
Abstract
Aims
To investigate the population pharmacokinetics and pharmacodynamics of doxorubicin and cyclophosphamide in breast cancer patients.
Patients and methods
Sixty-five female patients with early or advanced breast cancer received doxorubicin 60 mg/m2 over 15 minutes followed by cyclophos-phamide 600 mg/m2 over 15 minutes. The plasma concentration-time data of both drugs were measured, and the relationship between drug pharmacokinetics and neutrophil counts was evaluated using nonlinear mixed-effect modelling. Relationships were explored between drug exposure (the area under the plasma concentration-time curve [AUC]), toxicity and tumour response.
Results
Fifty-nine patients had complete pharmacokinetic and toxicity data. In 50 patients with measurable disease, the objective response rate was 60%, with complete responses in 6% of patients. Both doxorubicin and cyclophosphamide pharmacokinetics were associated with neutrophil toxicity. Cyclophosphamide exposure (the AUC) was significantly higher in patients with at least stable disease (n = 44) than in patients with progressive disease (n = 6; 945 μmol ∙ h/L [95% CI 889, 1001] vs 602 μmol ∙ h/L [95% CI 379, 825], p = 0.0002). No such correlation was found for doxorubicin. Body surface area was positively correlated with doxorubicin clearance; AST and patient age were negatively correlated with doxorubicin clearance; creatinine clearance was positively correlated with doxorubicinol clearance; and occasional concurrent use of carbamazepine was positively correlated with cyclophosphamide clearance.
Conclusions
The proposed inhibitory population pharmacokinetic-pharmacodynamic model adequately described individual neutrophil counts after administration of doxorubicin and cyclophosphamide. In this patient population, exposure to cyclophosphamide, as assessed by the AUC, might have been a predictor of the treatment response, whereas exposure to doxorubicin was not. A prospective study should validate cyclophosphamide exposure as a predictive marker for the treatment response and clinical outcome in this patient group.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Breast cancer is by far the most common malignancy in women in the Western world.[1] The combination of doxorubicin (Adriamycin®) and cyclophosphamide (AC) is a well established option in advanced breast cancer patients,[2,3] as well as in the neoadjuvant and adjuvant setting.[4,5] In the neoadjuvant setting, simultaneous AC was equivalent to doxorubicin and docetaxel (AD) with regard to the tumour response.[6] AC is currently used as part of multidrug schedules in advanced breast cancer,[7] as well as in the adjuvant setting with sequential trastuzumab.[8,9] Taxane-anthracycline-containing combination chemotherapy has been shown to result in an improved objective response rate and improved progression-free survival,[2] but any potential improvement in overall survival is still controversial. AC chemotherapy produced response rates of roughly 50% in advanced breast cancer patients, with complete remission in 3%[3] and 7%[2] of patients in phase III randomised trials. Generally, AC chemotherapy is better tolerated than AD chemotherapy, with febrile neutropenia occurring in 33% of AD-treated patients versus 10% of AC-treated patients in the study by Nabholtz et al.,[2] and in 32% versus 9% of patients in the study by Biganzoli et al.[3]
Current dosing strategies for anticancer drugs result in large interindividual variability in drug exposure. For doxorubicin, which undergoes extensive metabolism in the body and subsequent biliary elimination, liver impairment has been shown to result in decreased clearance.[10] The population pharmacokinetics of doxorubicin as a part of combination chemotherapy for solid tumours have been published previously.[11,12] Apart from 4-iodo-4-deoxydoxorubicin, for which a clear pharmacokinetic-pharmacodynamic relationship is suggested,[13] our understanding of the exposure-toxicity relationship of doxorubicin is still incomplete. Cyclophosphamide is a prodrug that undergoes extensive metabolic activation and inactivation, and there is still incomplete understanding of the role of cyclophosphamide and its metabolites in the efficacy and toxicity of cyclophosphamide treatment.[14] There are some data about the exposure-toxicity relationship of high-dose cyclophosphamide (1000–1500 mg/m2/day) given concurrently with thiotepa and carboplatin, where the first-course area under the plasma concentration-time curve (AUC) of the active metabolite 4-hydroxycyclophosphamide appeared to be predictive for the occurrence of veno-occlusive disease after multiple courses.[15] Another study in 19 women with metastatic breast cancer found a lower AUC of total cyclophosphamide in patients experiencing moderate, but transient, congestive heart failure than in patients who did not develop congestive heart failure.[16] Furthermore, the median duration of the tumour response was also more durable in patients with lower total cyclophosphamide AUC values than in those with higher AUC values (22 months vs 5.25 months, p = 0.008). The investigators suggested that a lower total cyclophosphamide AUC value is a consequence of enhanced cyclophosphamide activation to the active metabolites, leading to both greater tumour cytotoxicity and increased cardiac toxicity.[16] Physiology-based modelling approaches have recently been adapted to study the pharmacokinetic-pharmacodynamic relationship between (anticancer) drugs and toxicity. Friberg et al.[17] described a semimechanistic (pharmacokinetic-pharmacodynamic) model of anticancer drug-induced myelosuppression by using nonlinear mixed-effects modelling. The semimechanistic modelling approach typically uses drug-related and system-/(patient)-related parameters, and is capable of describing the complete time profile of haematological toxicity instead of only a summary measure such as the nadir value. Such physiology-based models are often preferred because they are more predictive, with parameters that refer to actual processes and conditions.[17] To our knowledge, the AC combination has not yet been studied for its pharmacokinetic-pharmacodynamic relationship. Especially in the setting of adjuvant treatment with AC, it is important to avoid unnecessary toxicity, and the occurrence of febrile neutropenia at a frequency of roughly 10% still exposes treated patients to a considerable risk. The objectives of this study were to assess the pharmacokinetics-pharmacodynamics of the AC treatment schedule in patients with mostly advanced breast cancer by using a population approach, and to use this model to identify predictors of drug toxicity and activity.
Methods
Patient Population, Blood Sampling and Bioanalysis
Twelve study centres participated in the trial (see Appendix). All centres had approval from their local medical ethics committees. Written informed consent was obtained from all patients. Sixty-five female patients with early or advanced breast cancer were recruited between 1998 and 2002. The inclusion criteria for study participation were histologically or cytologically verified breast cancer; performance status ≤2 on the WHO scale; life expectancy ≥3 months; absolute neutrophil count (ANC) ≥1500/μL; platelet count ≥100 000/μL; age ≥18 years; creatinine clearance (CLCR) ≥40 mL/min; serum bilirubin ≤26 μmol/L (1.5 mg/dL); ALT and AST ≤2 times the upper limit of normal (unless related to liver metastases, when ALT and AST may be 5 times the upper limit of normal); and a left ventricular ejection fraction (LVEF) within institutional normal limits. Patients were excluded if they had received prior chemotherapy for advanced disease or other (medical) conditions that might interfere with the current study, such as active uncontrolled infection. All patients received at least one cycle of doxorubicin 60 mg/m2 intravenously over 15 minutes followed by a 15-minute infusion of cyclophosphamide 600 mg/m2. This dose remained fixed for all cycles unless toxicity necessitated any dose reduction. Antiemetics (metoclopramide, ondansetron or granisetron) were used according to local guidelines. The cycles were repeated at 3-week intervals, and routine weekly haematological assessments were performed, including the haemoglobin level, leukocyte count, ANC, lymphocyte count and platelet count. Patients with progressive disease under study treatment were excluded from the study. Patients with at least stable disease under study treatment remained in the study and received further treatment at the discretion of the responsible investigator. The tumour response was assessed at every alternate treatment cycle in patients with measurable or evaluable disease. For measurable lesions, the sum of the products of the largest perpendicular diameter of all measurable lesions was assessed, and the response was classified as a complete response (disappearance of all known disease), a partial response (decrease by ≥50%), progressive disease (increase by ≥25%) or stable disease (none of the above). Pharmacokinetic-pharmacodynamic analysis for doxorubicin and cyclophosphamide, as well as assessment of haematological and nonhaematological toxicity, were performed during the first treatment cycle in all patients.
A limited sampling strategy was performed for pharmacokinetic analysis during the first cycle of AC treatment. The samples for doxorubicin analysis (n = 290) were collected in EDTA tubes prior to the start of the infusion, at the end of the infusion, and at 30 minutes, 1.5 and 24 hours after the end of the infusion, similar to a previously described limited sampling strategy.[18] Whole blood was centrifuged immediately after withdrawal for 5 minutes (3000 rpm, 4°C) and the plasma fraction was stored at −20°C until analysis. The plasma concentrations of doxorubicin were determined by a validated isocratic high-performance liquid chromatographic method with solid-phase extraction as the sample pre-treatment procedure, as described in detail elsewhere,[19] and with minor adaptations for the analysis of human plasma samples. The lower limit of quantification (LLQ) was in the range of 1.8–2.4 nmol/L. Bioanalysis of doxorubicin was performed in the network of EORTC-PAMM (European Organisation for Research and Treatment of Cancer-Pharmacology and Molecular Mechanisms) centres (see Appendix).
The samples for cyclophosphamide analysis (n = 250) were collected in 5mL heparin-containing tubes prior to the start of the infusion, at the end of the infusion, and at 1 and 24 hours after the start of the infusion, similar to a previously described limited sampling strategy.[20] Whole blood was centrifuged immediately after withdrawal for 5 minutes (3000 rpm, 4°C) and the plasma fraction was stored at −20°C until analysis. Cyclophosphamide in plasma was quantified with a previously described and validated capillary gas chromatography assay,[21] also within the network of EORTC-PAMM centres.
Bioanalysis of doxorubicin and cyclophosphamide took place after extensive cross-validation of all involved centres. This was a two-step process. First, all participating centres had to send their validation report of the assay to the coordinator of the Bioanalytical Steering Committee (Jos Beijnen at the Netherlands Cancer Institute, Amsterdam, The Netherlands) for reviewing purposes. Second, when the report of a site was considered appropriate, five blinded samples containing two blanks and three samples with spiked concentrations of cyclophosphamide and doxorubicin were sent to the participating bioanalytical centres. The spiked samples contained concentrations in the lower, middle and upper quartiles of the calibration curve of the respective assays. The centres had to achieve the minimal acceptance levels for successful validation of their assay, which meant that the deviations from the nominal values had to be <15%.
Basic Population Pharmacokinetic and Covariate Model
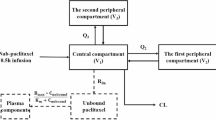
Population pharmacokinetic analysis of the concentration-time data of doxorubicin and cyclophosphamide was performed using the nonlinear mixed-effect modelling program (NONMEM) version V (double precision, level 1.1; GloboMax LLC, Hanover, MD, USA).[22] NONMEM uses a maximum likelihood criterion to simultaneously estimate the population values of fixed-effects parameters (e.g. drug clearance) and the values of the random-effects parameters (e.g. interindividual, interoccasion and residual variability). Log-transformed plasma drug concentrations were used together with the first-order conditional estimation method (FOCE) throughout data analysis. Drug concentrations below the LLQ were processed for data analysis as ‘zero’ values. Standard errors for all parameters were calculated using the COVARIANCE option of NONMEM, and individual Bayesian pharmacokinetic parameters were obtained with the POSTHOC option. The S-Plus (MathSoft Inc., Seattle, WA, USA)-based model-building aid Xpose 3.0 was used for graphical processing.[23] In the first step, an open two-compartment model with first-order elimination, including one peripheral compartment for doxorubicin, was used to describe the concentration-time data of doxorubicin and doxorubicinol, as described previously,[24] with adaptations for the fact that doxorubicin aglycones were not analysed in this study. The following pharmacokinetic parameters were estimated for doxorubicin: the volume of distribution of the central compartment (V1 in L) and the peripheral compartment (V2 in L) for doxorubicin; the volume of distribution of the central compartment (V3/fm in L) for doxorubicinol; doxorubicin clearance (CLDOX in L/h); doxorubicinol clearance (CLDOL/fm in L/h); and intercompartmental clearance between the central and peripheral compartments of doxorubicin (Q in L/h), in which fm is the fraction of the parent compound, doxorubicin, metabolised to doxorubicinol. The concentration-time data of cyclophosphamide were described by evaluating open one- and two-compartment models with linear elimination from the central compartment. The following pharmacokinetic parameters were estimated for cyclophosphamide: V1 and cyclophosphamide clearance (CLCYC in L/min).
Model selection was based on the minimum objective function value (OFV), equal to minus twice the log-likelihood, the precision of parameter estimates (according to the standard error values of the parameter estimates obtained by the COVARIANCE option of NONMEM) and the fit of the model to the data as approached by graphical plots. A proportional error model was used to estimate interindividual variability and residual error. For example, the interindividual variability of V1 was defined by equation 1:
where V1i represents the V1 of the ith individual, V1POP is the typical population value of V1, and ηI V1 is the interindividual random effect with mean zero and variance ω2. Intraindividual or residual variability was modelled as equation 2:
where Cij and Ĉij are the jth measured and model-predicted drug concentration of the ith individual, respectively, and εij is the residual random error with mean zero and variance σ2. Bayesian-derived estimates of the individual pharmacokinetic parameters for doxorubicin and cyclophosphamide were obtained from the final population pharmacokinetic model using the POSTHOC option in NONMEM, and were used as input variables for the pharmacokinetic-pharmacodynamic model.
The following covariates were tested for their correlation with CLDOX, CLDOL and CLCYC: bodyweight; body surface area (BSA); performance status according to the WHO criteria; patient age; presence of liver metastases; comedication with enzyme-inducing antiepileptic drugs (EIADs); ALT; AST; total bilirubin; CLCR (calculated according to the Cockcroft-Gault formula); and serum albumin level. Forward selection and backward elimination were used for this purpose, with the OFV as the main discriminator between different models. The difference in the OFV of hierarchical models approximated to a χ2 distribution with one degree of freedom. Covariates were entered individually into the basic population model by forward inclusion. Continuous covariates, such as bodyweight, were centred on their median values, as exemplified by V1 (equation 3):
where θ1 represents the V1 value of a (median) patient with a bodyweight of 70kg, and θ2 is the exponential factor for bodyweight to describe the correlation with V1. Binary covariates were coded as equation 4:
where θ3 represents the clearance value in non-users of EIADs, and θ4 is the proportional change in clearance in EIAD users (EIAD = 1). The difference in the OFV was evaluated after the introduction of a covariate into the model (forward inclusion) and the significance level was set at p < 0.01 (equivalent to a >6.7-point decrease in the OFV). All significant covariates were subsequently included in an intermediate multivariate model followed by a stepwise backward elimination procedure. Covariates remained in the model when the elimination of the covariate caused an OFV increase of >7.9 (p < 0.005).
Semiphysiological Modelling of Haematological Toxicity
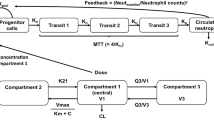
The semiphysiological model, as used in this study to describe drug-related neutrophil toxicity, was introduced by Friberg et al.[25] The model comprised a compartment representing the proliferating cells linked to a compartment representing the systemic circulation through three transit compartments, mimicking precursor cell maturation within the bone marrow. The chain of transit compartments allows description of the time delay between drug exposure, impaired cell proliferation or cell killing, and the resulting impact on circulating blood cells. The transition rate constant (ktr) between the compartments was supposed to be first-order and equal for all transitions. The average maturation time or mean transition time (MTT) represented the time a cell takes to pass from the proliferation stage to the circulation pool. MTT is calculated as equation 5:
where n is the number of transit compartments, which leads to MTT equalling (ktr)−4. The status of the proliferation compartment was dependent on the number of cells in that compartment and on the proliferation rate constant (kprol). The disappearance of peripheral blood cells from the circulation pool was given by the first-order rate constant (kcirc). At steady-state conditions, kprol and kcirc equal ktr. A feedback mechanism (FB) imitated the effect of the release of endogenous growth factors as a response to the decrease of cells in the circulation pool. This leads to increased cell proliferation and was modelled by a power function of the ratio between the baseline ANC (ANCbase) and the ANC at time t (ANCt), according to equation 6:
where γ constitutes the feedback constant. The function by which drug concentrations (doxorubicin and cyclophosphamide) affected the proliferation rate of circulating blood cells (Edrug) was modelled using either a linear function in which Edrug was represented by a slope factor (Slope) and drug concentration (Cdrug) [equation 7] or a maximum effect model in which Edrug was represented by the maximal drug effect (Emax), the drug concentration at half the maximal drug effect (EC50) and the Cdrug (equation 8):
The linear and Emax models were separately adapted to the concentration-time data of doxorubicin and cyclophosphamide to predict individual neutrophil counts. Subsequently, a model incorporating the effect of doxorubicin (EDOX) and cyclophosphamide (ECYC) was constructed according to equation 9:
where Edrug corresponds to (Slope · Cdrug) for the linear model and to (Emax · Cdrug)/(EC50 + Cdrug) for the Emax model, based on a previous publication by Sandstrom et al.[26] (equations 7, 8 and 9).
Logarithmic transformation of both blood count data was used throughout model-building together with the FOCE method. In order to test whether the data supported a relationship between pharmacokinetics and blood counts over a model of dose versus blood counts, the semiphysiological model was applied with individual pharmacokinetic parameters as input (supporting a pharmacokinetic-pharmacodynamic correlation) and with typical values of the pharmacokinetic parameters for all individuals as input (supporting a dose-pharmacodynamic relationship).
Data simulations using the population pharmacodynamic estimates and individual pharmacokinetic parameters were used to construct the typical neutrophil curve following a single dose of doxorubicin 60 mg/m2 and cyclophosphamide 600 mg/m2. For model evaluation, a posterior predictive check with simulation of 1000 datasets from the final pharmacokinetic-pharmacodynamic model was performed. The 25th and 75th percentiles of the simulated neutrophil curve were chosen for model evaluation purposes and compared with the real dataset.
Drug Exposure, Haematological Toxicity and Clinical Outcome
Drug exposure to doxorubicin and cyclophosphamide, as assessed by the AUC, was separately analysed for its correlation with the objective tumour response and haematological toxicity, using the Student’s t-test. As there was a short median follow-up period of only 3.8 months (and progression-free survival data in only 10 of 59 patients), no correlation analysis was performed for drug exposure and progression-free survival. The AUC was estimated by NONMEM and defined as the AUC from time zero to infinity after the first dose of chemotherapy. No cumulative AUC from all administered treatment cycles was used, because this would have resulted in a potential bias, as responding patients were more likely to receive higher cumulative chemotherapy doses than nonresponding patients.
Results
Patient Population and Treatment
Of the 65 patients who were included in the study, 2 received one AC cycle, 7 received two cycles, 4 received three cycles, 7 received four cycles, 3 received five cycles, 36 received six cycles, 3 received seven cycles and 3 received eight cycles. A total of 59 patients had complete pharmacokinetic and toxicity data for inclusion in the pharmacokinetic analysis. The remaining six patients had incomplete pharmacokinetic data. Of these 59 patients, 9 received adjuvant AC treatment and were not evaluable for the drug response, while the remaining 50 patients received first-line AC chemotherapy for metastatic breast cancer and, where available, for a treatment response. Of these 50 patients with measurable disease, 3 had a complete response (6%), 27 had a partial response (54%), 14 had stable disease (28%) and 6 (12%) had progressive disease, giving an overall response rate of 60%. The patient characteristics are outlined in table I. Dose delays and dose reductions due to haematological and nonhaematological toxicity are outlined in tables II and III.

Patient characteristics at the start of study treatment (n = 59)

Haematological toxicity from the first course of chemotherapy

Overall haematological and nonhaematological toxicity over all treatment cycles, leading to dose delay or reduction (excluding dose delays due to logistical reasons)
Basic Population Pharmacokinetic and Covariate Model
The population parameter estimates from the final models are presented in table IV. An open two-compartment model with first-order elimination, including one peripheral compartment for doxorubicin, was used to describe doxorubicin and doxorubicinol concentration-time data. CLDOX was estimated at 47.6 L/h. A one-compartment model was used to describe the concentration-time data of cyclophosphamide. The inclusion of a peripheral compartment (open two-compartment model) did not improve the data fit for cyclophosphamide as compared with a one-compartment model, and the results suggested overparameterisation. CLCYC was estimated at 4.25 L/h. No significant correlation was observed between the respective pharmacokinetic parameters of each drug. Goodness-of-fit plots between model-predicted and observed pharmacokinetic parameters supported the accuracy of the model (figure 1).

Population pharmacokinetic parameters

Goodness-of-fit plots of the final population pharmacokinetic model. Observed concentrations versus model-predicted concentrations (PRED) for (a) doxorubicin, (c) doxorubicinol and (e) cyclophosphamide. Observed concentrations versus individual Bayesian predicted concentrations (IPRED) for (b) doxorubicin, (d) doxorubicinol and (f) cyclophosphamide.
Covariate testing produced the following significant correlations between covariates and drug clearance (equations 10, 11 and 12):
Typically, a 0.2m2 increase in BSA led to a 16% increase in CLDOX, a 20-unit increase in AST led to a 15% decrease in CLDOX, a 10-year increase in patient age led to a 9% decrease in CLDOX, a 20 mL/min increase in CLCR led to a 13% increase in CLDOL/fm, and comedication with EIADs (carbamazepine in three patients, administered for definite epilepsy in two patients and for unspecified antiepileptic treatment in another patient) led to a 89% increase in CLCYC. Of these relationships, the correlation between the use of EIADs and increased CLCYC was the most significant, with a 20.3-point decrease in the OFV (p < 0.0001; table V). The three patients receiving carbamazepine comedication had CLCYL values between 5.9 L/h and 10.1 L/h, well above the population estimate of 4.23 L/h.

Correlation between pharmacokinetic parameters and covariates
Semiphysiological Modelling of Haematological Toxicity
The semiphysiological model with a linear model to describe EDOX and ECYC could be fitted to the neutrophil data. Population predictions and Bayesian individual predictions for neutrophils are plotted against the observed neutrophil data in figure 2. According to the precision of parameter estimates (standard error, interindividual variability), the linear Edrug model was more appropriate than the Emax model for both compounds. The estimates for the combined modelling of EDOX and ECYC (according to equation 9) for neutrophil concentration-time data are outlined in table VI. The higher slope value for doxorubicin compared with cyclophosphamide indicates a stronger impact of doxorubicin on neutrophil toxicity compared with cyclophosphamide. At the same time, considerable interindividual variability was found for the doxorubicin slope parameter (71.8%), markedly higher than for the cyclophosphamide slope parameter (27.4%). Platelet counts were not modelled, as thrombocytopenia was not dose limiting with the AC treatment schedule; besides one patient with grade 3 thrombocytopenia, only grade 1 thrombocytopenia was found in 24 of 50 patients.

Goodness-of-fit plots of the final population pharmacokinetic-pharmacodynamic model. Observed absolute neutrophil cell count (ANC) versus (a) model predictions (PRED) and (b) individual Bayesian predictions (IPRED).

Population pharmacodynamic parameters for absolute neutrophil counts (ANCs)
Combined pharmacokinetic-pharmacodynamic modelling of doxorubicin, cyclophosphamide and neutrophil data revealed that doxorubicin was the main component responsible for variability of neutrophil toxicity. Relative standard errors indicated that all parameters were accurately estimated. The MTT for neutrophils was 113 hours (4.7 days). Population estimates for the neutrophil nadir were 640/µL at day 10. The 25th and 75th percentiles for the 1000 simulated datasets for ANCs are plotted against individual observed neutrophil data in figure 3.

Observed absolute neutrophil count (ANC) and simulated 25th and 75th percentiles from 1000 simulated datasets after administration of doxorubicin 60 mg/m2 and cyclophosphamide 600 mg/m2.
The omission of patients receiving adjuvant AC (n = 9) from the study population did not produce significant or relevant differences in pharmacokinetic-pharmacodynamic parameter estimates. Furthermore, subgroup modelling of pharmacokinetic-pharmacodynamic parameters according to the disease state and metastatic site (e.g. bone only, visceral metastases) was not performed, because of small patient numbers. The use of individual pharmacokinetic parameters as input for the pharmacokinetic-pharmacodynamic model was superior to the use of typical values for the pharmacokinetic parameters, indicating that a true pharmacokinetic-pharmacodynamic relationship was present.
Drug Exposure, Haematological Toxicity and Clinical Outcome
Cyclophosphamide exposure (the AUC) was significantly higher in patients with at least stable disease (n = 44) than in patients with progressive disease (n = 6; 945 µmol · h/L [95% CI 889, 1001] vs 602 µmol · h/L [95% CI 379, 825], p = 0.0002) [table VII]. Similarly, patients with a complete response (1062 µmol · h/L [95% CI 790, 1334]) and patients with a treatment response (931 µmol · h/L [95% CI 867, 995]) had significantly higher cyclophosphamide AUC values than patients with progressive disease (p = 0.04 and 0.0006, respectively). As patients with stable disease had cyclophosphamide AUC values similar to those of patients with a partial response, the same correlations were not significant when responding patients were compared with patients with stable or progressive disease (p ≥ 0.3).

Derived pharmacokinetic parameters and objective tumour responsea
For doxorubicin, drug exposure, as assessed by the AUC, was not significantly higher in responding patients compared with nonresponding patients (table VII). Figure 4 depicts the relationship between the relative decrease of neutrophils and drug clearance. As with population modelling, these graphs do not suggest any significant relationship between neutrophil toxicity and drug clearance.

(a) Relative neutropenia as a function of doxorubicin clearance (CLDOX) [Pearson correlation coefficient 0.21, p = NS]. (b) Relative neutropenia as a function of cyclophosphamide clearance (CLCYC) [Pearson correlation coefficient 0.05, p = NS]. NS = not significant.
Discussion
The optimisation of doses and administration schedules for anticancer agents is desirable not only in the development of new drugs but also for established drugs and drug combinations. One important component in optimising cancer therapy is to describe the haematological toxicity of the drug, which is dose limiting for most cytotoxic drugs, as a function of its pharmacokinetic behaviour. The relationship between pharmacokinetic parameters and toxicity or activity is not well understood for doxorubicin and cyclophosphamide, despite their common use. Only recently has a correlation been described between exposure to doxorubicin and the treatment response, in that children with acute myeloid leukaemia and a complete response to induction doxorubicin had a significantly lower CLDOX than patients without a complete response (657 vs 513 mL/min/m2, p = 0.017).[27] For high-dose cyclophosphamide, some data have been published regarding the relationship between drug pharmacokinetics and toxicity. In the study by de Jonge et al.,[15] high exposure to high-dose cyclophosphamide was correlated with an increased risk of hepatic veno-occlusive disease, and in the study by McDonald et al.,[28] exposure to the active metabolite carboxyethylphosphoramide mustard was related to liver toxicity and mortality from high-dose cyclophosphamide treatment. In the studies by Ayash et al.[16] and Petros et al.,[29] lower systemic exposure to parent cyclophosphamide in patients treated with high-dose cyclophosphamide was correlated to an increased risk of cardiotoxicity (congestive heart failure, attributed to the parent drug’s faster clearance to potentially cardiotoxic metabolites). To our knowledge, however, no similar data have been published for standard-dose cyclophosphamide or for the relationship between myelotoxicity and the drug response. The establishment of any pharmacokinetic-pharmacodynamic relationship may allow optimisation of the AC regimen in breast cancer patients, with the potential for higher response rates and/or less drug-related toxicity.
We found an overall response rate of 60%, with a complete response in 6% of patients receiving AC treatment. Patients with a favourable treatment response had higher cyclophosphamide exposure than patients with progressive disease as the best treatment response. This might translate into higher activation of cyclophosphamide to 4-hydroxycyclophosphamide. Although only the intracellularly formed phosphoramide mustard fraction may be considered cytotoxic, it has been suggested that the 4-hydroxycyclophosphamide systemic concentration reflects the intracellular activation state of cyclosphosphamide and is thus a surrogate for the cyclophosphamide active drug concentration.[14] Although the bioactivation of cyclophosphamide is known to be dependent on various cytochrome P450 isoenzymes (most importantly, 2B6, 3A4, 2C9 and 2C19), little is known about the correlation and interindividual variability between cyclophosphamide and 4-hydroxycyclophosphamide. In an extended dataset of patients receiving high-dose cyclophosphamide at the Netherlands Cancer Institute, a positive correlation between exposure to cyclophosphamide and 4-hydroxycyclophosphamide was found (Pearson correlation coefficient 0.48; Ekhart C, personal communication). A positive correlation between cyclophosphamide and its active metabolite 4-hydroxycyclophosphamide would corroborate the positive correlation between cyclophosphamide exposure and the clinical outcome, as found in this study. Several other investigators could not demonstrate a consistent correlation between cyclophosphamide and 4-hydroxycyclophosphamide.[30–32] Certainly, the fact that 4-hydroxycyclophosphamide concentrations were not assessed in the present study must be taken into account when interpreting the data. The multicentre basis of the study prohibited sample handling that would allow assessment of the unstable metabolites of cyclophosphamide. The correlation between cyclosphosphamide exposure and the treatment response, as found in the present study, could have been corroborated by showing a similar correlation with progression-free survival. The follow-up of our patients was, however, very limited, and we had essentially no data on progression-free survival in responding patients.
The pharmacokinetic model provided an estimate of the population clearance of cyclophosphamide (4.23 L/h) that was in accordance with previously published data.[33] Cyclophosphamide clearance was significantly increased in three patients receiving carbamazepine comedication, consistent with induction of cyclophosphamide metabolism. A similar effect has previously been observed in a patient with a germ-cell tumour receiving high-dose cyclophosphamide (1500 mg/m2/day) together with thiotepa (120 mg/m2/day) and carboplatin for 4 days.[34] This patient, who was concurrently treated with phenytoin for tumour-related seizures, had markedly increased bioactivation of cyclophosphamide to 4-hydroxycyclophosphamide. For doxorubicin, drug clearance (47.6 L/h) was also comparable to what has been described in breast cancer patients (26 L/h/m2, corresponding to roughly 46 L/h)[35] but lower than what has been described in more general patient populations,[36] as well as in children with leukaemia,[27] potentially as a consequence of a gender effect.[35]
Covariate analysis found a significant negative impact of an increased AST level and higher patient age on CLDOX, and an increase of doxorubicinol clearance with higher CLCR (p ≤ 0.005 for all comparisons). Although doxorubicin dose adaptations are usually made depending on total bilirubin concentrations, Twelves et al.[10] have previously observed that increased AST levels were a better predictor of impaired CLDOX than total bilirubin. Similarly, we found no correlation between total bilirubin levels and CLDOX (p = 0.7). Total bilirubin levels were, however, essentially within the normal range (table I), and so any impact of increased total bilirubin on CLDOX may have been obscured. Additionally, we found that a 10-year increase in patient age led to a 9% decrease in CLDOX, similar to what was observed by Li and Gwilt.[37] in 56 patients with solid tumours who were pooled from four clinical studies. Most importantly, it has been argued that impaired CLDOX may be causative of increased cardiac toxicity in elderly patients.[37] The single patient with a >10% decrease in the LVEF was aged 59 years and was not found to have decreased CLDOX (63 L/h, 34.8 L/h/m2). Obviously, an analysis of the relationship between doxorubicin pharmacokinetics and cardiac toxicity would only be valid within a larger patient population.
Finally, BSA was a significant covariate of CLDOX (equation 10) [p < 0.005]. Interestingly, Rudek et al.[12] found a similar positive correlation between BSA and CLDOX in males (p < 0.001), but not in females (p = 0.61), in a group of 152 patients with solid tumours who were receiving various schedules of doxorubicin-based combination therapy. The same investigators found no correlation between patient age and CLDOX in men or in women. The broad range of patient ages that we observed (29–81 years) may have facilitated the identification of any correlation with CLDOX in our study. Overall, the highly significant correlation between BSA and CLDOX in our model suggests that BSA-guided dosing is justified in the studied patient population. The strongest correlation was seen between administration of EIADs and a higher CLDOX. This finding must, however, be interpreted with great caution as the subgroup of patients receiving EIADs was very small (n = 3).
The pharmacokinetic-pharmacodynamic model adequately described ANCs after administration of the AC regimen. Individual pharmacokinetic parameters of doxorubicin and cyclophosphamide were markedly superior to population-typical pharmacokinetic parameters in predicting neutropenia in this group of patients, indicating that there was some correlation between individual drug exposure and neutrophil toxicity, although no significant relationship could be detected with classical correlation analysis. One possible reason may be a considerable interindividual variability in haematological toxicity following AC administration. We found an MTT of 113 hours (4.7 days) for neutrophils, comparable to what has been found for other standard anticancer drugs, including paclitaxel, irinotecan and etoposide.[17] Neutrophil toxicity was comparable to that described in previous reports of standard-dose AC treatment in patients with advanced breast cancer, with 66% having severe (WHO grade 3 or 4) neutropenia as compared with 88% in the study by Nabholtz et al.[2] and 81% in the study by Biganzoli et al.[3] Finally, simulation studies confirmed the validity of the integrated pharmacokinetic-pharmacodynamic model for the AC regimen in this patient population, as the true individual ANCs essentially fell within the 25th and 75th percentiles of 1000 simulated datasets.
Clinical Implications
A clear relationship between the pharmacokinetics of doxorubicin and cyclophosphamide and the dose-limiting haematological toxicity of this commonly used regimen has been demonstrated and quantitated using a semimechanistic pharmacokinetic-pharmacodynamic model. A relationship between cyclophosphamide exposure and the treatment response was suggested. The establishment of a pharmacokinetic-pharmacodynamic relationship may allow optimisation of the AC regimen in breast cancer patients, with the potential for higher response rates and/or less drug-related toxicity.
Conclusion
The proposed inhibitory population pharmacokinetic-pharmacodynamic model adequately described individual neutrophil counts after administration of the AC regimen. In this patient population, exposure to cyclosphosphamide, as assessed by the AUC, might have been a predictor of the treatment response, whereas exposure to doxorubicin was not. A prospective study should validate cyclophosphamide exposure as a predictive marker of the treatment response and clinical outcome in this patient group.
References
Beslija S, Bonneterre J, Burstein H, et al. Second consensus on medical treatment of metastatic breast cancer. Ann Oncol 2007; 18: 215–25
Nabholtz JM, Falkson C, Campos D, et al. Docetaxel and doxorubicin compared with doxorubicin and cyclophos-phamide as first-line chemotherapy for metastatic breast cancer: results of a randomized, multicenter, phase III trial. J Clin Oncol 2003; 21(6): 968–75
Biganzoli L, Cufer T, Bruning P, et al. Doxorubicin and pacli-taxel versus doxorubicin and cyclophosphamide as first-line chemotherapy in metastatic breast cancer: the European Organization for Research and Treatment of Cancer 10961 multicenter phase III trial. J Clin Oncol 2002; 20(14): 3114–21
Jones SE, Clark G, Koleszar S, et al. Adjuvant chemotherapy with doxorubicin and cyclophosphamide in women with rapidly proliferating node-negative breast cancer. Clin Breast Cancer 2002; 3(2): 147–52
Fumoleau P, Chauvin F, Namer M, et al. Intensification of adjuvant chemotherapy: 5-year results of a randomized trial comparing conventional doxorubicin and cyclophosphamide with high-dose mitoxantrone and cyclophosphamide with filgrastim in operable breast cancer with 10 or more involved axillary nodes. J Clin Oncol 2001; 19(3): 612–20
Evans TR, Yellowlees A, Foster E, et al. Phase III randomized trial of doxorubicin and docetaxel versus doxorubicin and cyclophosphamide as primary medical therapy in women with breast cancer: an Anglo-Celtic Cooperative Oncology Group study. J Clin Oncol 2005; 23(13): 2988–95
Smith RE, Anderson SJ, Brown A, et al. Phase II trial of doxorubicin/docetaxel/cyclophosphamide for locally advanced and metastatic breast cancer: results from NSABP trial BP-58. Clin Breast Cancer 2002; 3(5): 333–40
Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 2005; 353(16): 1673–84
Slamon D, Eiermann W, Robert N, et al. Phase III randomized trial comparing doxorubicin and cyclophosphamide followed by docetaxel (AC-T) with doxorubicin and cyclophosphamide followed by docetaxel and trastuzumab (AC-T) with docetaxel, carboplatin and trastuzumab (TCH) in HER2 positive early breast cancer patients: BCIRG 006 study [abstract]. Breast Cancer Res Treat 2005; 94 Suppl. 1: 5
Twelves CJ, Dobbs NA, Gillies HC, et al. Doxorubicin pharma-cokinetics: the effect of abnormal liver biochemistry tests. Cancer Chemother Pharmacol 1998; 42(3): 229–34
Freyer G, Tranchand B, Ligneau B, et al. Population pharma-cokinetics of doxorubicin, etoposide and ifosfamide in small cell lung cancer patients: results of a multicentre study. Br J Clin Pharmacol 2000; 50(4): 315–24
Rudek MA, Sparreboom A, Garrett-Mayer ES, et al. Factors affecting pharmacokinetic variability following doxorubicin and docetaxel-based therapy. Eur J Cancer 2004; 40(8): 1170–8
Robert J, Armand JP, Huet S, et al. Pharmacokinetics and metabolism of 4′-iodo-4′-deoxy-doxorubicin in humans. J Clin Oncol 1992; 10(7): 1183–90
de Jonge ME, Huitema AD, Rodenhuis S, et al. Clinical pharma-cokinetics of cyclophosphamide. Clin Pharmacokinet 2005; 44(11): 1135–64
de Jonge ME, Huitema AD, Beijnen JH, et al. High exposures to bioactivated cyclophosphamide are related to the occurrence of veno-occlusive disease of the liver following high-dose chemotherapy. Br J Cancer 2006; 94(9): 1226–30
Ayash LJ, Wright JE, Tretyakov O, et al. Cyclophosphamide pharmacokinetics: correlation with cardiac toxicity and tumor response. J Clin Oncol 1992; 10(6): 995–1000
Friberg LE, Henningsson A, Maas H, et al. Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol 2002; 20(24): 4713–21
Ratain MJ, Robert J, van der Vijgh WJ. Limited sampling models for doxorubicin pharmacokinetics. J Clin Oncol 1991 May; 9(5): 871–6
van Asperen J, van Tellingen O, Beijnen JH. Determination of doxorubicin and metabolites in murine specimens by highperformance liquid chromatography. J Chromatogr B Biomed Sci Appl 1998; 712(1–2): 129–43
Egorin MJ, Forrest A, Belani CP, et al. A limited sampling strategy for cyclophosphamide pharmacokinetics. Cancer Res 1989 Jun 1; 49(11): 3129–33
Huitema ADR, Tibben MM, Kerbusch T. Simulataneous determination of N,N′,N″-triethylenethiphosphoramide, cyclophos-phamide and some of their metabolites in plasma using capillary gas chromatography. J Chromatogr B Biomed Sci Appl 1998; 716: 177–86
Beal SL, Sheiner LB. NONMEM user’s guide. San Francisco (CA): University of California NONMEM Project Group, 1998
Jonsson EN, Karlsson MO. Xpose — an S-Plus based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 1999; 58(1): 51–64
Joerger M, Huitema AD, Meenhorst PL, et al. Pharmacokinetics of low-dose doxorubicin and metabolites in patients with AIDS-related Kaposi sarcoma. Cancer Chemother Pharmacol 2005; 55(5): 488–96
Friberg LE, Freijs A, Sandstrom M, et al. Semiphysiological model for the time course of leukocytes after varying schedules of 5-fluorouracil in rats. J Pharmacol Exp Ther 2000; 295(2): 734–40
Sandstrom M, Lindman H, Nygren P, et al. Model describing the relationship between pharmacokinetics and hematologic toxicity of the epirubicin-docetaxel regimen in breast cancer patients. J Clin Oncol 2005; 23(3): 413–21
Palle J, Frost BM, Peterson C, et al. Doxorubicin pharmacokinetics is correlated to the effect of induction therapy in children with acute myeloid leukemia. Anticancer Drugs 2006; 17(4): 385–92
McDonald GB, McCune JS, Batchelder A, et al. Metabolism-based cyclophosphamide dosing for hematopoietic cell transplant. Clin Pharmacol Ther 2005; 78(3): 298–308
Petros WP, Broadwater G, Berry D, et al. Association of highdose cyclophosphamide, cisplatin, and carmustine pharma-cokinetics with survival, toxicity, and dosing weight in patients with primary breast cancer. Clin Cancer Res 2002; 8(3): 698–705
Belfayol-Pisante L, Guillevin L, Tod M, et al. Pharmacokinetics of cyclophosphamide (CP) and 4-OH-CP/aldophosphamide in systemic vasculitis. Fundam Clin Pharmacol 2000; 14(4): 415–21
Chen TL, Kennedy MJ, Anderson LW, et al. Nonlinear pharma-cokinetics of cyclophosphamide and 4-hydroxycyclophos-phamide/aldophosphamide in patients with metastatic breast cancer receiving high-dose chemotherapy followed by autolo-gous bone marrow transplantation. Drug Metab Dispos 1997; 25(5): 544–51
Sladek NE, Doeden D, Powers JF, et al. Plasma concentrations of 4-hydroxycyclophosphamide and phosphoramide mustard in patients repeatedly given high doses of cyclophosphamide in preparation for bone marrow transplantation. Cancer Treat Rep 1984; 68(10): 1247–54
de Jonge ME, Huitema AD, van Dam SM, et al. Population pharmacokinetics of cyclophosphamide and its metabolites 4-hydroxycyclophosphamide, 2-dechloroethylcyclophos-phamide, and phosphoramide mustard in a high-dose combination with thiotepa and carboplatin. Ther Drug Monit 2005; 27(6): 756–65
de Jonge ME, Huitema AD, van Dam SM, et al. Significant induction of cyclophosphamide and thiotepa metabolism by phenytoin. Cancer Chemother Pharmacol 2005; 55(5): 507–10
Dobbs NA, Twelves CJ, Gillies H, et al. Gender affects doxo-rubicin pharmacokinetics in patients with normal liver biochemistry. Cancer Chemother Pharmacol 1995; 36(6): 473–6
Mross K, Mayer U, Hamm K, et al. Pharmacokinetics and metabolism of iodo-doxorubicin and doxorubicin in humans. Eur J Clin Pharmacol 1990; 39(5): 507–13
Li J, Gwilt PR. The effect of age on the early disposition of doxorubicin. Cancer Chemother Pharmacol 2003; 51(5): 395–402
Acknowledgements
This study was part of the Biomed Project: Population Pharmacokinetic/Pharmacodynamic Analysis of Anticancer Drug Treatment (PL96-2333). The coordinator was Jan Schellens. The study was performed on behalf of the European Organisation for Research and Treatment of Cancer (EORTC)-Pharmacology and Molecular Mechanisms Group (PAMM) and New Drug Development Group (NDDG). Markus Joerger is supported by a fellowship grant funded by the European Society of Medical Oncology and by a research grant from the Swiss National Science Foundation (PBBSB-102331). The project was supported by a grant from the European Community (Biomed Project PL 96-2333). The authors have no conflicts of interest that are directly relevant to the content of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Joerger, M., Huitema, A.D.R., Richel, D.J. et al. Population Pharmacokinetics and Pharmacodynamics of Doxorubicin and Cyclophosphamide in Breast Cancer Patients. Clin Pharmacokinet 46, 1051–1068 (2007). https://doi.org/10.2165/00003088-200746120-00005
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200746120-00005