
Abstract
The current global population of 7.3 billion is estimated to reach 9.7 billion in the year 2050. Rapid population growth is driving up global food demand. Additionally, global climate change, environmental degradation, drought, emerging diseases, and salty soils are the current threats to global food security. In order to mitigate the adverse effects of these diverse agricultural productivity constraints and enhance crop yield and stress-tolerance in plants, we need to go beyond traditional and molecular plant breeding. The powerful new tools for genome editing, Transcription Activator-Like Effector Nucleases (TALENs) and Clustered Regulatory Interspaced Short Palindromic Repeats (CRISPR)/Cas systems (CRISPR-Cas9), have been hailed as a quantum leap forward in the development of stress-resistant plants. Plant breeding techniques, however, have several drawbacks. Hence, identification of transcriptional regulatory elements and deciphering mechanisms underlying transcriptional regulation are crucial to avoiding unintended consequences in modified crop plants, which could ultimately have negative impacts on human health. RNA splicing as an essential regulated post-transcriptional process, alternative polyadenylation as an RNA-processing mechanism, along with non-coding RNAs (microRNAs, small interfering RNAs and long non-coding RNAs) have been identified as major players in gene regulation. In this chapter, we highlight new findings on the essential roles of alternative splicing and alternative polyadenylation in plant development and response to biotic and abiotic stresses. We also discuss biogenesis and the functions of microRNAs (miRNAs) and small interfering RNAs (siRNAs) in plants and recent advances in our knowledge of the roles of miRNAs and siRNAs in plant stress response.

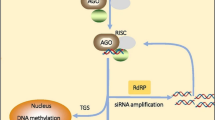
Graphical Abstract

Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Abiotic stress
- Biotic stress
- Alternative splicing
- Alternative polyadenylation
- microRNAs
- Small interfering RNAs
1 Introduction
Biotic and abiotic stresses, global climate change, and environmental pollution have a significant negative impact on crop yields. Together with rapid global population growth, these factors threaten global food security. The current world population of 7.3 billion is expected to reach 8.5 billion by 2030, 9.7 billion in 2050 and 11.2 billion in 2100, according to the most recent UN DESA report, “World Population Prospects: The 2015 Revision” [1]. Moreover, food demand is expected to increase by 59–98% between 2005 and 2050 [2]. In order to mitigate and control these diverse agricultural productivity constraints, extensive effort has been put into improving crop yield and stress-tolerance through traditional and molecular plant breeding. Traditional or conventional plant breeding is a time-consuming and labor-intensive approach. It is limited to the exchange of genes between fairly closely related species [3]. In order to overcome these hurdles, molecular breeding through gene manipulation has widely been used to develop new high-yielding, stress-tolerant crop varieties [4]. However, this technology has some limitations such as non-targeted and unanticipated effects [5]. Recently, new technologies such as Transcription Activator-Like Effector Nucleases (TALENs) and Clustered Regulatory Interspaced Short Palindromic Repeats (CRISPR)/Cas systems have emerged for genome editing [6]. However, harnessing the benefits of these technologies requires a complete understanding of the complexity of plant defense mechanisms and stress signaling pathways at the molecular level. This knowledge is crucial to enable development of high-yielding, stress-resistant crop plants with minimum yield penalty through selective genetic engineering and precise gene editing techniques [7].
Over the past 20 years, the field of gene expression profiling has undergone a dramatic revolution. Transcriptomics has witnessed remarkable success due to major advances in transcriptome sequencing and analysis technologies. A wide variety of molecular biology techniques have been used for expression profiling and transcription quantification. Traditional techniques such as northern blotting and in situ hybridization [8] and reverse transcription polymerase chain reaction (RT-PCR) [9] allow only single transcripts or small groups of transcripts to be analyzed at once. The real-time RT-PCR method is a medium-throughput and very sensitive technique for the detection of low-abundance mRNA. It has been widely used for absolute and relative gene expression quantification [10,11,12]. The development of microarrays in the mid-1990s revolutionized gene expression studies and provided a new tool for genome expression profiling by allowing large-scale analysis of thousands of genes simultaneously [13]. Microarrays have widely been employed to understand molecular mechanisms underlying plant development and response to a multitude of stresses [14,15,16,17]. More recently, next-generation sequencing (NGS) has essentially provided the second revolution since the development of microarrays. Through high-throughput sequencing, it has remarkably improved our understanding and knowledge of gene regulatory mechanisms and epigenetics [18, 19]. NGS-based RNA sequencing (RNA-seq) allows detection and quantification of known, novel and rare transcripts, genome annotation, and rearrangement detection to non-coding RNA discovery. Furthermore, it provides greater insights into biological pathways and molecular mechanisms that regulate cell fate, development, and disease progression [6, 18, 20].
Recent advances in transcriptomics technologies shed light on the dark intergenic regions between protein-coding genes traditionally referred to as transcriptional “noise,” “junk DNA,” or experimental artifact. In 2012, ENCODE (Encyclopedia of DNA Elements) declared that 80% of the human genome has a biochemical function. However, scientists of the ENCODE project recently got together in Potomac, MD, USA and claimed that ~50% is functional [21]. By contrast, Rands and Colleagues claim that 8.2% of the human genome is likely to be functional [22]. In comparison, only a tiny portion of the transcribed human genome (~1–2%) codes for proteins [23]. The number of protein-coding genes in the human genome is reported to be fewer than 20,000 genes and has continued to shrink [24]. According to the most recent estimate of the GENCODE annotation of the human genome, GENCODE release 24 (09.12.2015) corresponds to Ensembl 83, 84, and the human genome encompasses 19,815 protein-coding genes and 25,823 non-coding RNA genes (ncRNAs). Of these, 15,941 and 9,882 are long and small non-coding RNA genes, respectively. Moreover, many new non-coding RNA classes have been identified in the last few years and classified based on their distinct biogenesis pathways. Although little is known about plant genomics and plant genome composition, several recent studies have identified different types of non-coding RNAs with diverse functions in plants [25]. In this chapter, we discuss recent advances in our knowledge of the biological functions of mRNAs (with a particular focus on alternatively spliced mRNAs and polyadenylation) and ncRNAs (with a particular focus on miRNAs and siRNAs) in plant development and response to biotic and abiotic stresses. The purpose of this chapter is to provide an overview of important regulatory components, apart from pure mRNA expression, which has been studied for several decades.
2 Alternative Splicing, Alternative Polyadenylation, and Other Modifications of mRNA
The pre-mRNA containing introns can be alternatively spliced to generate multiple transcripts from a single gene through the differential use of splice sites that increase the transcriptome and proteome complexity of the cells and tissues [26, 27]. Eighty percent of the genes in plants and animals contain introns. Splicing, which is the removal of introns, is carried out by the spliceosome that surrounds the splice sites at each intron. The pre-mRNA (primary transcript) structure includes cis-elements such as the 5′ splice site, the branch-point that is close to the 3′end splice site, the polypyrimidine ring tract, and the 3′ splice site, which are required for splicing. The splice sites contain consensus sequences that are recognized by the spliceosome. The spliceosome, which is a complex ribonucleoprotein mega particle, consists of small nuclear ribonucleoproteins (snRNPs)- U1, U2, U4, U5, U6, and auxiliary factors, U2AF65 and U2AF35. Spliceosomes have a stronger affinity for some splice sites and a weaker affinity for others, and this phenomenon is important in alternative splicing (AS) [28]. The spliceosome recognizes these features and it applies two sequential trans-esterification reactions that ligate the selected exon sequences and remove the introns. The first step involves the nucleophilic attack by the 2′OH group of an important adenosine in the branch consensus site on the 5′ splice site, forming a branched RNA intermediate called intron lariat. After this, some of the snRNPs are released. In the second step, the 3′OH group of the upstream exon targets the 3′ splice site. This reaction results in the spliced mRNA and the intron lariat is removed and degraded [28]. The cis-regulatory sequences in the pre-mRNA (exon splicing enhancers (ESE), exonic splicing silencers (ESSs), intronic splicing enhancers (ISEs), and intronic splicing silencers (ISSs)) as well as the AS regulatory proteins (Ser/Arg-rich proteins (SRs), and heterogeneous nuclear ribonucleoproteins (hnRNPs)) are important regulators of the splicing process [29, 30]. The exon-exon junction complex (EJC) accumulates 24 nt upstream of the exon-exon junction for exportation of mRNA from the nucleus and for the cytoplasmic mRNA control [31]. The binding of the serine/arginine-rich (SR) family of splicing factors to ESE helps in the recruitment of the splicing components and prevents “exon skipping” [32]. The key studies on mRNA modification are summarized in Table 1.
2.1 Types of Alternative Splicing (AS)
Using different splice sites, the AS generates two or more mRNAs from the same pre-mRNA. Based on the type of AS, sequences of exons and introns are either included or excluded from the mRNA. The cassette exon is an exon that is included or excluded from the mRNA. Mutually exclusive exons refer to the splicing of the adjacent exon, causing only one of them to be included at a time in the mRNA. The alternative 5′ splice site involves the use of the distal or proximal 5′ splice site producing mRNAs of different size. The alternative 3′ splice site involves the exploitation of the 3′ proximal and distal splice sites, resulting in the production of mRNAs of different sizes. The final type of AS is the retention of an intron where the intron is retained or removed from the mRNA [27].
2.2 Coupling of Transcription with AS
More recently, it has become widely accepted that AS is a co-transcriptional event in which crosstalk is involved [33, 50]. The carboxy-terminal domain (CTD) involved in the coupling of transcription and processing steps is required for the recruitment of Ser/Arg-rich splicing factor 3 (SRSF3), which then inhibits the inclusion of alternative exons [51]. The mediator joins with the general transcription factors (GTFs) at promoters and specific TFs that are bound to gene enhancers, and recruits the negative splicing factor hnRNPL. This process causes hnRNPL to inhibit the inclusion of an alternative exon during splicing [52].
2.3 AS in Plants
AS is uncommon in unicellular eukaryotes and commonly found in multicellular eukaryotes and differs greatly among tissues and species [28, 53]. Only about 4% of the genes in budding yeast contain introns and AS is uncommon [54]. In comparison, the RNA structures containing exon-intron precincts, spliceosome components, and other splicing factors are commonly found in plants [27]. However, splicing in plants is unique due to their shorter introns compared to animals. Furthermore, intron retention is a common method of AS in plants and they contain more genes encoding Ser/Arg-rich (SR) proteins. The pre-mRNA of the spliceosomal proteins, particularly SR proteins, which have a key role in spliceosome assembly and splicing regulation, are extensively spliced. The availability of plant genome and transcript sequence data has allowed the global analysis of AS in many plant species. Genome-wide analysis of AS has been performed in model plants such as Arabidopsis [55], Brachypodium [56], and in crop plants such as rice [57] and soybean [58]. These studies have shown that plant genes have one or more alternative transcript isoforms (~20% of the genes) [59]. Studies have shown that nearly 61% of multiexonic genes in Arabidopsis and nearly 33% of rice genes are alternatively spliced [55, 57]. The AS of genes has been studied to understand their role in plant growth development, environmental changes, and stress responses [60,61,62]. The studies mentioned above and others support the importance of intron retention in plants.
2.4 Database Resources of Plant Spliceosomal Proteins and AS
There are several resources available in relation to plant spliceosomal proteins and AS. Many of them are based on the model plant, Arabidopsis, such as the Arabidopsis slicing-related genes (ASRG) [63], and The Arabidopsis Information Resource (TAIR) [64]. Others include the AS in plants (ASIP), which is available for Arabidopsis and rice [65].
2.5 Role in Plant Development
Whole transcriptome profiling using RNA-seq was useful in enhancing the understanding of the gene expression of key genes and the coordinated expression of related genes during early somatic embryogenesis in maize [66]. AS is important in photosynthesis as it generates two protein products from the Rubisco activase gene, which is a nuclear-encoded chloroplast protein that mediates light activation of ribulose 1,5-biphosphate carboxylase/oxygenase (Rubisco) [67]. Based on the cDNAs and ESTs of Arabidopsis and rice analyzed using genome-wide computational analysis, AS has been shown to be common during flowering [65]. The alteration from the vegetative to the reproductive developmental stages is regulated by the alternative processing of the FCA pre-RNA [68]. Further, AS of the transcripts controls the spatial and temporal production of the FCA protein that regulates flowering.
2.6 Role in Biotic and Abiotic Stress Response
The pre-mRNAs of spliceosomal proteins are severely affected by biotic and abiotic stresses leading to AS [69, 70]. For example, OSDREB2B was shown to be regulated by stress-inducible AS [71]. Furthermore, the AS of the NRR transcript (related to root growth) identified in rice, produced two 5′ co-terminal transcripts (NRRa and NRRb), and both products were shown to possess negative regulatory roles [72]. In another example, the TIR-NBS-LRR gene that is involved in tobacco mosaic virus (TMV) resistance produced two transcripts, NS and NL, through AS. Expression of both transcripts were shown to be required for complete resistance to the virus [73]. Similarly, the combined presence of RPS4 transcripts containing both full-length and truncated open reading frames was required to mediate disease resistance [74]. Abiotic stresses have been shown to affect the AS of the pre-mRNA in several SR genes [75]. The AS regulators such as the SR proteins, hnRNPs, and protein kinases have been suggested to play significant roles in stress responses [27]. They have been suggested to allow plants to react promptly in regulating splicing and gene expression.
2.7 Alternative Polyadenylation
The regulatory role of polyadenylation in eukaryotic gene expression involves alternative polyadenylation (APA) sites that produce different transcripts with altered coding capacity for proteins and/or RNA [34]. APAs have been reported in plants, in relation to flowering time control pathways [76], seed dormancy [77], and stress responses [78, 79]. It has been exhibited through global profiling methods that plants exploit APA for diversity generation in their transcriptomes. Through genome-wide analysis in Arabidopsis, the HLP1 protein was identified to regulate the pre-mRNA 3′-end processing and targets APA. It was enriched at transcripts involved in metabolism and flowering [76]. Another genome-wide study in Arabidopsis showed that the CPSF30 is associated with APA in response to oxidative stress [80]. The Plant APA is a recently developed database for the visualization and analysis of APA [81]. The role of AS in plant development and response to biotic and abiotic stresses is summarized in Table 2.
2.8 Modifications in mRNA
Recently, modifications of mRNA with N6-methyladenosine (m6A), 5-methylcytosine (m5C), and pseudouridine (ψ) have been revealed through technical advances. The m6A mRNA was the first internal mRNA modification to be identified. Due to its abundance it has been easily detected through bulk mRNA analysis, and NGS approaches have allowed the mapping of its locations [36]. Recently, transcriptome-wide m6A profiling of rice callus and leaf [35] and shoot [37] have been reported. There are, however, other RNA methylation events that have been found in other organisms and are thought to occur in plants. Subsequent studies will reveal their frequency of occurrence in plants and if they have any role in development or response to biotic/abiotic stresses.
2.9 Stress Response Mechanism and the Cytoplasmic RNA-Containing Granules
It is important in transcriptomics to study the mRNAs that are translated, degraded, or stored temporarily during stress [97]. Based on the environmental or developmental conditions, the messenger ribonucleoprotein complexes (mRNPs) are formed through transcribed mRNA. While the polysome-associated mRNAs are translated, the non-translated mRNAs are localized on either the mRNA processing body (PB) or stress granules (SG), which are cytoplasmic mRNP granules. The PB (identified in yeast and mammals) contains RNA decay machinery for destroying unwanted mRNA in the 5′-3′ direction. The SG store the non-translated mRNA that is stalled during initiation of translation and under stress conditions that cause the SG numbers to increase and accumulate. Several studies have suggested that SGs and PBs are an essential cytoplasmic structure that control gene expression during plant stress responses [98, 99].
3 microRNAs (miRNAs) and Small Interfering RNAs (siRNAs)
microRNAs (miRNAs, 19–25 nt) and small interfering RNAs (siRNAs, 21–22 nt) are small non-coding RNAs with important regulatory functions. Though miRNAs and siRNAs share a number of features in size, structure, and molecular function, they differ in biogenesis pathway and precursor structure [39, 49, 100, 101]. Both, miRNAs and siRNAs are capable of producing a gene silencing effect at the post-transcriptional and transcriptional (epigenetic regulation) levels [38, 47, 102]. In contrast to miRNAs that are derived from either double-stranded or hairpin-like (60–70 nt) RNA precursors in almost all eukaryotes [38], siRNAs are generated from long double-stranded RNAs [103]. The miRNAs are endogenous, encoded by the host genome, while siRNAs can be exogenous or endogenous in origin. The former is originally derived from the transcription of viruses, transposons, repetitive DNA sequences, or transgene trigger [104, 105]. The miRNAs have numerous targets and regulate the expression of large numbers of target mRNAs. In contrast, the siRNAs are specific and mostly regulate the same genes they originate from [106]. Another major difference between miRNAs and siRNAs is that siRNAs base-pair to their target gene and exert targeted gene knockdown through the siRNA-induced mRNA cleavage, translational repression, and DNA methylation, whereas the former are partially complementary to target mRNAs and mediate post-transcriptional gene regulation through either mRNA cleavage or translational repression [47, 106].
3.1 Biogenesis of miRNAs in Plants
miRNAs are evolutionary highly conserved RNA molecules [39]. In the nucleus, miRNAs are transcribed by RNA polymerase II into primary miRNA (pri-miRNA), which are capped and polyadenylated. Subsequently, the pri-miRNAs are processed by the ribonuclease III enzyme, Dicer like 1 (DCL1), in the Dicer family, to a smaller stem-loop structure called precursor miRNAs (pre-miRNAs) [38, 39]. The pre-miRNAs are further processed again by DCL1 into the mature miRNA: miRNA* duplexes that carry 5′ phosphates and 2-nt overhangs on their 3′ end that are not fully complementary [40]. Next, they are exported from the nucleus to the cytoplasm by HASTY, the Arabidopsis homolog of exportin 5 in animals [41, 42]. In the cytoplasm, the mature miRNA strand, the so-called guide strand, is subsequently incorporated into the RNA-induced silencing complex (RISC, or miRISC for miRNA-containing RISC), where it is loaded onto a member of the Argonaute protein family and guides effector RISC to the target mRNA. The miRNA*, which is derived from the other strand known as the passenger strand, can be either degraded or functional [43, 44]. The key studies on miRNA and siRNA biogenesis are summarized in Table 1.
3.2 Biogenesis of siRNAs in Plants
In contrast to miRNAs, siRNAs are derived from double-stranded RNAs originating from protein-coding genes, non-coding transcripts, and transposable elements with perfect base-pairing complementarity to target mRNAs [47]. The DCL2, DCL3, and DCL4 sequentially cleave the long duplex structure into 22-nt, 24-nt, and 21-nt siRNAs, respectively [39, 45, 46]. Short RNA duplexes are similar to the miRNA:miRNA* duplexes but are fully based-paired along the length. Once small RNA duplexes are generated, they are also loaded on an Argonaute protein. Next, the passenger strand is removed and the remainder forms the effector RNAi complex RISC (siRISC, which is loaded with siRNA [39]). Besides the RNA-dependent RNA polymerase 2 and 6 (RDR2, RDR6), SUPPRESSOR OF GENE SILENCING 3 (SGS3), and dsRNA-BINDING 4 (DRB4) are also implicated in siRNA biogenesis [107, 108].
Endogenous siRNAs in plants have been characterized based on their characteristics and biogenesis pathways into hairpin-derived siRNAs (hp-siRNAs, 21–24 nt), natural antisense siRNAs (nat-siRNAs, 21–24 nt), secondary siRNAs, and heterochromatic siRNAs (het-siRNAs, 24 nt) [47,48,49]. Secondary siRNAs could be subclassified into trans-acting siRNAs (tasiRNAs), phased siRNAs (phasiRNAs), and epigenetically activated siRNAs (easiRNAs) [45, 47].
3.3 Functions of miRNAs and siRNAs in Plants
A large number of miRNAs have been identified and characterized in plant genomes with diverse functions. Several miRNAs have been identified to have a crucial regulatory role in a wide range of biological processes in diverse plant species including but not limited to, cell signaling, differentiation, heterosis, DNA damage repair, hormone signaling, organ development, and response to biotic and abiotic stresses (reviewed in [47, 109, 110]).
The siRNA are widespread and numerous endogenous siRNAs have been discovered in plants by deep sequencing. The results of several studies revealed that siRNA is implicated in the morphological control of leaf [111]; developmental timing (temporal regulation) [112, 113]; hormone signaling [114]; fertility and reproductive function [115]; maintenance of genomic integrity, and developmental patterning [116].
Furthermore, both miRNAs and siRNAs, as part of multi-layered sophisticated defense mechanisms, play pivotal roles in regulating immune responses to environmental stresses [109, 116,117,118]. The role of miRNAs and siRNAs in plant development, and response to biotic and abiotic stresses is summarized in Table 2.
3.4 Role of miRNAs in Plant Stress Responses
Plants as sessile organisms are continuously and simultaneously challenged by multiple biotic and abiotic stressors. They have evolved sophisticated defense mechanisms and intricate regulatory networks to perceive their attackers. The miRNAs as critical regulators of gene expression, fine-tune defense responses by regulating the expression of their stress/defense-related target genes. Thousands of miRNAs responsive to biotic and abiotic stresses and their targets have been identified in diverse plant species using deep sequencing technologies and degradome sequencing, respectively [119].
3.5 miRNAs in Biotic Stress
miRNAs orchestrate plant adaptive response to pathogens as the key players in hormone signaling pathways and plant immunity [89, 95, 117, 120]. Thus far, numerous biotic stress-responsive miRNAs have been identified in different plants [93, 94, 120,121,122]. Recently, several miRNAs such as miR156, miR159, miR172, miR319, and miR393 were found to be responsive to Cucumber mosaic virus in tomato [93]. In response to the fungal pathogen Verticillium longisporum, several miRNAs including the miR319 family have been identified in oilseed rape (Brassica napus) [121]. Flagellin-22 triggered miR393 expression and conferred resistance to Pseudomonas syringae in Arabidopsis [95]. In line with this, several miRNAs have been identified to be differentially expressed in Arabidopsis in response to a bacterial pathogen, P. syringae pv. tomato, using deep sequencing [122]. The results of these studies indicate that miRNAs target genes that are related to hormone signaling pathways and negatively regulate their target genes to enhance plant resistance to bacterial infection. The miR393 was reported to target TIR1 (Transport Inhibitor Response 1) and its functional paralogs, AFB2 and AFB3 (Auxinsignaling F-Box proteins 2 and 3). Whereas miR160 and miR167 target ARF8, ARF10, ARF16, and ARF17 to repress auxin signaling. Therefore, miRNAs confer a high degree of resistance to the bacterial pathogen P. syringae through miRNA-mediated suppression of auxin signaling [95, 122]. A recent study found that there is a negative correlation between miR319 and its target TEOSINTE BRANCHED1/CYCLOIDEA/PROLIFERATING CELL FACTOR 4 (TCP4) in response to root-knot nematode (RKN, Meloidogyne incognita) invasion in tomato (Solanum lycopersicum var Castlemart) [94]. The TCP genes encode plant-specific transcription factors that positively regulate jasmonic acid (JA) biosynthesis genes and JA levels in plants. The expression of miR319b was repressed, while the expression of its target, TCP4, was increased under JA treatment. On the other hand, the expression levels of all miR319-targeted TCP genes were significantly decreased in transgenic tomato plants overexpressing miR319 [94]. The results of this study showed that miR319 negatively regulates RKN resistance and JA-mediated miR319 confers systemic resistance to RKN infection. Additionally, crosstalk between miRNAs and hormone signaling pathways was revealed.
Altogether, miRNAs are responsive to a broad range of biotic stresses and confer resistance to plants against pathogens through complex mechanisms such as miRNA-mediated hormone signaling and/or hormone-mediated miRNA regulation.
3.6 miRNAs in Abiotic Stress
Several studies have shown that miRNA expression is regulated in response to a wide array of abiotic stresses such as drought, salinity, cold, heat, heavy metals, nutrients, oxidation, hypoxia, and UV-B in an miRNA-, stress-, tissue-, and genotype-dependent manner (reviewed in [109, 118, 119, 123]). The miR319 miRNA family is one of the most conserved and ancient miRNA families in plants [83]. miR319 was found to be induced in response to not only different biotic stresses, e.g., bacteria, fungi, viruses, and nematodes [93, 94, 121, 122], but also to multiple abiotic stress factors such as drought, salinity, cold, and aluminum [82, 83, 124,125,126,127]. Constitutive expression of miR319 significantly increased salt and drought tolerance in transgenic creeping bentgrass (Agrostis stolonifera) [82, 83]. Hence, miR319 can be a general multi-stress responsive miRNA. A recent study in Arabidopsis revealed that three miRNAs from the miR319 family, i.e., miR319a/b and miR319b.2, are associated with several abiotic stresses [84]. Interestingly, miR319a and miR319b exhibited the same patterns of expression in response to copper, cadmium, and sulfur deficiency conditions as well as salt stress. Moreover, the expression of miR319b.2 was augmented in response to copper, cadmium, and sulfur deficiency stresses, whilst it was down-regulated in response to drought, heat, and salinity. Similarly, the expression levels of miRNA319a/b were increased under metal stresses. On the other hand, miRNA319a/b was notably up-regulated under salinity stress [84]. These results suggest that miRNAs appear to have a complex regulatory role and orchestrate defense responses to a wide range of abiotic stresses through different regulatory networks.
In addition to the miR319 family, the miR169 family is another highly conserved family that plays a critical role in response to abiotic stresses in several plant species. The results of several studies indicated that miR169 plays an important role in response to several abiotic stresses including drought, salt, cold, abscisic acid, nitrogen starvation, and phosphate deficiency [85, 86, 88, 128,129,130].
The miR169 was repressed under drought and phosphate deficiency in Arabidopsis and nitrogen-starvation in Arabidopsis and maize [85,86,87,88]. In contrast, miR169 was up-regulated in response to cold stress in different plant species [130]. Overexpression of miR169 enhanced drought tolerance in Solanum lycopersicum [122]. The miR169 family members are up-regulated in Arabidopsis, maize, and soybean under cold, drought, and salinity stresses [129]. Their results showed that stress-induced miR169 promotes early flowering by repressing the AtNF-YA transcription factor [129]. Conversely, a recent study in Solanum tuberosum exhibited that downregulation of miR169 enhanced drought resistance through over-expression of StNF-YA genes [90]. Nuclear factor Y (NF-Y) transcription factors are the main targets of miR169. NF-Y encodes a CCAAT-binding transcription factor [86]. These findings revealed that there is a negative correlation between the expression of miR169 and its target NF-YA genes, and the miR169 regulates negatively and/or positively their target expression at the post-transcriptional level to enhance stress tolerance in different plant species [90, 129]. Taken together, these results suggest that multi-stress responsive miR169 may orchestrate the expression of its target genes in a host- and stress-dependent manner. Different signaling pathways are mediated by miR169 and there is a complex crosstalk between the miR169 family members and their target transcription factors.
3.7 Role of siRNAs in Plant Stress Responses
Several studies have indicated that natural antisense transcripts (NATs) play a vital role in the regulation of defense signaling pathways and are involved in the response to different environmental stimuli by orchestrating corresponding NAT mRNAs [91, 96, 131].
3.8 siRNAs in Biotic Stress
nat-siRNAATGB2, the first endogenous siRNA, was specifically expressed in Arabidopsis thaliana leaves challenged with a virulent form of the bacterial pathogen Pseudomonas syringae pv. tomato (Pst) carrying effector avrRpt2 [96]. The nat-siRNAATGB2 and its antisense target PPRL were transcribed in the opposite direction and a negative correlation was observed between the nat-siRNA and its antisense target expression in response to P. syringae infection. nat-siRNAATGB2 induced resistance by repressing the expression of PPRL as a negative regulator of the RPS2-mediated resistance against Pst that triggered hypersensitive response (HR) and cell death by recognition of PstavrRpt2 effector [96]. A novel class of siRNAs known as long siRNAs (lsiRNAs, 30–40 nt) was discovered by Katiyar-Agarwal and colleagues in 2007. The results of this study revealed that lsiRNAs are stress-induced and expressed by a bacterial infection. AtlsiRNA-1 was remarkably and specifically over-expressed in response to Pst carrying effector avrRpt2. Overexpression of AtlsiRNA-1 repressed the expression of its target AtRAPmRNA and induced resistance by silencing AtRAP as a negative regulator of plant defense [132].
Using deep sequencing, 17,000 unique siRNAs corresponding to cis-NATs have been found in Arabidopsis thaliana in response to biotic stress in the form of bacterial infection and abiotic stresses in the form of cold, drought, and salt [72]. The results of these studies suggest that siRNAs are stress-induced and regulate defense response in plants through the reprogramming of gene expression.
3.9 siRNAs in Abiotic Stress
A large number of nat-siRNAs have been identified in rice (Oryza sativa cv. japonica) in response to cold, drought, and salt [133]. In Arabidopsis thaliana, the 21-nt nat-siRNAs repressed the expression of Δ1-pyrroline-5-carboxylate dehydrogenase (P5CDH), a stress-related gene, through mRNA cleavage under salt stress. Down-regulation of the P5CDH led to proline accumulation. Proline is as an osmoprotectant and ROS quencher that helps to tolerate salt stress, although under-expression of P5CDH instigated increased ROS production [91]. One study has indicated that four siRNAs were differentially expressed in response to cold, heat, salt, and drought in wheat (Triticum aestivum) [92]. The siRNA 002061_0636_3054.1 was significantly repressed by heat, salt, and drought stress; 005047_0654_1904.1 was strongly over-expressed in response to cold, whilst down-regulated in response to heat, salt, and drought stress; 080621_1340_0098.1 was faintly induced by cold and repressed by heat but not by either salt or drought stress; and 007927_0100_2975.1 was down-regulated by cold, salt, and drought stress [92]. Further, their results revealed that the four siRNAs were preferentially expressed in spikes and uniformly expressed in leaves and roots [92]. Therefore, the results of these studies exhibited that nat-siRNAs respond to biotic and abiotic stress conditions in a stress-specific and developmental stage-dependent manner.
4 Conclusions and Future Prospects
Alternative splicing, miRNAs, and siRNAs play critical regulatory roles in modulating gene expression during plant growth and development, in response to biotic and abiotic stresses, and plant adaptation to an ever-changing environment. Several small regulatory molecules have been identified to have versatile functions in food and feed crops. Manipulating expression levels of miRNAs and siRNAs in economically important crop plants can be an effective strategy to improve desirable traits, stress tolerance, and resiliency in response to environmental stress and pathogen attack in plants. Therefore, miRNAs and siRNAs can be used as new targets for developing trait-improved crop plants and improving plant tolerance to stresses. Two powerful genome editing tools, TALENs and CRISPR/Cas, can be used for targeted genome editing and knockdown/knockout of small RNAs.
In addition to the identification of small regulatory molecules and their transcriptional profiling, it is indispensable for scientific communities to understand the regulatory mechanisms of small RNAs that orchestrate cellular functions and adaptation to environmental stresses to minimize the unintended side effects in modified plants.
References
World Population Prospect: The 2015 Revision (2015) United Nations, Department of Economic and Social Affairs, Population Division. [Online] Available at: http://www.un.org/en/development/desa/news/population/2015-report.html. Accessed 7 Aug 2017
Valin H, Sands RD, van der Mensbrugghe D, Nelson GC, Ahammad H, Blanc E, Bodirsky B et al (2014) The future of food demand: understanding differences in global economic models. Agric Econ 45:51–67
Gilbert N (2014) Cross-bred crops get fit faster. Nature 513:292
Kissoudis C, van de Wiel C, RGF V, van der Linden G (2014) Enhancing crop resilience to combined abiotic and biotic stress through the dissection of physiological and molecular crosstalk. Front Plant Sci 5:207
Gepts P (2002) A comparison between crop domestication, classical plant breeding, and genetic engineering. Crop Sci 42:1780–1790
Nejat N, Cahill DM, Vadamalai G, Ziemann M, Rookes J, Naderali N (2015) Transcriptomics-based analysis using RNA-seq of the coconut (Cocos nucifera) leaf in response to yellow decline phytoplasma infection. Mol Gen Genomics 290:1899–1910
Nejat N, Mantri N (2017) Plant immune system: crosstalk between responses to biotic and abiotic stresses the missing link in understanding plant defence. Curr Issues Mol Biol 23:1
Parker RM, Barnes NM (1999) mRNA: detection by in situ and northern hybridization. Methods Mol Biol 106:247–283
Weis JH, Tan SS, Martin BK, Wittwer CT (1992) Detection of rare mRNAs via quantitative RT-PCR. Trends Genet 8:263–264
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193
Deepak SA, Kottapalli KR, Rakwal R, Oros G, Rangappa KS, Iwahashi H et al (2007) Real-time PCR: revolutionizing detection and expression analysis of genes. Curr Genomics 8:234–251
Nejat N, Vadamalai G, Dickinson M (2012) Expression patterns of genes involved in the defence and stress response of Spiroplasmacitri infected Madagascar Periwinkle Catharanthusroseus. Int J Mol Sci 13:2301–2313
Schena M, Shalon D, Davis RW, Brown PO (1995) Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–470
Harmer SL, Hogenesch JB, Straume M, Chang H-S, Han B, Zhu T et al (2000) Orchestrated transcription of key pathways in Arabidopsis by the circadian clock. Science 290:2110–2113
Mantri NL, Ford R, Coram TE, Pang ECK (2007) Transcriptional profiling of chickpea genes differentially regulated in response to high-salinity, cold and drought. BMC Genomics 8:303
Mantri NL, Ford R, Coram TE, Pang ECK (2010) Evidence of unique and shared responses to major biotic and abiotic stresses in chickpea. Environ Exp Bot 69:286–292
Zik M, Irish VF (2003) Global identification of target genes regulated by APETALA3 and PISTILLATA foral homeotic gene action. Plant Cell 15:207–222
Mardis ER (2008) Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet 9:387–402
Margulies M, Egholm M, Altman WE et al (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Mortazavi A, William BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628
ENCODE (2015) Research applications and users meeting. Bolger Center, Potomac
Rands CM, Meader S, Ponting CP, Lunter G (2014) 8.2% of the human genome is constrained: variation in rates of turnover across functional elements classes in the human lineage. PLoS Genet 10:e1004525
ENCODE Project Consortium (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489:57–74
Ezkurdia I, Juan D, Rodriguez JM, Frankish A, Diekhans M, harrow J, Vazquez J (2014) Multiple evidence strands suggest that there may be as few as 19000 human protein-coding genes. Hum Mol Genet 23:5866–5878
Nejat N, Mantri N (2017) Emerging roles of long non-coding RNAs in plant response to biotic and abiotic stresses. Crit Rev Biotechnol 20:1–13. https://doi.org/10.1080/07388551.2017.1312270
Isshiki M, Tsumoto A, Shimamoto K (2006) The serine/arginine-rich protein family in rice plays important roles in constitutive and alternative splicing of pre-mRNA. Plant Cell 18:146–158
Reddy AS (2007) Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu Rev Plant Biol 58:267–294
Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ (2013) Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14:153–165
Wang J, Smith PJ, Krainer AR, Zhang MQ (2005) Distribution of SR protein exonic splicing enhancer motifs in human protein-coding genes. Nucleic Acids Res 33:5053–5062
Wang Z, Burge CB (2008) Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 14:802–813
Le Hir H, Andersen GR (2008) Structural insights into the exon junction complex. Curr Opin Struct Biol 18:112–119
Schaal TD, Hertel KJ, Reed R, Maniatis T (2005) Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc Natl Acad Sci U S A 102:5002–5007
Dujardin G, Lafaille C, Petrillo E, Buggiano V, LIG A, Fiszbein A, MAG H, Moreno NN, Muñoz MJ, Alló M, Schor IE (2013) Transcriptional elongation and alternative splicing. Biochim Biophys Acta 1829:134–140
Xing D, Li QQ (2011) Alternative polyadenylation and gene expression regulation in plants. Wiley Interdiscip Rev RNA 2:445–458
Li Y, Wang X, Li C, Hu S, Yu J, Song S (2014) Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol 11:1180–1188
Gilbert WV, Bell TA, Schaening C (2016) Messenger RNA modifications: form, distribution, and function. Science 352:1408–1412
Shen L, Liang Z, Gu X, Chen Y, ZWN T, Hou X, Cai WM, Dedon PC, Liu L, Yu H (2016) N 6-Methyladenosine RNA modification regulates shoot stem cell fate in arabidopsis. Dev Cell 38:186–200
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bologna NG, Voinnet O (2014) The diversity, biogenesis, and activities of endogenous silencing small RNAs in Arabidopsis. Annu Rev Plant Biol 65:473–503
Jones-Rhoades WM, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 57:19–53
Bollman KM, Aukerman MJ, Park MY, Hunter C, Berardini TZ, Poethig RS (2003) HASTY, the Arabidopsis ortholog of exportin 5/MSN5, regulates phase change and morphogenesis. Development 130:1493–1504
Park MY, Wu G, Gonzalez-Sulser A, Vaucheret H, Poethig RS (2005) Nuclear processing and export of microRNAs in Arabidopsis. Proc Natl Acad Sci U S A 102:3691–3696
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136:215–233
Meister G (2013) Argonaute proteins: functional insights and emerging roles. Nat Rev Genet 14:447–459
Liu YX, Wang M, Wang XJ (2014) Endogenous small RNA clusters in plants. Genomics Proteomics Bioinformatics 12:64–71
Nagano H, Fukudome A, Hiraguri A, Moriyama H, Fukuhara T (2014) Distinct substrate specificities of Arabidopsis DCL3 and DCL4. Nucleic Acids Res 42:1845–1856
Borges F, Martienssen RA (2015) The expanding world of small RNAs in plants. Nat Rev Mol Cell Biol 16:727–741
Chapman EJ, Carrington JC (2007) Specialization and evolution of endogenous small RNA pathways. Nat Rev Genet 8:884–896
Vazquez F (2006) Arabidopsis endogenous small RNAs: highways and byways. Trends Plant Sci 11:460–468
Fujiwara N, Masuda S, Shiki T (2012) mRNA biogenesis in the nucleus and its export to the cytoplasm. INTECH Open Access Publisher, London
de la Mata M, Kornblihtt AR (2006) RNA polymerase II C-terminal domain mediates regulation of alternative splicing by SRp20. Nat Struct Mol Biol 13:973–980
Huang Y, Li W, Yao X, Lin QJ, Yin JW, Liang Y, Heiner M, Tian B, Hui J, Wang G (2012) Mediator complex regulates alternative mRNA processing via the MED23 subunit. Mol Cell 45:459–469
Lareau LF, Green RE, Bhatnagar RS, Brenner SE (2004) The evolving roles of alternative splicing. Curr Opin Struct Biol 14:273–282
Barrass JD, Beggs JD (2003) Splicing goes global. Trends Genet 19:295–298
Marquez Y, Brown JW, Simpson C, Barta A, Kalyna M (2012) Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis. Genome Res 22:1184–1195
Sablok G, Gupta PK, Baek JM, Vazquez F, Min XJ (2011) Genome-wide survey of alternative splicing in the grass Brachypodiumdistachyon: a emerging model biosystem for plant functional genomics. Biotechnol Lett 33:629–636
Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, Zhuang R, Lu Z, He Z, Fang X, Chen L (2010) Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res 20:646–654
Shen Y, Zhou Z, Wang Z, Li W, Fang C, Wu M, Ma Y, Liu T, Kong LA, Peng DL, Tian Z (2014) Global dissection of alternative splicing in paleopolyploid soybean. Plant Cell 26:996–1008
Barbazuk WB, Fu Y, McGinnis KM (2008) Genome-wide analyses of alternative splicing in plants: opportunities and challenges. Genome Res 18:1381–1392
Ali GS, Reddy ASN (2008) Regulation of alternative splicing of pre-mRNAs by stresses. Curr Top Microbiol Immunol 326:257–275
James AB, Syed NH, Bordage S, Marshall J, Nimmo GA, Jenkins GI, Herzyk P, Brown JW, Nimmo HG (2012) Alternative splicing mediates responses of the Arabidopsis circadian clock to temperature changes. Plant Cell 24:961–981
Kumar S, Asif MH, Chakrabarty D, Tripathi RD, Trivedi PK (2011) Differential expression and alternative splicing of rice sulphate transporter family members regulate sulphur status during plant growth, development and stress conditions. Funct Integr Genomics 11:259–273
Wang BB, Brendel V (2004) The ASRG database: identification and survey of Arabidopsis thaliana genes involved in pre-mRNA splicing. Genome Biol 5:1
Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, Li D, Meyer T, Muller R, Ploetz L, Radenbaugh A (2007) The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36:D1009–D1014
Wang BB, Brendel V (2006) Genomewide comparative analysis of alternative splicing in plants. Proc Natl Acad Sci 103:7175–7180
Salvo SA, Hirsch CN, Buell CR, Kaeppler SM, Kaeppler HF (2014) Whole transcriptome profiling of maize during early somatic embryogenesis reveals altered expression of stress factors and embryogenesis-related genes. PLoS One 9:e111407
Zhang Z, Komatsu S (2000) Molecular cloning and characterization of cDNAs encoding two isoforms of ribulose-1, 5-biosphosphate carboxylase/oxygenaseactivase in rice (Oryzasativa L.) J Biochem 128:383–389
Razem FA, El-Kereamy A, Abrams SR, Hill RD (2006) The RNA-binding protein FCA is an abscisic acid receptor. Nature 439:290–294
Kaashyap M, Ford R, Bohra A, Kuvalekar A, Mantri N (2017) Improving salt tolerance of chickpea using modern genomics tools and molecular breeding. Curr Genomics 18:557–567. https://doi.org/10.2174/1389202918666170705155252
Nakaminami K, Matsui A, Shinozaki K, Seki M (2012) RNA regulation in plant abiotic stress responses. Biochim Biophys Acta 1819:149–153
Matsukura S, Mizoi J, Yoshida T, Todaka D, Ito Y, Maruyama K, Shinozaki K, Yamaguchi-Shinozaki K (2010) Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol Gen Genomics 283:185–196
Zhang YM, Yan YS, Wang LN, Yang K, Xiao N, Liu YF, YP F, Sun ZX, Fang RX, Chen XY (2012) A novel rice gene, NRR responds to macronutrient deficiency and regulates root growth. Mol Plant 5:63–72
Dinesh-Kumar SP, Baker BJ (2000) Alternatively spliced N resistance gene transcripts: their possible role in tobacco mosaic virus resistance. Proc Natl Acad Sci 97:1908–1913
Zhang XC, Gassmann W (2003) RPS4-mediated disease resistance requires the combined presence of RPS4 transcripts with full-length and truncated open reading frames. Plant Cell 15:2333–2342
Egawa C, Kobayashi F, Ishibashi M, Nakamura T, Nakamura C, Takumi S (2006) Differential regulation of transcript accumulation and alternative splicing of a DREB2 homolog under abiotic stress conditions in common wheat. Genes Genet Syst 81:77–91
Zhang Y, Gu L, Hou Y, Wang L, Deng X, Hang R, Chen D, Zhang X, Zhang Y, Liu C, Cao X (2015) Integrative genome-wide analysis reveals HLP1, a novel RNA-binding protein, regulates plant flowering by targeting alternative polyadenylation. Cell Res 25:864
Cyrek M, Fedak H, Ciesielski A, Guo Y, Sliwa A, Brzezniak L, Krzyczmonik K, Pietras Z, Kaczanowski S, Liu F, Swiezewski S (2016) Seed dormancy in Arabidopsis is controlled by alternative polyadenylation of DOG1. Plant Physiol 170:947–955
Motion GB, Amaro TM, Kulagina N, Huitema E (2015) Nuclear processes associated with plant immunity and pathogen susceptibility. Brief Funct Genomics 14:243–252
Tao P, Huang X, Li B, Wang W, Yue Z, Lei J, Zhong X (2014) Comparative analysis of alternative splicing, alternative polyadenylation and the expression of the two KIN genes from cytoplasmic male sterility cabbage (Brassica oleracea L. var. capitata L.) Mol Gen Genomics 289:361–372
Thomas PE, Wu X, Liu M, Gaffney B, Ji G, Li QQ, Hunt AG (2012) Genome-wide control of polyadenylation site choice by CPSF30 in Arabidopsis. Plant Cell 24:4376–4388
Wu X, Zhang Y, Li QQ (2016) Plant APA: a portal for visualization and analysis of alternative polyadenylation in plants. Front Plant Sci 7:889
Zhou M, Li D, Li Z, Hu Q, Yang C, Zhu L, Luo H (2013) Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol 161:1375–1391
Zhou M, Luo H (2014) Role of microRNA319 in creeping bentgrass salinity and drought stress response. Plant Signal Behav 9:e28700
Barciszewska-Pacak M, Milanowska K, Knop K, Bielewicz D, Nuc P et al (2015) Arabidopsis microRNA expression regulation in a wide range of abiotic stress responses. Front Plant Sci 6:410
Hsieh LC, Lin SI, Shih AC, Chen JW, Lin WY, Tseng CY, Li WH, Chiou TJ (2009) Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol 151:2120–2132
Li WX, Oono Y, Zhu J, HeX J, JM W, Iida K et al (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20:2238–2251
Xu Z, Zhong S, Li X, Li W, Rothstein SJ, Zhang S, Bi Y, Xie C (2011) Genome-wide identification of microRNAs in response to low nitrate availability in maize leaves and roots. PLoS One 6:e28009
Zhao M, Ding H, Zhu JK, Zhang F, Li WX (2011) Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol 190:906–915
Zhang W, Gao S, Zhou X, Chellappan P, Chen Z, Zhou X, Zhang X et al (2011) Bacteria-responsive microRNAs regulate plant innate immunity by modulating plant hormone networks. Plant Mol Biol 75:93–105
Yang J, Zhang N, Zhou X, Si H, Wang D (2016) Identification of four novel stu-miR169s and their target genes in Solanum tuberosum and expression profiles response to drought stress. Plant Syst Evol 302:55–66
Borsani O, Zhu JH, Verslues PE, Sunkar R, Zhu JK (2005) Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 123:1279–1291
Yao Y, Ni Z, Peng H, Sun F, Xin M, Sunkar R et al (2010) Non-coding small RNAs responsive to abiotic stress in wheat (Triticumaestivum L.) Funct Integr Genomics 10:187–190
Feng JL, Liu SS, Wang MN, Lang QL, Jin CZ (2014) Identification of microRNAs and their targets in tomato infected with Cucumber mosaic virus based on deep sequencing. Planta 240:1335–1352
Zhao W, Li Z, Fan J, Hu C, Yang R, Qi X, Chen H et al (2015) Identification of jasmonic acid-associated microRNAs and characterization of the regulatory roles of the miR319/TCP4 module under root-knot nematode stress in tomato. J Exp Bot 66:4653–4667
Navarro L, Dunoyer P, Jay F, Arnold B, Dharmasiri N, Estelle M, Voinnet O, Jones JD (2006) A plant miRNA contributes to antibacterial resistance by repressing auxinsignaling. Science 312:436–439
Katiyar-Agarwal S, Morgan R, Dahlbeck D, Borsani O, Villegas A, Zhu JK, Staskawicz BJ, Jin HL (2006) A pathogen-inducible endogenous siRNA in plant immunity. Proc Natl Acad Sci USA 103:18002–18007
Urano K, Kurihara Y, Seki M, Shinozaki K (2010) ‘Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr Opin Plant Biol 13:132–138
Goeres DC, Van Norman JM, Zhang W, Fauver NA, Spencer ML, Sieburth LE (2007) Components of the Arabidopsis mRNA decapping complex are required for early seedling development. Plant Cell 19:1549–1564
Weber C, Nover L, Fauth M (2008) Plant stress granules and mRNA processing bodies are distinct from heat stress granules. Plant J 56:517–530
Mattick JS, Makunin IV (2006) Non-coding RNA. Hum Mol Genet 15:R17–R29
Zamore PD, Haley B (2005) Ribo-genome: the big world of smallRNAs. Science 309:1519–1524
Rogers K, Chen X (2013) Biogenesis, turnover, and mode of action of plant MicroRNAs. Plant Cell 25:2383–2399
Eamens A, Smith NA, Curtin SJ, Wang MB, Waterhouse PM (2009) The Arabidopsis thaliana double-stranded RNA binding protein DRB1 directs guide strand selection from microRNA duplexes. RNA 15:2219–2235
Carthew R, Sontheimer EJ (2009) Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655
Pontier D, Yahubyan G, Vega D, Bulski A, Saez-Vasquez J, Hakimi MA, Lerbs-Mache S, Colot V, Lagrange T (2005) Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev 19:2030–2040
Mack GS (2007) MicroRNA gets down to business. Nat Biotechnol 25:631–638
Hiraguri A et al (2005) Specific interactions between Dicer-like proteins and HYL1/DRB-family dsRNA-binding proteins in Arabidopsis thaliana. Plant Mol Biol 57:173–188
Yoshikawa M, Peragine A, Park M-Y, Poethig RS (2005) A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev 19:2164–2175
Mantri N, Baskar N, Ford R, Pang ECK, Pardeshi V (2013) The role of micro-ribonucleic acids in legumes with a focus on abiotic stress response. Plant Genome 6:3
Wahid F, Shehzad A, Khan T, Kim YY (2010) MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochem Biophys Acta 1803:1231–1243
Adenot X, Elmayan T, Lauressergues D, Boutet S, Bouche N, Gasciolli V, Vaucheret H (2006) DRB4-dependent TAS3 trans-acting siRNAs control leaf morphology through AGO7. Curr Biol 16:927–932
Poething RS (2009) Small RNAs and developmental timing in plants. Curr Opin Genet Dev 19:374–378
Talmor-Neiman M, Stav R, Klipcan L, Buxdorf K, Baulcombe DC, Arazi T (2006) Identification of trans-acting siRNAs in moss and an RNA-dependent RNA polymerase required for their biogenesis. Plant J 48:511–521
Zubko E, Meyer P (2007) A natural antisense transcript of the Petunia hybrida Sho gene suggests a role for an antisense mechanism in cytokinin regulation. Plant J 52:1131–1139
Ron M, Saez MA, Williams LE, Fletcher JC, McCormick S (2010) Proper regulation of a sperm-specific cis-nat-siRNA is essential for double fertilization in Arabidopsis. Genes Dev 2010(24):1010–1021
Lelandais-Brière C, Sorin C, Declerck M, Benslimane A, Crespi M, Hartmann C (2010) Small RNA diversity in plants and its impact in development. Curr Genomics 11:14–23
Khraiwesh B, Zhu JK, Zhu J (2012) Role of miRNAs and siRNAs in biotic and abiotic stress responses of plants. Biochim Biophys Acta 1819:137–148
Sunkar R, Li YF, Jagadeeswaran G (2012) Functions of microRNAs in plant stress responses. Trends Plant Sci 17:196–203
Zhang B (2015) MicroRNA: a new target for improving plant tolerance to abiotic stress. J Exp Bot 66:1749–1761
Baldrich P, Sun Segundo B (2016) MicroRNAs in rice innate immunity. Rice (N Y) 9:6. https://doi.org/10.1186/s12284-016-0078-5
Shen D, Suhrkamp I, Wang Y, Liu S, Menkhaus J, Verreet JA, Fan L, Cai D (2014) Identification and characterization of microRNAs in oilseed rape (Brassica napus) responsive to infection with the pathogenic fungus Verticillium longisporum using Brassica AA (Brassica rapa) and CC (Brassica oleracea) as reference genomes. New Phytol 204:577–594
Zhang X, Zou Z, Gong P, Zhang J, Ziaf K, Li H, Xiao F, Ye Z (2011) Over-expression of microRNA169 confers enhanced drought tolerance to tomato. Biotechnol Lett 33:403–409
Kumar R (2014) Role of microRNAs in biotic and abiotic stress responses in crop plants. Appl Biochem Biotechnol 174:93–115
Chen L, Wang T, Zhao M, Tian Q, Zhang WH (2012) Identification of aluminum-responsive microRNAs in Medicagotruncatula by genome-wide highthroughput sequencing. Planta 235:375–386
Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019
Thiebaut F, Rojas CA, Almeida KL, Grativol C, Domiciano GC, Lamb CRC, Engler Jde A, Hemerly AS, Ferreira PCG (2012) Regulation of miR319 during cold stress in sugarcane. Plant Cell Environ 35:502–512
Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryzasativa. J Exp Bot 61:4157–4168
Luan M, Xu M, Lu Y, Zhang L, Fan Y, Wang L (2015) Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene 555:178–185
Xu MY, Zhang L, Li WW, Hu XL, Wang MB, Fan YL et al (2014) Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J Exp Bot 65:89–101
Zhang J, Xu Y, Huan Q, Chong K (2009) Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics 10:449
Jin H, Vacic V, Girke T, Lonardi S, Zhu JK (2008) Small RNAs and the regulation of cis-natural antisense transcripts in Arabidopsis. BMC Mol Biol 9:6
Katiyar-Agarwal S, Gao S, Vivian-Smith A, Jin H (2007) A novel class of bacteria-induced small RNAs in Arabidopsis. Genes Dev 21:3123–3134
Zhang X, Xia J, Lii YE, Barrera-Fiqueroa BE, Zhou X, Gao S et al (2012) Genome-wide analysis of plant nat-siRNAs reveals insights into their distribution, biogenesis and function. Genome Biol 13:R20
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Nejat, N., Ramalingam, A., Mantri, N. (2018). Advances in Transcriptomics of Plants. In: Varshney, R., Pandey, M., Chitikineni, A. (eds) Plant Genetics and Molecular Biology. Advances in Biochemical Engineering/Biotechnology, vol 164. Springer, Cham. https://doi.org/10.1007/10_2017_52
Download citation
DOI: https://doi.org/10.1007/10_2017_52
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-91312-4
Online ISBN: 978-3-319-91313-1
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)