
Abstract
Warfarin is one of the most widely prescribed oral anticoagulants. However, optimal use of the drug has been hampered by its >10-fold interpatient variability in the doses required to attain therapeutic responses. Pharmacogenetic polymorphism of cytochrome P450 (CYP) may be associated with impaired elimination of warfarin and exaggerated anticoagulatory responses to the drug in certain patients.
Clinically available warfarin is a racemic mixture of (R)- and (S)-warfarin, and the (S)-enantiomer has 3 to 5 times greater anticoagulation potency than its optical congener. Both enantiomers are eliminated extensively via hepatic metabolism with low clearance relative to hepatic blood flow. CYP2C9 is almost exclusively responsible for the metabolism of the pharmacologically more active (S)-enantiomer.
Several human allelic variants of CYP2C9 have been cloned, designated as CYP2C9*1 (reference sequence or wild-type allele), CYP2C9*2, CYP2C9*3and CYP2C9*4, respectively. The allelic frequencies for these variants differ considerably among different ethnic populations. Caucasians appear to carry the CYP 2C9*2 (8 to 20%) and CYP2C9*3 (6 to 10%) variants more frequently than do Asians (0% and 2 to 5%, respectively).
The metabolic activities of the CYP2C9 variants have been investigated in vitro. The catalytic activity of CYP2C9*3 expressed from cDNA was significantly less than that of CYP2C9*1. Human liver microsomes obtained from individuals heterozygous for CYP2C9*3 showed significantly reduced (S)-warfarin 7-hydroxylation as compared with those obtained from individuals genotyped as CYP2C9*1.
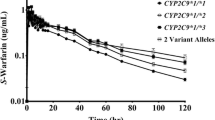
The influence of the CYP2C9*3 allele on the in vivo pharmacokinetics of (S)-warfarin has been studied in Japanese patients. Patients with the homozygous CYP2C9*3 genotype, as well as those with the heterozygous CYP2C9*1/*3 genotype, had significantly reduced clearance of (S)-warfarin (by 90 and 60%, respectively) compared with those with homozygous CYP2C9*1.
The maintenance dosages of warfarin required in Japanese patients with heterozygous and homozygous CYP2C9*3 mutations were significantly lower than those in patients with CYP2C9*1/*1. In addition, 86% of British patients exhibiting adequate therapeutic responses with lower maintenance dosages of warfarin (<1.5 mg/day) had either the CYP2C9*2 or CYP2C9*3 mutation singly or in combination, whereas only 38% of randomly selected patients receiving warfarin carried the corresponding mutations. Furthermore, the former group showed more frequent episodes of major bleeding associated with warfarin therapy.
These data indicate that the CYP2C9*3 allele may be associated with retarded elimination of (S)-warfarin and the resulting clinical effects. However, relationships between CYP2C9 genotype, enzyme activity, metabolism of probe substrates, dosage requirements and bleeding complications should be interpreted with caution, and further studies are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Warfarin is the most widely used oral anticoagulant throughout the world despite a narrow therapeutic index and frequent bleeding complications during treatment.[1] For the prevention and treatment of thromboembolic disorders it has been recommended that anticoagulation responses of an International Normalised Ratio (INR) ranging from 2 to 3 should be considered optimal or target responses, except for prosthetic heart valves, for which an INR of 2.5 to 3.5 is recommended.[1] However, it is not easy for clinicians to estimate the optimal initial dosage of the drug to attain such a narrow therapeutic range of INR for each patient, because there is wide variation in the anticoagulant response to warfarin between and even within patients. Therefore, numerous attempts have been made to clarify possible causative factors for the variability of the pharmacokinetics and pharmacodynamics of warfarin.
In this article we summarise not only in vitro but also in vivo data that are relevant to the influence of the pharmacogenetics of cytochrome P450 (CYP) on warfarin elimination. We also discuss possible pitfalls that must be considered when extrapolating in vitro data obtained from cDNA-expressed CYP isoform systems to in vivo pharmacokinetics. In this context, we discuss possible reasons to explain why in vitro studies clearly demonstrating reduced enzyme activity of CYP2C9 variants toward certain probe substrate might fail to predict the influence on the overall elimination kinetics of the corresponding probe substrate in in vivo studies.
1. Pharmacokinetics of Warfarin Enantiomers
The clinically available form of warfarin is a racemic mixture of equal amounts of (R)- and (S)-enantiomers that have distinct pharmacological activity. (S)-Warfarin has been shown to have an anticoagulation effect 3 to 5 times more potent than that of its optical congener in humans,[2,3] implying that (S)-warfarin is largely associated with anticoagulation responses elicited by the administration of racemic warfarin. Previous studies[4,5] have shown that (S)-warfarin is metabolised mainly to its major metabolite, (S)-7-hydroxy-warfarin, and to a much lesser extent to (S)-6-hydroxy-warfarin; these reactions are mediated almost exclusively by CYP2C9 in humans. In addition, recent advances in molecular biology have revealed that this CYP isoform has numerous allelic variants associated possibly with altered catalytic activities.[6] Therefore, pharmacogenetic polymorphism of CYP2C9 and resultant alterations in its in vitro enzyme kinetics - maximum rate of metabolism (Vmax), Michaelis-Menten constant (Km) and intrinsic clearance (CLint) [Vmax/Km] - have been studied extensively to elucidate the possible effect of genetic polymorphism of CYP2C9 on the interpatient variability of (S)-warfarin elimination, and ultimately on the anticoagulation response.
Although (S)-warfarin has 3 to 5 times greater anticoagulation effect than (R)-warfarin, caution must be exercised in attributing the clinical response of the drug solely to the pharmacologically more active (S)-enantiomer. (S)-Warfarin has greater systemic clearance (CL) and shorter elimination half-life (t1/2β) than its optical congener: CL 0.31 vs 0.15 L/h/70kg (5.2 vs 2.5 ml/min/70kg) and t1/2β 24 vs 40 hours.[2,3] Thus, plasma concentrations of (R)-warfarin at steady state would be on average 94% higher than those of (S)-warfarin. Assuming that only unbound drug can reach its site of action, the anticoagulation response to warfarin should be considered as a function of plasma unbound (Cu), rather than total, concentrations of warfarin enantiomers. Both warfarin enantiomers are extensively bound to plasma protein, and no enantioselective differences have been observed in their binding (plasma unbound fractions of 1%).[7] Consequently, at steady state, (S)-warfarin would be expected to be associated with 60 to 70% of overall anticoagulation response to warfarin.
In this context, we analysed the anticoagulation response to racemic warfarin as a function of Cu of (S)-warfarin [i.e. Cu(S)]. According to basic pharmacokinetic principles, the average Cu(S) at steady state is determined by the dose rate and the unbound oral clearance for (S)-warfarin [i.e. CLpo,u(S)] as follows:
where Cu(S)ss is the average Cu of (S)-warfarin at steady state, D is the daily dose of racemic warfarin and τ is the administration interval (e.g. 24 hours). Previous studies have shown that the bioavailability of warfarin after oral administration is almost complete and that more than 98% of the warfarin dose absorbed is eliminated by hepatic metabolism in humans.[8] Therefore, according to the physiological pharmacokinetic model,[9] CLpo,u for (S)-warfarin is largely equal to its hepatic intrinsic clearance (CLint,h), representing the sum of the individual hepatic enzyme activities associated with (S)-warfarin metabolism. As a result, either CLpo,u(S) or CLint,h(S) is the sole pharmacokinetic parameter that determines Cu(S)ss and thereby would be responsible, at least in part, for interpatient variability in the anticoagulant responses to racemic warfarin. Because the metabolism of (S)-warfarin is mediated almost exclusively by CYP2C9, in vivo CLint,h or CLpo,u for (S)-warfarin would also be relevant parameters for CYP2C9 activity. In addition, because (S)-7-hydroxy-warfarin is excreted into urine largely in an unchanged form (being minimally glucuronidated),[10] the in vivo formation clearance (CLf) of (S)-7-hydroxy-warfarin may also serve as another useful and more specific in vivo parameter for hepatic CYP2C9 activity.
In contrast, total (bound plus unbound) plasma concentrations of (S)-warfarin are dependent not only on CLint,h(S) but also on the status of plasma protein binding of the drug:
where fub is the unbound fraction of (S)-warfarin in the blood. Since Cu(S) can be related to the anticoagulation response, total plasma concentrations of (S)-warfarin or racemic warfarin and their clearances may not necessarily be reliable parameters for analysing the interpatient variability of anticoagulant effects elicited by the administration of racemic warfarin. In this context, we discuss the pharmacogenetics of warfarin elimination and its clinical implications as a function of Cu of (S)-warfarin or CLpo,u(S).
Although many factors such as age, sex, race and disease state may influence interpatient variability in the anticoagulation response to warfarin, our premise that hepatic CYP2C9 activity is one of the prime determinants of this variability is supported by a number of drug interaction studies in which warfarin was involved. Previous studies[11–15] have shown that certain anti-inflammatory agents (e.g. phenylbutazone), uricosuric agents (e.g. sulfinpyrazone, benzbromarone, bucolome) and antifungal agents (e.g. fluconazole) exaggerate the anticoagulation effects of warfarin to clinically hazardous levels via inhibition of metabolism by (S)-warfarin. Subsequent studies[13–16] have clearly demonstrated that these drugs are potent inhibitors of CYP2C9. These data appear compatible with our assumption that the expression of functionally defective CYP2C9 variants may be associated with apparently exaggerated sensitivity to warfarin, whereas it would not explain the reason for certain patients exhibiting apparent resistance to the anticoagulation effect of the drug.
Although the pharmacologically less active (R)-warfarin is also metabolised extensively, the hepatic metabolism of this enantiomer involves the net results of catalysis of multiple isoforms of CYP (CYP1A2, CYP2C19, CYP3A) and ketoreductases.[4,5] Therefore, reduction of the catalytic activity of one of the enzymes involved in the metabolism of (R)-warfarin would lead to only a small change in Cu of (R)-warfarin. Indeed, previous studies have shown that coadministration of enoxacin,[17] cimetidine,[18] omeprazole,[19] ticlopidine[20] or diazepam[21] with racemic warfarin gave rise to an enantioselective metabolic inhibition of (R)-warfarin metabolism and selectively elevated Cu of (R)-warfarin, whereas no significant changes in the anticoagulant activity were observed. These data indicate that the influence of (R)-warfarin on the overall anticoagulation effect of warfarin is insignificant, if any.
2. Variants of Cytochrome P450 (CYP) 2C9 and Their Allelic Frequencies in Different Ethnic Groups
2.1 Coding-Region Single Nucleotide Polymorphisms
CYP2C9 is one of the major isoforms of the CYP2C subfamily, and accounts for about 20% of constitutively expressed hepatic CYP in humans.[22] This CYP isoform is principally responsible for the metabolism of a number of clinically important drugs, such as tolbutamide, (S)-warfarin, phenytoin, fluoxetine, losartan, torasemide, irbesartan, sulfamethoxazole, glipizide, tetrahydrocannabinol and numerous nonsteroidal anti-inflammatory drugs (NSAIDs).[6,16,23–25]
Although gross gene deletion and single nucleotide deletions or changes that create aberrant splicing or frame shifts have been reported for other polymorphic CYP isoforms (e.g. CYP2D6 and CYP2C19),[6,26] all allelic variants reported for CYP2C9 are single nucleotide polymorphisms (SNPs) leading to single amino acid changes.[6,27–43] According to a recent proposal by Nebert,[44] we refer to SNPs located within the coding region as cSNPs and those located inside or in the immediate vicinity of genes as perigenic SNPs (pSNPs).
At present, 2 cSNPs have been confirmed in randomly selected populations of more than 2 different ethnic backgrounds. The Human Cytochrome P450 Allele Nomenclature Committee has assigned the Arg144/Ile359, Cys144/Ile359, Arg144/Leu359 and Arg144/ Thr359 alleles of CYP2C9 as CYP2C9*1 (reference sequence or wild-type), CYP2C9*2, CYP2C9*3 and CYP2C9*4, respectively.[26] Recently, another possible cSNP (Arg144/Glu360 allele) was reported.[27] Table I shows the allelic frequencies for the 6 unique CYP2C9 alleles reported from different ethnic populations.[27–43] Among the distinct CYP2C9 variants, CYP2C9*3 has been detected in all ethnic groups so far studied (i.e. Caucasian, African-American and Asian), but its allelic frequencies are quite different across the ethnic groups: 6 to 10% in Caucasians and Canadian Native Indians, 1.7 to 5% in Asians and 0.5 to 1.5% in African-Americans. Ethnic differences in allelic frequency have also been observed for CYP2C9*2. The CYP2C9*2 allele was detected in 8 to 19.1% of Caucasians and 1 to 3.6% of African-Americans and Canadian Native Indians, no CYP2C9*2 alleles were detected in Asians (Chinese and Japanese).

Allelic frequencies of cytochrome P450 (CYP) 2C9 variants among different ethnic populations
Caution must be exercised in interpreting the genotyping data for CYP2C9*2 and CYP2C9*3 using different methods of the polymerase chain reaction (PCR) followed by restriction enzyme digestion. Yasar et al.[37] studied the validity of 3 commonly used PCR-based genotyping methods in 430 Swedish people and found that one of them may give erroneous results for both CYP2C9*2 and CYP2C9*3 alleles, probably because of co-amplification of other genes in the CYP2C locus. The other 2 methods gave completely concordant results with each other.
CYP2C9*4 has been detected so far in only 1 Japanese patient who was shown to have a reduced phenytoin clearance compared with those who had CYP2C9*1/*1.[28] The allelic frequency of this variant in Japanese would be very low, because we found no such variant in our Japanese study population (n = 86; table I) [H. Takahashi, unpublished data]. No information is available on whether this variant has been confirmed in randomly genotyped Caucasian or African-American populations. Although another new variant, Asp/Glu360, was detected in 5 of 110 African-Americans (CYP2C9*5, 2.3%),[27] it remains to be investigated whether this CYP2C9 variant will also be confirmed in other ethnic groups. At present, 7 other new CYP2C9 variant alleles (i.e., from CYP2C9*6 to CYP2C9*12) are registered at the web site of the Human Cytochrome P450 (CYP) Allele Nomenclature Committee.[26]
2.2 Noncoding-Region Single Nucleotide Polymorphisms
Two distinct pSNPs in intron 2[36,40] and 7 pSNPs in the 5′-noncoding region[45] of CYP2C9 have been reported. Of the pSNPs in intron 2, one is a T/C transition and the other is an A/T transition at 73 and 12 base pairs downstream of exon 2, respectively. The former mutation was discovered in 22.8% of German Caucasians[36] but was shown to be extremely rare in Japanese (0.4%: H. Takahashi et al., unpublished data). The latter was reported by Wang et al.[40] in Chinese but little is known about its allelic frequencies in other ethnic populations. With respect to the pSNPs in the 5′-flanking region, their functional influences are under investigation.[45] It is becoming increasingly clear that pSNPs may also have a great effect on the phenotype, and several established cases were summarised in a recent review.[44] Therefore, further studies are required to clarify whether the pSNPs of CYP2C9 might have any clinical implications.
3. Metabolic Activities of CYP2C9 Variants In Vitro
3.1 cDNA Expression Systems
Considerable interest has arisen as to whether any single amino acid-substituted CYP2C9 variants play a significant role in interpatient variability in the metabolic activity of (S)-warfarin and other CYP2C9 substrates. Table II summarises the in vitro catalytic activities of CYP2C9*2 and CYP2C9*3 for (S)-warfarin 7-hydroxylation as compared with that of CYP2C9*1 in different cDNA expression systems using yeast, HepG2 cells, insect cells and lymphoblasts. These data indicate that caution must be exercised in comparing the in vitro enzyme kinetic parameters for the wild-type and variant CYP2C9s obtained from different expression systems and measured under different experimental conditions. For instance, there were 7-, 3- and 9-fold differences in Km, Vmax and Vmax/Km (CLint) for (S)-warfarin 7-hydroxylation among the expressed CYP2C9*1 (wild-type) reported from different laboratories.[29,46–52]

In vitro enzyme kinetic parameters for (S)-warfarin 7-hydroxylation by cDNA-expressed CYP2C9*1, CYP2C9*2 and CYP2C9*3 obtained from different expression systems
3.1.1 CYP2C9*2
Controversial data have been reported regarding the influence of the Cys144/Ile359 mutation (CYP2C9*2) on the in vitro catalytic activities of (S)-warfarin 7-hydroxylation as compared with CYP2C9*1, despite the fact that comparisons were made using comparable expression systems and experimental conditions in various laboratories. Rettie et al.[47,52] and Yamazaki et al.[49] demonstrated that the Km values of CYP2C9*2 obtained from HepG2 cells, insect cells and yeast were largely similar to the corresponding values of CYP2C9*1, whereas their CLint values for (S)-warfarin 7-hydroxylation were 85, 32 and 59% less than the corresponding values obtained from the wild-type allele (CYP2C9*1). Yamazaki et al.[49] also showed that the CLint of yeast-expressed CYP2C9*2 for (S)-flurbiprofen 4′-hydroxylation and diclofenac 4′-hydroxylation were 65 and 35% lower than that of CYP2C9*1, respectively. Tracy et al.[53] reported that the velocity of (S)-flurbiprofen 4′-hydroxylation by CYP2C9*2 at a near-Vmax substrate concentration was about 65% less than that of CYP2C9*1 in a HepG2 cell expression system. In contrast, Kaminsky and Zhang[5] reported that the CYP2C9*2 variant affected none of the enzyme kinetics (metabolic activity, regioselectivity and stereoselectivity) for multiple pathways of warfarin hydroxylation in a yeast expression system. Sullivan-Klose et al.[29] reported no remarkable differences between CYP2C9*2 and CYP2C9*1 regarding CLint for tolbutamide hydroxylation (another CYP2C9 substrate), and Chang et al.[54] reported similar results for the 4-hydroxylation of cyclophosphamide and ifosfamide in a yeast expression system.
Crespi and Miller[51] found that the differences in in vitro catalytic activity between CYP2C9*1 and CYP2C9*2 were strongly dependent on the ratio of NADPH-CYP reductase to CYP2C9 in an expression system using lymphoblastoid cells, such that the difference was more pronounced at higher ratios of NADPH-CYP reductase to CYP2C9. In addition, they found that the expressed CYP2C9*1 exhibited a fairly comparable turnover number to that obtained from human liver microsomes at high ratios of NADPH-CYP reductase to CYP. As a result, they concluded that an excess of NADPH-CYP reductase over CYP2C9 in the expression system would be required to simulate in vivo CYP2C9 activity. On the basis of these findings, they claimed that the CYP2C9*2 variant should have a markedly reduced metabolic activity in vivo. Because the ratios of NADPH-CYP reductase to CYP2C9 might have differed among the different expression systems used in the previous reports, the apparent discordance regarding the effect of CYP2C9*2 mutation on enzyme activity should be viewed with caution.
In addition, Crespi and Miller[51] suggested that the CYP2C9*2 mutation may disturb the interaction of CYP2C9 protein with NADPH-CYP reductase (i.e. Vmax and Vmax/Km) rather than that with substrates (i.e. Km), because they found no substantial differences in Km values between CYP2C9*1 and CYP2C9*2. Their contention may be compatible with a recently proposed substrate recognition site (SRS) model for CYP2C subfamilies. At present, 6 putative SRSs have been inferred for CYP2C9 based on comparative analysis of amino acids and coding nucleotide sequences for human CYP2C subfamilies and for bacterial CYP isoforms of which SRSs were identified by x-ray crystallography.[55] According to this model, residue 144 is not located at or in the vicinity of the known SRSs.
3.1.2 CYP2C9*3
With regard to the influence of the Arg144/Leu359 mutation (CYP2C9*3) on the in vitro activity of the enzyme, all reports[29,46,48–50,52] agree that the catalytic activity of cDNA-expressed CYP2C9*3 is markedly reduced, not only for (S)-warfarin 7-hydroxylation but also for other CYP2C9 substrates (e.g. tolbutamide and phenytoin) irrespective of the expression system used (i.e. yeast and insect cells) [table II]. CLint values for (S)-warfarin 7-hydroxylation obtained from cDNA-expressed CYP2C9*3 were reduced to 4 to 17% of that obtained from CYP2C9*1. Most of the studies demonstrated that CYP2C9*3 had a greater Km (lesser affinity) and a lower Vmax (lesser capacity) for (S)-warfarin 7-hydroxylation than the corresponding values for CYP2C9*1, indicating that both the affinity and capacity of CYP2C9 were reduced by the single amino acid change associated with the CYP2C9*3 mutation.
Since residue 359 of CYP2C9 was suggested to be located close to a putative SRS (SRS5),[55] a single amino acid substitution at this position from Ile to Leu appears to result in a drastic decrease in the catalytic efficiency. Nonetheless, controversy remains as to whether this variant of CYP2C9 would have altered stereo- and regioselectivity for the metabolism of warfarin enantiomers compared with the wild-type CYP2C9*1.[5,46] A recent in vitro study[50] using the yeast expression system indicated that the Ile/Leu359 mutation significantly decreased the catalytic activity for 7 chemically diverse CYP2C9 substrates [4 NSAIDs, phenytoin, (S)-warfarin and tolbutamide], whereas the extent of reduction was substrate-dependent and varied among the substrates: Vmax/Km for diclofenac 4′-hydroxylation was reduced 3.4-fold, but that for piroxicam 5′-hydroxylation was reduced 34-fold.
3.1.3 New Variants
Using site-directed mutagenesis coupled with cDNA expression, Ieiri et al.[56] reported that CYP2C9 with the Ile/Thr359 mutation, which was originally cloned from a patient with a low in vivo clearance for phenytoin,[28] had a 3- to 5-fold reduced CLint for diclofenac 4′-hydroxylation as compared with CYP2C9*1. The magnitude of reduction observed in the patient with the CYP2C9 Ile/Thr359 variant for the in vivo clearance of phenytoin[28] was largely comparable to that observed in another patient genotyped as having heterozygous CYP2C9*3.[57,58] This variant was recently designated as CYP2C9*4.[26]
At present, there is a paucity of information regarding the effects of the Asp/Glu360 mutation on the in vivo disposition of any CYP2C9 substrates. As mentioned in section 2.2, pSNPs, as found in intron 2 of CYP2C9, may give rise to a great effect on the phenotype of the related genes.[44] In addition, the strategy used for cSNPs (site-directed mutagenesis coupled with cDNA expression) may not be applicable to pSNPs, except for splice-site mutations. The clinical implications of pSNPs of CYP2C9 should be studied using in vivo metabolic activity of CYP2C9 substrates as a phenotypic marker, or by linkage disequilibrium analysis with other defective genes.
3.2 Human Liver Microsome Systems
Yamazaki et al.[49] recently reported the effects of the CYP2C9*3 mutation on the in vitro catalytic activity of human liver microsomes. They demonstrated that human liver microsomes prepared from 3 different individuals genotyped as being heterozygous for the Leu359 allele (CYP2C9*1/*3) exhibited reduced catalytic rates for CYP2C9-related reactions [racemic and (S)-warfarin 7-hydroxylation and tolbutamide methyl hydroxylation], but not for a CYP2C19-related reaction [(S)-mephenytoin 4′-hydroxylation] and a CYP3A4-related reaction (testosterone 6β-hydroxylation) as compared with individuals with the homozygous wild-type CYP2C9*1 allele (CYP2C9*1/*1). In contrast, they found no remarkable differences in the catalytic activity for tolbutamide or (S)-warfarin between microsomes obtained from individuals with CYP2C9*1/*2 and CYP2C9*1/*1.
Conversely, Inoue et al.,[59] Bhasker et al.[60] and Gill et al.[24] reported no appreciable differences in the catalytic activity for tolbutamide and other CYP2C9 substrates (i.e. phenytoin, torasemide, diclofenac and sulfamethoxazole) between human liver microsomes prepared from Caucasian liver donors with either CYP2C9*1/*2 or CYP2C9*1/*3 and those prepared from donors with CYP2C9*1/*1. Such apparent discrepancies regarding the presence or absence of a CYP2C9 variant-related gene-dose effect in different human liver microsome samples may be attributable to the fact that these data[49,59,60] were not corrected for variable expression levels of CYP2C9 protein in each of the microsomal samples. Nonetheless, further studies are required to draw definite conclusions on the relationship between distinct CYP2C9 genotypes and catalytic activity in human liver microsomes.
4. Relationship of CYP2C9 Genotype to Elimination of CYP2C9 Substrates, and Its Clinical Implications
4.1 Warfarin
Table III summarises the effects of different CYP2C9 genotypes on the in vivo elimination of warfarin and other CYP2C9 substrates.

Effects of genetic polymorphism of cytochrome P450 (CYP) 2C9 on the in vivo pharmacokinetics and pharmacodynamics of warfarin and other CYP2C9 substrates
We were the first to report that patients with heterozygous or homozygous CYP2C9*3 mutations exhibited 63 to 66% and 90% reductions in the CLpo,u for (S)-warfarin compared with those with homozygous CYP2C9*1 (wild-type).[42,48] In addition, we found that the CLf to (S)-7-hydroxy-warfarin in these groups of patients was reduced to a similar extent as the CLpo,u for (S)-warfarin. Our data are consistent with the idea that the CYP2C9*3 mutation might produce clinically significant changes in anticoagulation response. Indeed, we found that the median maintenance dosage of racemic warfarin (1.75 mg/day) that was required to attain therapeutic anticoagulant effects in 3 Japanese patients with CYP2C9*3/*1 was reduced by 40% compared with the corresponding value (3 mg/day) obtained from patients with CYP2C9*1/*1. Furthermore, the maintenance dosage required by a patient with the CYP2C9*3/*3 genotype was only 0.4 mg/day, indicating an approximately 90% reduction in warfarin dosage requirement.
Aithal et al.[62,63] reported data that were largely consonant with ours. The median maintenance dosage of racemic warfarin for 10 British patients with the CYP2C9*1/*3 genotype (2.5 mg/day) was reduced by 40% compared with patients with the CYP2C9*1/*1 genotype (4.25 mg/day). Steward et al.[61] also reported that a patient who could tolerate no more than 0.5 mg/day of racemic warfarin was genotyped as having homozygous CYP2C9*3. Another study performed with British patients showed that those with CYP2C9*1/*3 attained an INR of 2.5 with a maintenance dosage of the drug (3.97 mg/day) that was approximately 20% less than that given to patients with CYP2C9*1/*1 (5.01 mg/day).[33]
In contrast to the data on the CYP2C9*3 mutation, those regarding the correlation between the CYP2C9*2 variant and alteration in in vivo systemic clearance of (S)-warfarin in humans are less conclusive. Because the CYP2C9*2 allele has never been detected in Asians, as we and others have reported, we cannot comment on this issue on the basis of our own data. However, the effects of the CYP2C9*2 mutation on the maintenance dosage requirement for racemic warfarin have been studied by 4 groups of investigators in Caucasians and African-Americans. Furuya et al.[32] and Taube et al.[33] reported that patients with the CYP2C9*1/*2 and CYP2C9*2/*2 genotypes required on average 15 to 20% and 40%, respectively, lower dosages of racemic warfarin than those with the CYP2C9*1/*1 genotype (wild-type). Aithal et al.[62,63] and Freeman et al.[39] also reported that patients with the CYP2C9*1/*2, CYP2C9*2/*2, CYP2C9*1/*3 or CYP2A6*1/*2 genotypes required on average 15 to 30% lower maintenance dosages of racemic warfarin than those with the CYP2C9*1/*1 genotype. These data imply that the CYP2C9*2 variant may be associated with reduced in vivo elimination of (S)-warfarin, albeit to a lesser extent than that elicited by the CYP2C9*3 variant. At present, it remains unclear whether patients with homozygous CYP2C9*2 variant (i.e. CYP2C9*2/*2) or with combined heterozygous variants of CYP2C9*2 and CYP2C9*3 (i.e. CYP2C9*2/*3) have a significantly altered elimination of (S)-warfarin compared with those having the homozygous wild-type CYP2C9.
Aithal et al.[62] reported that genetic polymorphisms of CYP2C9 are associated not only with warfarin dosage requirement but also with the risk of bleeding complications and the probability of having unexpectedly elevated INR at the time of induction of warfarin therapy. They compared the allelic frequencies of CYP2C9*2 and CYP2C9*3, the numbers of patients with INR greater than 4 at the time of warfarin induction and the incidence of bleeding complications during follow-up periods between patients who required a daily warfarin dosage of 1.5mg or less (the low dosage group) and those who required a wide range of dosages (the randomly selected control group). In all patients, INRs were kept in the range of 2.0 to 3.0. They found that approximately 80% of patients in the low dosage group carried at least one of the CYP2C9 variants (CYP2C9*2 or *3), whereas only 40% of patients in the control group did, indicating that genotyping of CYP2C9 may have the potential to identify individuals who require lower dosages of warfarin among the general patient population. In addition, a greater proportion of patients in the low dosage group (>4) had supratherapeutic INRs at induction, and patients in the low dosage group had a 4-fold increased frequency of major bleeding episodes during the follow-up period as compared with those in the control group. Margaglione et al.[64] also reported that patients carrying CYP2C9*2 and/or CYP2C9*3 variants required lower maintenance dosages of warfarin to acquire the target anticoagulation response and had a higher bleeding complication rate than those carrying CYP2C9*1/*1.
Because warfarin has a narrow therapeutic index, CYP2C9*2- and/or CYP2C9*3-associated reduction in CLpo,u for (S)-warfarin might have led to a greater variability in INR and thereby more frequent bleeding episodes in these studies. The data indicate that the pharmacogenetics of warfarin have significant clinical implications, not only to improve the effectiveness but also to avoid the untoward adverse reactions of warfarin.
On the other hand, Taube et al.[33] reported somewhat contradictory results, despite the fact that the study was conducted in patients who were considered largely similar to those of Aithal et al.[62] Although Taube et al.[33] confirmed that patients with the CYP2C9*2 or CYP2C9*3 variants required lower maintenance dosages of warfarin for attaining therapeutic anticoagulation response, they found that those with the CYP2C9*2 or CYP2C9*3 allele did not appear to have greater odds ratios for developing severely exaggerated or unstable anticoagulation responses during long term therapy compared with those with CYP2C9*1/*1. Interpatient variability of anticoagulation response to warfarin is probably associated not only with the CYP polymorphism-associated alteration in the pharmacokinetics of (S)-warfarin but also with interpatient variability in the sensitivity to the drug (pharmacodynamics) that may be under control of distinct genetic, racial and dietary parameters. Therefore, further studies in different ethnic populations are necessary to clarify the relation between bleeding complications and CYP2C9 genotypes.
4.2 Other CYP2C9 Substrates
Because a number of other therapeutically important drugs are metabolised by CYP2C9, it is of interest to review the effects of the known CYP2C9 variants on the in vivo disposition of these drugs.
Sullivan-Klose et al.[29] reported that 2 patients with either CYP2C9*1/*3 or CYP2C9*3/*3 genotype exhibited a 60 to 80% lower in vivo CLpo for tolbutamide than those with the CYP2C9*1/*1 genotype. Their data appear fairly comparable to those observed for (S)-warfarin in our previous study.[42,48] With regard to phenytoin, a 30% higher mean plasma concentration of the drug at 12 hours after administration was observed in Turkish patients with either heterozygous CYP2C9*3 or CYP2C9*2 mutations than in patients with CYP2C9*1/*1.[38] This finding agrees with the data reported previously[57,58] that Japanese epileptic patients with the heterozygous CYP2C9*3 mutation had a 30 to 40% lower Vmax value for phenytoin than those with the homozygous CYP2C9*1 genotype. Kidd et al.[25] also reported that a patient homozygous for CYP2C9*3 had an 80% lower CLpo for phenytoin and glipizide (both are CYP2C9 substrates), but not for nifedipine (a CYP3A substrate) and chlorpheniramine (a CYP2D6 substrate), as compared with the control individuals with CYP2C9*1/*1.
It has been reported that a healthy individual who had minimal to undetectable plasma concentrations of the active metabolite (E-3174) of losartan after receiving 50mg of the drug orally on 3 occasions was homozygous for CYP2C9*3.[65] In vitro studies showed that the conversion of losartan to E-3174 is mediated partly by CYP3A4, but almost exclusively by CYP2C9.[67] Collectively, the reported studies are consistent with the idea that CYP2C9*3, and to a lesser extent CYP2C9*2, confers a significant reduction in the in vivo elimination of many CYP2C9 substrates.
In contrast to the preceding discussion, Shimamoto et al.[66] reported no appreciable differences in CLpo for diclofenac (a CYP2C9 substrate) and the CLf to 4′-hydroxy-diclofenac between individuals with CYP2C9*1/*1 and those with the heterozygous CYP2C9*3 mutation. A possible explanation for this aberrant observation is given in section 5.2.
As to the newly recognised Ile/Thr359 variant (CYP2C9*4), Imai et al.[28] reported that a Japanese epileptic patient heterozygous for this variant had a 40% lower CLint of phenytoin than the population mean, using the Bayesian method applied to a single plasma phenytoin concentration obtained from the patient. The estimated CLint for the patient was fairly similar to that obtained from patients with heterozygous CYP2C9*3 reported by Mamiya et al.,[58] suggesting that patients with CYP2C9*3 or CYP2C9*4 may require lower maintenance dosages not only for phenytoin but also for warfarin.
5. Extrapolation of CYP2C9 Enzyme Activity In Vitro to Substrate Disposition In Vivo
5.1 Warfarin
Because there is evidence to suggest that the pharmacogenetics of CYP2C9 affect in vivo elimination of warfarin and thereby its therapeutic responses (section 4.1),[42,48,62,63] attempts have been made to develop an in vitro method that would enable quantitative prediction of the effect associated with the genetic polymorphisms of CYP2C9 on the in vivo pharmacokinetics of CYP2C9 substrates. Thus, we conducted a study in which the in vitro catalytic activities of (S)-warfarin 7-hydroxylation obtained from cDNA-expressed CYP2C9*1, CYP2C9*3 and a mixture of equal amounts of CYP2C9*3 and CYP2C9*1 (the in vitro models for CYP2C9*1/*1, CYP2C9*3/*3 and CYP2C9*1/*3, respectively) were compared with the CLpo,u (i.e. CLint,h) for (S)-warfarin obtained from patients with the corresponding CYP2C9 genotypes.[48] The validity of our in vitro model for CYP2C9*1/*3 was further confirmed by a study showing that the Ile359 and Leu359 variants of mRNA in 3 human livers genotyped as CYP2C9*1/*3 were expressed in relatively similar amounts.[49]
The results clearly showed that the mean in vitro CLint (Vmax/Km) values obtained from the recombinant CYP2C9*3 and the mixture of CYP2C9*3 and CYP2C9*1 were 94 and 65% lower than that obtained from the recombinant CYP2C9*1. The in vivo study showed that the median CLpo,u values for (S)-warfarin obtained from the patients with CYP2C9*3/*3 and CYP2C9*1/*3 were reduced by 90 and 66%, respectively, as compared with that obtained from those with CYP2C9*1/*1. Consequently, there was a significant correlation between the in vitro CLint for (S)-warfarin 7-hydroxylation and the in vivo CLpo,u for (S)-warfarin in terms of the effect of the CYP2C9*3 allele. To our knowledge, no data are so far available for other CYP2C9 substrates regarding the direct comparison between in vitro enzyme kinetics and in vivo drug disposition.
5.2 Pitfalls in In Vitro-to-In Vivo Extrapolation for Other CYP2C9 Substrates
Reductions in the in vitro catalytic activity for a certain CYP substrate, whether with a cDNA-expressed variant allele or with human liver microsomes containing the corresponding variant proteins, have often been extrapolated uncritically into in vivo clearance of the corresponding CYP substrates. However, there are several pitfalls in this naive extrapolation.
For an ideal in vivo probe drug for CYP2C9, fh [hepatic clearance (CLh) expressed as a fraction of systemic clearance] and fm (CYP2C9-mediated clearance expressed as a fraction of CLh) should be close to 1. In addition, the hepatic extraction ratio (Eh), defined as CLh divided by the hepatic blood (or plasma) flow, should be less than 0.3 if the probe drug is to be given intravenously.[13,14] Obviously, changes observed in the in vitro enzyme activity associated with genetic polymorphism of CYP2C9 will not be reflected in its in vivo kinetics unless fh for the probe drug is close to 1. In addition, if Eh for a probe drug is greater than 0.3, its hepatic clearance may be affected not only by enzyme activity but also by blood flow rate, particularly after intravenous administration. Finally, fm for the metabolic pathway associated with genetic polymorphism should be large enough to dominate the overall hepatic metabolism of the probe drug.
The 5 commonly used substrates have fh values close to 1 (100%) and have very small Eh (Eh < 0.03), except for diclofenac, indicating that they are typical metabolically cleared drugs with capacity-limited hepatic clearance (table IV). Furthermore, all CYP2C9 probe substrates except diclofenac have fm,CYP2C9 ranging from 55 to 90%,[68–71] indicating that CYP2C9 is largely responsible for overall hepatic elimination of the respective drugs. These data suggest that in vitro data for these CYP2C9 substrates, except for diclofenac, may be extrapolated to in vivo situations. In contrast, because CLh for diclofenac is much greater than for other CYP2C9 substrates [18 L/h (300 ml/min)], its hepatic elimination may not necessarily be dependent solely on the rate of hepatic metabolism (Eh = 0.35) if the drug is administered intravenously. However, this does not pose a practical limitation as long as diclofenac is given orally. A more serious problem for the in vitro-to-in vivo extrapolation of diclofenac is that the fm,CYP2C9 value for diclofenac 4′-hydroxylation was estimated to be only 33%[66] (table IV), indicating that the contribution of CYP2C9 to the overall CLint,h is small and that other unidentified CYP or nonCYP isoform(s) dominate the CLh of diclofenac. On the basis of these considerations, diclofenac would be a useful CYP2C9 probe substrate to study the effects of CYP2C9 variants on the catalytic activity of the enzyme in in vitro experiments performed with a cDNA-expressed system where only a single CYP isoform is involved in the catalytic reaction, but not necessarily so in in vivo situations where multiple enzymes may be involved in the catalytic reaction.

Pharmacokinetic parameters for commonly used in vitro probe substrates for cytochrome P450 (CYP) 2C9
In this context, the reason why Shimamoto et al.[66] failed to find significant differences in CLpo of diclofenac between individuals with CYP2C9*1/*1 and those with CYP2C9*1/*3 might be explained by the fact that the quantitative contribution of CYP2C9 to the overall in vivo hepatic elimination of diclofenac is rather small (i.e. fm,CYP2C9 = 33%). Nevertheless, they found no significant differences even in CLf to 4′-hydroxy-diclofenac between the corresponding groups. Although this finding is difficult to explain, the use of heterozygous, rather than homozygous, CYP2C9*3 individuals in the in vivo pharmacokinetic study, and a relatively smaller effect of the CYP2C9*3 mutation on in vitro diclofenac 4′-hydroxylation in the cDNA expression system as compared with other CYP2C9 substrates,[50] or a combination of these reasons, might have made the influence of CYP2C9*3 on in vivo diclofenac elimination ambiguous.
6. Conclusion
The genetic polymorphism of CYP2C9 is associated with wide interpatient variability in the hepatic metabolism of the pharmacologically more active (S)-enantiomer of warfarin and consequently with variable anticoagulation response. At present, 5 CYP2C9 variants [Arg144/Ile359 (CYP2C9*1, the wild-type genotype), Cys144/Ile359 (CYP2C9*2), Arg144/Leu359 (CYP2C9*3), Arg144/Thr359 (CYP2C9*4) and Arg144/Asp360] have been confirmed in randomly selected populations. Considerable interethnic differences exist in the allelic frequencies of the respective variants; Caucasians appear to have greater allelic frequencies than African-Americans and Asians for CYP2C9*2 and CYP2C9*3.
The relationship between in vitro enzyme activity of cDNA-expressed CYP2C9 variants and the in vivo disposition of warfarin has been studied extensively. With regard to the CYP2C9*2 mutation, there remains controversy as to whether the cDNA-expressed CYP2C9*2 (Cys144/Ile359 variant) has an altered in vitro CLint for (S)-warfarin 7-hydroxylation compared with that of CYP2C9*1 (wild-type), because Vmax changes depending on the experimental conditions (i.e. the ratio of NADPH-CYP reductase to CYP). Although no data are available regarding the in vivo clearance of (S)- or racemic warfarin in individuals with the CYP2C9*2 allele, patients with CYP2C9*1/*2 reportedly require a 20% lower maintenance dosage of racemic warfarin to attain therapeutic anticoagulation responses compared with those with CYP2C9*1/*1. Together, these observations suggest that the CYP2C9*2 mutation might confer a small reduction in the in vivo metabolic activity of CYP2C9.
As to the CYP2C9*3 mutation, all reports consistently demonstrate that this mutation, even in heterozygous form, confers a substantial reduction not only in in vitro CLint for (S)-warfarin 7-hydroxylation in cDNA-expressed systems and human liver microsomes, but also in in vivo CLpo,u for (S)-warfarin and CLf for (S)-warfarin 7-hydroxylation. There was an excellent correlation between the in vivo CLpo,u for (S)-warfarin in patients with CYP2C9*1/*1 (wild-type), CYP2C9*3/*3 and CYP2C9*1/*3 and the in vitro CLint for recombinant CYP2C9*1 and CYP2C9*3 enzymes and their mixture. It was also demonstrated that genetic polymorphisms of CYP2C9 (CYP2C9*3 and/or CYP2C9*2) may be associated with increased risks of bleeding complications and with excessive anticoagulation responses at the time of induction of warfarin therapy. However, further studies are necessary to confirm these observations. The clinical implications of the newly discovered variants (CYP2C9*4 and CYP2C9*5) remain to be studied.
Caution must be exercised in extrapolating in vitro data to in vivo situations in an attempt to predict the influence of CYP2C9 variants on in vivo pharmacokinetics of alleged CYP2C9 substrates. Among the CYP2C9 substrates discussed in this article, diclofenac may not be a suitable substrate for in vitro to in vivo extrapolation because the quantitative contribution of CYP2C9 to its overall metabolic clearance is rather small and the effect of the CYP2C9*3 mutation on the in vitro metabolism of the drug is limited relative to that of other CYP2C9 substrates.
References
Hirsh J, Dalen JE, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest 1998; 114: 445S–69S
O’Reilly RA. Studies on the optical enantiomorphs of warfarin in man. Clin Pharmacol Ther 1974; 16: 348–54
Breckenridge A, Orme M, Wesseling H, et al. Pharmacokinetics and pharmacodynamics of the enantiomers of warfarin in man. Clin Pharmacol Ther 1974; 15: 424–30
Rettie AE, Korzekwa KR, Kunze KL, et al. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: a role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem Res Toxicol 1992; 5: 54–9
Kaminsky LS, Zhang Z-Y. Human P450 metabolism of warfarin. Pharmacol Ther 1997; 73: 67–74
Goldstein JA, de Morais SMF. Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics 1994; 4: 285–99
Wilting J, van der Giesen WF, Janssen LHM, et al. The effect of albumin conformation on the binding of warfarin to human serum albumin: the dependence of the binding of warfarin to human serum albumin on the hydrogen, calcium, and chloride ion concentrations as studied by circular dichroism, fluorescence and equilibrium dialysis. J Biol Chem 1980; 255: 3032–7
Porter RS, Sawyer WT. Warfarin. In: Evans WE, Schentag JJ, Jusko WJ, editors. Applied pharmacokinetics: principles of therapeutic drug monitoring. Vancouver (WA): Applied Therapeutics Inc, 1992: 31 (1)-(46)
Wilkinson GR, Shand DG. A physiological approach to hepatic drug clearance. Clin Pharmacol Ther 1975; 18: 377–90
Takahashi H, Kashima T, Kimura S, et al. Determination of unbound warfarin enantiomers in human plasma and 7- hydroxywarfarin in human urine by chiral stationary-phase liquid chromatography with ultraviolet or fluorescence and on-line circular dichroism detection. J Chromatogr B 1997; 701:71–80
Toon S, Low LK, Gibaldi M, et al. The warfarin-sulfinpyrazone interactions: stereochemical considerations. Clin Pharmacol Ther 1986; 39: 15–24
Chan E, McLachlan A, O’Reilly R, et al. Stereochemical as pects of warfarin drug interactions: use of a combined pharmacokinetic-pharmacodynamic model. Clin Pharmacol Ther 1994; 56: 286–94
Takahashi H, Sato T, Shimoyama Y, et al. Potentiation of anti coagulant effect of warfarin caused by enantioselective metabolic inhibition by the uricosuric agent benzbromarone. Clin Pharmacol Ther 1999; 66: 569–81
Takahashi H, Kashima T, Kimura S, et al. Pharmacokinetic in teraction between warfarin and a uricosuric agent, bucolome: application of in vitro approaches to predicting in vivo reduction of (S)-warfarin clearance. Drug Metab Dispos 1999; 27: 1179–86
Kunze KL, Trager WF. Warfarin-fluconazole interaction III: a rational approach to management of a metabolically based drug interaction. Drug Metab Dispos 1996; 24: 429–35
Miners JO, Birkett DJ. Cytochrome P4502C9: an enzyme of major importance in human drug metabolism. BrJ Clin Pharmacol 1998; 45: 525–38
Toon S, Hopkins KJ, Garstang FM, et al. Enoxacin-warfarin interaction: pharmacokinetic and stereochemical aspects. Clin Pharmacol Ther 1987; 42: 33–41
Niopas I, Toon S, Aarons L, et al. The effect of cimetidine on the steady-state pharmacokinetics and pharmacodynamics of warfarin in humans. Eur J Clin Pharmacol 1999; 55: 399–404
Unge P, Svedberg LE, Nordgren A, et al. A study of the inter action of omeprazole and warfarin in anticoagulated patients. Br J Clin Pharmacol 1992; 34: 509–12
Gidal BE, Sorkness CA, McGill KA, et al. Evaluation of a potential enantioselective interaction between ticlopidine and warfarin in chronically anticoagulated patients. Ther Drug Monit 1995; 17: 33–8
Abernethy DR, Kaminsky LS, Dickinson TH. Selective inhibition of warfarin metabolism by diltiazem in humans. J Pharmacol Exp Ther 1991; 257; 411–5
Shimada T, Yamazaki H, Miura M, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 1994; 270: 414–23
Bourrie M, Meunier V, Berger Y, et al. Role of cytochrome P-4502C9 in irbesartan oxidation by human liver microsomes. Drug Metab Dispos 1999; 27: 288–96
Gill HJ, Tjia JF, Kitteringham NR, et al. The effect of genetic polymorphisms in CYP2C9 on sulphamethoxazole N-hydr-oxylation. Pharmacogenetics 1999; 9: 43–53
Kidd RS, Straughn AB, Meyer MC, et al. Pharmacokinetics of chlorpheniramine, phenytoin, glipizide and nifedipine in an individual homozygous for the CYP2C9*3 allele. Pharmacogenetics 1999; 9: 71–80
Oscarson M, Ingelman-Sunberg M, Daly AK, et al. Human cytochrome P450 (CYP) genes. Available from: URL: http://www.imm.ki.se/CYPalleles/ [Accessed 2001 July 10]
Schwarz UI, Choo EF, Dresser GK, et al. Identification of anew CYP2C9 variant in African-Americans. Clin Pharmacol Ther 2000; 67: 169
Imai J, Ieiri I, Mamiya K, et al. Polymorphism of the cytochrome P450 (CYP) 2C9 gene in Japanese epileptic patients: genetic analysis of the CYP2C9 locus. Pharmacogenetics 2000; 10: 85–9
Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics 1996; 6: 341–9
London SJ, Daly AK, Leathart JBS, et al. Lung cancer risk in relation to the CYP2C9*1/CYP2C9*2 genetic polymorphism among African-American and Caucasians in Los Angeles county, California. Pharmacogenetics 1996; 6: 527–33
Stubbins MJ, Harries LW, Smith G, et al. Genetic analysis of the human cytochrome P450 CYP2C9 locus. Pharmacogenetics 1996; 6: 429–39
Furuya H, Fernandez-Salguero P, Gregory W, et al. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics 1995; 5: 389–92
Taube J, Halsall D, Baglin T. Influence of cytochrome P-450 CYP2C9 polymorphisms on warfarin sensitivity and risk of over-anticoagulation in patients on long-term treatment. Blood 2000; 96: 1816–9
Gaedigk A, Leeder JS, Sellers EM, et al. Cytochrome P450 CYP2C9allele frequencies in Canadian native Indians (CNI). Clin Pharmacol Ther 2000; 67: 168
Brockmöller J, Rost KL, Gross D, et al. Phenotyping of CYP2C19 with enantiospecific HPLC-quantification of R- and 5-mephenytoin and comparison with the intron 4/exon 5 G→A splice site mutation. Pharmacogenetics 1995; 5: 80–8
Cascorbi I, Ackermann E, Sachse C, et al. A novel CYP2C9 intron 2 T/C transition and linkage to mutations Leu359 and Cys 144. Clin Pharmacol Ther 1998; 65: 198
Yasar Ü, Eliasson E, Dahl ML, et al. Validation of methods for CYP2C9 genotyping: frequencies of mutant alleles in a Swedish population. Biochem Biophys Res Commun 1999; 254: 628–31
Aynacioglu AS, Brockmöller J, Bauer S, et al. Frequency of cytochrome P450 CYP2C9 variants in a Turkish population and functional relevance for phenytoin. Br J Clin Pharmacol 1999; 48: 409–15
Freeman BD, Zehnbauer BA, McGrath S, et al. Cytochrome P450 polymorphisms are associated with reduced warfarin dose. Surgery 2000; 128: 281–5
Wang SL, Huang JD, Lai MD, et al. Detection of CYP2C9 polymorphism based on the polymerase chain reaction in Chinese. Pharmacogenetics 1995; 5: 37–42
Nasu K, Kubota T, Ishizaki T. Genetic analysis of CYP2C9poly-morphism in a Japanese population. Pharmacogenetics 1997; 7: 405–9
Takahashi H, Kashima T, Nomizo Y, et al. Metabolism of warfarin enantiomers in Japanese patients with heart disease having different CYP2C9 and CYP2C19 genotypes. Clin Pharmacol Ther 1998; 63: 519–28
Kimura M, Ieiri I, Mamiya K, et al. Genetic polymorphism of cytochrome P450s, CYP2C19, and CYP2C9 in a Japanese population. Ther Drug Monit 1998; 20: 243–7
Nebert DW. Suggestions for the nomenclature of human alleles: relevance to ecogenetics, pharmacogenetics and molecular epidemiology. Pharmacogenetics 2000; 10: 279–90
Shintani M, Ieiri I, Inoue K, et al. Genetic polymorphism and functional characterization in 5′-flanking region of human cytochrome P450 (CYP) 2C9 gene: in vitro and in vivo studies. Xenobio Metab Dispos 2000; 15; S307
Haining RL, Hunter AP, Veronese ME, et al. Allelic variants of human cytochrome P450 2C9: baculovirus-mediated expression, purification, structural characterization, substrate stereoselectivity, and prochiral selectivity of the wild-type and I359Lmutant forms. Arch Biochem Biophys 1996; 333: 447–58
Rettie AE, Wienkers LC, Gonzalez FJ, et al. Impaired (S)-war-farin metabolism catalyzed by the R144C allelic variant of CYP2C9. Pharmacogenetics 1994; 4: 39–42
Takahashi H, Kashima T, Nomoto S, et al. Comparisons be tween in-vitro and in-vivo metabolism of (5)-warfarin: catalytic activities of cDNA-expressed CYP2C9, its Leu359 variant and their mixture versus unbound clearance in patients with the corresponding CYP2C9 genotypes. Pharmacogenetics 1998; 8: 365–73
Yamazaki H, Inoue K, Chiba K, et al. Comparative studies on the catalytic roles of cytochrome P450 2C9 and its Cys- and Leu-variants in the oxidation of warfarin, flurbiprofen, and diclofenac by human liver microsomes. Biochem Pharmacol 1998; 56: 243–51
Takahashi K, Tainaka H, Kobayashi K, et al. CYP2C9 Ile359 and Leu359 variants: enzyme kinetic study with seven substrates. Pharmacogenetics 2000; 10: 95–104
Crespi CL, Miller VP. The R144C change in the CYP2C9*2 allele alters interaction of the cytochrome P450 with NADPH: cytochrome P450 oxidoreductase. Pharmacogenetics 1997; 7: 203–10
Rettie AE, Haining RL, Bajpai M, et al. A common genetic basis for idiosyncratic toxicity of warfarin and phenytoin. Epilepsy Res 1999; 35: 253–5
Tracy TS, Rosenbluth BW, Wrighton SA, et al. Role of cytochrome P450 2C9 and an allelic variant in the 4′-hydroxyla- tion of (R)- and (S)-flurbiprofen. Biochem Pharmacol 1995; 49; 1269–75
Chang TKH, Yu L, Goldstein JA, et al. Identification of the polymorphically expressed CYP2C19 and the wild-type CYP2C9-Ile359 allele as low-Km catalysts of cyclophosphamide and ifosfamide activation. Pharmacogenetics 1997; 7: 211–21
Gotoh O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem 1992; 267: 83–90
Ieiri I, Tainaka H, Morita T, et al. Catalytic activity of three variants (Ile, Leu, and Thr) at amino acid residue 359 in human CYP2C9 gene and simultaneous detection using singlestrand conformation polymorphism analysis. Ther Drug Monit 2000; 22: 237–44
Odani A, Hashimoto Y, Otsuki Y, et al. Genetic polymorphism of the CYP2C subfamily and its effect on the pharmacokinetics ofphenytoin in Japanese patients with epilepsy. Clin Pharmacol Ther 1997; 62: 287–92
Mamiya K, Ieiri I, Shimamoto J, et al. The effects of genetic polymorphisms of CYP2C9 and CYP2C19 on phenytoin metabolism in Japanese adult patients with epilepsy: studies in stereoselective hydroxylation and population pharmacokinetics. Epilepsia 1998; 39: 1317–23
Inoue K, Yamazaki H, Imiya K, et al. Relationship between CYP2C9 and 2C19 genotypes and tolbutamide methyl hydroxylation and S-mephenytoin 4′-hydroxylation activities in livers of Japanese and Caucasian populations. Pharmacogenetics 1997; 7: 103–13
Bhasker CR, Miners JO, Coulter S, et al. Allelic and functional variability of cytochrome P4502C9. Pharmacogenetics 1997; 7: 51–8
Steward DJ, Haining RL, Henne KR, et al. Genetic association between sensitivity to warfarin and expression of CYP2C9*3. Pharmacogenetics 1997; 7: 361–7
Aithal GP, Day CP, Kesteven PJL, et al. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999; 353: 717–9
Aithal GP, Day CP, Kesteven PJL, et al. Warfarin dose requirement and CYP2C9 polymorphisms — Authors’ reply. Lancet 1999; 353: 1972–3
Margaglione M, Colaizzo D, D’Andrea G, et al. Genetic modulation of oral anticoagulation with warfarin. Thromb Haemost 2000; 84: 775–8
McCrea JB, Cribb A, Rushmore T, et al. Phenotypic and genotypic investigations of a healthy volunteer deficient in the conversion of losartan to its active metabolite E-3174. Clin Pharmacol Ther 1999; 65: 348–52
Shimamoto J, Ieiri I, Urae A, et al. Lack of differences in diclofenac (a substrate for CYP2C9) pharmacokinetics in healthy volunteers with respect to the single CYP2C9*3 allele. Eur J Clin Pharmacol 2000; 56: 65–8
Stearns RA, Chakravarty PK, Chen R, et al. Biotransformation of losartan to its active carboxylic acid metabolite in human liver microsomes. Drug Metab Dispos 1995; 23: 207–15
Chan E, McLachlan AJ, Pegg M, et al. Disposition of warfarin enantiomers and metabolites in patients during multiple dosing with rac-warfarin. Br J Clin Pharmacol 1994; 37: 563–9
Dickinson RG, Hooper WD, Patterson M, et al. Extent of urinary excretion of p-hydroxyphenytoin in healthy subjects given phenytoin. Ther Drug Monit 1985; 7: 283–9
Brogden RN, Heel RC, Pakes GE, et al. Glipizide: a review of its pharmacological properties and therapeutic uses. Drugs 1979; 18: 329–53
Veronese ME, Miners JO, Rees DLP, et al. Tolbutamide hydroxylation in humans: lack of bimodality in 106 healthy subjects. Pharmacogenetics 1993; 3: 86–93
Benet LZ, Oie S, Schwartz JB. Design and optimization of dosage regimens: pharmacokinetic data. In: Hardman JG, Limbird LE, editors. Goodman & Gilman’s the pharmacological basis of therapeutics. 9th ed. New York: McGraw-Hill, 1996: 1707–92
Acknowledgements
The authors thank Dr Ichiro Ieiri, Tottori University, for his helpful discussion during the preparation of the article. This paper was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (12670703) and Japanese Research Foundation for Clinical Pharmacology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Takahashi, H., Echizen, H. Pharmacogenetics of Warfarin Elimination and its Clinical Implications. Clin Pharmacokinet 40, 587–603 (2001). https://doi.org/10.2165/00003088-200140080-00003
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200140080-00003