
Abstract
Background: Vinflunine is a new-generation microtubule inhibitor, which is currently registered in Europe and in some countries elsewhere as an intravenous formulation for the second-line treatment of transitional urothelial cell carcinoma. On the basis of favourable non-clinical results, the clinical development of an oral formulation was initiated.
Objective: The absolute oral bioavailability was investigated in patients through two consecutive trials: the first trial used soft gelatin capsules filled with solubilized vinflunine (SLCaps), while the second study investigated hard gelatin capsules containing vinflunine as a formulated powder (HPCaps).
Study Design: Each pharmacokinetic trial was conducted according to a randomized cross-over design. Patients received 120 mg/m2 of either oral (SLCaps or HPCaps) or intravenous vinflunine on day 1, followed by the alternate dosing route after a 2-week washout period. Blood samples were collected over 168 hours. A pharmacokinetic analysis was conducted for each patient and route of dosing to derive the absolute oral bioavailability of SLCaps and HPCaps.
Results: A total of 12 and 22 patients were enrolled, for SLCaps and HPCaps, respectively. Vinflunine absorption was rapid for both oral formulations. Blood concentrations peaked at 2.5 hours following oral intake with food, and then decreased similarly to the intravenous profile. The mean absolute bioavailability was high, at 58.3 ± 14.4% (SLCaps) and 57.3 ± 11% (HPCaps), with limited inter-individual variability (coefficient of variation = 25% and 19% for SLCaps and HPCaps, respectively). Neither sequence nor period effects were detected. The gastro-intestinal tolerance was satisfactory. The main drug-related adverse events were asthenia, fatigue, constipation and neutropenia, mostly of grade 1 or 2. No grade 4 and no drug-related serious adverse events were reported.
Conclusion:– The high bioavailability and low inter-individual variability are favourable pharmacokinetic properties, which could be valuable for further clinical development of oral vinflunine.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vinflunine ditartrate is a microtubule-interacting agent, which is a new semi-synthetic vinca alkaloid derivative. The superacid chemistry made possible the selective introduction of two fluorine atoms at the 20′ position on the catharanthine moiety of vinorelbine, which was previously inaccessible by classical chemistry.[1] Vinflunine, or 20′,20′-difluoro-3′,4′-dihydrovinorelbine, was selected from a series of derivatives on the basis of its activity in primary pharmacological screening.[2] High levels of in vivo antitumour activity against experimental tumour models were also confirmed over vinorelbine.[3]
Vinflunine has been developed as an intravenous solution. It is currently registered in Europe and in some countries elsewhere for use in a dosing schedule of 280 or 320 mg/m2 once every 3 weeks, given as a 20-minute intravenous infusion, for the second-line treatment of transitional cell carcinoma of the urothelium. During clinical studies, intravenous vinflunine demonstrated linear pharmacokinetics over the dose range examined (30–400 mg/m2), with an average total blood clearance of 40 L/h, a large volume of distribution (over 2400 L), and a terminal half-life (t1/2z) of about 40 hours.[4,5]
In parallel to the development of the intravenous form of vinflunine, the preclinical development of an oral formulation has been initiated. Vinflunine was first administered orally to mice xenografted with P388 leukemia. In this model, vinflunine exhibited similar and marked antitumoral activity whether given by intravenous, intraperitoneal or oral routes.[3] The optimal dose for the oral route was 2-fold that used for intraperitoneal or intravenous administration. Those results, suggesting that vinflunine has the property to cross physiological barriers and is well absorbed in mice, indicated the feasibility of the oral route in humans.
More and more oral anticancer drugs are available for patient treatment. It was recently reported that over 20 oral anti-neoplastic drugs are currently used in the US and Europe.[6] Oral chemotherapy provides several advantages, including convenience for the patient, due to the ease of administration (compared with intravenous chemotherapy) and the reduced need for hospitalization.[7] This makes oral chemotherapy particularly suitable for a fractionated regimen that requires frequent dosing and prolonged treatment duration. However, patients’ and physicians’ preference for oral chemotherapy is conditioned by its efficacy and tolerance, which must be at least equivalent to those of the corresponding intravenous treatment.[6] In order to achieve this goal for a new anticancer compound candidate for development as an oral therapy, the pharmacokinetic behaviour has to be explored. The evaluation of the absolute bioavailability is particularly pivotal. Low bioavailability values are often associated with high intra- or inter-individual variability, which make cytotoxic compounds difficult to manage. It is therefore necessary to assess the human oral absorption and bioavailability as early as possible during the process of drug development, in order to ensure that both the rate and the extent of absorption are maximal in patients.
We report here the clinical investigation of the oral bioavailability of vinflunine compared with intravenous administration through two consecutive phase I trials. The first study was performed using a preliminary soft-gelatin capsule ‘liquid’ formulation of solubilized vinflunine (SLCaps) in order to assess the feasibility of oral chemotherapy with vinflunine. In the second study, a hard-gelatin capsule ‘powder’ formulation (HPCaps) of vinflunine was similarly evaluated.
The primary objective of both trials was to evaluate the absolute oral bioavailability of vinflunine SLCaps and HPCaps in comparison with intravenous dosing, using a cross-over study design. The secondary objective was to document the safety profile of a single administration of SLCaps or HPCaps.
Patients and Methods
Patient Selection
The study protocols were reviewed by an independent ethics committee and by the National Privacy and Ethics Committee, and studies were performed in accordance with the principles stated in the Declaration of Helsinki.
Male or female patients aged between 18 and 75 years, having signed informed written consent, were eligible to participate in the studies, provided that they had a confirmed malignant solid tumour for which no effective standard therapy was available. If they had progressive disease after conventional therapy, a maximum of three lines of chemotherapy and a minimum of 4 weeks between the last drug administration and study entry was accepted. Previous treatment with nitroso-ureas or mitomycin C was admitted with a maximum of two lines of chemotherapy and at least a 6-week delay before study entry. Prior radiotherapy had to be stopped at least 2 weeks or 3 weeks before study entry with less than 20% and 30% bone marrow reserve, respectively. Performance status (the WHO definition) was to be ≤1, with an estimated life expectancy of at least 3 months. Normal cardiac function, as assessed by ECG, was necessary. Normal renal and hepatic parameters and adequate haematopoietic function were required.
The exclusion criteria included pregnancy or lactation, as well as unwillingness or inability to avoid pregnancy. Also excluded were patients presenting with symptomatic brain involvement or any other major underlying medical condition (serious active infection, diabetes mellitus, unstable cardiovascular condition). Concurrent treatment with another investigational drug within the previous 30 days, and administration of intravenous antibiotics for active infection or use of drugs known to modulate cytochrome P450 (CYP) 3A4 enzymatic activity were additional causes of exclusion. Furthermore, participation in the studies was not permitted for patients presenting with any alteration of the gastro-intestinal tract likely to impact on absorption (malabsorption syndrome, extensive surgery of the stomach or the small bowel, major alteration of gastro-intestinal transit).
Study Design
The two trials were open-label phase I pharmacokinetic studies for patients with various solid tumours. The first one, using vinflunine SLCaps, was conducted in one single centre, while two clinical centres participated in the second trial using HPCaps. Both trials were conducted according to a cross-over design. Eligible patients were randomized to receive either single oral or single intravenous administration of vinflunine on day 1, followed by the alternate formulation on day 15 after a 2-week washout period.
Pharmacokinetic and safety evaluations were performed for 15 days after both administrations. One week after the end of the study observation period, patients could optionally resume treatment with intravenous vinflunine, according to the investigator’s decision.
Pre-Treatment and Follow-Up Assessments
Baseline tumour evaluation, physical examination with vital signs and performance status, chest x-ray, ECG, biochemistry and complete haematology, including a blood differential cell count, as well as a full medical history, were undertaken at study entry. Patients were monitored during the treatment period with a physical examination, weight and height measurement, haematology and biochemistry, and screening for adverse events. An additional ECG was performed on day 15 of the study. The National Cancer Institute (NCI)/Common Toxicity Criteria (CTC) grading scales were used for safety assessment (CTC v.2.0, 1999, for SL Caps; CTC v. 3.0, 2003, for HP Caps).
Study Drug Administration
The same dose level of vinflunine 120 mg/m2 was selected for all intravenous and oral administrations during the study period (day 1 and day 15). This dose level was chosen on the basis of the clinical knowledge of the intravenous form of vinflunine, for which the safety of doses up to 400 mg/m2 once every 3 weeks was evaluated in a dose escalation study[4] and for which a recommended weekly dose was established at 120 or 150 mg/m2 in pre-treated or chemonaive patients, respectively.[8,9]
Oral vinflunine was supplied as two different forms: for the first trial, oral vinflunine was supplied as SLCaps containing solubilized vinflunine ditartrate (20% W/W, expressed as base). The adequate number of SLCaps to be administered was determined from the total dose scheduled, according to the patient’s body surface area (BSA). For each patient, the vinflunine SLCaps were prepared extemporaneously just before the administration. For the second trial, oral vinflunine was supplied as HPCaps in two strengths. Red HPCaps (size 3 and size 1) were used, containing 20 mg or 75 mg of vinflunine ditartrate expressed as base, and a sufficient quantity of excipients. The adequate number of capsules was determined according to the patient’s BSA. Oral vinflunine was administered in the presence of a physician or a nurse at a dose of 120 mg/m2, immediately after intake of a standard breakfast. Capsules had to be rapidly swallowed together with a glass of water without chewing or sucking them.
Intravenous vinflunine was supplied for both trials as a pyrogen-free, sterile parenteral dosage form in 50 mg vials: neutral glass containing 50 mg of ditartrate vinflunine expressed as base as 25 mg/mL in a volume of 2 mL of water for injection. Intravenous vinflunine was administered by infusion in NaCl 0.9% over a 20-minute period with an electric syringe device, after flushing the vein with 250 mL of normal saline. The intravenous line was also rinsed with normal saline just after the end of dosing. As was done with the oral form, intravenous vinflunine was administered immediately after a standard breakfast.
Pharmacokinetic Assessments
Vinflunine pharmacokinetics were evaluated on days 1 and 15 according to a 13-timepoint sampling scheme. The total administered dose, exact time of oral administration, start and end of intravenous infusion, and actual time of blood sampling were recorded. For intravenous administration, blood samples were collected from the contralateral side to the infusion at pre-dose (just before the start of infusion), 10 minutes (mid-infusion), 20 minutes (end of infusion), 40 minutes, then at 1, 1.5, 3, 6, 9, 24, 48, 96 and 168 hours after the start of infusion. For oral administration, initial blood samples were obtained at pre-dose (just before administration), 15 minutes, 30 minutes and 45 minutes after drug intake, then according to the same schedule as intravenous administration.
At each sampling time, two tubes of 1.5 mL of blood was collected, using Venoject®-type heparinized glass tubes, by venipuncture or via an indwelling venous line. The two tubes were then inverted several times to mix blood with heparin and were immediately stored frozen until bioanalysis.
Blood concentrations of vinflunine were quantified using a fully validated high-performance liquid chromatography (HPLC)/UV bioanalytical method[10] with a lower limit of quantification of 2 ng/mL. The within-study precision (coefficient of variation [CV]) and accuracy (mean absolute bias) were lower than 8.9% and 9%, respectively.
Pharmacokinetic Analysis
Patients were evaluable for pharmacokinetics if complete blood sampling was available after both oral and intravenous administrations, and if no vomiting occurred within the 3 hours after oral dosing. The pharmacokinetic analysis was carried out through a model-independent approach using the Kinetica (v. 4.4.1.) program (Thermo Electron Corp., Philadelphia, PA, USA) and the following parameters were obtained for each individual patient:
The maximum blood concentration (Cmax) was estimated directly from the experimental data, and the time to reach Cmax (tmax) was observed. The observed area under the blood concentration-time curve (AUC) values were calculated from time zero to the last quantifiable concentration (AUClast) or to infinite time (AUC∞), according to the mixed linear log-linear trapezoidal rule. The t1/2z was calculated as (equation 1):

where λz is the least-square regression slope of the final part of the log-linear curve. The total blood clearance (CLtot) was obtained from (equation 2):

The terminal volume of distribution (VZ) was calculated as (equation 3):

The absolute oral bioavailability factor (F), was obtained as equation 4:

Statistical Analysis
Analyses were carried out using SAS® 8.2. software (SAS Institute Inc., Cary, NC, USA). Individual values of pharmacokinetic parameters were tabulated per route of administration, and descriptive statistics including mean and CV values were calculated. Pharmacokinetic parameters (AUC∞ and t1/2z) were compared between routes of administration by an analysis of variance (ANOVA) with sequence, period, patient (sequence) and route factors. The comparison was carried out using log-transformed parameters and the α-risk was set at 5%.
Results
Demographics
A total of 12 and 22 patients were enrolled in the vinflunine SLCaps and the HPCaps trials, respectively. Age, WHO performance status and sex were well balanced between the oral-intravenous and the intravenous-oral treatment sequences among all patients included.
In the SLCaps trial, the most common tumour types were pancreas, non-small cell lung, and head and neck cancers. In the HPCaps trial, the most common tumour types were non-small cell lung cancer, transitional cell carcinoma of the urothelium and oesophageal cancer. Patient characteristics and primary tumour sites are summarized in table I.

Demographic data and primary tumour sites
Pharmacokinetics
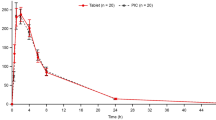
All 12 enrolled patients were evaluable for pharmacokinetics in the vinflunine SLCaps trial, while 2 out of 22 enrolled subjects were not evaluable in the HPCaps trial: one patient did not complete both the intravenous and oral dosing regimens, as he withdrew from the study after he received one single dose of vinflunine; and a bioanalytical interference precluded the quantification of drug concentrations in blood from the other patient. Mean blood concentration-time profiles for oral and intravenous administration are plotted in figure 1. Except for the early phase, which was specific to both routes, intravenous and oral administrations resulted in similar blood concentration profiles. The pharmacokinetic parameters are reported in table II.

Main pharmacokinetic parameters of vinflunine after a 20 minute intravenous infusion or oral intake of either soft gelatin capsule (SLCaps) or hard gelatin capsule (HPCaps) formulations, at the dose-level of 120 mg/m2

a) Mean (SD) blood concentration-time profiles of vinflunine after intravenous and oral administrations: () intravenous administration vs soft gelatin capsules (SLCaps) [n = 12]; (b) intravenous administration vs hard gelatin capsules (HPCaps) [n = 20].
The Cmax values after oral intake were similar (255 ± 121 and 235 ± 71 ng/mL) and were reached simultaneously for both SLCaps and HPCaps, since tmax was 2.5 ± 1.4 hours and 2.5 ± 1.5 hours, respectively. The t1/2z values, in accordance with the observed blood concentration profiles, were similar for the intravenous and the oral formulations. The mean t1/2z values were 32.9 ± 7.1 hours/29.2 ± 7.4 hours (intravenous/oral) for the SLCaps study, and 34.3 ± 5.5 hours/33 ± 8.0 hours (intravenous/oral) for the HPCaps trial. The mean exposure (AUC∞) values reached after both SLCaps and HPCaps dosing were 3528 ± 1746 and 3272 ± 839 ng · h/mL, respectively, while the AUC∞ values were 5919 ± 2446 and 5795 ± 1570 ng · h/mL, respectively, after intravenous dosing in each study. The observed AUC∞ values are plotted in figure S-1 in the Supplemental Digital Content (SDC; available online at http://links.adisonline.com) for oral versus intravenous administration in individual patients in both trials.
The mean absolute bioavailability values of oral vinflunine, calculated on an intra-patient basis, were 58.3 ± 14.4% and 57.3 ± 11% for SLCaps and HPCaps, respectively. The analysis of variance in AUC∞ evidenced neither sequence nor period effects.
A potential correlation between oral vinflunine disposition and the BSA of the patients was investigated. The SLCaps and HPCaps results were pooled, as no difference was observed in the oral bioavailability of both these formulations. As shown in figure 2, no relationship was evidenced between clearance of oral vinflunine (CL/F) and the BSA of the patients.

Individual oral clearance values (CL/F) of vinflunine 120 mg/m2 given by the oral route, either as soft gelatin capsules (SLCaps) or hard gelatin capsules (HPCaps), as a function of the body surface area (BSA) of each patient.
Clinical Results: Safety
The safety profile of vinflunine SLCaps or HPCaps was comparable to that of intravenous administration. The main drug-related adverse events were asthenia, fatigue, constipation and neutropenia. Most of those adverse events were graded 1 or 2, except for two patients who experienced grade 3 fatigue in the SLCaps/intravenous study, and one patient with grade 3 asthenia and another with grade 3 anaemia in the HPCaps/intravenous study. No drug-related serious adverse event was recorded. Two vomiting episodes were recorded in the first study, one after SLCaps dosing and one after intravenous dosing.
Discussion
The interest of researchers, patients and physicians in treatment of cancer by oral administration is still growing. Vinflunine is currently available for intravenous use, and the results reported here in cancer patients show that its absolute oral bioavailability is high, reaching about 58%. The achievement of high oral bioavailability is pivotal, since pharmacokinetic variability is generally inversely proportional to the bioavailability factor. Large pharmacokinetic variability in patients should be avoided, especially for anticancer agents with a narrow therapeutic range of drug exposure. The reported inter-patient variability in drug exposure was particularly low in the vinflunine HPCaps trial, where the CVs were 26% and 27% for the oral and intravenous routes, respectively. The higher variability observed with SLCaps, with CVs of 41% and 49% for the oral and intravenous routes, respectively, is likely to relate to the smaller number of subjects and to some extreme pharmacokinetic values reported for two patients. It is clear that the inter-individual variability in the two studies was similar for both dosing routes, showing that the oral absorption process did not induce more variability as compared with the intravenous route. Indeed, the previously reported inter-individual variability was between 10% and 40%.[4] The other main pharmacokinetic parameters measured in the current studies (CLtot, t1/2z and Vz) were also comparable to previously reported results.[11]
The absolute bioavailability and inter-patient variability of vinflunine HPCaps also compares favourably with those of other vinca alkaloid derivatives. Vinorelbine oral pharmacokinetics have been extensively documented, with bioavailability factors ranging from 38% to 43%,[12,13] and associated inter-individual exposure variability of 43% and 37%, respectively. Beside this, it was shown that liquid-filled gelatin capsules resulted in better absorption of vinorelbine compared with powder-filled hard-gelatin capsules.[14] The HPCaps trial demonstrated that this does not hold true for vinflunine, making powder-formulated hard gelatin capsules of vinflunine — and not only liquid-filled capsules — suitable for oral route dosing.
The evaluation of oral vinflunine bioavailability was performed according to a robust experimental procedure where a common dose was selected for both dosing routes, and a randomized cross-over design was used instead of separate, parallel groups of patients. The comparison of oral and reference intravenous results obtained on two occasions was made possible since the day-to-day variability of vinflunine disposition is low, being reported to be only 8%.[15] Moreover, should a differential effect of one of the treatment sequences be encountered, it would have been detected by the analysis of the cross-over design. An even more sophisticated method to totally avoid the day-to-day variations in pharmacokinetics would be to administer oral and intravenous vinflunine simultaneously to patients. This would require both the use of an intravenous micro-dose of a 14C-radiolabelled drug and access to the highly demanding method of accelerated mass spectrometry to assess circulating tracer concentrations.[16] Considering the low inter-occasion variability of vinflunine, this approach would not be significantly more accurate in the evaluation of bioavailability. On the basis of the knowledge gained about intravenous vinflunine, it could be also possible to deduce and quantify the impact of all pre-systemic processes involved in oral dosing. After the oral ingestion of a drug, the fraction of the total dose that reaches the systemic circulation may be limited by pre-hepatic factors (such as partial drug dissolution, incomplete absorption through the gut wall, intestinal metabolism or drug efflux back into the gut lumen) and then by hepatic first-pass metabolism. Knowing the extent of hepatic clearance from the pharmacokinetics of intravenous vinflunine, the liver could be estimated to metabolize about 33% of the total oral dose (the hepatic first-pass effect). If the first-pass effect was the only limitation of the systemic absorption of the parent compound, the maximal oral bioavailability theoretically ever achievable for vinflunine would then be 67% (i.e. 100% = 33%). This means that all pre-hepatic parameters would limit the actual oral bioavailability by less than 10% only (67% = 58% = 9%). This result is of particular interest in view of the potential involvement of drug efflux transporters, of which vinca alkaloids are commonly known to be substrates.[17] For vinflunine, there should be minimal, if any, impact on oral absorption by P-glycoprotein (P-gp) or other efflux transporters, as the calculated amount of the drug that is unabsorbed prior to the hepatic first-pass effect is only 10% of the total dose, this 10% fraction being probably accounted for by diverse pre-hepatic limitation factors. The minor role of P-gp efflux in the absorption process is confirmed by the fact that inter-patient variability is similar for the oral and intravenous routes. This is also in line with in vitro results, which suggest that vinflunine may only be a weak P-gp substrate as compared with other vincas.[17]
The absorption phase was rapid, with a mean tmax of 2.5 hours in both studies. Moreover, the comparability of the observed t1/2z between the oral and the intravenous dosing suggested that no flip-flop phenomenon was involved between the absorption and the elimination phase. Both the rapid rate and the extent of absorption suggest that both oral formulations tested were adequate for further clinical development. Pharmacokinetic grounds imply that no further improvement of bioavailability would be achievable, even by pharmaceutical or formulation optimizations. Those properties also suggest that vinflunine is likely to behave like a Biopharmaceutics Classification System (BCS) class I compound.[18] As no strong relationship was evidenced between CL/F and the BSA of the enrolled patients, it would be possible for future studies to give oral vinflunine according to a flat, fixed dosing regimen, rather than according to the BSA as was done in the current study.
The tolerance of vinflunine given at 120 mg/m2 in either the oral/intravenous or intravenous/oral sequence on day 1 and day 15 was acceptable. No unexpected adverse event was encountered with the oral route compared with the current or previous clinical experience of the intravenous form. Only three patients experienced grade 3 adverse events, consisting of asthenia/fatigue and anaemia. Longer durations of treatment with the oral forms is needed to better characterize their tolerance.
Conclusion
The high bioavailability (about 58%), and the low inter-individual variability we report here for oral vinflunine are favourable pharmacokinetic properties. The similarity of bioavailability results between vinflunine SLCaps and HPCaps suggests that either liquid or solid ‘powder’ formulations of vinflunine might be selected for further clinical development. The results of these pharmacokinetic trials may help in designing future dose-escalation studies of oral vinflunine.
References
Fahy J, Duflos A, Ribet JP, et al. Vinca alkaloids in superacidic media: a method for creating a new family of antitumor derivatives. J Am Chem Soc 1997; 119: 8576–7
Fahy J, Hellier P, Breillout F, et al. Vinflunine: discovery and synthesis of a novel microtubule inhibitor. Semin Oncol 2008; 35 (3 Suppl. 3): S3–5
Kruczynski A, Colpaert F, Tarayre JP, et al. Preclinical in vivo antitumor activity of vinflunine, a novel fluorinated vinca alkaloid. Cancer Chemother Pharmacol 1998; 41(6): 437–47
Bennouna J, Fumoleau P, Armand JP, et al. Phase I and pharmacokinetic study of the new vinca alkaloid vinflunine administered as a 10-min infusion every 3 weeks in patients with advanced solid tumours. Ann Oncol 2003; 14(4): 630–7
Lobert S, Puozzo C. Pharmacokinetics, metabolites, and preclinical safety of vinflunine. Semin Oncol 2008; 35 Suppl. 3: S28–33
Banna GL, Collova E, Gebbia V, et al. Anticancer oral therapy: emerging related issues. Cancer Treat Rev 2010; 36(8): 595–605
Liu G, Franssen E, Fitch MI, et al. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol 1997; 15(1): 110–5
Delord JP, Stupp R, Pinel MC, et al. Phase I study of vinflunine given as a 10 minute intravenous (IV) infusion on a weekly schedule in patients (pts) with advanced solid tumours [abstract no. 441]. Proc Am Soc Clin Oncol 2001; 20 [online]. Available from URL: http://www.asco.org [Accessed 2012 Mar 2]
Vermorken JB, Stupp R, Nguyen L, et al. Phase I study of IV vinflunine given on a weekly schedule in previously untreated patients (pts) with advanced solid tumours [abstract no. 887]. Proc Am Soc Clin Oncol 2003; 22 [online]. Available from URL: http://www.asco.org [Accessed 2012 Mar 2]
Zorza G, Pellerin D, Fortune V, et al. A simple and sensitive high-performance liquid chromatographic method for the determination of vinflunine and 4-O-deacetylvinflunine from human blood. Ther Drug Monit 2010; 32(6): 734–40
Frampton JE, Moen MD. Vinflunine. Drugs 2010; 70(10): 1283–93
Marty M, Fumoleau P, Adenis A, et al. Oral vinorelbine pharmacokinetics and absolute bioavailability study in patients with solid tumors. Ann Oncol 2001; 12(11): 1643–9
Bourgeois H, Vermorken J, Dark G, et al. Evaluation of oral versus intravenous dose of vinorelbine to achieve equivalent blood exposures in patients with solid tumours. Cancer Chemother Pharmacol 2007; 60(3): 407–13
Zhou XJ, Zhou-Pan XR, Favre R, et al. Relative bioavailability of two oral formulations of navelbine in cancer patients. Biopharm Drug Dispos 1994; 15(7): 577–86
Javlor 25 mg/mL concentrate for solution for infusion. Summary of product characteristics. Boulogne: Pierre Fabre Medicament, 2009
Lappin G, Stevens L. Biomedical accelerator mass spectrometry: recent applications in metabolism and pharmacokinetics. Expert Opin Drug Metab Toxicol 2008; 4(8): 1021–33
Etievant C, Barret JM, Kruczynski A, et al. Vinflunine (20′, 20′-difluoro-3′, 4′-dihydrovinorelbine), a novel vinca alkaloid, which participates in P-glycoprotein (Pgp)-mediated multidrug resistance in vivo and in vitro. Invest New Drugs 1998; 16(1): 3–17
Amidon GL, Lennernas H, Shah VP, et al. A theoretical basis for a bio-pharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res 1995; 12(3): 413–20
Acknowledgements
Joël Bougaret, Maud Brandely-Talbot and Pierre Ferré are employed by the Institut de Recherche Pierre Fabre (Boulogne-Billancourt, France), which initiated and funded this study. Jean-Pierre Delord, Jaafar Bennouna and Loïc Mourey have no conflicts of interest that are directly related to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Delord, JP., Bennouna, J., Mourey, L. et al. Vinflunine Oral Pharmacokinetics and Absolute Bioavailability of Soft and Hard Gelatin Capsules. Clin Pharmacokinet 51, 357–364 (2012). https://doi.org/10.2165/11599300-000000000-00000
Published:
Issue Date:
DOI: https://doi.org/10.2165/11599300-000000000-00000