6,143 m2/g, second only to MOF-210, reported at almost the same time [85]) (see Fig 17). We recently reported the synthesis of a MOF, which had been output by our bottom-up generator, whose crystal structure and methane adsorption agreed well with our computational predictions [57], but had been previously synthesized by Lin et al. under the name NOTT-107 [86]. In the future, we anticipate that more MOFs generated in silico will be subsequently synthesized experimentally, but it is worth expanding on the notion that significant insight can be obtained from large-scale screening even without going to the final synthesis step.

A new organic linker (a) was designed and the resulting crystal, the MOF NU-100, (b) was predicted computationally. NU-100 was then synthesized (c), and the measured crystal structure and gas adsorption properties were in excellent agreement with the computational predictions. Figure adapted and reprinted with permission from [81]. Copyright 2010 Nature Publishing Group
5.1.2 Discovering Performance Limits and Structure–property Relationships
A natural question when designing a new MOF for a particular application is what is the best possible performance outcome (e.g., what is the highest possible methane storage density at 35 bar and 298 K? [15, 16, 60]). This is a question for which generating and screening hypothetical MOFs on a large scale is ideal. However, it is difficult to know whether the best structure from any particular set of hypothetical MOFs represents the limit of the class of MOFs as a whole. Occasionally it is easier to address a question that appears at first more ambitious: what are the performance limits for any material whatsoever (i.e., beyond just MOFs)? When considering all physical arrangements of matter, any discovered performance limit will necessarily be an upper bound on what is possible to achieve with MOFs. This abstract notion is, in fact, an important consideration when creating a database of hypothetical MOFs. One can, for example, choose to use exotic building blocks to generate a more diverse database but at the risk of inadvertently creating hypothetical materials that are unlikely to be synthesized (or entirely unphysical). The benefit of a more diverse database is that it is more likely to span the full range of possible properties (including perhaps inadvertently unobtainable properties), which is helpful for determining fundamental performance limits (see Fig. 18).

A database of computationally generated materials might contain structures that correspond to i) MOFs that have already been or can be synthesized, (ii) physically plausible materials that are not MOFs (e.g., no inorganic building blocks), and (iii) unphysical structures (i.e., thermodynamically unstable, artifically raised binding interactions). In the process of mapping the properties of the hypothetical MOFs, three possible cases can result: (1) the hypothetical MOF with the best performance exceeds that of all physically realizable materials and thus reveals an upper bound on what is possible with real materials, (2) the span of property values of hypothetical MOFs contains all realizable MOFs, but a non-MOF material is potentially better, and (3) the properties exhibited by the hypothetical MOFs are a subset within the space of MOFs. It may not always be possible to know which case is most applicable in a given scenario, but it is a function of both the particular hypothetical MOF database and property being considered
Beyond discovering performance limits, the large data sets generated from simulating gas adsorption in thousands of hypothetical MOFs can be mined to find new structure–property relationships. Structure–property relationships are useful in establishing design rules for MOFs that can guide subsequent synthesis efforts. However, when experimental data from only a handful of MOFs is considered, it can be difficult to resolve the trends. As with the performance limits, it is not necessary for every hypothetical MOF to be a good candidate for chemical synthesis in order to be useful in identifying structure–property trends.
For both performance limits and structure–property relationships to yield accurate insights, not only must the set of hypothetical MOFs considered be large and diverse, but the model used to calculate gas adsorption must also be accurate. Below, we describe the commonly used classical atomistic simulation method to predict gas adsorption behavior in MOFs.
5.2 The Simulation Model
Gas adsorption properties of MOFs are usually calculated using Monte Carlo methods, which use random moves to sample a statistical mechanical ensemble and determine average quantities such as the equilibrium uptake and enthalpy [87, 88]. Typically the MOF framework is assumed to be rigid, but the gas molecules can move and take on many possible configurations. For each configuration, the energy is calculated using a classical interaction potential. For simulations of CH4, CO2, and N2 adsorption (pertinent to the examples described later in this chapter) the interaction energies between non-bonded atoms were computed through a Lennard-Jones (LJ) plus Coulomb potential:
where i and j are interacting atoms, r ij is the distance between atoms i and j, ε ij and σ ij are the LJ well depth and diameter, respectively, q i and q j are the partial charges of the interacting atoms, and ε 0 is the dielectric constant.
The LJ parameters for the gas molecules are often taken from the TraPPE force field, which stands for transferable potentials for phase equilibria [89, 90]. The TraPPE force field was developed to reproduce vapor–liquid coexistence curves for pure components of various classes of molecules. For framework atoms in MOFs, the LJ parameters are usually taken from rather general force fields, such as the Universal Force Field (UFF) [91]. Partial charges for the framework atoms have in the past (when only a handful of MOFs were being investigated) been derived from quantum chemistry calculations [46, 92]. For large databases it is impractical to use quantum chemistry-based methods, and so we applied a semi-empirical charge equilibration method that estimates partial charges based on known ionization energies [47, 93].
Adsorption isotherms are typically calculated using grand canonical Monte Carlo (GCMC) simulations. In this method an adsorbate phase at constant temperature T, volume V, and chemical potential μ is allowed to equilibrate with a gas phase (which is not simulated). The number of molecules N in the adsorbate phase is allowed to fluctuate so that the chemical potentials of the two phases are equal. For more details on our simulation method, the reader is referred to [35].
5.3 Selected Gas Storage and Separations Applications
5.3.1 Natural Gas Storage in Vehicles
Natural gas is an abundant fuel that can be used to power transportation vehicles [94]. It is less expensive and generates less CO2 per mile travelled than liquid petroleum-based fuels [16]. However, it is a challenge to store natural gas in sufficient quantities in light-duty vehicles in a compact form. Compressed natural gas (CNG) has less than a third of the volumetric energy density of gasoline, and in the United States CNG tanks are pressurized to 3,600 psi, resulting in fuel tanks that are costly, heavy, and require capitally intensive compression equipment to refill [16, 95]. An alternative to storing natural gas at high pressures is to use a porous adsorbent that can store methane at similar concentrations to CNG tanks but at reduced pressures. Over a decade ago, the Department of Energy put forth a methane storage target for adsorbent materials aimed at vehicular natural gas storage: 180 v(STP)/v at 35 bar and 298 K. This target has since been met, but given the high concentration of methane in CNG tanks (~260 v(STP)/v), there is significant interest in an even better adsorbent [16].
Both to find a MOF that could outperform existing adsorbents and to learn about the underlying structure–property relationships of methane storage in porous materials, we generated and screened a database of 137,953 hypothetical MOFs from 102 building blocks [57]. From this screening effort we found, in addition to a promising MOF for methane storage (that we subsequently synthesized), several revealing structure–property relationships (see Fig. 19). We found that volumetric methane storage density increased linearly with volumetric surface area, but that there was an optimal gravimetric surface area in the 2,000–3,000 m2/g range. This latter observation ran counter to conventional wisdom that higher BET surface areas were always better for gas storage. We also found that the best MOFs in our database for storing methane at 35 bar shared a remarkably narrow range of void fraction values. The void fraction is the fraction of empty space within a porous material, and the highest densities of methane were found in MOFs that had a void fraction of almost exactly 80% (see Fig. 19c).

Structure–property relationships obtained from the database of hypothetical MOFs. Reprinted with permission from [57]. Copyright 2012 Nature Publishing Group
It was also possible to correlate the effects of choosing specific building blocks with methane storage ability. For example, we found that MOFs in our database with short alkyl functional groups (i.e., methyl, ethyl, and propyl groups) were found in over 75% of MOFs with methane storage capacities above 205 v(STP)/v (see Fig. 19). This observation is relatively straightforward to test experimentally by incorporating specific functional groups into new MOFs that may or may not be in our generated database.
From the mountain of generated data, there are potentially many patterns within that may be hard to detect. Rather than merely plotting combinations of variables against each other and discovering patterns by visual inspection, rigorous data mining techniques can be used. Although data mining is common in large-scale drug and catalyst design, these methods have not yet been significantly applied to MOFs. This is likely because, until very recently, there were no databases of MOFs of suitable size.
Fernandez et al. recently used data mining techniques to investigate systematically our database of 137,953 hypothetical MOFs to determine the relative importance of various properties for predicting methane storage [69] (see Fig. 20). They applied a range of established data mining algorithms (e.g., multi-linear regression, decision trees, supported vector machines) to see which were the most suitable for extracting trends in MOF-based gas adsorption data.

A comparison of GCMC simulation data (“Actual”) to data mining-based predictions using a multi-linear regression (MLR) fit (“Predicted”). The MLR fit used a training set of 10,000 MOFs to estimate methane storage at (a) 35 bar and (b) 100 bar on the remaining 127,953 MOFs based on three structure variables: the void fraction, dominant pore diameter, and gravimetric surface area. Reprinted with permission [69]. Copyright 2013 American Chemical Society
Fernandez et al. found that the methane storage at 35 bar, U 35bar, or at 100 bar, U 100bar, could be estimated reasonably well by knowledge of just the void fraction (VF), dominant pore diameter (DP, which is the diameter that corresponds to the tallest peak in a pore size distribution of a porous material), and gravimetric surface area (Sg):
Using supported vector machines, they were able to identify combinations of parameters that suggest there are MOFs with greater methane storage capacities than those present in the database itself.
This preliminary illustration indicates that data mining is likely to become commonplace in future MOF research. Such systematic methods will be particularly helpful for problems where computational simulations are very costly, such as in MOF-based catalysis or low pressure CO2 capture where water and other trace gases can play a significant role.
5.3.2 Carbon Dioxide Separation and Capture
Due to both rising global greenhouse gas emissions [96] and an increased worldwide demand for natural gas [97], there is significant interest in the development of porous materials to separate carbon dioxide (CO2) from mixtures of gases, such as the exhaust of fossil-fuel-based power plants (flue gas) and gases that are rich sources of methane (CH4).
Porous materials like MOFs can be used to separate CO2 from these mixtures via pressure-swing adsorption (PSA) or vacuum-swing adsorption (VSA), where the material is exposed to impure gas at a high(er) pressure and then regenerated by lowering (i.e., releasing or “swinging”) the pressure. The effectiveness of a MOF for either PSA or VSA depends on how well it adsorbs CO2 at the higher pressure and then how easily it releases the CO2 at the lower pressure.
Creating a MOF that is optimal for a CO2 separation process requires that we are able to synthesize a structure with pores that selectively bind CO2 either much more strongly, or more weakly, than other gases in the mixture. This, in turn, requires that we determine (independent of our synthesis capabilities) what the optimal shape, size, and chemistry of the pores ought to be for CO2 separation. To address this need we used the same database of 137,953 hypothetical MOFs as was used for methane storage screening, but instead ran molecular simulations of CO2 and N2 adsorption (as well as CH4 adsorption, but at lower pressures than in the earlier work that focused on compressed methane storage in vehicles). The objective was solely to determine structure–property relationships, which were unclear at the time, rather than find a particular MOF candidate to synthesize [84].
In this study we considered every MOF in the database in four distinct cases corresponding to separating CO2 from either N2 or CH4 at pressures and compositions selected for their industrial relevance, namely: (1) natural gas purification using PSA, (2) landfill gas separation using PSA, (3) landfill gas separation using VSA, and (4) flue gas separation using VSA. See Table 1 for gas phase mixture compositions and pressures that approximate each of these four cases (at temperatures of 298 K).
With this large dataset we were able to observe sharply defined correlations between the properties of the MOFs, such as the pore diameter, surface area, pore volume, and chemical functionality, and their usefulness for CO2 separations in each of the four cases (see Fig. 21). As with the case of high pressure methane storage, we observed what could be described as structure–property domains whose boundaries arise from either the limited diversity of our database or from fundamental physical limits (as discussed earlier).

A sample of structure–property relationships derived from simulated CO2, CH4, and N2 adsorption in over 130,000 hypothetical MOFs. Clear relationships can be discerned between (a) CO2 working capacity (ΔN1) and surface area, (b) CO2 uptake (N1) at 2.5 bar and CO2 heat of adsorption (Q st), and (c) selectivity of CO2 over N2 (α 12 ads) and maximum pore diameter. Q st values are determined from CO2 adsorption at the lowest simulated pressure, 0.01 bar. Each plot is divided into 50 × 50 regions that are represented by a filled circle if more than 25 structures exist within the region. The color of each circle represents the average (d) helium void fraction of all structures in that plot region. Figure obtained from [84] and reprinted with permission from The Royal Society of Chemistry
Our work complements several recently reported large-scale computational screening efforts focused on CO2 separations [98–102]. Lin et al. [100] screened hundreds of thousands of hypothetical zeolite and zeolitic imidazolate framework structures for their application to CO2 capture from flue gas [100]. In this study, each structure was measured on its ability to reduce the “parasitic load,” which is the amount of energy a fossil fuel-based power plant would need to spend on capturing CO2 (instead of delivering electrical power) (see Fig. 22). Similarly, Haldoupis et al. [98] computationally screened ~500 MOFs for their ability to separate CO2 from N2, which was the largest set of predictions for CO2 adsorption in MOFs at the time it was reported. In their work, Henry’s constants were correlated with pore diameters, but similar comparisons with other structural characteristics (e.g., surface area, void fraction) or other adsorption properties (e.g., working capacity, selectivity) were not reported. Wu et al. [102] recently examined 105 MOFs for CO2/N2 separations and discovered that simultaneously increasing Q st values while decreasing the void fraction was a useful design rule for increasing the selectivity.

(a) Six example hypothetical zeolites that Lin et al. [100] found performed well for CO2 capture. Materials shown as ball and stick (O,red; Si,tan), with colored surfaces indicating the local free energies of binding CO2. An important focus in this screening study was the (b) parasitic energy that each porous material could potentially reduce. The green line indicates the parasitic energy load of existing monoethanolamine CO2 capture technology and the black line indicates a minimum parasitic energy threshold. Reprinted with permission from [100]. Copyright 2012 Nature Publishing Group
6 Conclusions
Large-scale computational methods have great potential to accelerate the development of new materials. MOFs provide a potentially ideal platform for applying computational crystal engineering methods due to their predictable structures and predictable gas adsorption behaviors. We have shown how large-scale simulations can reveal structure–property insights in MOFs for important gas storage and separation applications such as natural gas storage in vehicles and CO2 separation and capture.
In the future we will likely see experimentally synthesized MOFs that were designed and optimized entirely in silico. This will depend, however, not only on the ability to generate hypothetical MOFs that can be readily synthesized but also on further improvements in our simulation models used to predict gas adsorption behavior.
It is also exciting to consider the possibility of high throughput computational methods working in tandem with high throughput robotic synthesis equipment. This would enable researchers to spend less time attempting to discover promising new materials serendipitously and more time testing material hypotheses and creating the next generation of MOFs.
References
Kitagawa S, Kitaura R, Noro S (2004) Functional porous coordination polymers. Angew Chem Int Ed 43:2334
Zhou H-C, Long JR, Yaghi OM (2012) Introduction to metal-organic frameworks. Chem Rev 112:673
Yaghi OM, Li G, Li H (1995) Selective binding and removal of guests in a microporous metal-organic framework. Nature 378:703
Li H, Eddaoudi M, O’Keeffe M, Yaghi OM (1999) Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 402:276
Subramanian S, Zaworotko MJ (1995) Porous solids by design: [Zn(4,4′-bpy)2(SiF6)] n ·xDMF, a single framework octahedral coordination polymer with large square channels. Angew Chem Int Ed Engl 34:2127
Robson R (2000) A net-based approach to coordination polymers. J Chem Soc Dalton Trans 3735
Spokoyny AM, Kim D, Sumrein A, Mirkin CA (2009) Infinite coordination polymer nano- and microparticle structures. Chem Soc Rev 38:1218
Kitaura R, Seki K, Akiyama G, Kitagawa S (2003) Porous coordination-polymer crystals with gated channels specific for supercritical gases. Angew Chem Int Ed 42:428
Uemura T, Yanai N, Kitagawa S (2009) Polymerization reactions in porous coordination polymers. Chem Soc Rev 38:1228
Chui SS-Y, Lo SM-F, Charmant JPH, Orpen AG, Williams ID (1999) A chemically functionalizable nanoporous material [Cu3(TMA)2(H2O)3]n. Science 283:1148
Noro S, Kitagawa S, Kondo M, Seki K (2000) A new, methane adsorbent, porous coordination polymer [{CuSiF6(4,4′-bipyridine)2} n ]. Angew Chem Int Ed 39:2081
Czaja AU, Trukhan N, Müller U (2009) Industrial applications of metal-organic frameworks. Chem Soc Rev 38:1284
Farha OK, Eryazici I, Jeong NC, Hauser BG, Wilmer CE, Sarjeant AA, Nguyen ST, Snurr RQ, Yazaydin AÖ, Hupp JT (2012) Metal-organic framework materials with ultrahigh surface areas: is the sky the limit? J Am Chem Soc 134:15016
Millward AR, Yaghi OM (2005) Metal-organic frameworks with exceptionally high capacity for storage of carbon dioxide at room temperature. J Am Chem Soc 127:17998
Düren T, Sarkisov L, Yaghi OM, Snurr RQ (2004) Design of new materials for methane storage. Langmuir 20:2683
Wilmer CE, Farha OK, Krungleviciute V, Eryazici I, Sarjeant AA, Yildirim T, Snurr RQ, Hupp JT (2013) Gram-scale, high-yield synthesis of a robust metal-organic framework for methane storage. Energy Environ Sci 6:1158
Rosi NL, Eckert J, Eddaoudi M, Vodak DT, Kim J, O’Keeffe M, Yaghi OM (2003) Hydrogen storage in microporous metal-organic frameworks. Science 300:1127
Murray L, Dinca M, Long J (2009) Hydrogen storage in metal-organic frameworks. Chem Soc Rev 38:1294
Li J-R, Kuppler RJ, Zhou H-C (2009) Selective gas adsorption and separation in metal-organic frameworks. Chem Soc Rev 38:1477
Li J-R, Sculley J, Zhou H-C (2012) Metal-organic frameworks for separations. Chem Rev 112:869
Guo H, Zhu G, Hewitt IJ, Qiu S (2009) “Twin Copper Source” growth of metal-organic framework membrane: Cu3(BTC)2 with high permeability and selectivity for recycling H2. J Am Chem Soc 131:1646
Finsy V, Ma L, Alaerts L, De Vos DE, Baron GV, Denayer JFM (2009) Separation of CO2/CH4 mixtures with the MIL-53(Al) metal-organic framework. Microporous Mesoporous Mater 120:221
Maes M, Alaerts L, Vermoortele F, Ameloot R, Couck S, Finsy V, Denayer JFM, De Vos DE (2010) Separation of C5-hydrocarbons on microporous materials: complementary performance of MOFs and zeolites. J Am Chem Soc 132:2284
Nicolau MPM, Bárcia PS, Gallegos JM, Silva JAC, Rodrigues AE, Chen B (2009) Single- and multicomponent vapor-phase adsorption of xylene isomers and ethylbenzene in a microporous metal-organic framework. J Phys Chem C 113:13173
Gu Z-Y, Jiang D-Q, Wang H-F, Cui X-Y, Yan X-P (2010) Adsorption and separation of xylene isomers and ethylbenzene on two Zn–terephthalate metal−organic frameworks. J Phys Chem C 114:311
Seo J, Whang D, Lee H, Jun S, Oh J, Jeon Y, Kim K (2000) A homochiral metal-organic porous material for enantioselective separation and catalysis. Nature 404:982
Bradshaw D, Prior TJ, Cussen EJ, Claridge JB, Rosseinsky MJ (2004) Permanent microporosity and enantioselective sorption in a chiral open framework. J Am Chem Soc 126:6106
Düren T, Snurr RQ (2004) Assessment of isoreticular metal-organic frameworks for adsorption separations: a molecular simulation study of methane/n-butane mixtures. J Phys Chem B 108:15703
Watanabe T, Keskin S, Nair S, Sholl DS (2009) Computational identification of a metal organic framework for high selectivity membrane-based CO2/CH4 separations: Cu(hfipbb)(H2hfipbb)0.5. Phys Chem Chem Phys 11:11389
Liu B, Yang Q, Xue C, Zhong C, Chen B, Smit B (2008) Enhanced adsorption selectivity of hydrogen/methane mixtures in metal-organic frameworks with interpenetration: a molecular simulation study. J Phys Chem C 112:9854
Yaghi OM, O’Keeffe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J (2003) Reticular synthesis and the design of new materials. Nature 423:705
Ockwig NW, Delgado Friedrichs O, O’Keeffe M, Yaghi OM (2005) Reticular chemistry: occurrence and taxonomy of nets and grammar for the design of frameworks. Acc Chem Res 38:176
Banerjee R, Phan A, Wang B, Knobler C, Furukawa H, O’Keeffe M, Yaghi OM (2008) High-throughput synthesis of zeolitic imidazolate frameworks and application to CO2 capture. Science 319:939
Sumida K, Horike S, Kaye SS, Herm ZR, Queen WL, Brown CM, Grandjean F, Long GJ, Dailly A, Long JR (2010) Hydrogen storage and carbon dioxide capture in an iron-based sodalite-type metal-organic framework (Fe-BTT) discovered via high-throughput methods. Chem Sci 1:184
Getman RB, Bae Y-S, Wilmer CE, Snurr RQ (2011) Review and analysis of molecular simulations of methane, hydrogen, and acetylene storage in metal-organic frameworks. Chem Rev 112:703
Snurr RQ, Yazaydin AO, Dubbeldam D, Frost H (2010) In: MacGillivray LR (ed) Metal–organic frameworks: design and application. Wiley, Hoboken, p 313
Walton KS, Millward AR, Dubbeldam D, Frost H, Low JJ, Yaghi OM, Snurr RQ (2008) Understanding inflections and steps in carbon dioxide adsorption isotherms in metal-organic frameworks. J Am Chem Soc 130:406
Peng Y, Srinivas G, Wilmer CE, Eryazici I, Snurr RQ, Hupp JT, Yildirim T, Farha OK (2013) Simultaneously high gravimetric and volumetric methane uptake characteristics of the metal-organic framework NU-111. Chem Commun 49:2992
Sese L (1995) Feynman–Hibbs potentials and path integrals for quantum Lennard–Jones systems: theory and Monte Carlo simulations. Mol Phys 85:931
Wang Q, Johnson JK (1999) Molecular simulation of hydrogen adsorption in single-walled carbon nanotubes and idealized carbon slit pores. J Chem Phys 110:577
Farha OK, Wilmer CE, Eryazici I, Hauser BG, Parilla PA, O’Neill K, Sarjeant AA, Nguyen ST, Snurr RQ, Hupp JT (2012) Designing higher surface area metal-organic frameworks: are triple bonds better than phenyls? J Am Chem Soc 134:9860
Frost H, Snurr RQ (2007) Design requirements for metal-organic frameworks as hydrogen storage materials. J Phys Chem C 111:18794
Muller E, Rull L, Vega L, Gubbins K (1996) Adsorption of water on activated carbons: a molecular simulation study. J Phys Chem 100:1189
Ramachandran CE, Chempath S, Broadbelt LJ, Snurr RQ (2006) Water adsorption in hydrophobic nanopores: Monte Carlo simulations of water in silicalite. Microporous Mesoporous Mater 90:293
Paranthaman S, Coudert F-X, Fuchs AH (2010) Water adsorption in hydrophobic MOF channels. Phys Chem Chem Phys 12:8124
Yazaydin AÖ et al (2009) Screening of metal-organic frameworks for carbon dioxide capture from flue gas using a combined experimental and modeling approach. J Am Chem Soc 131:18198
Wilmer CE, Kim K-C, Snurr RQ (2012) An extended charge equilibration method. J Phys Chem Lett 3:2506
Xiang SC, Zhou W, Zhang ZJ, Green MA, Liu Y, Chen BL (2010) Open metal sites within isostructural metal-organic frameworks for differential recognition of acetylene and extraordinarily high acetylene storage capacity at room temperature. Angew Chem Int Ed 49:4615
Chen B, Ockwig NW, Millward AR, Contreras DS, Yaghi OM (2005) High H2 adsorption in a microporous metal-organic framework with open metal sites. Angew Chem 117:4823
Getman RB, Miller JH, Wang K, Snurr RQ (2011) Metal alkoxide functionalization in metal−organic frameworks for enhanced ambient-temperature hydrogen storage. J Phys Chem C 115:2066
Haldoupis E, Nair S, Sholl DS (2010) Efficient calculation of diffusion limitations in metal organic framework materials: a tool for identifying materials for kinetic separations. J Am Chem Soc 7258
Eddaoudi M, Kim J, Rosi N, Vodak D, Wachter J, O’Keeffe M, Yaghi OM (2002) Systematic design of pore size and functionality in isoreticular MOFs and their application in methane storage. Science 295:469
Zhao D, Timmons DJ, Yuan D, Zhou H-C (2011) Tuning the topology and functionality of metal-organic frameworks by ligand design. Accounts Chem Res 44:123
Wang Z, Cohen SM (2009) Postsynthetic modification of metal-organic frameworks. Chem Soc Rev 38:1315
Karagiaridi O, Bury W, Sarjeant AA, Stern CL, Farha OK, Hupp JT (2012) Synthesis and characterization of isostructural cadmium zeolitic imidazolate frameworks via solvent-assisted linker exchange. Chem Sci 3:3256
Takaishi S, DeMarco EJ, Pellin MJ, Farha OK, Hupp JT (2013) Solvent-assisted linker exchange (SALE) and post-assembly metallation in porphyrinic metal-organic framework materials. Chem Sci 4:1509
Wilmer CE, Leaf M, Lee C- Y, Farha OK, Hauser BG, Hupp JT, Snurr RQ (2012) Large-scale screening of hypothetical metal-organic frameworks. Nat Chem 4:83
Cook TR, Zheng Y-R, Stang PJ (2013) Metal-organic frameworks and self-assembled supramolecular coordination complexes: comparing and contrasting the design, synthesis, and functionality of metal-organic materials. Chem Rev 113:734
Gould SL, Tranchemontagne D, Yaghi OM, Garcia-Garibay MA (2008) Amphidynamic character of crystalline MOF-5: rotational dynamics of terephthalate phenylenes in a free-volume, sterically unhindered environment. J Am Chem Soc 130:3246
Ma S, Sun D, Simmons JM, Collier CD, Yuan D, Zhou HC (2008) Metal-organic framework from an anthracene derivative containing nanoscopic cages exhibiting high methane uptake. J Am Chem Soc 130:1012
Amirjalayer S, Schmid R (2008) Conformational isomerism in the isoreticular metal organic framework family: a force field investigation. J Phys Chem C 112:14980
Rowsell JLC, Yaghi OM (2006) Effects of functionalization, catenation, and variation of the metal oxide and organic linking units on the low-pressure hydrogen adsorption properties of metal-organic frameworks. J Am Chem Soc 128:1304
Farha OK, Malliakas CD, Kanatzidis MG, Hupp JT (2010) Control over catenation in metal-organic frameworks via rational design of the organic building block. J Am Chem Soc 132:950
Ryan P, Broadbelt LJ, Snurr RQ (2008) Is catenation beneficial for hydrogen storage in metal-organic frameworks? Chem Commun 4132
Eddaoudi M, Moler DB, Li H, Chen B, Reineke TM, O’Keeffe M, Yaghi OM (2001) Modular chemistry: secondary building units as a basis for the design of highly porous and robust metal-organic carboxylate frameworks. Accounts Chem Res 34:319
Tranchemontagne DJ, Mendoza-Cortés JL, O’Keeffe M, Yaghi OM (2009) Secondary building units, nets and bonding in the chemistry of metal-organic frameworks. Chem Soc Rev 38:1257
Deng H, Doonan CJ, Furukawa H, Ferreira RB, Towne J, Knobler CB, Wang B, Yaghi OM (2010) Multiple functional groups of varying ratios in metal-organic frameworks. Science 327:846
Bae Y-S, Snurr RQ (2011) Development and evaluation of porous materials for carbon dioxide separation and capture. Angew Chem Int Ed 50:11586
Fernandez M, Woo TK, Wilmer CE, Snurr RQ (2013) Large-scale quantitative structure–property relationship (QSPR) analysis of methane storage in metal-organic frameworks. J Phys Chem C 117:7681
Bauer S, Serre C, Devic T, Horcajada P, Marrot J, Férey G, Stock N (2008) High-throughput assisted rationalization of the formation of metal organic frameworks in the iron(III) aminoterephthalate solvothermal system. Inorg Chem 47:7568
Mellot Draznieks C, Newsam JM, Gorman AM, Freeman CM, Férey G (2000) De novo prediction of inorganic structures developed through automated assembly of secondary building units (AASBU method). Angew Chem Int Ed 39:2270
Kirkpatrick S, Gelatt C, Vechhi M (1983) Optimization by simulated annealing. Science 220:671
Delgado Friedrichs O, Dress AWM, Huson DH, Klinowski J, Mackay AL (1999) Systematic enumeration of crystalline networks. Nature 400:644
Wells AF (1977) Three dimensional nets and polyhedra. Wiley, New York
O’Keeffe M, Peskov MA, Ramsden SJ, Yaghi OM (2008) The Reticular Chemistry Structure Resource (RCSR) database of, and symbols for, crystal nets. Accounts Chem Res 41:1782
Hyde ST, Delgado Friedrichs O, Ramsden SJ, Robins V (2006) Towards enumeration of crystalline frameworks: the 2D hyperbolic approach. Solid State Sci 8:740
Ramsden SJ, Robins V, Hyde ST (2009) Three-dimensional Euclidean nets from two-dimensional hyperbolic tilings: kaleidoscopic examples. Acta Crystallogr A 65:81
McColm GL, Clark WE, Eddaoudi M, Wojtas L, Zaworotko M (2011) Crystal engineering using a “Turtlebug” algorithm: a de novo approach to the design of binodal metal-organic frameworks. Cryst Growth Des 11:3686
Thomas JM, Klinowski J (2007) Systematic enumeration of microporous solids: towards designer catalysts. Angew Chem Int Ed 46:7160
Bureekaew S, Schmid R (2013) Hypothetical 3D-periodic covalent organic frameworks: exploring the possibilities by a first principles derived force field. CrystEngComm 15:1551
Farha OK, Yazaydın AÖ, Eryazici I, Malliakas CD, Hauser BG, Kanatzidis MG, Nguyen ST, Snurr RQ, Hupp JT (2010) De novo synthesis of a metal-organic framework material featuring ultrahigh surface area and gas storage capacities. Nat Chem 2:944
Chakrabarty R, Mukherjee PS, Stang PJ (2011) Supramolecular coordination: self-assembly of finite two- and three-dimensional ensembles. Chem Rev 111:6810
Sikora BJ, Wilmer CE, Greenfield ML, Snurr RQ (2012) Thermodynamic analysis of Xe/Kr selectivity in over 137000 hypothetical metal-organic frameworks. Chem Sci 3:2217
Wilmer CE, Farha OK, Bae Y-S, Hupp JT, Snurr RQ (2012) Structure–property relationships of porous materials for carbon dioxide separation and capture. Energy Environ Sci 5:9849
Furukawa H, Ko N, Go YB, Aratani N, Choi SB, Choi E, Yazaydin AÖ, Snurr RQ, O’Keeffe M, Kim J, Yaghi OM (2010) Ultrahigh porosity in metal-organic frameworks. Science 329:424
Lin X et al (2009) High capacity hydrogen adsorption in Cu(II) tetracarboxylate framework materials: the role of pore size, ligand functionalization, and exposed metal sites. J Am Chem Soc 131:2159
Metropolis N, Ulam S (1949) The Monte Carlo method. J Am Stat Assoc 44:335
Leach AR (2001) Molecular modelling: principles and applications. Prentice Hall, Upper Saddle River
Martin MG, Siepmann JI (1998) Transferable potentials for phase equilibria. 1. United-atom description of N-alkanes. J Phys Chem B 102:2569
Martin MG, Siepmann JI (1999) Novel configurational-bias Monte Carlo method for branched molecules. Transferable potentials for phase equilibria. 2. United-atom description of branched alkanes. J Phys Chem B 103:4508
Rappé AK, Casewit CJ, Colwell KS, Goddard WA III, Skiff WM (1992) UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J Am Chem Soc 114:10024
Breneman CM, Wiberg KB (1990) Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J Comput Chem 11:361
Rappé AK, Goddard WA III (1991) Charge equilibration for molecular dynamics simulations. J Phys Chem 95:3358
Flynn PC (2002) Commercializing an alternate vehicle fuel: lessons learned from natural gas for vehicles. Energy Policy 30:613
Brown RN (2005) Compressors: selection and sizing. Elsevier, Oxford
Chu S (2009) Carbon capture and sequestration. Science 325:1599
Tagliabue M, Farrusseng D, Valencia S, Aguado S, Ravon U, Rizzo C, Corma A, Mirodatos C (2009) Natural gas treating by selective adsorption: material science and chemical engineering interplay. Chem Eng J 155:553
Haldoupis E, Nair S, Sholl DS (2012) Finding MOFs for highly selective CO2/N2 adsorption using materials screening based on efficient assignment of atomic point charges. J Am Chem Soc 134:4313
Wu D, Wang C, Liu B, Liu D, Yang Q, Zhong C (2012) Large-scale computational screening of metal-organic frameworks for CH4/H2 separation. Aiche J 58:2078
Lin L-C, Berger AH, Martin RL, Kim J, Swisher JA, Jariwala K, Rycroft CH, Bhown AS, Deem MW, Haranczyk M, Smit B (2012) In silico screening of carbon-capture materials. Nat Mater 11:633
Krishna R, van Baten JM (2011) In silico screening of metal-organic frameworks in separation applications. Phys Chem Chem Phys 13:10593
Wu D, Yang Q, Zhong C, Liu D, Huang H, Zhang W, Maurin G (2012) Revealing the structure–property relationships of metal-organic frameworks for CO2 capture from flue gas. Langmuir 28:12094
Acknowledgements
The authors thank Profs. Omar Farha and Joseph Hupp for stimulating discussions and the Defense Threat Reduction Agency (HDTRA – 10 – 1 – 0023) for financial support.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Berlin Heidelberg
About this chapter
Cite this chapter
Wilmer, C.E., Snurr, R.Q. (2013). Large-Scale Generation and Screening of Hypothetical Metal-Organic Frameworks for Applications in Gas Storage and Separations. In: Atahan-Evrenk, S., Aspuru-Guzik, A. (eds) Prediction and Calculation of Crystal Structures. Topics in Current Chemistry, vol 345. Springer, Cham. https://doi.org/10.1007/128_2013_490
Download citation
DOI: https://doi.org/10.1007/128_2013_490
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-05773-6
Online ISBN: 978-3-319-05774-3
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)