
Abstract
Percutaneous coronary intervention (PCI) promotes thrombosis by inducing extreme vascular injury. The concomitant presence of dysfunctional endothelium, vulnerable plaque, and endothelial erosion promotes further thrombotic risk. Platelet adhesion to newly exposed collagen and von Willebrand factor by specific receptors and binding of thrombin generated by tissue factor to protease-activated receptors (PARs) cause initial platelet activation. Following activation, adenosine diphosphate (ADP) is released from dense granules and thromboxane A2 is generated by cyclooxygenase-1 (COX-1). Although both thromboxane A2 and ADP amplify platelet activation and aggregation, continuous ADP-P2Y12 receptor signaling is essential for sustained activation of the GPIIb/IIIa receptor and stable thrombus generation. Simultaneously, platelet activation exposes the phosphatidylserine surface providing binding sites for coagulation factors and the generation of thrombin. Thrombin converts fibrinogen to fibrin and activates factor XIII that cross-links the fibrin network, stabilizes the platelet-fibrin clot at the site of vascular injury, and impairs myocardial blood supply. Therefore, the rationale for antithrombotic therapy during and following PCI is to prevent thrombus formation within the target lesion and also in nontarget vessels by attenuating platelet activation and aggregation and arresting coagulation processes. Since clot formation involves multiple pathways including platelet activation and aggregation and coagulation, simultaneous blockade of these pathways is essential to prevent periprocedural and post-PCI ischemic event occurrences. Optimal inhibition of these pathways is essential for maximizing antithrombotic effects and minimizing bleeding risk and is critically dependent on individual patient risk.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Antiplatelet
- Antithrombotic
- Platelet
- Aspirin
- Clopidogrel
- Ticagrelor
- Prasugrel
- Glycoprotein inhibitor
- Heparin
- Bivalirudin
Inova Heart and Vascular Institute provides the full spectrum of diagnostic, therapeutic, surgical, and interventional cardiac services including a nationally recognized program for treating atrial fibrillation, heart and lung transplantation, innovative minimally invasive surgical techniques, robot-assisted surgery, and implantation of artificial heart devices. Yearly we perform more than 4000 cardiac diagnostic investigations, 5000 cardiac catheterizations, 170 TAVR procedures, and 500 CABG surgeries.
Professor Paul A. Gurbel is an interventional cardiologist at Inova Heart and Vascular Institute. He specializes in the catheter-based treatment of patients with complex cardiovascular disease and he has performed over 15,000 percutaneous cardiac and peripheral procedures. He is currently the Director of the Inova Center for Thrombosis Research and Drug Development, Professor of Medicine at the Johns Hopkins University School of Medicine, and Adjunct Professor of Medicine at Duke University School of Medicine. For over 15 years his research has focused on defining the effects of antiplatelet agents, developing antiplatelet agents, and understanding the relation of platelet reactivity to ischemic event occurrence in patients undergoing stenting. Dr. Udaya S Tantry is the Director of the Thrombosis Research Laboratory.
Platelet Function in Percutaneous Coronary Intervention
-
Percutaneous coronary intervention (PCI) promotes thrombosis by inducing extreme vascular injury. The concomitant presence of dysfunctional endothelium, vulnerable plaque, and endothelial erosion promotes further thrombotic risk.
-
Platelet adhesion to newly exposed collagen and von Willebrand factor by specific receptors and binding of thrombin generated by tissue factor to protease-activated receptors (PARs) cause initial platelet activation.
-
Following activation, adenosine diphosphate (ADP) is released from dense granules and thromboxane A2 is generated by cyclooxygenase-1 (COX-1) . Although both thromboxane A2 and ADP amplify platelet activation and aggregation, continuous ADP-P2Y12 receptor signaling is essential for sustained activation of the GPIIb/IIIa receptor and stable thrombus generation.
-
Simultaneously, platelet activation exposes the phosphatidylserine surface providing binding sites for coagulation factors and the generation of thrombin. Thrombin converts fibrinogen to fibrin and activates factor XIII that cross-links the fibrin network, stabilizes the platelet-fibrin clot at the site of vascular injury, and impairs myocardial blood supply [1, 2].
-
Therefore, the rationale for antithrombotic therapy during and following PCI is to prevent thrombus formation within the target lesion and also in nontarget vessels by attenuating platelet activation and aggregation and arresting coagulation processes. Since clot formation involves multiple pathways including platelet activation and aggregation and coagulation, simultaneous blockade of these pathways is essential to prevent periprocedural and post-PCI ischemic event occurrences (Fig. 12.1).
-
Optimal inhibition of these pathways is essential for maximizing antithrombotic effects and minimizing bleeding risk and is critically dependent on individual patient risk.

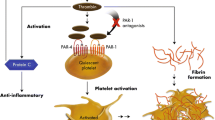
Antiplatelet and antithrombotic agents in percutaneous coronary intervention (PCI). During PCI, at the site of vascular injury, exposure of the subendothelial matrix leads to adhesion and activation of platelets and subsequent release of secondary agonists, TxA2 and ADP. These two locally generated secondary agonists play a critical role in the sustained activation of GPIIb/IIIa receptors and stable platelet aggregation. Simultaneously, platelet activation exposes the phosphatidylserine surface providing binding sites for coagulation factors and the generation of large amounts of thrombin. Thrombin converts fibrinogen to fibrin and activates factor XIII which cross-links the fibrin network, and stabilizes the platelet-fibrin clot at the site of vascular injury. Since clot formation involves multiple pathways including platelet activation, aggregation, and coagulation, simultaneous blockade of these pathways is essential to prevent periprocedural and post-PCI ischemic events. Antiplatelet strategies include (a) inhibition of platelet cyclooxygenase-1 enzyme by aspirin; (b) inhibition of the P2Y12 receptor by clopidogrel, prasugrel, ticagrelor, or cangrelor; and (c) inhibition of activated GPIIb/IIIa receptors by abciximab, eptifibatide, and tirofiban. Major antithrombotic agents include (a) indirect thrombin inhibitors such as heparin, and low-molecular-weight heparins; (b) direct thrombin inhibitors such as bivalirudin and dabigatran; and (c) direct Xa inhibitors such as rivaroxaban and apixaban. Key: AT antithrombin, ADP adenosine diphosphate, TxA 2 thromboxane-A2, vWF von Willebrand factor, TF tissue factor, TP thromboxane A2 receptor, PAR-1 protease-activated receptor-1, GP glycoprotein, Factor II prothrombin, Factor IIa thrombin. Adapted from Gurbel PA et al. JACC Heart Fail. 2014;2:1–14 [2]
Antiplatelet Agents
Aspirin
-
Aspirin remains the bedrock of antiplatelet treatment strategies in patients undergoing PCI. The antithrombotic property of aspirin is primarily attributed to irreversible acetylation of the platelet COX-1 enzyme. Subsequently, the generation of TxA2- and TxA2-induced platelet aggregation is inhibited.
-
The optimal aspirin dose remains controversial. In the Clopidogrel Optimal Loading Dose Usage to Reduce Recurrent Events–Organization to Assess Strategies in Ischemic Syndromes (CURRENT OASIS-7) trial, aspirin 300–325 mg daily as compared to aspirin 75–100 mg daily was associated with increased 30-day gastrointestinal bleeding, but there was no significant differences in the outcome of cardiovascular death, MI, or stroke and no differences in major bleeding observed between groups (Fig. 12.2) [3].
-
Current revascularization guidelines recommend immediate treatment with an initial loading dose of non-enteric-coated aspirin 150–300 mg (or 80–150 mg IV) followed by a lifelong maintenance dose of 75–100 mg per day [4,5,6] (Table 12.1).
-
The most common side effect of aspirin treatment is gastrointestinal intolerance.

Primary outcome in CURRENT OASIS 7 trial : invasive cohort. Adapted from Mehta et al. Lancet. 2010;376:1233–43 [3]
P2Y12 Receptor Blockers
The most widely used oral P2Y12 receptor blockers are the thienopyridines (clopidogrel and prasugrel), and ticagrelor (Fig. 12.3). The European Commission issued marketing authorization for cangrelor, an intravenous P2Y12 receptor blocker, in March 2015.

Metabolism and mechanism of action of P2Y12 inhibitors
Clopidogrel
-
Clopidogrel, a second-generation thienopyridine, remains the most widely prescribed oral P2Y12 receptor blocker. Following absorption, nearly ~85% of clopidogrel is hydrolyzed to an inactive carboxylic metabolite and the remaining 15% is rapidly and extensively metabolized by CYP450-dependent two-step process in liver to a highly unstable active metabolite R-130964. Plasma concentrations of the parent compound are below the detection limit beyond 2 h post-dosing. The active metabolite binds specifically and irreversibly to the platelet P2Y12 receptor during passage through the hepatic circulation and inhibits the P2Y12 receptor for the life span of platelets.
-
Results of the earlier landmark trials strongly influenced the widely implemented strategy of dual-antiplatelet therapy for the PCI patient as the standard of care. In the CURRENT OASIS-7 trial a strategy of double-dose clopidogrel (600 mg on day 1, 150 mg on days 2–7, then 75 mg daily) was compared to standard-dose clopidogrel (300 mg on day 1, then 75 mg daily) in patients with ACS. In an analysis of 78% of patients who underwent PCI, double-dose clopidogrel therapy was associated with a 14% reduction in the rate of the primary outcome, 46% reduction in the secondary outcome of definite stent thrombosis, and 41% more CURRENT defined major bleeding (Fig. 12.2) [3].
-
The presence of single-nucleotide polymorphisms (SNPs) of the gene encoding CYP450 2C19, particularly the loss-of-function (LoF) allele (CYP2C19*2, *3, *4, and *5), has been shown to be independently associated with reduced clopidogrel active metabolite generation, reduced inhibition of ADP-induced platelet aggregation, and increased post-PCI ischemic events. Measurements of ex vivo platelet function indicative of P2Y12 receptor activity demonstrated a slow onset of action, wide response variability, and an absence of inhibition (resistance) in ~30% of patients undergoing PCI treated with a 300 mg clopidogrel added to aspirin therapy [5].
-
In multiple studies of patients undergoing PCI, high on-treatment platelet reactivity during clopidogrel therapy was associated with an increased risk of ischemic event occurrence.
-
Currently available evidence supports the concept of a threshold for on-treatment platelet reactivity to ADP in patients treated with dual-antiplatelet therapy that may be used to stratify patient risk for ischemic/thrombotic events following PCI, including stent thrombosis.
-
Pharmacodynamic studies have demonstrated that therapy with potent P2Y12 receptor blockers such as prasugrel or ticagrelor is an optimal strategy to overcome high on-treatment platelet reactivity and genetic polymorphisms [7].
-
Selective, but not routine, platelet function testing or genetic testing may be considered in determining an antiplatelet strategy in patients with a history of stent thrombosis and in patients prior to undergoing high-risk PCI.
Prasugrel
-
Prasugrel, a third-generation thienopyridine, is rapidly absorbed after oral administration with modest intra- and inter-recipient variability.
-
Prasugrel is extensively hydrolyzed by intestinal and plasma esterases to an inactive short-lived thiolactone metabolite that is further metabolized to the pharmacologically active metabolite, R-138727, mainly by hepatic CYP3A4 and CYP2B6 in a one-step oxidation process (Fig. 12.3).
-
Prasugrel is associated with a more rapid onset of action and greater active metabolite generation resulting in less response variability, a lower prevalence of non-responsiveness, and greater inhibition of ADP-induced platelet aggregation compared with clopidogrel.
-
In the TRITON-TIMI 38 trial , in ACS patients undergoing planned PCI, prasugrel (60 mg load/10 mg daily maintenance) plus aspirin treatment (75–162 mg/day) was associated with a 19% reduction in the primary composite endpoint of cardiovascular death, nonfatal MI, and nonfatal stroke at a median 14.5-month follow-up compared with clopidogrel (300 mg load/75 mg daily maintenance) plus aspirin treatment.
-
However, these benefits were associated with significantly increased key safety end points of TIMI major bleeding, including life-threatening and fatal bleeding in patients treated with prasugrel as compared to clopidogrel (2.4% vs. 1.8%; p < 0.03) (Fig. 12.4) [8].
-
Prasugrel is not recommended in patients with active pathological bleeding or a history of TIA or stroke.
-
In patients ≥75 years of age, prasugrel is generally not recommended because of increased risk of fatal and intracranial bleeding and uncertain benefit.
-
It is recommended not to start prasugrel therapy in patients likely to undergo urgent CABG. When possible, prasugrel should be discontinued at least 7 days before any surgery [4].
-
In the Comparison of Prasugrel at the Time of Percutaneous Coronary Intervention or as Pretreatment at the Time of Diagnosis in Patients with Non-ST Elevation Myocardial Infarction (ACCOAST) trial, NSTE-ACS patients with positive troponin levels, scheduled to undergo coronary angiography within 2–48 h after randomization, were treated with either prasugrel 30 mg loading dose pre-angiography and 30 mg at PCI (pretreatment group) or 60 mg at PCI. The rate of the primary efficacy composite endpoint of cardiovascular death, MI, stroke, urgent revascularization, or glycoprotein IIb/IIIa bailout through day 7 did not differ between the treatment groups (HR, 95% CI = 1.02, 0.84–1.25; p = 0.81), but the key safety endpoint of TIMI major bleeding (CABG or non-CABG) was increased in pretreated patients (HR, 95% CI = 1.90, 1.19–3.02; p = 0.006). These results suggest that pretreatment with prasugrel in NSTE ACS patients is not beneficial in reducing ischemic risk but is associated with elevated bleeding risk [9].

TRITON TIMI 38 trial outcomes (Key: MI myocardial infarction, TIMI Thrombolysis in Myocardial Infarction, HR hazard ratio, CI confidence interval, CV cardiovascular, CABG coronary artery bypass graft) [8]
Ticagrelor
-
Ticagrelor (AZD6140), a cyclopentyltriazolopyrimidine derivative, is an oral, reversibly binding, direct-acting P2Y12 inhibitor.
-
In stable coronary artery disease (CAD) patients, ticagrelor therapy was associated with a rapid onset of action, a greater level of inhibition that persisted during maintenance therapy, and a more rapid offset of pharmacodynamic action compared with clopidogrel [10].
-
In a prespecified analysis of PLATO trial involving ACS patients in whom an invasive strategy was planned (72% of total patients), ticagrelor (180 mg loading/90 mg bid) versus clopidogrel (300–600 mg loading dose/75 mg per day) was associated with a significant reduction in the primary efficacy endpoint of CV death, MI, or stroke (event rate at 360 days = 9.0% vs. 10.7%, HR = 0·84, 95% CI = 0·75–0·94; p = 0·0025). Similarly, there were significant reductions in the secondary key endpoints of all-cause death plus MI plus stroke (9.4% vs. 11.2%; p = 0.0016), all-cause death (3.9% vs. 5.0%; p = 0.013), and MI (5.3% vs. 6.6%; p = 0.0023) in favor of ticagrelor therapy (Fig. 12.5) [11].
-
There were no differences in TIMI major bleeding (7.9% vs. 7.9%, p = 1.00), or TIMI non-CABG-related major bleeding (2.8% vs. 2.2%, p = 0.08) in patients treated with ticagrelor vs. clopidogrel.
-
The ticagrelor benefit remained significant (vs. clopidogrel) whether or not patients were given standard or higher loading doses of clopidogrel, and in those already on clopidogrel at the start of the study.
-
The PLATO trial demonstrated a significant reduction in mortality associated with ticagrelor therapy. An absence of clinical benefit associated was, however, noted among the North American patient population enrolled in the PLATO trial. This has been attributed to the concomitant use of high-dose aspirin (aspirin >100 mg/day). Therefore, 75–81 mg per day aspirin is recommended in all patients treated with ticagrelor.
-
Ticagrelor therapy is associated with side effects including dyspnea, which is rarely severe enough to cause discontinuation of treatment, and bradycardia.
-
When possible, ticagrelor should be discontinued at least 5 days before surgery [4].
-
In the ATLANTIC study , prehospital treatment with ticagrelor in STEMI patients was associated with a similar proportion of patients without ≥70% resolution of ST-segment elevation before PCI or the proportion of patients without TIMI flow grade 3 at initial angiography in the infarct-related artery as compared to in-hospital treatment. Similarly no differences in 30-day major adverse cardiovascular events or bleeding events were also observed [12].
-
The TWILIGHT (Ticagrelor With Aspirin or Alone in High-risk Patients After Coronary Intervention ; ClinicalTrials.gov NCT02270242) study will determine the benefit of ticagrelor monotherapy alone versus ticagrelor plus low-dose aspirin for 12 months in reducing bleeding among high-risk patients undergoing PCI who have completed a 3-month course of aspirin plus ticagrelor.

PLATO trial outcomes: invasive cohort. Adapted from Cannon et al. Lancet. 2010;375:283–93 [11]. (Key: MI myocardial infarction, HR hazard ratio, CI confidence interval, CABG coronary artery bypass graft)
Cangrelor
-
Cangrelor is a parenterally administered adenosine triphosphate (ATP) analog with a short half-life (3–6 min), with rapid onset/offset of action, and dose-dependent and predictable pharmacodynamic effects.
-
Cangrelor directly, reversibly, and competitively inhibits binding of ADP to the P2Y12 receptor.
-
In patients with stable CAD or ACS undergoing PCI in the CHAMPION-PHOENIX trial , a bolus and infusion of cangrelor therapy versus a loading dose of 600 mg or 300 mg of clopidogrel was associated with a significantly reduced primary endpoint of death, MI, ischemia-driven revascularization, or stent thrombosis at 48 h [4.7% vs. 5.9%, odds ratio (95% CI) = 0.78 (0.66–0.93), p = 0.005]. The primary safety endpoint of severe bleeding at 48 h was similar between the treatment groups [0.16% vs. 0.11% odds ratio (95% CI) = 1.50 (0.53–4.22), p = 0.44]. The rate of stent thrombosis was lower in the cangrelor group compared with clopidogrel group [0.8% vs. 1.4%, odds ratio (95%CI) = 0.62 (0.43–0.90), p = 0.01]. Furthermore, the benefits associated with cangrelor were consistent across the subgroups of Stable angina, n = 6138, NSTE-ACS, n = 2810 and STEMI, n = 1991 p interaction in patients receiving clopidogrel 300 mg LD or 600 mg LD: p = 0.62. GUSTO severe bleeding was similar between groups [13].
-
Cangrelor has been recommended in Europe to be co-administered with aspirin, for the reduction of thrombotic cardiovascular events in adult patients with coronary artery disease undergoing PCI who have not received an oral P2Y12 inhibitor prior to the PCI procedure and in whom oral therapy with P2Y12 inhibitors is not feasible or desirable.
Glycoprotein (GP) IIb/IIIa Inhibitors
-
The GPIIb/IIIa receptor , a member of the integrin family of receptors, is the most abundant platelet glycoprotein receptor (~80,000 per platelet).
-
Platelet activation by various agonists and stimuli induces a conformational change in GPIIb/IIIa that markedly enhances its affinity for fibrinogen. The pharmacological agents that directly block the binding of fibrinogen to the GPIIb/IIIa receptor are more effective in inhibiting platelet aggregation than any oral antiplatelet strategy.
-
In addition to inhibition of platelet aggregation, GPIIb/IIIa inhibitors also induce platelet disaggregation and may attenuate microembolization, and release of vasoconstrictors [14].
-
All of the GPIIb/IIIa inhibitors have been associated with an increase in bleeding as compared to treatment with heparin alone. However, GPIIb/IIIa inhibitors are frequently incorrectly dosed, and overdosing has been associated with increased bleeding. Moreover, in current practice the activated clotting time target during the time of GPIIb/IIIa inhibitor use for PCI is lower than in earlier studies.
-
Efficacy and increased safety have been reported with use of GPIIb/IIIa inhibitors in conjunction with heparin at activating clotting time (ACT) levels of 200–250 s [15].
-
Most of the clinical trials demonstrating a favorable net clinical efficacy of GP IIb/IIIa inhibitor therapy predated the era of early invasive therapy, PCI with uniform or near-uniform stenting, and thienopyridine pretreatment. These older studies supported the upstream use of a GP IIb/IIIa inhibitor in combination with aspirin and an anticoagulant in high-risk patients.
-
The efficacy of glycoprotein (GP) IIb/IIIa inhibitor therapy has been established particularly among high-risk patients undergoing PCI with elevated cardiac biomarkers, and diabetes. Most often, in the current era of PCI, GPIs are used for “bailout” when visible thrombus is present in the target vessel.
-
According to guidelines, upstream use of GPI (vs. in-lab use) can be considered only in high-risk patients undergoing transfer for primary PCI and routine upstream use of GP IIb/IIIa inhibitor in NASTE-ACS patients undergoing angiography is not recommended.
-
Following the development of fast-acting, potent oral P2Y12 receptor blockers, such as prasugrel and ticagrelor, the use of GPIs in high-risk patients waned and now is more limited in current interventional practice as compared to two decades ago.
-
Moreover, cangrelor, an intravenous P2Y12 receptor antagonist with very fast onset and offset of action, represents a new strategy of modulating peri-PCI platelet reactivity. The characteristics and recommended dosing of three commercially available GP IIb/IIIa inhibitors, abciximab, eptifibatide, and tirofiban, are given in Table 12.2 .
Duration of Dual-Antiplatelet Therapy
-
The optimal duration of DAPT after stenting is not yet clearly defined. The risk for recurrent thrombotic event occurrences following stenting is high during the first 3 months and thrombotic events continue to increase for at least 3 years.
-
Complete stent endothelialization, the most desired outcome, has been observed within a month with bare-metal stent (BMS) implantation, whereas drug-eluting stent (DES) implantation has been associated with highly suppressed early healing and poor endothelial cell coverage that may persist for years.
-
In addition, recent randomized clinical trials (RCT’s) of longer duration DAPT suggested a continued reduction of thrombotic events at about 3 years in patients treated with prolonged DAPT and this event reduction was mostly observed in non-culprit lesion vessels. In this line, the duration of DAPT appears dependent on stent type (BMS vs. earlier generation DES vs. newer generation DES vs. biodegradable stents), presence or absence of prior MI, balance between ischemic and bleeding risk, and cost versus benefit.
-
There are numerous trials that have investigated the duration of DAPT in patients treated with bare-metal stents and drug-eluting stents including newer generation stents. Since none of these trials are powered for ischemic endpoints; all were open label and the time for stenting to randomization varied among these trials.
-
Numerous meta-analyses have also addressed the duration of DAPT. Based on the available evidence, it is recommended that DAPT be administered for at least 1 month after BMS implantation in stable CAD, for 6 months after new-generation DES implantation in stable CAD, and for up to 1 year in patients after ACS, irrespective of revascularization strategy [4].
-
In the DAPT study, 9961 patients treated with standard thienopyridine therapy (clopidogrel or prasugrel) and aspirin for 12 months and who were without any ischemic or bleeding events were randomly assigned to receive DAPT or aspirin alone for another 18 months. The prolonged DAPT therapy was associated with a 71% relative (1% absolute) reduction in stent thrombosis (p < 0.001), a 53% relative (2.0% absolute) reduction in MI (p < 0.001), a 29% relative (1.6% absolute) reduction in major adverse cardiovascular and cerebrovascular events (p < 0.001), and a 1.0% absolute increase in GUSTO moderate or severe bleeding (p = 0.001) (Fig. 12.6) [16].
-
Trials of prolonged or extended DAPT suggest that the benefit/risk ratio of prolonged DAPT may be more favorable for those with prior MI, with an absolute decrease in ischemic events of ≈1% to 2% at the cost of an absolute increase in bleeding events of ≈1% over the course of several years of prolonged or extended therapy (median durations of therapy: 18–33 months) [17].
-
A new risk score (the “DAPT score”), derived from the DAPT study, may be useful for decisions about whether to continue (prolong or extend) in patients treated with coronary stent implantation (Table 12.3) [18].
-
The PEGASUS-TIMI 54 study provided some evidence regarding the long-term efficacy of ticagrelor therapy in the setting of post-MI and post-PCI. Here 21,162 patients (83% underwent PCI), 1–3 years post-MI, were treated with 90 mg bid ticagrelor, 60 mg bid ticagrelor, or placebo in addition to low-dose aspirin for a median duration of 33 months. Prolonged ticagrelor therapy was associated with 14–15% reduction in the primary efficacy endpoint of CV death, MI, or stroke, but 2.3–2.7-fold increased risk for clinically significant bleeding. However, the 60 mg dose was associated with a better safety and tolerability profile with numerically lower rates of bleeding and other side effects such as dyspnea. In light of this, the European Medicines Agency in October 2016 recommended a 60 mg twice-daily dose when an extended treatment (for up to 3 years) is required for patients with a history of MI of at least 1 year earlier and a high risk of an atherothrombotic event [19].

DAPT trial outcomes [16]. (Key: HR hazard ratio, MACCE major adverse cardiovascular and cerebrovascular events, MI myocardial infarction, BARC bleeding academic research consortium)
Antithrombotics
Indirect Thrombin Inhibitors
-
Unfractionated heparin (UFH) is a heterogeneous mixture of polysaccharide molecules. The pentasaccharide sequence of UFH binds to antithrombin and enhances the inhibition of thrombin and also factor Xa.
-
UFH binds plasma proteins strongly, leading to unpredictable levels of free heparin in the circulation. UFH therefore exhibits significant variability in antithrombotic effect and requires close monitoring.
-
Most of the benefits of UFH are short term. Its other disadvantages include the need for continuous intravenous administration and the infrequent but serious complication of immunogenic heparin -induced thrombocytopenia. Despite this, UFH is the standard of care for prevention of thrombus generation in the setting of PCI in all patients.
-
Low-molecular-weight heparins (LMWHs) were developed with the goal of providing improved anticoagulation over that of UFH .
-
LMWHs have less direct effect on thrombin, less plasma binding, better bioavailability, less platelet activation, and more effect on factor Xa, and a lower risk of immune-mediated thrombocytopenia than UFH .
-
LMWH therapy can be administered subcutaneously on a weight basis and does not require dose adjustments or monitoring. Patients with renal insufficiency require lower dosing of LMWHs, since LMWH is mainly cleared by the kidneys.
-
In the STEEPLE trial , the primary endpoint of 48-h non-CABG-related bleeding was lower with low-dose enoxaparin (0.5 mg/kg) but not with the higher dose (0.75 mg/kg) as compared to UFH , whereas major bleeding was decreased with similar efficacy with both doses as compared to UFH in stable CAD patients undergoing PCI.
-
The enoxaparin low-dose therapy was stopped prematurely because of a nonsignificant trend towards excess mortality not related to ischemic events and not confirmed at 1 year of follow-up [20].
-
In recent studies, enoxaparin therapy did not demonstrate increased benefit over UFH when pre-randomization anticoagulation was not consistent with the study treatment or when there was a post-randomization crossover.
-
In the ATOLL trial , enoxaparin (0.5 mg/kg) did not significantly reduce the primary composite endpoint of death, MI, procedure failure, or major bleeding as compared to UFH (p = 0.069) and there was no indication for higher incidence of bleeding with enoxaparin versus UFH in patients undergoing primary PCI.
-
In the per-protocol analysis of the ATOLL trial, that included 87% of patients, enoxaparin was superior to UFH in reducing the primary endpoint (relative risk 0.76, p = 0.012), mortality (RR = 0.46, p = 0.05), and major bleeding (RR = 0.46, p = 0.0002) [21].
-
Based on these favorable results, enoxaparin with or without GPI should be considered as an alternative to UFH for primary PCI according to European guidelines [4].
Fondaparinux
-
Fondaparinux, an indirect factor Xa inhibitor , is a synthetic pentasaccharide that binds (reversibly with high affinity) to antithrombin III, thereby catalyzing the antithrombin III-mediated inhibition of factor Xa.
-
Fondaparinux is not preferred during PCI due to the risk of catheter thrombosis [4].
-
In NSTE-ACS patients undergoing PCI in the OASIS 5 trial (6239 out of 22,078 patients), fondaparinux 2.5 mg subcutaneous once-daily dose as compared to enoxaparin was associated with significantly lower major bleeding (including access-site complications) at 9 days (2.3% vs. 5.1%, HR = 0.45, p < 0.001). Catheter thrombus formation, however, was observed more frequently with fondaparinux (0.9% vs. 0.4%) and was abolished by injection of an empirically determined bolus of UFH at the time of PCI.
-
Therefore, a single-bolus UFH (85 IU/kg, or 60 IU/kg in the case of concomitant use of GP IIb/IIIa receptor inhibitors) is indicated during PCI in patients with NSTEMI treated with fondaparinux.
Direct Thrombin Inhibitors
-
Direct thrombin inhibitors are small molecules that bind to thrombin (both fluid phase and fibrin bound) and block thrombin-induced conversion of fibrinogen to fibrin and activation of FV, FVII, and FIX. They have limited interaction with plasma proteins and cells, making dosing and bioavailability much more predictable.
-
The major direct thrombin inhibitors available are dabigatran, argatroban, and bivalirudin of which bivalirudin is widely used during coronary intervention.
-
In the ISAR-REACT-3 trial , among stable CAD patients undergoing PCI and pretreated with clopidogrel, bivalirudin (bolus 0.75 mg/kg; infusion 1.75 mg/kg/h) showed similar net clinical outcomes as compared with UFH , but higher-than-recommended dosage of UFH (140 IU/kg) was attributed to excess major bleeding [22]. A lower dose of UFH (100 IU/kg) was associated with similar major bleeding as compared to bivalirudin and a trend towards less ischemic events in the UFH arm [23].
-
Therefore, UFH is the standard anticoagulant treatment for elective PCI and bivalirudin should be considered in patients at high risk of bleeding.
-
In the ACUITY trial, moderate- to high-risk patients (n = 13,819) with NSTE-ACS managed with contemporary pharmacotherapy and undergoing an early invasive strategy were randomized to UFH or enoxaparin plus planned GPI, bivalirudin plus planned GPI, or bivalirudin monotherapy.
-
Bivalirudin monotherapy met non-inferiority criteria with respect to the 30-day primary ischemic endpoint (death, MI, or unplanned revascularization), with a significantly lower risk of major bleeding [24]. Among patients who underwent PCI (n = 7789) (57% received PCI through femoral access), there was no difference in the primary ischemic endpoint or stent thrombosis, but bivalirudin monotherapy was associated with a significant reduction in major bleeding, minor bleeding, and transfusion requirements (Fig. 12.7) [25].
-
In the ISAR-REACT 4 trial , the safety and efficacy of bivalirudin monotherapy versus UFH plus GPI in NSTE-ACS patients undergoing PCI through femoral access and pretreated with clopidogrel were assessed. This trial provided further evidence in favor of bivalirudin with similar primary ischemic endpoint and significantly lower major bleeding that was attributed to lower access-site bleeding [26]. It should be noted that most of the evidence in support of bivalirudin was derived from trials where it was compared to UFH plus GPI, a combination which is no longer routinely implemented in the current practice.
-
In the HORIZON-AMI trial , among patients with STEMI undergoing primary PCI through femoral access (93%), bivalirudin plus provisional GPI was found to be superior to UFH plus routine GPIs with respect to 30-day major bleeding (4.9% vs. 8.3%, RR = 0.60, p = 0.001) and 30-day net adverse clinical events including all-cause death, reinfarction, repeat revascularization, definite stent thrombosis, stroke, or major bleeding (9.2% vs. 121.1%, RR = 0.76, p = 0.005). The clinical benefit of bivalirudin therapy persisted for 3 years. However, a higher incidence of stent thrombosis was observed during the first 24 h in the bivalirudin arm (1.3% vs. 0.3%, p < 0.001), but no difference was observed at 30 days. Pre-randomization use of UFH and 600 mg loading dose of clopidogrel were independent predictors of lower risk of acute and subacute stent thrombosis [27].
-
The recent EUROMAX trial compared a strategy of prehospital bivalirudin therapy with UFH or LMWH with optional use of GPIs (69%) in 2218 STEMI patients, with frequent use of radial access (47%) and pretreatment with P2Y12 inhibitors (98%). Prehospital use of bivalirudin was associated with significantly lower 30-day primary endpoint of death or non-CABG major bleeding as compared to UFH group (5.1% vs. 8.5%, RR = 0.60, p < 0.001) that was driven by a significant reduction in major bleeding (2.6% vs. 6.0%, RR = 0.43, p < 0.001). Similar to HORIZON-AMI, 30-day stent thrombosis was more frequent in the bivalirudin group (1.6 vs. 0.5%, RR = 2.89, p = 0.002) that was solely driven by a difference during the first 24 h and was paralleled by a trend towards a higher rate of re-infarction (1.7% vs. 0.9%, RR = 1.93, p = 0.08) despite the use of prasugrel and ticagrelor in more than half of patients. Again, the mortality benefit observed in the HORIZON-AMI trial was not demonstrated in the EUROMAX trial [28].
-
The HEAT-PPCI trial compared bivalirudin (n = 905) with UFH alone (n = 907) in STEMI patients who were planned to undergo primary PCI. In this trial, GPI use was allowed only for bailout (15%), and prasugrel or ticagrelor, and radial access was frequently used (89% and 80% of patients, respectively). Bivalirudin therapy was associated with higher rates of the 30-day primary composite endpoint of all-cause death, cerebrovascular accidents, recurrent infarction, and urgent target-vessel revascularization (8.7% vs. 5.7%, HR = 1.52, p = 0.01), and stent thrombosis (3.4% vs. 0.9%, RR = 3.91, p = 0.001). Bivalirudin had a similar primary safety endpoint of major BARC 3–5 bleeding (3.5% vs. 3.1%, p = 0.59) and similar mortality rate (5.1% vs. 4.3%) as compared to UFH therapy.
-
Finally, the results of these trials further reinforced the higher risk of stent thrombosis associated with bivalirudin therapy as compared to UFH without systematic use of GPIs while there were small differences in major bleeding [29]. These concerns were reflected in the recent European guidelines that downgraded the recommendation for the use of bivalirudin in primary PCI from Class I A to Class IIa A [4].

ACUITY trial outcomes [24]. (1) Composite ischemia endpoint = death from any cause, myocardial infarction, or unplanned revascularization for ischemia. (2) Major bleeding = not related to CABG. (3) Net clinical outcome endpoint = the composite ischemia endpoint or major bleeding. (Key: Bival bivalirudin, GPI glycoprotein inhibitor)
Non-vitamin K Oral Anticoagulants
Factor Xa Inhibitors
-
Direct factor Xa inhibitors apixaban, rivaroxaban, darexaban, and otamixaban have been, or, are currently being, investigated in patients with ACS, either in the acute phase during intervention or in the secondary prevention after the acute event.
-
In the preplanned interim analysis of the TAO trial , otamixaban did not reduce the rate of ischemic events relative to unfractionated heparin plus eptifibatide but did increase bleeding in patients with NSTE-ACS undergoing planned early PCI [30].
-
In the landmark ATLAS-ACS 2 TIMI 51 trial that enrolled patients with recent ACS, low-dose rivaroxaban (2.5 mg bid, 25% of the total dose used for atrial fibrillation), added to aspirin and clopidogrel, reduced major cardiovascular adverse events (9.1% vs. 10.7%, p = 0.02), and cardiovascular death (2.7% vs. 4.1%, p = 0.002) and all-cause death (2.9% vs. 4.5%, p = 0.002), but with an increased risk of non-CABG major bleeding (1.8% vs. 0.6%, p < 0.001) and intracranial hemorrhage (0.4% vs. 0.2%, p = 0.04) but not the risk of fatal bleeding. In this study, time from index event to randomization was 4.7 days and ~60% of patients underwent PCI or CABG for the index event [31].
-
The APPRAISE trial (a phase III trial) that compared 5 mg bid apixaban (full dose) added to DAPT vs. DAPT alone in high-risk ACS patients was prematurely stopped due to excess bleeding risk in the absence of benefit with respect to ischemic outcomes [32].
-
In the WOEST trial , 573 patients undergoing PCI were randomized to receive clopidogrel plus oral anticoagulant (double therapy) or clopidogrel plus aspirin plus oral anticoagulant (triple therapy) for 30 days after BMS placement (35%) and 1 year for DES placement (65%). The primary endpoint of any TIMI bleeding was significantly lower in the dual-therapy arm (19.5% vs. 44.9%, HR = 0.49, p < 0.001). Furthermore, dual therapy was associated with similar rates of MI, stroke, target-vessel revascularization, or stent thrombosis but lower all-cause death [33].
-
In the PIONEER AF-PCI trial , in patients with atrial fibrillation undergoing PCI with stenting, the administration of either 15 mg once daily rivaroxaban plus a P2Y12 inhibitor or 2.5 mg twice-daily rivaroxaban was associated with a lower rate of clinically significant bleeding than was standard therapy with a vitamin K antagonist plus DAPT and a similar rate of the efficacy endpoint of CV death, MI, or stroke [34].
-
Based on encouraging results from above trials, the safety of 2.5 mg bid rivaroxaban was compared to 100 mg daily aspirin on top of clopidogrel or ticagrelor in patients with MI in the GEMINI-ACS trial and >84% of patients were stented. Randomized therapy was started a median of 5.5 days after the index event and continued a median of 291 days. The primary endpoint of TIMI non-CABG clinically significant bleeding and the composite exploratory ischemic endpoint (cardiovascular death, MI, stroke, or definite stent thrombosis) were similar between groups. In a post hoc analysis, rivaroxaban was associated with numerically higher occurrence of the primary and ischemic composite endpoints in the first 30 days, but thereafter safety and efficacy appeared the same. There was numerically more ISTH and BARC 3 bleeding with rivaroxaban [35]. GEMINI-ACS was not powered for ischemic outcomes, and conclusions about the comparative efficacy of aspirin versus low-dose rivaroxaban cannot be made, but there was no signal of an antithrombotic benefit of rivaroxaban over aspirin [36].
-
In summary, evidence supports the benefit of addition of low-dose Xa inhibitor in high-risk ACS patients treated with aspirin and clopidogrel who were stabilized after an index event. Far more robust evidence is required to support the addition of a low-dose oral Xa inhibitor on top of DAPT with ticagrelor or prasugrel or to replace aspirin with a Xa inhibitor in high-risk ACS patients undergoing PCI or stabilized after PCI.
Summary of Current Evidence
-
Ischemic events during and following PCI are strongly influenced by platelet function and coagulation; simultaneous blockade of these pathways is essential to reduce ischemic events. Optimal inhibition of these pathways is needed for maximizing total antithrombotic effects. Minimizing bleeding risk is also a critical goal in the treatment of the PCI patient.
-
Aspirin remains the bedrock oral antiplatelet agent. The totality of evidence supports dual-antiplatelet therapy with aspirin plus a P2Y12 receptor blocker as the standard of care during and following PCI.
-
Both of the newer oral P2Y12 inhibitors, prasugrel and ticagrelor, are associated with a faster onset of action, greater platelet inhibition, and lower on-treatment platelet reactivity than clopidogrel. These superior pharmacodynamic properties have translated into lower ischemic outcomes as compared to clopidogrel in the treatment of the ACS/PCI patient. However, greater non-CABG-related major bleeding was associated with prasugrel and ticagrelor therapy.
-
The new intravenous P2Y12 receptor blocker, cangrelor, is associated with a faster onset and offset of effect and represents a new strategy of modulating peri-PCI platelet reactivity. It has been associated with lower ischemic event rates than clopidogrel loading at the time of PCI. The clinical efficacy of cangrelor has never been evaluated in patients treated with prasugrel or ticagrelor.
-
The pharmacological agents that directly block the binding of fibrinogen to the GPIIb/IIIa receptor (GPIIb/IIIa inhibitors) are highly effective and more potent in inhibiting platelet aggregation than cangrelor. The increased use of the P2Y12 inhibitors with a rapid onset of potent pharmacodynamic effects (prasugrel, ticagrelor, or cangrelor) may challenge the role of GPIIb/IIIa inhibitors in the treatment of the PCI patient. There have been no head-to-head clinical studies of cangrelor versus GPIs.
-
The optimal duration of DAPT in patients treated with PCI remains controversial. Based on the available evidence, it is recommended that DAPT be administered for at least 1 month after BMS implantation in stable CAD, for 6 months after new-generation DES implantation in stable CAD, and for up to 1 year in patients after ACS.
-
Recent randomized trials in patients treated with new-generation coronary artery stents have suggested shorter duration DAPT. However these trials were underpowered.
-
The DAPT and PEGASUS trials compared the efficacy of long-term (>12 months) P2Y12 inhibitor therapy on top of aspirin. Both trials demonstrated enhanced efficacy of long-term DAPT at the expense of greater bleeding.
-
In addition to antiplatelet therapy, anticoagulation is recommended in ACS patients undergoing PCI. Current choices are heparin, UFH , and bivalirudin .
-
Evidence supports the benefit of adding low-dose Xa inhibitor in high-risk ACS patients who are stabilized after the index event and treated with aspirin and clopidogrel.
-
Stronger evidence is required to support the addition of a low-dose oral Xa inhibitor on top of DAPT with ticagrelor or prasugrel or to replace aspirin with a low-dose Xa inhibitor in high-risk ACS patients undergoing PCI.
Change history
25 March 2021
The book was published with aerrors on page 171. It stated “Furthermore, the benefits associated with cangrelor were consistent across the subgroups of stable angina (n = 1991), NSTEMI (n = 2810), and STEMI (n = 6138) (interaction, p = 0.98), and whether the patient received clopidogrel 300 mg LD or 600 mg LD (interaction, p = 0.61).”
References
Tantry US, Etherington A, Bliden KP, Gurbel PA. Antiplatelet therapy: current strategies and future trends. Future Cardiol. 2006;2:343–66.
Gurbel PA, Tantry US. Antiplatelet and anticoagulant agents in heart failure: current status and future perspectives. JACC Heart Fail. 2014;2:1–14.
Mehta SR, Tanguay JF, Eikelboom JW, et al. CURRENT-OASIS 7 trial investigators. Double-dose versus standard-dose clopidogrel and high-dose versus low-dose aspirin in individuals undergoing percutaneous coronary intervention for acute coronary syndromes (CURRENT-OASIS 7): a randomised factorial trial. Lancet. 2010;376:1233–43.
Windecker S, Kolh P, Alfonso F, et al. 2014 ESC/EACTS Guidelines on myocardial revascularization: The Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS)Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur Heart J. 2014;35:2541–619.
Roffi M, Patrono C, Collet JP, et al. Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J. 2016;37:267–315.
Steg PG, James SK, Atar D, et al. ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation. Eur Heart J. 2012;33:2569–619.
Tantry US, Bonello L, Aradi D, et al. Working Group on On-Treatment Platelet Reactivity. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol. 2013;62:2261–73.
Wiviott SD, Braunwald E, McCabe CH, et al. TRITON-TIMI 38 Investigators. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–15.
Montalescot G, Bolognese L, Dudek D, et al. ACCOAST Investigators. Pretreatment with prasugrel in non-ST-segment elevation acute coronary syndromes. N Engl J Med. 2013;369:999–1010.
Gurbel PA, Bliden KP, Butler K, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120:2577–85.
Cannon CP, Harrington RA, James S, et al. PLATelet Inhibition and Patient Outcomes Investigators. Comparison of ticagrelor with clopidogrel in patients with a planned invasive strategy for acute coronary syndromes (PLATO): a randomised double-blind study. Lancet. 2010;375:283–93.
Montalescot G, van ‘t Hof AW, Lapostolle F, et al. ATLANTIC Investigators. Prehospital ticagrelor in ST-segment elevation myocardial infarction. N Engl J Med. 2014;371:1016–27.
Bhatt DL, Stone GW, Mahaffey KW, et al. CHAMPION PHOENIX Investigators. Effect of platelet inhibition with cangrelor during PCI on ischemic events. N Engl J Med. 2013;368:1303–13.
Lefkovits J, Plow EF, Topol EJ. Platelet glycoprotein IIb/IIIa receptors in cardiovascular medicine. N Engl J Med. 1995;332:1553–9.
Hanna EB, Rao SV, Manoukian SV, Saucedo JF. The evolving role of glycoprotein IIb/IIIa inhibitors in the setting of percutaneous coronary intervention strategies to minimize bleeding risk and optimize outcomes. JACC Cardiovasc Interv. 2010;3:1209–19.
Mauri L, Kereiakes DJ, Yeh RW, et al. DAPT study Investigators. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med. 2014;371:2155–66.
Levine GN, Bates ER, Bittl JA, et al. 2016 ACC/AHA Guideline Focused Update on Duration of Dual Antiplatelet Therapy in Patients With Coronary Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2016;68:1082–115.
Yeh RW, Secemsky EA, Kereiakes DJ, et al. DAPT Study Investigators. Development and validation of a prediction rule for benefit and harm of dual antiplatelet therapy beyond 1 year after percutaneous coronary intervention. JAMA. 2016;315:1735–49.
Bonaca MP, Bhatt DL, Cohen M, et al. PEGASUS-TIMI 54 Steering Committee and Investigators. Long-term use of ticagrelor in patients with prior myocardial infarction. N Engl J Med. 2015;372:1791–800.
Montalescot G, White HD, Gallo R, et al. Enoxaparin vs. unfractionated heparin in elective percutaneous coronary intervention. N Engl J Med. 2006;355:1006–17.
Collet J-P, Huber K, Cohen M, et al. A direct comparison of intravenous enoxaparin with unfractionated heparin in primary percutaneous coronary intervention (from the ATOLL trial). Am J Cardiol. 2013;112:1367–72.
Kastrati A, Neumann FJ, Mehilli J, et al. Bivalirudin vs. unfractionated heparin during percutaneous coronary intervention. N Engl J Med. 2008;359:688–96.
Schulz S, Mehilli J, Neumann FJ, et al. Intracoronary Stenting and Antithrombotic Regimen: Rapid Early Action for Coronary Treatment (ISAR-REACT) 3A Trial Investigators. ISAR-REACT 3A: a study of reduced dose of unfractionated heparin in biomarker negative patients undergoing percutaneous coronary intervention. Eur Heart J. 2010;31:2482–91.
Stone GW, McLaurin BT, Cox DA, et al. Bivalirudin for patients with acute coronary syndromes. N Engl J Med. 2006;355:2203–16.
Stone GW, White HD, Ohman EM, et al. Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) Trial Investigators. Bivalirudin in patients with acute coronary syndromes undergoing percutaneous coronary intervention: a subgroup analysis from the Acute Catheterization and Urgent Intervention Triage strategy (ACUITY) trial. Lancet. 2007;369:907–19.
Kastrati A, Neumann F-J, Schulz S, et al. Abciximab and heparin vs. bivalirudin for non-ST-elevation myocardial infarction. N Engl J Med. 2011;365:1980–9.
Stone GW, Witzenbichler B, Guagliumi G, et al. HORIZONS-AMI Trial Investigators. Bivalirudin during primary PCI in acute myocardial infarction. N Engl J Med. 2008;358:2218–30.
Steg PG, van ‘t Hof AW, Hamm CW, et al. Bivalirudin started during emergency transport for primary PCI. N Engl J Med. 2013;369:2207–17.
Shahzad A, Kemp I, Mars C, et al. for the HEAT-PPCI trial investigators. Unfractionated heparin versus bivalirudin in primary percutaneous coronary intervention (HEAT-PPCI): an open-label, single centre, randomized controlled trial. Lancet. 2014;384:1848.
Steg PG, Mehta SR, Pollack CV Jr, Investigators TAO. Anticoagulation with otamixaban and ischemic events in non-ST-segment elevation acute coronary syndromes: the TAO randomized clinical trial. JAMA. 2013;310:1145–55.
Mega JL, Braunwald E, Wiviott SD, et al., the AACSTIRivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med. 2012;366:9–19.
Alexander JH, Lopes RD, James S, et al. Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med. 2011;365:699–708.
Dewilde WJ, Oirbans T, Verheugt FW, et al. Use of clopidogrel with or without aspirin in patients taking oral anticoagulant therapy and undergoing percutaneous coronary intervention: an open label, randomised, controlled trial. Lancet. 2013;381:1107–15.
Gibson CM, Mehran R, Bode C, et al. Prevention of bleeding in patients with atrial fibrillation undergoing PCI. N Engl J Med. 2016;375:2423–34.
Ohman EM, Roe MT, Steg PG, et al. Clinically significant bleeding with low-dose rivaroxaban versus aspirin, in addition to P2Y12 inhibition, in acute coronary syndromes (GEMINI-ACS-1): a double-blind, multicentre, randomised trial. Lancet. 2017;389:1799–808.
Gurbel PA, Tantry US. GEMINI-ACS-1: toward unearthing the antithrombotic therapy cornerstone for acute coronary syndromes. Lancet. 2017;89:1773–5.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Gurbel, P.A., Tantry, U.S. (2018). Percutaneous Coronary Intervention: Adjunctive Pharmacology. In: Myat, A., Clarke, S., Curzen, N., Windecker, S., Gurbel, P.A. (eds) The Interventional Cardiology Training Manual. Springer, Cham. https://doi.org/10.1007/978-3-319-71635-0_12
Download citation
DOI: https://doi.org/10.1007/978-3-319-71635-0_12
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-71633-6
Online ISBN: 978-3-319-71635-0
eBook Packages: MedicineMedicine (R0)