
Abstract
Eptifibatide is a truncated derivative of the naturally occurring rattlesnake venom protein known as barbourin. It is a cyclic heptapeptide that mimics the tertiary structure found in the parent compound which allows it to bind receptors with the KGD (Lys-Gly-Asp) peptide recognition sequence. Specifically, eptifibatide is a competitive antagonist for the activated platelet glycoprotein IIb/ IIIa receptor. Its mechanism of action involves preventing the binding and cross-linking of fibrinogen to the platelet surface. This binding site for fibrinogen is associated with five Ca2+ ions that help maintain the tertiary structure of the receptor and affect the affinity of other ligands such as eptifibatide.
Arterial injury induced by percutaneous coronary interventions (PCI) such as balloon angioplasty and stenting, and the spontaneously occurring disease process known as the acute coronary syndrome (ACS), share a common underlying pathophysiology. In both situations, disruption of integrity of the arterial wall initiates a cascade of platelet activation, adhesion and aggregation. Ultimately, this process may proceed to arterial thrombosis unless controlled or modified. Advances in understanding how the platelet plays a pivotal role in this process have significantly enhanced therapy for patients with ACS and have resulted in important reductions in thrombotic complications from PCI procedures. Central to these advances has been evolving understanding of platelet-inhibiting pharmaceutical agents such as eptifibatide.
The development of a rational administration regimen for eptifibatide parallels the growth in the understanding of the underlying mechanisms of platelet receptor functions. The binding of eptifibatide to the receptor involves displacement of receptor-associated Ca2+ from the activated binding site. Early in the clinical development of eptifibatide, this was poorly appreciated and resulted in an underestimation of the appropriate doses for this agent. Through a series of small clinical trials and laboratory studies, deficiencies in the early administration regimens were identified and a more effective dose schedule was determined. Modelling of the drug based on its two-compartment pharmacokinetics further defined the role of a newer double-bolus initiation of therapy verses the original single-bolus approach. In a large-scale clinical trial using this double-bolus followed by infusion regimen in PCI procedures, clinical efficacy was shown to be significantly improved over placebo and the earlier, low-dose regimens used in the original trials of eptifibatide.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1. Background
1.1 Pathophysiology of Acute Coronary Syndromes and Percutaneous Coronary Interventions
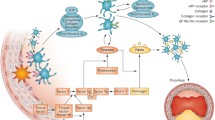
The pathophysiology of the acute coronary syndromes within the atherosclerotic artery can be viewed as a logical extension of the normal haemostatic response to vessel wall injury. The mechanisms of haemostasis are critical to the maintenance of an intact vasculature and therefore react rapidly to injury to the vascular wall. Damaged endothelial wall exposes the subendothelial substrates such as collagen and a variety of different adhesive proteins to the circulating blood. Platelets derived from the blood pool initially deposit at the site of injury and initiate the formation of a primary haemostatic plug.[1]The platelets become activated in the process of vessel wall adhesion, setting off a cascade of platelet aggregation with fibrinogen cross-linking. In the normal reaction to vessel injury, this process results in sealing of the vessel wall, but the process of platelet aggregation abates before pathological thrombosis of the vessel lumen occurs. The pathological response produces a critical or total occlusion of the injured vessel and results in acute coronary syndromes.[2]
Atherosclerotic vessels are different from normal vessels in that their cellular architecture has been altered by atherosclerotic plaques.[3]The initiating cause of these plagues remains a mystery, although a multitude of risk factors such as tobacco smoking, diabetes, hypertension, hyperlipidaemia, hyperhomocystineaemia and a family history of atherosclerosis have been identified as being associated with the disease process. As these plaques grow and expand into the vascular lumen, they have a tendency to form a relatively unstable pool of lipoid material in their core. With erosion of the surface endothelial layer and fissure formation into this subendothelial lipid pool, this highly thrombogenic material becomes exposed to the circulating blood and triggers a thrombotic response.
Spontaneous fissuring and rupture of lipid-laden atherosclerotic plagues initiates an over-stimulation or exaggeration of the normal haemostatic response. With atherosclerotic debris in addition to the usual subendothelial initiators of vascular haemostasis accentuating the thrombotic reaction, acute coronary occlusion becomes likely. When plaque disruption occurs spontaneously, it is manifested clinically as one of the acute coronary syndromes. Iatrogenically, plaques are fissured during percutaneous coronary intervention (PCI) as a direct result of mechanical injury.[4,5]Many of the challenges in developing safe and effective PCI procedures have evolved from efforts to minimise and control the thrombotic response during this iatrogenic injury to the vessel wall. Adverse clinical events such as myocardial infarctions, revascularisation and death occur in 10–15% of patients within 30 days of undergoing PCI in closely monitored clinical trials. This has provided, and continues to provide, a rich clinical environment to test pharmaceutical agents that might be effective in reducing this morbidity and mortality. Central to controlling the multitude of complications that can occur within this environment is therapy to inhibit platelet aggregation.[6]
1.2 Platelet Glycoprotein IIb/IIIa Receptors
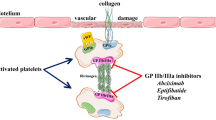
Normal platelets contain 50 000–80 000 glycoprotein (GP) IIb/IIIa receptors within their surface membranes. In an inactivated state, these receptors will not cross-link with fibrinogen or support aggregation. When a platelet is activated, for example when exposed to a damaged arterial wall, the GPIIb/IIIa receptor undergoes a conformational change that permits binding with fibrinogen, von Willebrand factor and vitronectin. This expression of activated binding sites and the potential to block this final common pathway of platelet aggregation and subsequent thrombosis has been the focus of extensive pharmaceutical development.
Initial attempts to control thrombosis from PCI-induced injury in the coronary artery focused on the use of antithrombin agents such as heparin and warfarin (coumadin). This was partially a function of a lack of clearly effective antiplatelet medications beyond aspirin, and partially due to a lack of appreciation of the pivotal role the platelet plays in arterial thrombosis. Platelets activate and subsequently aggregate with cross-linking with fibrinogen in response to a wide variety of physiological pathways. Although activation is possible from many stimuli, the final common pathway for aggregation appears to be an attachment via an activated GPIIb-IIIA complex with fibrinogen.[7]
Research into a rare bleeding disorder known as Glanzmann’s thrombasthenia helped elucidate the mechanism of platelet aggregation.[8,9]Patients with this disorder have platelets that will not react to the stimuli that aggregate normal platelets. A mutation in the gene that codes for parts of the membrane integrin GPIIb/IIIa is present, and the patients manifest no receptors for fibrinogen on their cell surface. This suggested a link or association between the GPIIb/IIIa receptor and the binding with fibrinogen that occurs during aggregation. Collier was able to induce a condition similar to Glanzmann’s thrombasthenia with an anti-GPIIb/IIIa monoclonal antibody that prevented platelet aggregation in animal models.[10]This pioneered a new pharmacological strategy of receptor inhibition to prevent platelet-mediated arterial thrombosis.
1.3 Overview of the Glycoprotein IIb/IIIa Receptor Antagonists
There are presently two classes of GPIIb/IIIa receptor antagonists widely available for clinical use. The first class, the monoclonal antibody, has one commercially available compound, abciximab, although others are in development. This class is a direct extension of early research on the GPIIb/IIIa receptor. A Fab fragment of an anti-GPIIb/IIIa murine monoclonal antibody was developed that could irreversibly bind with the GPIIb/IIIa receptor and render it unable to bind with fibrinogen. While tested in humans in a limited fashion, the potential for immune reactions and thrombocytopenia prompted the development of a human-murine chimaeric version with the same binding properties as the original murine Fab fragment.[11]This chimaeric agent, originally referred to as c7E3, overcame many of the antigenic concerns with the original murine antibody and has become known as abciximab. The first large-scale trial to test a GPIIb/IIIa inhibitor in the setting of PCI was the EPIC (Evaluation of c7E3 for the Prevention of Ischemic Complications) trial. This was a 2099-patient phase III study using abciximab during PCI. It demonstrated a statistically significant 35% relative reduction in the composite endpoint of death, non-fatal myocardial infarction or the need for emergency revascularisation within 30 days of the index procedure.[12]This pivotal trial provided the proof-of-principle that GPIIb/IIIa receptor antagonists could improve outcome during PCI.
The other class of GPIIb/IIIa receptor antagonist is the short-acting agents with competitive binding to the target receptor. Tirofiban and eptifibatide represent the most widely available examples of this group of pharmaceuticals. These agents trace their origins to venom derived from viper snakes. Both are relatively small molecules, with a molecular size of 1000Da for eptifibatide compared with 50 000Da for abciximab,[13]and appear to act by interfering with fibrinogen binding using either the amino acid sequence RGD (Arg-Gly-Arg)[14]in the case of tirofiban or KGD (Lys-Gly-Asp)[15]with eptifibatide. Both share relatively short half-lives measured in hours and are non-immunogenic compared with abciximab. Therapeutic trials with these agents demonstrated effectiveness in blocking the GPIIb/IIIa receptors, but the early clinical results were less robust than trials with abciximab. This has stimulated further research, a better understanding of platelet mechanisms and ultimately a refinement of the administration regimen for eptifibatide. This further understanding of the pharmacokinetics and pharmacodynamics of eptifibatide is the subject of the present review.
2. Origins of Eptifibatide
Eptifibatide was developed as an analogue of barbourin, a 73-amino-acid disintegrin from the venom of the southeastern pigmy rattlesnake Sistrurus barbouri.[15]Unlike other disintegrins, which interfere with binding to RGD-containing ligands, barbourin interferes with a KGD sequence. The activity of barbourin appears to depend not only on the KGD motif, but also the tertiary structure that enhances the binding affinity to the GPIIb/IIIa receptor. Barbourin as a therapeutic agent appeared problematic since its molecular size raised the potential problems of antigenicity and rapid elimination.
Small analogues of barbourin were synthesised that retained the binding characteristics of barbourin with less risk of antigenicity and a more favourable pharmacodynamic profile.[16]From this series of synthetic analogues, the drug eptifibatide was identified as a potential therapeutic agent. Structurally, it is a heptapeptide sequence that has been cyclised with disulfide bridging. This maintains the tertiary structure required to preserve the KGD peptide recognition sequence.[17]
3. Characteristics of Eptifibatide
Eptifibatide binds rapidly to platelet binding sites with full platelet inhibition within 15 minutes.[18]The binding site of the GPIIb/IIIa receptor is known to have an association with five Ca2+ions[19]that compete with eptifibatide for receptor occupancy. This results in a 50% effective concentration (IC50) for eptifibatide that ranges from 100 nmol/L at low extracellular ionised calcium concentrations of 40–50 µmol/L to 500 nmol/L at a typical physiological concentration of ionised calcium (1.1 mmol/L).[20]The affinity of eptifibatide for the receptor, as measured by its dissociation constant KD, is 120 nmol/L.[21]The binding of eptifibatide to the GPIIb/IIIa receptor can be quantified by using a GPIIIa-specific antibody and fluorescence technique that detects the receptor bound by the antagonist.[22]
Approximately 25% of eptifibatide is protein-bound under physiological conditions, whereas the rest remains free and pharmacologically active in the circulation.[23]The primary route of excretion is through the renal system from the plasma compartment, with no apparent active metabolites. Elimination times are dependent on overall renal function, with an elimination half-life of 1–3 hours.[21,23,24]The pharmacokinetics of eptifibatide can be modelled by a two-compartment model with first-order elimination.[25]
4. Early Clinical Experience
4.1 Phase II Trials
Two early phase II trials in interventional cardiology formed the basis for the original administration regimens of eptifibatide. Based on the need for rapid onset of action, a bolus and infusion regimen was initially identified as a reasonable approach to initiate eptifibatide therapy. The first phase II trial performed on eptifibatide,[26]known as IMPACT (Integrelin to Minimize Platelet Aggregation and Prevent Coronary Thrombosis), tested the initial safety and pharmacodynamic-kinetic properties of eptifibatide. In this placebo-controlled trial, a regimen of a 90 µg/kg bolus followed by an infusion of 1 µg/kg/min for either 4 or 12 hours was evaluated (see table I). Platelet aggregometry was done using citrated blood samples.

Details of clinical trials with eptifibatide
Platelet pharmacodynamics demonstrated a mean platelet inhibition of 86%. The 95% CIs were wide, raising apprehension that this dosage might not be adequate for the full spectrum of patients. This wide interpatient variability also raised concerns about the potential for inadequate platelet-inhibitory effect early after initiation of therapy. This period of time would correspond to that time during the PCI procedure when platelet activation from vessel wall trauma would be expected to be most intense. Unfortunately, platelet aggregation data were only available for baseline, 1 hour after administration of the bolus and at termination of infusion.[26]
Although the IMPACT trial was not powered to show efficacy, the 30-day outcomes showed a statistically non-significant difference in composite endpoints (urgent/emergency need for intervention, coronary artery bypass surgery, myocardial infarction or death) with 12.2% in the placebo arm, 9.6% with 4 hours of eptifibatide and 4.1% with 8 hours of eptifibatide. At the same time, the results of the phase III trial of abciximab in high-risk angioplasty were available.[12]These were very favourable for the use of abciximab, but only when it was given as a bolus followed by a 12-hour infusion. Bolus abciximab, despite its long biological half-life, was not nearly as efficacious as a bolus plus infusion of abciximab.[12]Given the prolonged biological activity of abciximab, it was felt that a 24-hour infusion of eptifibatide might provide similar antiplatelet coverage immediately after PCI. The concept of an 18–24-hour eptifibatide infusion for enhanced efficacy was therefore established.
The second phase II trial was a placebo-controlled dose-escalating trial[18]performed to examine the safety and pharmacokinetic-pharmacodynamic properties of eptifibatide in a population with coronary artery disease undergoing PCI. Dosages ranged from a bolus of 90–180 µg/kg and infusion rates of 0.5–1.0 µg/kg/min, as noted in table I. The procedures performed were primarily balloon angioplasty (85%) with the rest either directional or rotational atherectomy. These procedures were performed on a background of aspirin and heparin adequate to increase the activated clotting time (ACT) to 300–350 seconds. Infusions were continued for 18–24 hours. Platelet inhibition was measured using citrated blood samples and light transmittance platelet aggregometry. The 180 µg/kg bolus was reported to provide 80–90% platelet inhibition in 75% of the patients that was maintained throughout the 2-hour duration of the standard procedure of the time. The 0.75 µg/kg/min infusion provided ongoing antiplatelet effect, with >80% inhibition in 75% of the patients. In contrast, the 0.50 µg/kg/min infusions provided 60–70% inhibition in 75% of the patients. Since only 73 patients were involved, with 19 receiving placebo and 54 active drug, differences in clinical outcome could not be examined. Concern was raised about whether the higher 180 µg/kg bolus might set the stage for haemorrhagic complications; the 0.5 µg/kg/min infusion might allow initial clinical protection during the procedure, but due to lesser degree of inhibition prevent later haemorrhagic complications.
4.2 Phase III Trial
The safety and pharmacodynamic results of the initial phase II trials of eptifibatide, along with the positive clinical outcome of the phase III EPIC trial of abciximab,[12]resulted in a phase III eptifibatide trial, known as IMPACT II, in patients undergoing PCI.[27]This trial was different from the initial abciximab trial, since abciximab was only tested in high-risk PCI patients whereas eptifibatide was to be tested in the full spectrum of risk groups undergoing PCI. Similar to the phase II trials, most of the patients in IMPACT II underwent balloon angioplasty, although 25% underwent an atherectomy-type intervention. Elective stenting was still not available and only under emergency situations such as vessel closure were stents used.
The 4010 patients enrolled in IMPACT II were randomised to one of three regimens: placebo, 135 µg/kg bolus plus 0.5 µg/kg/min infusion over a 20–24-hour period, or 135 µg/kg bolus followed by a 0.75 µg/kg/min infusion also for 20–24 hours (table I). The procedures were done on a background of aspirin 325mg and heparin to elevate the ACT to the 300–350 second range. The primary endpoint was again a composite of occurrence within 30 days of death, myocardial infarction, urgent or emergency repeat coronary intervention (either percutaneous or surgical), or placement of an intracoronary stent into the index artery.
The overall results of this trial appeared to show a treatment effect of eptifibatide, but it was not statistically significant on the intention-to-treat analysis (table II). In addition, there was no evidence of a dose effect. The results of the lower-dose arm were actually slightly better than those of the higher-dose arm. Safety analysis of the three arms also showed very little or no enhanced bleeding hazard at the higher dose. In addition, there also appeared to be treatment benefit regardless of pre-procedural risk stratification. In other words, both low-risk and high-risk patients appeared to benefit equally from this type of therapy.[27]

Results of phase III trials of eptifibatide in percutaneous coronary interventions (PCI)
The cause of this unexpected outcome was not initially recognised. Questions were raised about a possible difference in mechanism of binding between the abciximab-type receptor binders and the small-molecule receptor blockers such as eptifibatide. Another possibility was that the high specificity of the eptifibatide molecule for only the GPIIb/IIIa receptor was not enough, and the apparent enhanced efficacy seen from abciximab was derived from some of its nonspecific binding to other surface receptors such as vitronectin.[31]Perhaps the biological life of platelet inhibition was also too short with the reversibly bound eptifibatide, and the activated platelets were not adequately inhibited at the end of the 20–24-hour infusion.
In retrospect, it is now known that both the regimens used in IMPACT II were effectively low dosages that provided serum concentrations of eptifibatide at the low end of the dose-response curve.[32]Rather than testing the outcomes of two significantly different dose levels, the IMPACT II trial ended up testing two relatively low-dose regimens against placebo. Despite this, sustained benefits were seen at 6 months of follow-up.[27]
5. Dosage Adjustment and Optimisation
5.1 Calcium Binding and Eptifibatide Binding Interactions
Calcium binding within the GPIIb/IIIa complex was known about at the time of the IMPACT II trial, although its important interaction with eptifibatide binding had originally been under-appreciated. At physiological levels of calcium, five Ca2+ ions are associated with the GPIIb/IIIa structure.[19,33]This calcium is integral to maintaining the tertiary structure of the glycoprotein structure and its biological function. Part of this calcium is displaced when binding peptide ligands are introduced, a ‘displacement hypothesis’[34]that predicted the important inverse relationship between receptor affinity for eptifibatide and calcium concentrations.[35]
Blood samples collected from patients during the early trials of all the various GPIIb/IIIa receptor antagonists were routinely anticoagulated with citrate. This bound ionised calcium in the sample and resulted in effective calcium concentrations of 40–50 µmol/L. At this concentration of ionised calcium, the GPIIb/IIIa receptor may lose its Ca2+ ions[36,37]and dissociation of the complex may occur.[38]Unlike the non-competitive antibody-based receptor antagonists such as abciximab, competitive inhibitors such as eptifibatide appear to compete with Ca2+ at the receptor level.[34]Reduction of the ambient Ca2+ concentration would enhance apparent eptifibatide binding to the GPIIb/IIIa binding site.[39]
Phillips and colleagues analysed the effect of Ca2+ concentration on the GPIIb/IIIa receptor and eptifibatide binding.[20]Platelet-rich plasma (PRP) samples were either anticoagulated with 3.8% sodium citrate or with PPACK (Phe-Pro-Arg-chloromethyl ketone). PPACK anticoagulates without altering the Ca2+ concentrations. The platelets were then challenged with 20 µmol/L adenosine diphosphate (ADP) or thrombin receptor agonist peptide (TRAP) to induce aggregation with different concentrations of eptifibatide. The results are shown in table III. A 4-fold higher eptifibatide concentration was required to inhibit aggregation by 50% (EC50) in PPACK compared with citrated blood. The more potent TRAP-induced aggregation required a 7.5-fold higher eptifibatide concentration to inhibit aggregation by 50% between PPACK and citrated blood samples. The receptor occupancy data measured the amount of eptifibatide bound to unstimulated platelets. This confirmed earlier findings that suggested binding competition between Ca2+ and eptifibatide. A significantly lower EC50 was seen with the calcium-chelating citrate. EDTA (ethylenediaminetetraacetic acid), another calcium-chelating agent, when used in place of citrate caused similar apparent enhancement of eptifibatide-induced platelet inhibition. Likewise, the use of heparin rather than PPACK as a non-chelating anticoagulant did not alter eptifibatide-induced platelet inhibition. This later finding points towards the chelation of Ca2+, rather than an isolated effect of citrate on the receptor binding site, as the putative mechanism for the apparent calcium-dependent binding of eptifibatide.[20]

Effect of calcium-binding anticoagulant (citrate) compared with non-binding anticoagulant (PPACK) on platelet function and responsiveness to eptifibatide[20]
Based on this work with the calcium/eptifibatide interactions, Phillips et al.[20]estimated the true platelet inhibitory levels that eptifibatide produced in the IMPACT II trial. The combination of an initial 135 µg/kg bolus followed by either a 0.5 or 0.75 µg/kg/min infusion would produce steady-state eptifibatide concentrations in the 350–500 nmol/L range. Based on citrated platelet samples, this should have produced 70–80% inhibition of ADP-induced platelet aggregation. Extrapolating the results of platelet inhibition without the use of calcium-chelating anticoagulants, the IMPACT II steady-state eptifibatide concentration would have inhibited ADP-induced aggregation by <30%. Although this level of inhibition was markedly less than originally planned, it still showed efficacy and probably contributed to the relatively few safety concerns related to bleeding raised by this trial compared with the earlier trials of abciximab.[12]
5.2 Phase II Trials of Higher Single Bolus plus Infusion of Eptifibatide
Two further pharmacokinetic and pharmacodynamic studies prospectively evaluated the need for greater eptifibatide concentrations to adequately inhibit platelet function.
A dose-escalating study known as the PRIDE (Platelet Receptor Occupancy with Integrilin — Dynamic Evaluation) trial[28]evaluated the effects of three different eptifibatide regimens on platelet inhibition during PCI (table I). One arm repeated the higher IMPACT II dose of 135 µg/kg bolus followed by a 0.75 µg/kg/min infusion. The two other arms evaluated higher-dose regimens of 180 µg/kg bolus followed by 2 µg/kg/min infusion or 250 µg/kg bolus followed by 3 µg/kg/min infusion. Studies of platelet aggregation stimulated by 20 µmol/L ADP measured on PRP samples anticoagulated with the non-chelating agent PPACK demonstrated that the original IMPACT II dose failed to maintain at least 80% platelet inhibition. The two higher-dose arms in the PRIDE trial did maintain >80% inhibition, with very profound inhibition in the highest dose group (figure 1). A Michaelis-Menten model of the relationship was developed, based on the measured eptifibatide concentrations and receptor occupancy determinations (figure 2). This model suggested that the steady-state eptifibatide concentration threshold required to achieve a goal of 80% receptor occupancy was 1650 µg/L (95% CI 1406–1967 µg/L).[28]

Platelet aggregation in response to stimulation with 20 µmol/L ADP as a function of time and eptifibatide regimen in the PRIDE trial. Numbers in the key represent the bolus (µg/kg) + infusion (µg/kg/min) dosage of eptifibatide; the dosage used in IMPACT II was 135 µg/kg + 0.75 µg/kg/min (reproduced from Tcheng et al.,[28]with permission). ADP = adenosine diphosphate; IMPACT = Integrelin to Minimize Platelet Aggregation and Prevent Coronary Thrombosis; PRIDE = Pilot Study of Receptor Occupancy with Integrilin — A Dynamic Evaluation.

Model of eptifibatide plasma concentration and competitive occupation of platelet glycoprotein IIb/IIIa receptors (estimated mean values and 95% CIs). The cross-hairs denote the estimated eptifibatide concentration required to achieve 80% receptor occupancy (1650 µg/L, 95% CI 1406–1967 µg/L) [reproduced from Tcheng et al.,[28]with permission].
This goal of >80% platelet inhibition is based on earlier work done in canines by Folts. In the Folts model of occlusion and reperfusion, >80% inhibition of platelet aggregation prevented platelet-induced thrombosis.[40–42]From this canine work, the goal of >80% has been the standard of inhibition to achieve. Since bleeding times start to be significantly prolonged at 90% platelet inhibition,[43]dosage limitation has been considered prudent for safety reasons.
An additional observation from the PRIDE study is noted in figure 3, which depicts the plasma eptifibatide concentration over 24 hours for the 180µg + 2 µg/kg/min eptifibatide regimen. A transient decrease in drug concentration to <1650 µg/L was observed within the first few hours after the bolus. For an agent being used during PCI, this nadir was cause for concern, since it occurred during the peak time of coronary intervention and raised the possibility of poor drug coverage during exactly the time period that the best platelet inhibition was required. Since this was a relatively small trial, efficacy could not be tested. Likewise, safety appeared to be acceptable, but again the number of patients enrolled was too small for confidence.[28]

Population-based estimate of plasma eptifibatide concentration over time for the eptifibatide regimen of 180 µg/kg bolus followed by 2 µg/kg/min infusion. The eptifibatide concentration (1650 µg/L) required for 80% receptor occupancy is indicated as a broken line (reproduced from Tcheng et al.,[28]with permission).
The second pharmacokinetic/dynamic study to evaluate a higher dose of eptifibatide was known as the PERIGEE (Precise Evaluation of Response to Integrilin Given for Elimination of Cardiac Events).[29]This was a substudy of the very large (10 948 patients) trial of eptifibatide in acute coronary syndromes known as the PURSUIT (Platelet Glycoprotein IIb/IIIa in Unstable Angina: Receptor Suppression Using Integrilin [eptifibatide] Therapy) trial.[44] The PERIGEE study provided pharmacology data as a surrogate for the much larger PURSUIT trial that provided the safety and efficacy data. In this study, the 180 µg/kg bolus followed by a 2.0 µg/kg/min infusion of eptifibatide was compared against placebo. The patient population was not specifically undergoing PCI, as in the PRIDE trial, but rather they were being treated for up to 72 hours for acute coronary syndromes. This provided an opportunity to test the safety of this higher dosage of eptifibatide during longer exposure to the drug.
The PURSUIT trial tested the hypothesis that inhibition of platelet activity with eptifibatide would reduce the frequency of adverse outcomes in patients with acute coronary syndromes who did not have persistent ST-segment elevation. Patients were enrolled worldwide to receive up to 72 hours of eptifibatide using a regimen of 180 µg/kg bolus followed by a 2 µg/kg/min infusion or the equivalent placebo. Local practices for adjunctive medications and the use of invasive approaches to patient management were left to the discretion of the treating physician. The primary endpoint related to efficacy was the composite of death from any cause or nonfatal myocardial infarction at 30 days. Eptifibatide therapy caused a 1.5% absolute reduction in the primary efficacy endpoint (eptifibatide 14.2%, placebo 15.7%; p = 0.04). The primary safety endpoints included evaluation of bleeding complications and platelet counts. By 30 days, there were more red-cell transfusions noted in the eptifibatide-treated group (eptifibatide 11.6%, placebo 9.2%; relative risk 1.3, 95% CI 1.1–1.4), although stroke (eptifibatide 0.7%, placebo 0.8%; p = 0.41) and thrombocytopenia (eptifibatide 6.8%, placebo 6.7%) were not increased in the eptifibatide-treated patients. These results suggested that the higher infusion rate of eptifibatide (2 µg/kg/min) was efficacious in the acute coronary syndrome population, and its use could be extended out to 72 hours without apparent significant safety concerns.
The 100 patients enrolled in the PERIGEE substudy of PURSUIT underwent multiple sampling of their blood for platelet function. This provided the pharmacological foundation and support for the overall PURSUIT trial. Blood was anticoagulated with either citrate or PPACK. Platelet aggregation was tested using standard platelet aggregometry with stimulation by both TRAP and ADP. As was expected from prior work, the samples anticoagulated in the calcium-chelating citrate showed signs of more profound platelet inhibition than the samples drawn simultaneously and anticoagulated in PPACK. TRAP appeared to be a more powerful stimulus of platelet aggregation than did 20 µmol/L ADP using the latter 180 µg/kg bolus with the 2 µg/kg/min infusion produced the target level of platelet inhibition (>80%) in >80% of the patients during the whole 72-hour treatment period.[29]This extended the findings of the PRIDE study that the 2 µg/kg/min infusion gave adequate platelet inhibition at steady-state from the 24-hour period out to 72 hours.
Further analysis of the median platelet aggregation curves from PERIGEE confirmed the finding seen in the PRIDE study (figure 3) that a transient loss of platelet inhibition occurred at the 1-hour point after the bolus was given. With the usual patient variability, it could be expected that some patients would experience less than optimal platelet inhibition during this period. The PURSUIT trial treated patients with acute coronary syndromes, and such transient changes may not be clinically important. In contrast, for the patient undergoing PCI, this 1-hour time point could quite possibly fall into the period of maximal vessel wall injury and platelet activation. Under this scenario, the lack of adequate platelet inhibition might be a factor that significantly affects outcome.
5.3 Phase II Trials of Double-Bolus Administration of Eptifibatide
Returning to the basic pharmacokinetics of eptifibatide, with its relatively short half-life, single compartment of distribution, predictable clearance and lack of active metabolites, one could predict that when given as a single-bolus and infusion, a nadir of activity might appear at about 1 hour after administration. Based on the results from the initial PRIDE study and the PERIGEE trial, the steady-state concentrations of eptifibatide obtained with the higher infusion dose of 2 µg/kg/min appeared reasonable, with concurrent evidence of efficacy and safety, making it difficult to consider alterations in the infusion rate. The alternative was to consider the effects of adding a second bolus to the initiation of therapy in order to maintain the drug concentration during the early part of the infusion around the 1-hour timepoint. This became the focus of a study known as the PRIDE substudy, which was performed at several PRIDE investigative sites. This substudy provided pharmacokinetic and pharmacodynamic analysis on several different regimens that incorporated a double-bolus approach to eptifibatide therapy.
The PRIDE substudy[25]was performed as a dose-confirmation study evaluating the pharmacodynamics and pharmacokinetics of two single-bolus and two double-bolus regimens of eptifibatide therapy during the performance of elective PCI. Stenting was the primary mode of PCI. The administration strategy included either a single eptifibatide bolus or a double bolus separated by 30 minutes and then a continuous infusion of up to 24 hours of eptifibatide (table I). The dosage regimens were: 180 µg/kg bolus and 2 µg/kg/min infusion with or without a second bolus of 90 µg/kg after 30 minutes, or 250 µg/kg bolus and 3 µg/kg/min infusion with or without a second bolus of 125 µg/kg after 30 minutes. Serial blood samples anticoagulated with PPACK were then drawn for platelet aggregometry, GPIIb/IIIa receptor occupancy and eptifibatide serum concentrations.
Results from this trial demonstrated profound platelet inhibition at these higher dosages of eptifibatide, with >90% inhibition of platelet aggregation to 20 µmol/L ADP seen in most patients across all groups by 1 hour. The use of a double-bolus approach appeared to overcome the apparent partial recovery of platelet function seen in the single-bolus groups. Despite using relatively large dosages of eptifibatide, bleeding times had returned to ≤30 minutes in all groups within 4 hours of termination of infusion. There was significant patient variability within each group with regard to platelet activity and response to therapy. Given the constraints and difficulty in obtaining high quality data in a large group of patients, the data were analysed and then modelled using a population pharmacokinetic approach.[45]
The eptifibatide concentrations measured in the study population were comparable to those measured in the dose-escalation PRIDE study. For a given eptifibatide dosage, patients undergoing interventional procedures tended to have a higher plasma concentration than healthy study subjects, but a lower plasma concentration than patients with unstable angina.[46]The volume of distribution ranged from 0.203–0.229 L/kg, distribution half-life (t½ α) from 0.22–0.52 hours and terminal half-life (t½ β) from 2.57–2.71 hours, consistent with first-order kinetics.[25]The t½ β values were similar to those previously noted in healthy subjects (t½ β 2.4 hours) and in patients with unstable angina (t½ β 2.8 hours).[46]
Building on the patient data collected during the PRIDE trial and later PRIDE substudy, a population approach analysis was used, fitting the data to a two-compartment model (Appendix). The population pharmacokinetic parameters derived from this model are summarised in table IV. Using the 8-hour plasma eptifibatide concentration after an initial 180 µg/kg bolus and 8 hours of 2 µg/kg/min, the model predicted a concentration of 1619 µg/L, very close to the mean observed eptifibatide concentration in patients of 1645 µg/L. This steady-state concentration is also important since it is approximately the threshold (1650 µg/L) eptifibatide plasma concentration seen in prior analysis of the PRIDE trial that achieved approximately 80% platelet GPIIb/IIIa receptor occupancy and >80% inhibition of ex vivo platelet aggregation to 20 µmol/L ADP.

Estimated eptifibatide pharmacokinetic parameters in a population of patients undergoing percutaneous coronary intervention (reproduced from Gilchrist et al.,[25]with permission)
Figure 4 shows the eptifibatide concentration-time profiles estimated from the population pharmacokinetic model. This demonstrates the rapid decrease in eptifibatide concentration after the standard single-bolus therapy with 180 µg/kg bolus and 2 µg/kg/min infusion. From soon after the initiation of the therapy, eptifibatide concentrations are below the 1650 µg/L concentration threshold for adequate eptifibatide effectiveness. At 6 hours the eptifibatide concentration is still prediced to be <1500 µg/L. By using a second bolus of eptifibatide, the eptifibatide concentrations appear to remain above the threshold value for effectiveness during the first hour. The higher-dose regimens using an initial bolus of 250 µg/kg were also considered. Although no safety concerns were noted based on the small sample that received these relatively large dosages of eptifibatide, the profound platelet inhibition produced by these concentrations raised concerns about potential bleeding problems if applied to a larger population under less controlled conditions.

Pharmacokinetic modelling of eptifibatide using an initial eptifibatide regimen of 180 µg/kg bolus and 2 µg/kg/min infusion either alone or followed by a second eptifibatide bolus of 180 µg/kg at 5, 10, 15 or 30 minutes after the initial bolus (reproduced from Gilchrist et al.,[25]with permission).
Further evaluation with the population model was then performed for potential variations of the 180 µg/kg bolus and 2 µg/kg/min infusion using a variety of different timing intervals for a second 180 µg/kg bolus. Figure 4 shows pharmacokinetic plots for the single bolus and several different intervals for the second bolus of eptifibatide. In order to cover the nadir of eptifibatide concentration that has been observed within the first hour, it appeared that a second bolus of 180 µg/kg administered 10 minutes after the first bolus along with the 2 µg/kg/min infusion would give the best coverage. The estimated eptifibatide concentrations would therefore be expected to maintain >80% inhibition of platelet aggregation with this double-bolus regimen throughout the early period of eptifibatide therapy. This early period is when endothelial damage would be expected to occur, with the potential for enhanced platelet activity that is thought to be responsible for the adverse outcomes from PCIs. With intense and consistent platelet inhibition during this critical period of the PCI procedure, it was hoped that outcomes could be further enhanced. The double-bolus calculations from the PRIDE trial and its substudy became the foundation for the dosages selected in later phase III/IV trials of eptifibatide.
6. Recent Clinical Trials
During the period of time between the early 1990s, when the first phase II studies were conducted on eptifibatide, and the mid-1990s, when the second round of phase II studies was completed after the interaction between ionised calcium and receptor occupancy was better understood, the field of interventional cardiology had changed markedly. The US FDA had approved eptifibatide to be marketed at the dosage tested in IMPACT II for the PCI indication. Subsequently, pharmacokinetic data, and pharmacodynamic data obtained without calcium-chelating anticoagulants clearly suggested that the FDA-approved dose was a marked underestimate of effective dose. Stenting as a PCI procedure was rare during the IMPACT II trial, and was primarily performed to rescue a failed balloon angioplasty procedure with a first-generation stent. By the later 1990s, stenting was the dominant form of PCI, with all other modes of PCI relegated to a very small proportion of the total volume of procedures. In addition, the use of adjunctive therapies was changing. Heparin use was seen as a hazard for bleeding from the earlier PCI trials with GPIIb/IIIa inhibitors,[47]and heparin dosages in common use had decreased from a target ACT of >300 seconds to one of between 200 and 300 seconds. The use of thienopyridine agents (clopidogrel and ticlopidine) was also recognised as important and had replaced the use of warfarin in the post-stent regimens of most practitioners of interventional cardiology.
Based on the available data, the cardiology community was left torn between using abciximab, with its solid clinical outcomes data but high monetary cost, or eptifibatide, with less impressive clinical outcomes data but a cost of one-third to one-quarter that of abciximab. Despite the compelling data seen with the use of abciximab, routine use of this agent was not the standard of care in the US in 1995, and its use on a worldwide basis was even less. In addition, due to its high cost, many physicians were reserving it for situations where the procedure was ‘going bad’, the so-called ‘bail-out’ treatment that had never been formally examined as a viable approach to therapy.[48]In order to examine many of these issues and concerns, the ESPRIT (Enhanced Suppression of the Platelet IIb/IIIa Receptor with Integrilin Therapy) trial was designed.[49]
The ESPRIT study design planned to randomised 2 400 patients undergoing elective stent PCI to either eptifibatide given as two 180 µg/kg boli administered 10 minutes apart along with a 2 µg/kg/min infusion for 16–24 hours, or to a placebo arm that received no eptifibatide as an initial therapy (table I). This trial was done on a background of aspirin, pretreatment with thienopyridine and heparin therapy to raise the ACT to the 200–300 second range. The primary endpoint was the composite of death, myocardial infarction, urgent target vessel revascularisation and ‘bail-out’ antithrombotic GPIIb/IIIa inhibitor therapy within 48 hours after randomisation. Secondary analysis extended evaluation of the composite endpoint to 30 days, 6 months, and 1 year.[30]
Enrolment in this trial continued from June 3 1999 until February 4 2000 when, after 2064 patients had been enrolled, the safety monitoring board recommended termination of randomisation because of a 43% relative risk reduction in the irreversible endpoints of death or myocardial infarction at 48 hours (95% CI 19–61%, p = 0.0017) in the active treatment arm with eptifibatide.[30] This early termination because of an overwhelming positive treatment effect was similar to that in the early abciximab trials, where safety monitoring boards terminated trials due to positive treatment effects. With follow-up out to 1 year completed, the ESPRIT trial showed a very positive treatment effect, with a reduction in the primary composite of death, myocardial infarction or revascularisation from 22.1% in the placebo arm to 17.5% in the eptifibatide arm (hazard ratio 0.76, 95% CI 0.63–0.93; p = 0.007). This treatment effect was seen across the major patient subsets and broad clinical presentations.[50,51]
The ESPRIT trial is a prime example of the importance of understanding the pharmacokinetics and pharmacodynamics of a pharmaceutical agent before applying it to a large patient population. With the data contributed by this trial, the present practice of interventional cardiology has shifted to the point that the majority of procedures in the US are performed with eptifibatide and the market for abciximab has contracted, primarily because of its cost disadvantage compared with eptifibatide. In addition, data from the ESPRIT trial suggested that the ‘bail-out’ approach to choosing whom to treat with platelet inhibitors was not advantageous, and as a result this practice has markedly decreased in many institutions.
Although the ESPRIT study appears to confirm the safety and efficacy results that were suggested by the smaller phase II trials, there continues to be some controversy about its widespread applicability. It was performed only in patients undergoing elective stent PCI, and it could be argued that its results may not be generalisable to the complete spectrum of coronary artery patients. The population tested was selected in order to have a placebo arm as a comparator, and may have been somewhat low-risk for complications. The highly positive results of prior inhibitor trials in high-risk patients made it unethical to expose these patients to placebo and thus they were not used in the trial. Although high-risk patients were not enrolled, prior attempts to identify or stratify subgroups prospectively as high or low risk have not appeared to be predictors of benefit during PCI with GPIIb/IIIa inhibitors[52–54]and, as such, it is reasonable to assume that the benefits seen in this lower risk population subset do extend to high risk groups.
7. Remaining Questions
Despite the clinical use of GPIIb/IIIa receptor inhibitors in PCI for almost a decade and the multiple clinical trials with these agents, fundamental questions related to their pharmacokinetics and pharmacodynamics remain.
Perhaps the most basic is the duration of therapy required to obtain a maximal effect. From the initial abciximab trials in angioplasty it appeared that a single bolus of drug would not be adequate[12]and the concept that ‘longer was better’ emerged as a therapeutic theme. This was somewhat shaken by the results of abciximab in acute coronary syndromes, where prolonged infusions were found to be a hazard over placebo.[55]In addition, if one considers that the serum free half-life of abciximab is measured in minutes, an initial bolus may have left little drug available for the rapid expression of GPIIb/IIIa receptors that would be stimulated by the ongoing angioplasty procedure.[56]If one looks at the composite endpoints in the latest ESPRIT trial, with the study population divided by length of therapy, there appears to be a similar magnitude of benefit for eptifibatide whether it was given for less than 6 hours or for any other interval up to the full 18–24 hours.[57]There did not appear to be an incremental benefit to treatment by length of infusion. Either this is an artefact of post-hoc analysis inherent in any secondary analysis, or it may suggest that the most powerful effects of the GPIIb/IIIa inhibiting agents when combined with a stent procedure occur with profound platelet inhibition at the actual time of the procedure. Could it be that, at least for interventional cardiology indications, once the vessel has been successfully opened further GPIIb/IIIa inhibition adds little benefit?
The nature of cardiac enzyme release during the perioperative period surrounding the PCI procedure also continues to raise questions.[58]When most of the endpoints of interventional trials are examined, the predominant events are increases in cardiac enzymes. PCI procedures are becoming so safe that mortality is rare and the trial results are driven by what are in many cases asymptomatic rises in cardiac enzymes after the procedure. Although it appears clear that cardiac enzyme elevation is a marker of continued hazard for long-term adverse events,[59]how we can prevent these and whether this will translate into long-term benefit is unclear. Stents and newer pharmacological agents have improved the PCI procedure, but neither have eliminated the problem of enzyme release. Eptifibatide treatment does reduce the incidence of enzyme release, as seen in the previously reviewed trials, but does not eliminate the risk. In addition, it appears that up to one-third of the patients with increased enzymes do not manifest the enzyme rise until more than 6 hours after the procedure, well beyond the expected timescale from a pure thrombotic event at the time of the PCI given the usual time course of enzyme kinetics.[57]This suggests that other mechanisms may be in play, and indicates that downstream microembolism of the capillary beds, perhaps with atheromatous debris, may be a target for future pharmacological interventions.[60–62]
Finally, interactions with adjunctive therapies at the time of interventional procedures continue to raise questions. Although the trend over the years has been to use lower target ranges for heparin therapy, especially in combination with GPIIb/IIIa agents, recent analysis has suggested that ACT levels significantly higher than present practice might further improve outcomes.[63]Newer direct thrombin inhibitors[64]and the low molecular weight heparins are now coming into widespread use, although their interactions with eptifibatide therapy remain to be defined.
8. Conclusion
The development of eptifibatide for PCI has mirrored advances in understanding of the pharmacokinetics and pharmacodynamics of the drug. Basic receptor interactions with Ca2+ that were initially poorly understood led to an early overestimate of the in vivo potency of eptifibatide and significant underdosing in an early large clinical trial. Correction of this underdosing and pharmacokinetic modelling with a two-compartment model and population techniques indicated an administration regimen that would adequately suppress platelet activity, both during steady-state infusion periods and during the early acute phases of PCI when platelet activity may be enhanced.
The proof-of-principle seen in the smaller pharmacokinetic-pharmacodynamic studies and the pharmacological investigations was extended by the large-scale testing of the present double-bolus regimen of eptifibatide in the ESPRIT trial. With an appropriate administration regimen, the beneficial outcomes of this therapy appear to reach similar magnitudes to those seen in earlier trials with the monoclonal antibody abciximab therapy. Despite these studies, questions about pathophysiology and the appropriate use of eptifibatide therapy continue to evolve and drive future research.
References
Collier BS. Platelets in cardiovascular thrombosis and thrombolysis. In: Fozzard HA, Haber E, Jennings RB, et al., editors. The heart and cardiovascular system: scientific foundations. Vol 1. 2nd ed. New York: Raven Press, 1991: 219–73
Chesebro JH, Fuster V. Thrombosis in unstable angina. N Engl J Med 1992 7 16; 327(3): 192–4
Fuster V, Lewis A. Conner Memorial Lecture: mechanisms leading to myocardial infarction: insights from studies of vascular biology [published erratum appears in Circulation 1995 Jan 1; 91 (1): 256]. Circulation 1994 Oct 1; 90(4): 2126–46
Faxon D, Weber V, Haudenschild C, et al. Acute effects of transluminal angioplasty in three experimental models of atherosclerosis 1982 Mar 1; 2(2): 125–33
Steele P, Chesebro J, Stanson A, et al. Balloon angioplasty: natural history of the pathophysiological response to injury in a pig model. Circ Res 1985 Jul 1; 57(1): 105–12
White HD. Unmet therapeutic needs in the management of acute ischemia. Am J Cardiol 1997; 80(4A): 2B–10B
Phillips DR, Charo IF, Parise L, et al. The platelet membrane glycoprotein IIb-IIIa complex. Blood 1988; 71: 831–43
Nurden AT, Caen JP. An abnormal platelet glycoprotein pattern in three cases of Glanzmann thrombasthenia. Br J Haematol 1974; 28: 253–60
Phillips DR, Jenkins CSP, Luscher EF, et al. Molecular differences of exposed surface proteins on thrombasthenic platelet plasma membranes. Nature 1975; 257: 599–600
Coller BS. Platelets and thrombolytic therapy. N Engl J Med 1990; 322: 33–42
Tcheng J, Ellis S, George B, et al. Pharmacodynamics of chimeric glycoprotein IIb/IIIa integrin antiplatelet antibody Fab 7E3 in high-risk coronary angioplasty. Circulation 1994 Oct 1; 90(4): 1757–64
Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. The EPIC Investigation. N Engl J Med 1994; 330: 956–61
Knight DM, Wagner C, Jordan R, et al. The immunogenicity of the 7E3 murine monoclonal fab antibody fragment variable region is dramatically reduced in humans by substitution of human for murine constant regions. Mol Immunol 1995; 32(16): 1271–81
Pytela R, Pierschbacher MD, Ginsberg MH, et al. Platelet membrane glycoprotein IIb/IIIa: member of a family of Arg-Gly-Asp-specific adhesion receptors. Science 1986 Mar 28; 231(4745): 1559–62
Scarborough R, Rose J, Hsu M, et al. Barbourin: a GPIIb-IIIaspecific integrin antagonist from the venom of Sistrurus m. barbouri. J Biol Chem 1991 May 25; 266(15): 9359–62
Scarborough R, Naughton M, Teng W, et al. Design of potent and specific integrin antagonists: peptide antagonists with high specificity for glycoprotein IIb-IIIa. J Biol Chem 1993 Jan 15; 268(2): 1066–73
Phillips DR, Scarborough RM. Clinical pharmacology of eptifibatide. Am J Cardiol 1997; 80(4A): 11B–20B
Harrington RA, Kleiman N, Kottke-Marchant K, et al. Immediate and reversible platelet inhibition after intravenous administration of a peptide glycoprotein IIb/IIIa inhibitor during percutaneous coronary intervention. Am J Cardiol 1995 Dec; 76: 1222–7
Rivas GA, Gonzalez-Rodriguez J. Calcium binding to human platelet integrin GPIIb/IIIa and to its constituent glycoproteins: effects of lipids and temperature. Biochem J 1991 May 15; 276 (Pt 1): 35–40
Phillips DR, Teng W, Arfsten A, et al. Effect of Ca2+ on GP IIb-IIIa interactions with integrilin: enhanced GP IIb-IIIa binding and inhibition of platelet aggregation by reductions in the concentration of ionized calcium in plasma anticoagulated with citrate. Circulation 1997; 96(5): 1488–94
Scarborough R. Eptifibatide. Drugs Future 1998; 23(6): 585–90
Kouns W, Wall C, White M, et al. A conformation-dependent epitope of human platelet glycoprotein IIIa. J Biol Chem 1990 Nov 25; 265(33): 20594–601
Kleiman N. Pharmacokinetics and pharmacodynamics of glycoprotein IIb-IIIa inhibitors. Am Heart J 1999; 138(4): 263–75
Charo IF, Scarborough RM, du Mee CP, et al. Pharmacodynamics of the GpIIb-IIIa antagonist Integrelin: phase I clinical studies in normal healthy volunteers. Circulation 1992; 86(4): I–260
Gilchrist IC, O’Shea JC, Kosoglou T, et al. Pharmacodynamics and pharmacokinetics of higher-dose, double-bolus eptifibatide in percutaneous coronary intervention. Circulation 2001; 104(4): 406–11
Tcheng JE, Harrington RA, Kottke-Marchant K, et al. Multicenter, randomized, double-blind, placebo-controlled trial of the platelet integrin glycoprotein IIb/IIIa blocker integrelin in elective coronary intervention. Circulation 1995 Apr 15; 91(8): 2151–7
Randomised placebo-controlled trial of effect of eptifibatide on complications of percutaneous coronary intervention: IMPACT-II. Lancet 1997; 349 (9063): 1422-8
Tcheng JE, Talley JD, O’Shea JC, et al. Clinical pharmacology of higher dose eptifibatide in percutaneous coronary intervention (the PRIDE study). Am J Cardiol 2001; 88(10): 1097–102
Tardiff BE, Jennings LK, Harrington RA, et al. Pharmacodynamics and pharmacokinetics of eptifibatide in patients with acute coronary syndromes: prospective analysis from PURSUIT. Circulation 2001; 104(4): 399–405
Novel dosing regimen of eptifibatide in planned coronary stent implantation (ESPRIT): a randomised, placebo-controlled trial. Lancet 2000 Dec 16; 356 (9247): 2037-44
Jordan RE, Wagner CL, Mascelli MA, et al. Preclinical development of c7E3 Fab: a mouse/human chimeric monoclonal antibody fragment that inhibits platelet function by blockade of GP IIb/IIIa receptors with observations on the immunogenicity of c7E3 Fab in humans. In: Horton MA, editor. Adhesion receptors as therapeutic targets. Boca Raton (FL): CRC Press, 1996: 281–305
Phillips DR, Scarborough RM. Clinical pharmacology of eptifibatide. Am J Cardiol 1997; 80(4A): 11B–20B
Cierniewski CS, Haas T, Smith JW, et al. Characterization of cation-binding sequences in the platelet integrin GPIIb-IIIa (alpha IIb beta 3) by terbium luminescence. Biochemistry 1994 Oct 11; 33(40): 12238–46
D’Souza SE, Haas T, Piotrowicz RS, et al. Ligand and cation binding are dual functions of a discrete segment of the integrin beta 3 subunit: cation displacement is involved in ligand binding. Cell 1994; 79(4): 659–67
Hu DD, Barbas III CF, Smith JW. An allosteric Ca2+ binding site on the beta3-integrins that regulates the dissociation rate for RGD ligands. J Biol Chem 1996 Sep 6; 271(36): 21745–51
Shattil SJ, Brass LF. The interaction of extracellular calcium with the platelet membrane glycoprotein IIb-IIIa complex. Nouv Rev Fr Hematol 1985; 27(4): 211–7
Johnston GI, Heptinstall S. Identity of saturable calcium-binding sites on blood platelets and their involvement in platelet aggregation. Thromb Haemost 1988 Feb 25; 59(1): 54–61
Fujimura K, Phillips D. Calcium cation regulation of glycoprotein IIb-IIIa complex formation in platelet plasma membranes. J Biol Chem 1983 Sep 10; 258(17): 10247–52
Collen D, Lu HR, Stassen JM, et al. Antithrombotic effects and bleeding time prolongation with synthetic platelet GPIIb/IIIa inhibitors in animal models of platelet-mediated thrombosis. Thromb Haemost 1994; 71(1): 95–102
Yasuda T, Gold HK, Fallon JT, et al. Monoclonal antibody against the platelet glycoprotein (GP) IIb/IIIa receptor prevents coronary artery reocclusion after reperfusion with recombinant tissue-type plasminogen activator. J Clin Invest 1988; 81: 1284–91
Gold HK, Coller BS, Yasuda T, et al. Rapid and sustained coronary artery recanalization with combined bolus injection of recombinant tissue-type plasminogen activator and monoclonal anti-platelet GP IIb/IIIa antibody in a dog model. Circulation 1988; 77: 670–7
Folts JD. An in vivo model of experimental arterial stenosis, intimai damage, and periodic thrombosis. Circulation 1991; 83 (6 Suppl. 4): 3–14
Coller BS, Folts JD, Smith S, et al. Abolition of in vivo platelet thrombus formation in primates with monoclonal antibodies to the platelet GPIIb/IIIa receptor: correlation with bleeding time, platelet aggregation and blockade of GPIIb/IIIa receptors. Circulation 1989; 80: 1766–74
Inhibition of platelet glycoprotein IIb/IIIa with eptifibatide in patients with acute coronary syndromes: The PURSUIT Trial Investigators. Platelet glycoprotein IIb/IIIa in unstable angina: receptor suppression using integrilin therapy. N Engl J Med 1998; 339(7): 436–43
Sheiner L. The population approach to pharmacokinetic data analysis: rationale and standard data analysis methods. Drug Metab Rev 1984; 15(1–2): 153–71
Van Wart S, Kosoglou T, Tcheng JE, et al. Eptifibatide pharmacokinetics in patients undergoing coronary angioplasty [abstract]. Clin Pharmacol Ther 1998; 63: 232
Mandak JS, Blankenship JC, Gardner LH, et al. Modifiable risk factors for vascular access site complications in the IMPACT II trial of angioplasty with versus without eptifibatide: Integrilin to Minimize Platelet Aggregation and Coronary Thrombosis. J Am Coll Cardiol 1998; 31(7): 1518–24
Jacobs AK, Kip KE, Williams DO, et al. Current use of IIb/IIIa receptor antagonists during coronary intervention: the NHLBI 1997–1998 Dynamic Registry [abstract]. Circulation 1998; 98: I–16
O’Shea JC, Madan M, Cantor WJ, et al. Design and methodology of the ESPRIT trial: evaluating a novel dosing regimen of eptifibatide in percutaneous coronary intervention. Am Heart J 2000; 140(6): 834–9
O’Shea JC, Hafley GE, Greenberg S, et al. Platelet glycoprotein IIb/IIIa integrin blockade with eptifibatide in coronary stent intervention: the ESPRIT trial: a randomized controlled trial. JAMA 2001; 285(19): 2468–73
O’Shea JC, Buller CE, Cantor WJ, et al. Long-term efficacy of platelet glycoprotein IIb/IIIa integrin blockade with eptifibatide in coronary stent intervention. JAMA 2002; 287: 618–21
Blankenship JC, Sigmon KN, Pieper KS, et al. Effect of eptifibatide on angiographic complications during percutaneous coronary intervention in the IMPACT (Integrilin to Minimize Platelet Aggregation and Coronary Thrombosis) II trial. Am J Cardiol 2001; 88(9): 969–73
Singh M, Reeder GS, Ohman E, et al. Does the presence of thrombus seen on a coronary angiogram affect the outcome after percutaneous coronary angioplasty?: an angiographic trials pool data experience. J Am Coll Cardiol 2001; 38(3): 624–30
Thel M, Califf R, Tcheng J, et al. Clinical risk factors for ischemic complications after percutaneous coronary interventions: results from the EPIC trial. Am Heart J 1999; 137(2): 264–73
Effect of glycoprotein IIb/IIIa receptor blocker abciximab on outcome in patients with acute coronary syndromes without early coronary revascularisation: the GUSTO IV-ACS randomised trial. Lancet 2001; 357: 1915-24
Kleiman NS, Raizner AE, Jordan R, et al. Differential inhibition of platelet aggregation induced by adenosine diphosphate or a thrombin receptor-activating peptide in patients treated with bolus chimeric 7E3 Fab: implications for inhibition of the internal pool of GPIIb/IIIa receptors. J Am Coll Cardiol 1995; 26: 1665–71
Gilchrist IC, Pacchiana CM, Cantor WJ, et al. Temporal spectrum of ischemic complications with percutaneous coronary intervention: the ESPRIT experience. Circulation 2000; 102(II): 640–1
Abdelmeguid AE, Topol EJ. The myth of the myocardial ‘infarctlet’ during percutaneous coronary revascularization procedures. Circulation 1996 Dec 15; 94(12): 3369–75
Califf RM, Abdelmeguid AE, Kuntz RE, et al. Myonecrosis after revascularization procedures. J Am Coll Cardiol 1998; 31(2): 241–51
Gibson CM, Cohen DJ, Cohen EA, et al. Effect of eptifibatide on coronary flow reserve following coronary stent implantation (an ESPRIT substudy): Enhanced Suppression of the Platelet IIb/IIIa Receptor with Integrilin Therapy. Am J Cardiol 2001; 87(11): 1293–5
Gibson C, Murphy S, Marble S, et al. Relationship of creatine kinase-myocardial band release to thrombolysis in myocardial infarction perfusion grade after intracoronary stent placement: an ESPRIT substudy. Am Heart J 2002; 143(1): 106–10
Topol EJ, Yadav JS. Recognition of the importance of embolization in atherosclerotic vascular disease. Circulation 2000 Feb 8; 101(5): 570–80
Chew DP, Bhatt DL, Lincoff A, et al. Defining the optimal activated clotting time during percutaneous coronary intervention: aggregate results from 6 randomized, controlled trials. Circulation 2001; 104(22): 961–6
Roe MT, Granger CB, Puma J, et al. Comparison of benefits and complications of hirudin versus heparin for patients with acute coronary syndromes undergoing early percutaneous coronary intervention. Am J Cardiol 2001; 88(12): 1403–6
Acknowledgements
The author has received grant and research funding indirectly from: Millennium Pharmaceuticals, Inc. (formerly Cor Therapeutics, Inc.) and Schering-Plough Pharmaceuticals, Inc. (eptifibatide); Eli Lilly, Inc. (abciximab); and Merck, Inc. (tirofiban). None of these companies had editorial input into this article.
Author information
Authors and Affiliations
Corresponding author
9. Appendix: Formulae for Fitting Plasma Concentration-Time Data to Biexponential Equations
9. Appendix: Formulae for Fitting Plasma Concentration-Time Data to Biexponential Equations
9.1 Single Bolus Groups
(Equation 1):
C(t) is concentration at sampling time t, ti is time of infusion, α and β are the rate constants of the distribution and elimination phases, A and B are the coefficients of the exponential terms for α and β, and TSTAR = t — ti for t > ti and TSTAR = 0 for t ≤ti.
9.2 Double-Bolus Groups
(Equation 2):
TBOL = t − tb for t > tb and TBOL = 0 for t < tb where tb is time of second bolus administration.
Rights and permissions
About this article
Cite this article
Gilchrist, I.C. Platelet Glycoprotein IIb/IIIa Inhibitors in Percutaneous Coronary Intervention. Clin Pharmacokinet 42, 703–720 (2003). https://doi.org/10.2165/00003088-200342080-00001
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200342080-00001