
Abstract
Purpose
A capsule formulation of the tyrosine kinase inhibitor dovitinib (TKI258) was recently studied in a phase 3 renal cell carcinoma trial; however, tablets are the planned commercial formulation. Therefore, this randomized 2-way crossover study evaluated the bioequivalence of dovitinib tablet and capsule formulations in pretreated patients with advanced solid tumors, excluding breast cancer.
Methods
In this 2-part study, eligible patients received dovitinib 500 mg once daily on a 5-days-on/2-days-off schedule. During the 2-period bioequivalence phase, patients received their initial formulation (capsule or tablet) for 3 weeks before being switched to the alternative formulation in the second period. Patients could continue to receive dovitinib capsules on the same dosing schedule during the post-bioequivalence phase.
Results
A total of 173 patients were enrolled into the bioequivalence phase of the study (capsule → tablet, n = 88; tablet → capsule, n = 85), and 69 patients had evaluable pharmacokinetics (PK) for both periods. PK analyses showed similar exposure and PK profiles for the dovitinib capsule and tablet formulations and supported bioequivalence [geometric mean ratios: AUClast, 0.95 (90 % CI 0.88–1.01); C max, 0.98 (90 % CI 0.91–1.05)]. The most common adverse events, suspected to be study drug related, included diarrhea (60 %), nausea (53 %), fatigue (45 %), and vomiting (43 %). Of 168 patients evaluable for response, 1 achieved a partial response, and stable disease was observed in 32 % of patients.
Conclusions
Dovitinib capsules and tablets were bioequivalent, with a safety profile similar to that observed in other dovitinib studies of patients with heavily pretreated advanced solid tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Tyrosine kinases have known roles in tumorigenesis and tumor survival in multiple human malignancies, and the expression of some tyrosine kinases on solid tumors has been directly linked to abnormal tumor cell growth [1, 2]. Additionally, several tyrosine kinases, including vascular endothelial growth factor receptors (VEGFRs), fibroblast growth factor receptors (FGFRs), and platelet-derived growth factor receptors (PDGFRs), have known roles in tumor angiogenesis [3]. Thus, broad inhibition of tyrosine kinases may provide strong antitumor effects in multiple tumor types.
Dovitinib (TKI258; Novartis Pharmaceuticals Corporation) is an orally available tyrosine kinase inhibitor of FGFR, VEGFR, PDGFRβ, colony stimulating factor 1 receptor, cKIT, RET, tropomyosin receptor kinase A, and Fms-like tyrosine kinase 3. In cancer cell lines and xenograft models, dovitinib exhibited dual antitumor activity through antiproliferative and antiangiogenic effects [4–7]. Promising preclinical and clinical activity observed for dovitinib in various tumor types, including FGFR pathway–amplified and hormone receptor–positive breast cancer and endometrial cancer [8–13], has supported the continued development of dovitinib for multiple solid tumor indications. Dovitinib has previously been evaluated alone or in combination with other agents in several clinical studies, including a phase 2 trial of patients with endocrine-resistant breast cancer [14].
Initial clinical studies of dovitinib were conducted using an anhydrate-salt clinical service form (CSF) capsule of the drug. New monohydrate-salt final market image (FMI) formulations of dovitinib in capsule and tablet form have been developed, both of which were previously shown to have bioavailability comparable to that of the CSF capsule formulation [15, 16]. The FMI capsule formulation of dovitinib has been studied in a phase 3 trial of patients with pretreated metastatic renal cell carcinoma [17]; however, tablets are the planned commercial formulation for the drug. Here, we report on a bioequivalence study comparing dovitinib FMI capsules with dovitinib FMI tablets in patients with advanced solid tumors [registered clinical trial identifier NCT01421004 (www.clinicaltrials.gov)]. Since preliminary analysis of a phase 2 study suggested that the risk/benefit of dovitinib monotherapy may not be favorable for the treatment of heavily pretreated breast cancer [12], the current study excluded patients with breast cancer.
Materials and methods
Eligibility
Patients aged ≥18 years with a histopathologically confirmed diagnosis of an advanced solid tumor, excluding breast cancer, and who had progressed after standard chemotherapy were eligible for this study. All patients had acceptable blood chemistry, cardiac and liver function, and an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of ≤2. Patients with known or suspected brain metastases, history of another malignancy that was significant or required active intervention at the time of enrollment, history of venous thromboembolism events in the last 6 months, and those receiving full-dose warfarin or antiplatelet therapy were not eligible. Patients could not have received anticancer immunotherapy, hormonal therapy, or chemotherapy, radiotherapy, or major surgery within 4 weeks of starting the study drug or small-molecule inhibitor–targeted therapy within 2 weeks of starting the study drug [except nitrosourea or mitomycin C (within 6 weeks)]. Patients expected to receive inhibitors or inducers of CYP1A1/2 or CYP3A4, or H2 blockers or antacids or proton-pump inhibitors immediately prior to or during the bioequivalence phase of the study were excluded. Informed consent was obtained from all individual participants included in the study.
Study design and treatment
This open-label, multicenter, 1:1 randomized, 2-way crossover study evaluating the bioequivalence of dovitinib FMI capsules and FMI tablets consisted of 2 parts: a 2-period bioequivalence phase and a post-bioequivalence phase (Fig. 1). In both phases of the study, patients received dovitinib 500 mg orally once daily on a 5-days-on/2-days-off dosing schedule. In the first part of the bioequivalence phase, patients received their initial formulation [capsules (5 × 100 mg) or tablets (2 × 250 mg)] for 3 weeks. In the second part (week 4), patients were switched to the alternative formulation for 1 week on the same dosing schedule. In the post-bioequivalence phase, ongoing patients in either bioequivalence-phase treatment sequence continued to receive dovitinib FMI capsules on the same dosing schedule, starting 3–9 days after the last dose of dovitinib in the bioequivalence phase. Dovitinib treatment in the post-bioequivalence phase continued until unacceptable toxicity, disease progression, or discontinuation for any other reason. For patients unable to tolerate dovitinib 500 mg, dose adjustments to 400 mg (first dose reduction) or 300 mg (second dose reduction) were permitted; however, reductions due to an adverse event (AE) could not be re-escalated.

Study design. PK pharmacokinetics
The primary endpoint of this study was dovitinib pharmacokinetics (PK). Secondary endpoints were safety and tolerability and overall response rate (ORR). The protocol and all amendments were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Assessments
PK analysis was conducted using a noncompartmental analysis method on full PK profiles of the dovitinib capsule and tablet formulations. Blood samples for dovitinib plasma concentrations were collected during the 72 h following the day 19 (week 3 day 5) and day 26 (week 4 day 5) doses in the bioequivalence phase. PK measurements included log-transformed parameters (last observed area under the curve [AUClast] and maximal concentration [C max]) and secondary PK parameters [AUC from 0 to 24 h (AUC0–24), last observed plasma concentration (C last), terminal elimination rate constant (λz), half-life (T 1/2) apparent total clearance (CL/F), and apparent volume of distribution (Vz/F)].
AEs were recorded throughout the study and at least 30 days after the end of treatment. Physical condition, vital signs, height, weight, ECOG PS, complete blood count, blood chemistry, urinalysis, and thyroid and cardiac function were regularly monitored. AEs were summarized by system organ class, preferred term, and maximum grade according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03 [18].
The antitumor activity of dovitinib was assessed by local review of ORR according to the Response Evaluation Criteria in Solid Tumors version 1.1 [19]. Disease status was assessed at screening, post-bioequivalence phase week 4 day 5 ± 7 days and every 8 weeks thereafter, and at the end of treatment (if not previously assessed ≤6 weeks prior to the end of treatment).
Statistical methods
Noncompartmental PK analysis of each formulation was conducted for the PK analysis set, which consisted of patients who provided ≥2 evaluable PK profiles following dovitinib 500-mg capsule and tablet administration, did not vomit within 4 h after receiving dovitinib on PK blood collection days, received ≥7 of the first 10 scheduled dovitinib 500-mg doses, received 4 consecutive days of dosing at 500 mg prior to PK blood collections, and did not have any major protocol deviations. Using a 30 % intrapatient coefficient of variation, 48 patients were planned for the bioequivalence phase of the study to provide ≥90 % power to demonstrate bioequivalence. A linear mixed-effects model was used to fit AUClast and C max, which included formulation, period, and treatment sequence as fixed factors and patient within sequence as a random factor. Bioequivalence was determined based on whether the 90 % confidence intervals (CIs) of the geometric mean ratios for these log-transformed parameters were completely contained within the range of 0.80–1.25 for the tablet vs capsule. Descriptive statistics were used to summarize demographics and baseline characteristics for all randomized patients (full analysis set) and AEs for patients who received ≥1 dovitinib dose (safety set). Cumulative efficacy of dovitinib was summarized for the bioequivalence and post-bioequivalence phases using the full analysis set.
Results
Patient characteristics and disposition
A total of 173 patients were randomized 1:1 to the bioequivalence-phase treatment sequences [capsule → tablet (n = 88) and tablet → capsule (n = 85)]. Assuming a 40 % nonevaluable rate, the planned enrollment was approximately 80 patients in order to obtain 48 evaluable patients for the bioequivalence test. However, the study was overenrolled (from 80 to 173 patients) due to an initial lower-than-expected bioequivalence evaluability rate and an acceleration in the recruitment rate caused by activation of multiple sites late in the study. Patient demographics and baseline characteristics were well balanced between the 2 treatment sequences (Table 1). The median age was 60 (range 19–89) years, with the majority of patients being white (71 %) and having stage IV disease at study entry (94 %) and an ECOG PS of 1 at baseline (64 %). Primary sites included colon (28 %), lung (10 %), rectum (8 %), pancreas (8 %), and ovary (6 %). Patients were heavily pretreated, with most (90 %) receiving >1 antineoplastic therapy (2 regimens, ≈20 %; 3 regimens, ≈20 %; >3 regimens, ≈45 %).
Of the 173 randomized patients, 5 did not receive study drug due to AEs (n = 4) or withdrawal of consent (n = 1). Eighty patients completed the bioequivalence phase of the study (Table 2). AEs were the most common reason for not completing the bioequivalence phase because even a 1-day treatment interruption could make a patient nonevaluable for PK analysis. However, approximately half of the patients who discontinued treatment in the bioequivalence phase continued treatment in the post-bioequivalence phase. At the time of the final analysis, all patients in the post-bioequivalence phase had discontinued the study. Patients still receiving treatment at the end of this study were permitted to participate in an extension study evaluating the long-term safety/tolerability of dovitinib in patients with solid tumors (NCT02116803); at least 1 patient with thymoma receiving dovitinib 300 mg once daily remains on dovitinib for >4 years.
PK assessments
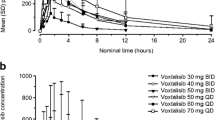
A total of 69 patients (40 %) met the criteria for inclusion in the PK analysis set. For the 104 patients excluded from the PK analysis set, dose interruption, principally due to AEs, and incorrect dosing were the most common reasons for nonevaluability. Dovitinib exposure was similar between the tablet and capsule formulations (Table 3). The 90 % CI for estimated geometric mean ratios (tablet/capsule) for the primary PK parameters fell within the bounds for bioequivalence [AUClast geometric mean ratio, 0.95 (90 % CI 0.88–1.01); C max geometric mean ratio, 0.98 (90 % CI 0.91–1.05)]. Additionally, geometric means for the secondary PK parameters (Table 3) and plasma PK profiles (Fig. 2) were similar for the 2 dovitinib formulations. Primary PK results from 2 sensitivity analyses using only the first 48 randomized patients or excluding 17 patients who were improperly randomized were similar to those using the full PK analysis set, with 90 % CIs for primary PK parameters for both sensitivity analysis subsets also falling within the bounds for bioequivalence.

Plasma PK profiles of dovitinib FMI capsule and tablet formulations. FMI final market image, PK pharmacokinetics, SD standard deviation
Safety and tolerability
A total of 168 patients who received ≥1 dose of dovitinib were included in the safety set. The median duration of exposure in the safety set was 7.6 weeks, with 41 patients (24 %) exposed to dovitinib for ≥16 weeks. Dose adjustments or interruptions were reported for 65 patients (39 %), most frequently due to AEs (28 %) or dosing errors (15 %).
Diarrhea (60 %), nausea (53 %), fatigue (45 %), and vomiting (43 %) were the most common AEs (any grade) suspected to be related to the study drug (Table 4). With the exception of fatigue (13 %), vomiting (6 %), and diarrhea (5 %), most grade 3/4 AEs occurred in <5 % of patients. Grade 3/4 laboratory abnormalities included increased γ-glutamyltransferase (13 %), lipase (9 %), triglycerides (9 %), alkaline phosphatase (8 %), and lymphocytes (5 %), as well as decreased lymphocytes (9 %). Treatment discontinuations due to AEs, regardless of study drug relationship, occurred in 57 patients (34 %), most commonly due to gastrointestinal toxicity or fatigue. Eleven deaths reported during the study or 30-day follow-up period were due to disease progression (n = 8) and AEs [myocardial infarction, respiratory failure, and sepsis (n = 1 each)], and no deaths during the study were suspected to be related to the study drug.
Efficacy
Analysis of dovitinib antitumor activity in these patients with heavily pretreated advanced solid tumors showed that 1 patient with penile squamous cell carcinoma who had been previously treated with 3 lines of therapy, including a platinum agent, achieved a partial response. Fifty-five patients (32 %) had stable disease, and 48 patients (28 %) had progressive disease. Of the 55 patients with stable disease, 21 remained on treatment for ≥120 days. Of patients continuing dovitinib treatment after discontinuing this study, 1 patient with thymoma remained on treatment for ≈4 years. Unknown response was reported for 69 patients (40 %), most commonly due to discontinuation prior to the first scheduled tumor assessment.
Discussion
The targeted inhibition of tyrosine kinases—based on their known roles in tumor development, angiogenesis, and survival—is expected to provide potent antitumor effects in multiple tumor types [1–3]. A capsule formulation of the tyrosine kinase inhibitor dovitinib was studied in a phase 3 trial in renal cell carcinoma. However, since a tablet formulation is planned for commercialization, the phase 1 randomized crossover study presented here was conducted to assess the bioequivalence of capsule and tablet formulations of dovitinib in patients with pretreated advanced solid tumors, excluding breast cancer.
Primary analysis of the PK parameters AUClast and C max demonstrated bioequivalence of the 2 dovitinib FMI formulations. Indeed, dovitinib exposure was similar between capsules and tablets, with tablet/capsule geometric mean ratios of 0.95 (90 % CI 0.88–1.01) for AUClast and 0.98 (90 % CI 0.91–1.05) for C max. Secondary PK analysis results were also similar between the 2 formulations, further supporting bioequivalence.
The safety profile of the FMI capsule and tablet formulations on a 5-days-on/2-days-off dosing schedule was similar to that observed in other dovitinib studies in heavily pretreated patients with advanced solid tumors [12, 17]. Diarrhea, nausea, fatigue, and vomiting were the most common treatment-related AEs, and no new safety concerns were found.
Tumor assessments showed that the best overall response achieved by patients treated with dovitinib capsule and tablet formulations was typically stable disease, which was reported for about one-third of patients. Definitive conclusions regarding the activity of dovitinib in any particular solid tumor indication are limited in this study due to the broad disease characteristics of the patient population and lack of tumor screening for features associated with responsiveness to the drug (e.g., FGFR expression status [13, 14]). However, the level of disease stabilization and the partial response observed in 1 patient provide intriguing signals that support further exploration.
In conclusion, this study demonstrated that dovitinib FMI capsules and FMI tablets were bioequivalent. Additionally, results from the large patient population in this study indicate that dovitinib was tolerable, with a safety profile similar to that reported in previous clinical studies of dovitinib in patients with pretreated advanced solid tumors. The results of this study support further development of the planned commercial tablet form of dovitinib, which is now being used in clinical studies, including a phase 2 trial of pretreated postmenopausal patients with advanced human epidermal growth factor receptor 2–negative/hormone receptor–positive breast cancer [14].
References
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Vlahovic G, Crawford J (2003) Activation of tyrosine kinases in cancer. Oncologist 8:531–538
Dvorak HF (2003) Rous-whipple award lecture. how tumors make bad blood vessels and stroma. Am J Pathol 162:1747–1757
Man WY, Mak JP, Poon RY (2014) Dovitinib induces mitotic defects and activates the G2 DNA damage checkpoint. J Cell Mol Med 18:143–155
Huynh H, Chow PK, Tai WM, Choo SP, Chung AY, Ong HS, Soo KC, Ong R, Linnartz R, Shi MM (2012) Dovitinib demonstrates antitumor and antimetastatic activities in xenograft models of hepatocellular carcinoma. J Hepatol 56:595–601
Lee SH, Lopes de Menezes D, Vora J, Harris A, Ye H, Nordahl L, Garrett E, Samara E, Aukerman SL, Gelb AB, Heise C (2005) In vivo target modulation and biological activity of CHIR-258, a multitargeted growth factor receptor kinase inhibitor, in colon cancer models. Clin Cancer Res 11:3633–3641
Lamont FR, Tomlinson DC, Cooper PA, Shnyder SD, Chester JD, Knowles MA (2011) Small molecule FGF receptor inhibitors block FGFR-dependent urothelial carcinoma growth in vitro and in vivo. Br J Cancer 104:75–82
Angevin E, Lopez-Martin JA, Lin CC, Gschwend JE, Harzstark A, Castellano D, Soria JC, Sen P, Chang J, Shi M, Kay A, Escudier B (2013) Phase I study of dovitinib (TKI258), an oral FGFR, VEGFR, and PDGFR inhibitor, in advanced or metastatic renal cell carcinoma. Clin Cancer Res 19:1257–1268
Escudier B, Grunwald V, Ravaud A, Ou YC, Castellano D, Lin CC, Gschwend JE, Harzstark A, Beall S, Pirotta N, Squires M, Shi M, Angevin E (2014) Phase II results of dovitinib (TKI258) in patients with metastatic renal cell cancer. Clin Cancer Res 20:3012–3022
Kim KB, Chesney J, Robinson D, Gardner H, Shi MM, Kirkwood JM (2011) Phase I/II and pharmacodynamic study of dovitinib (TKI258), an inhibitor of fibroblast growth factor receptors and VEGF receptors, in patients with advanced melanoma. Clin Cancer Res 17:7451–7461
Scheid C, Reece D, Beksac M, Spencer A, Callander N, Sonneveld P, Kalimi G, Cai C, Shi M, Scott JW, Stewart AK (2012) A phase 2, multicenter, nonrandomized, open-label study of dovitinib (TKI258) in patients with relapsed or refractory multiple myeloma with or without t(4;14) translocation. ASH Annual Meeting Abstracts: Abstract 4055
André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, Turner N, Rugo H, Smith JW, Deudon S, Shi M, Zhang Y, Kay A, Porta DG, Yovine A, Baselga J (2013) Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res 19:3693–3702
Konecny GE, Kolarova T, O’Brien NA, Winterhoff B, Yang G, Qi J, Qi Z, Venkatesan N, Ayala R, Luo T, Finn RS, Kristof J, Galderisi C, Porta DG, Anderson L, Shi MM, Yovine A, Slamon DJ (2013) Activity of the fibroblast growth factor receptor inhibitors dovitinib (TKI258) and NVP-BGJ398 in human endometrial cancer cells. Mol Cancer Ther 12:632–642
André F, Neven P, Musolino A, Latini L, Campone M, Cortes J, Barrios C, Squires M, Zhang Y, Deudon S, Gogov S, Blackwell K (2013) Dovitinib, a receptor tyrosine kinase inhibitor in combination with fulvestrant in postmenopausal endocrine resistant human epidermal growth factor receptor 2 negative (HER2-)/hormone receptor-positive (HR +) breast cancer: a phase II, randomized, double blind, placebo-controlled study. San Antonio Breast Cancer Symposium Abstracts: Abstract OT2-6-04
Sharma S, Britten CD, Mortimer J, Kulkarni S, Quinlan M, Liu A, Scott JW, George D (in press) The effect of formulation and food consumption on the bioavailability of dovitinib (TKI258) in patients with advanced solid tumors. Cancer Chemother Pharmacol
Sharma S, Ramanathan RK, George DJ, Quinlan M, Kulkarni S, Homsi A, Scott JW, Infante JR (2013) A randomized, crossover phase I study to assess the effects of formulation and meal consumption on the bioavailability of dovitinib (TKI258). J Clin Oncol 31:suppl; abstr 2593
Motzer RJ, Porta C, Vogelzang NJ, Sternberg CN, Szczylik C, Zolnierek J, Kollmannsberger C, Rha SY, Bjarnason GA, Melichar B, De Giorgi U, Grunwald V, Davis ID, Lee JL, Esteban E, Urbanowitz G, Cai C, Squires M, Marker M, Shi MM, Escudier B (2014) Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol 15:286–296
National Institutes of Health and National Cancer Institute (2009) National cancer institute common terminology criteria for adverse events version 4.0
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Acknowledgments
This study was supported by Novartis Pharmaceuticals Corporation. Additional support was provided by the Institute for Drug Development, Cancer Therapy and Research Center at University of Texas Health Science Center San Antonio in San Antonio, TX Cancer Center Support Grant P30CA054174. We thank Peter J. Simon, Ph.D., and Amanda L. Kauffman, Ph.D., from ArticulateScience (funded by Novartis Pharmaceuticals Corporation) and Sudipta Ridhurkar from Novartis Healthcare Pvt Ltd for providing medical editorial assistance with this manuscript.
Funding
Funding for this study and editorial assistance for the manuscript was provided by Novartis Pharmaceuticals Corporation.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sarantopoulos, J., Goel, S., Chung, V. et al. Randomized phase 1 crossover study assessing the bioequivalence of capsule and tablet formulations of dovitinib (TKI258) in patients with advanced solid tumors. Cancer Chemother Pharmacol 78, 921–927 (2016). https://doi.org/10.1007/s00280-016-3122-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-016-3122-7