
Abstract
Sirolimus (previously known as rapamycin), a macrocyclic lactone, is a potent immunosuppressive agent. Sirolimus was recently approved by the US Food and Drug Administration, on the basis of 2 large, double-blind, prospective clinical trials, for use in kidney transplant recipients at a fixed dosage of 2 or 5 mg/day in addition to full dosages of cyclosporin and prednisone. However, despite the fixed dosage regimens used in these pivotal trials, pharmacokinetic and clinical data show that sirolimus is a critical-dose drug requiring therapeutic drug monitoring to minimise drug-related toxicities and maximise efficacy.
Assays using high performance liquid chromatography coupled to mass spectrometry, although cumbersome, are the gold standard for evaluating other commonly used assays, such as liquid chromatography with ultraviolet detection, radioreceptor assay and microparticle enzyme immunoassay.
Sirolimus is available in oral solution and tablet form. It has poor oral absorption and distributes widely in tissues, displaying not only a wide inter- and intrapatient variability in drug clearance, but also less than optimal correlations between whole blood concentrations and drug dose, demographic features or patient characteristics. Furthermore, the critical role of the cytochrome P450 3A4 system for sirolimus biotransformation leads to extensive drug-drug interactions, among which are increases in cyclosporin concentrations. Thus, sirolimus is now being used to reduce or eliminate exposure to cyclosporin or corticosteroids. The long elimination half-life of sirolimus necessitates a loading dose but allows once daily administration, which is more convenient and thereby could help to improve patient compliance.
This review emphasises the importance of blood concentration monitoring in optimising the use of sirolimus. The excellent correlation between steady-state trough concentration (at least 4 days after inception of, or change in, therapy) and area under the concentration-time curve makes the former a simple and reliable index for monitoring sirolimus exposure. Target trough concentration ranges depend on the concomitant immunosuppressive regimen, but a range of 5 to 15 μg/L is appropriate if cyclosporin is being used at trough concentrations of 75 to 150 μg/L. Weekly monitoring is recommended for the first month and bi-weekly for the next month; thereafter, concentration measurements are necessary only if warranted clinically.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Sirolimus (Wyeth-Ayerst, Princeton, NJ, USA), formerly known as rapamycin, is a macrocyclic lactone (fig. 1). Sirolimus is a fermentation product of Streptomyces hygroscopicus, which was discovered in the soil of Rapa Nui (Easter Island).[1] Although first developed as an antifungal agent, the drug was later found to have potent antiproliferative and immunosuppressive properties.[2,3]

Chemical structure of sirolimus: (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22, 23,24,25,26,27,32,33,34,34a-hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone.
Sirolimus displays a synergistic action with, and a distinctive mechanism of action from, that of calcineurin inhibitors.[4,5] It inhibits both generation of the costimulatory signal during G0 to G1 activation of lymphocytes, and the post-transduction events after cytokine stimulation during G1.[6]
Previous reviews have primarily concentrated on the pharmacokinetics of sirolimus rather than on therapeutic monitoring, mainly because the clinical data on the correlations between dose, blood concentrations, acute rejection prophylaxis and toxicity are evolving and, therefore, are not yet well published. On the basis of our experience in the University of Texas-Houston transplant programme, we have tried in this review to present more information on therapeutic monitoring and emphasise the importance of blood concentration monitoring, as well as presenting current approaches to formulation and dosage selection.
1. Chemical Characteristics
Sirolimus is a white crystalline solid with empirical formula C56H89NO14, which melts at 183 to 185°C, is readily fat soluble and practically insoluble in water. The ultraviolet absorption spectrum shows a major peak at 277nm and minor peaks at 267 and 288nm.[1] The compound is sensitive to light and temperature, and blood samples, particularly after extraction, must be shielded to prevent degradation. However, a recent report suggests that the drug in the blood resists the degradative effects of freeze-thaw cycles.[7]
2. Analytical Assays
Sirolimus blood concentrations of 5 to 20 µg/L display potent immunosuppression in large animal models and in clinical practice.[8–10] Parent drug concentrations seem to represent the critical indices for clinical effects, since there is no evidence that metabolites have more than modest biological activity (at most <10% of that of sirolimus).[11] Thus, the methods used for therapeutic drug monitoring must meet the following criteria: (i) specificity for parent compound rather than metabolites; (ii) detection sensitivity of 2 µg/L; (iii) readily available analytical equipment; (iv) reliable extraction procedures and measurement techniques; and (v) sample turnaround time within 24 hours. A panel of experts[12] recommended that the analytical assay should also have the following performance characteristics: (i) interday coefficient of variation (%CV) of ≤10% at 5 µg/L and ≤5% at 40 µg/L concentrations; (ii) detection range of 1 to 75 µg/L; and (iii) slope of 0.9 to 1.1 and standard deviation of the estimate <5 µg/L when compared with a validated assay.
Because 95% of drug is sequestered in red blood cells,[13] the preferred matrix for sirolimus measurement is whole blood. Blood samples collected in tubes containing EDTA as the anticoagulant are stable up to 24 hours at room temperature, 7 days at 2 to 8°C and 3 months at -20°C.[12]
2.1 High Performance Liquid Chromatography with Detection by Tandem Mass Spectroscopy
High performance liquid chromatography with detection by tandem mass spectroscopy (LC/MS-MS) specifically measures sirolimus with a sensitivity of 0.25 µg/L, linearity over the range of 0.25 to 250 µg/L, and an interday %CV of 19 to 1.4% (table I).[14,15] Unfortunately, MS technology has the characteristics of being labour-intensive, technically difficult and expensive, which hinders its routine clinical application, and it has only recently become more available.

Assay methodologies for quantification of sirolimus
2.2 Liquid Chromatography with Ultraviolet Detection
Liquid chromatography with ultraviolet detection (LC/UV) specifically measures sirolimus in whole blood at the 278nm wavelength (table I).[16,19] Comparison of 385 paired determinations by LC/UV and LC/MS-MS showed a correlation coefficient of 0.943, regression line slope of 1.024, and standard error of estimate of 2.87.[20] In this large analysis, the majority of disparate samples showed values within 4 µg/L of one another, with no obvious bias of LC/UV values to be either greater or less than those obtained by LC/MS-MS (fig. 2). In a smaller cohort of 55 blood samples containing sirolimus 5 to 80 µg/L, LC/UV showed higher values than LC/MS-MS by 3 to 5.7 µg/L.[21] The LC/UV method has been reported to have a sensitivity of 1.0 µg/L, linearity over the 1.0 to 50 µg/L range and an interday %CV of 5.6 to 17%.[16,19] Furthermore, LC/UV methodology is more readily available and has successfully been used for therapeutic monitoring in clinical laboratories.[22]

Correlation between high performance liquid chromatography with ultraviolet detection (LC/UV) and high performance liquid chromatography with detection by tandem mass spectroscopy (LC/MS-MS) measurements of sirolimus. (a) Correlation plot with dotted lines showing 95% confidence limits; and (b) the ratio of difference between estimates by each method as a function of drug concentration (from Kahan et al.,[20] with permission).
2.3 Radioreceptor Assay
Radioreceptor assay (RRA), also called immunophilin-binding assay, is based on the affinity of parent drug and some metabolites for a 52kD immunophilin binding protein.[23] The assay displays a sensitivity of 1.0 µg/L, linearity over the 2.5 to 40 µg/L range and %CV of 5.9 to 12.9% (table I).[17] Although Goodyear et al.[23] reported an excellent correlation between the results of RRA and LC/UV (r = 0.977, slope of regression line 1.09), Davis et al.[17] observed poorer correlation coefficients among paired comparison of values from 51 blood samples in nonthrombocytopenic and thrombocytopenic patients (r = 0.706 and r = 0.806, respectively). The 10 to 26% cross-reactivity of the binding assay for sirolimus metabolites,[24] as detected by the deviation of RRA results from LC/UV values, did not afford a better prediction of thrombocytopenia.[25] Although RRA has the potential for semiautomation, it has not been developed into a clinical assay.
2.4 Microparticle Enzyme Immunoassay
An automated immunoassay using the IMx™ analyser (Abbott, N. Chicago, IL, USA) was used for concentration measurements in the phase III clinical trials, but was then withdrawn from further use. The assay overestimates LC results by 42.5% as a result of cross-reactivity with sirolimus metabolites.[26] It shows an interday %CV of 8.4 to 11% and a sensitivity of 3.0 µg/L (table I).[18] The slope of the regression line of IMx™ compared with LC values is 1.39, which is well outside the range recommended for a clinical assay. Although the method has the advantage of ready availability, easy methodology and fast turnaround time, it does not meet the suggested criteria for therapeutic drug monitoring, and clinical development is not expected.
3. Pharmacokinetics
The pharmacokinetic parameters of sirolimus are summarised in table II.

Summary of sirolimus pharmacokinetic parameters in stable renal transplant recipients concomitantly treated with cyclosporin and prednisone (unless otherwise noted)
3.1 Absorption
The absolute bioavailability of sirolimus is unknown. Based on the concentration data after a small group of haemodialysis patients received the drug intravenously (B.D. Kahan and J. Zimmerman, unpublished observations) versus the concentrations in stable renal transplant patients given similar doses orally, the apparent oral bioavailability has been estimated to be about 15%.[30] Escalating single oral dose studies (3 to 21 mg/m2) in the latter group receiving, in addition, cyclosporin and corticosteroids showed peak whole blood concentrations (Cmax) of 14 to 344 µg/L, time to peak concentration (tmax) of 0.5 to 3 hours (mean ± SD, 1.4 ± 1.2 hours), only modest correlations (r = 0.59) between dose and Cmax or area under the concentration-time curve (AUC),[29] but a good correlation (r2 = 0.85) of trough concentration prior to the dose (Cmin ss) to AUC.[27,31] Thus, Cmin ss provides a simple, useful index for therapeutic monitoring. Of note, analysis of a cohort of 150 kidney transplant recipients treated de novo with sirolimus, cyclosporin and steroids revealed no association between sex, age, bodyweight or ethnicity and observed or dose-adjusted Cmax, Cmin ss or AUC, all of which showed significant intraindividual variability (%CV of 42.4 to 82.0%).[20]
The current marketed form of sirolimus is an oil-based liquid available in 60 and 150ml bottles and 1, 2 and 5mg sachets containing 1 mg/ml of the drug. The tablet formulation, which offers greater palatability and convenience of administration, is expected to be available in mid-2001. In a phase IIB trial, the 2 forms showed equal efficacy[33] and similar pharmacokinetic behaviour to the liquid preparation, except for an earlier and higher peak concentration of the liquid formulation, with equal overall exposure during the 24-hour dosage interval.[34]
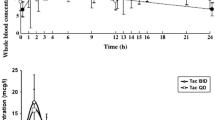
Pharmacokinetic studies in 21 stable renal transplant patients were performed on the last dose of oral liquid formulation prior to, and at 2, 4 and 8 weeks after, conversion to the tablet form on a milligram/milligram ratio.[35] There was no significant difference in the AUC values before versus 2, 4 and 8 weeks after conversion, namely, 256 versus 205, 226 and 224 µg · h/L, respectively. Although Cmin ss values were not significantly different between studies after administration of liquid versus tablet forms, Cmax values were significantly lower with the tablet form (37.1 versus 25.3, 24.9 and 26.7 µg/L, respectively; p < 0.05; fig. 3). Of note, the mean %CV of the dose-corrected AUC values was lower when patients received the tablet than the liquid formulation.[35]

Pharmacokinetic profiles of mean values of whole blood sirolimus concentrations of 21 patients over 12 hours after administration of the last dose of the liquid formulation of sirolimus and at 2, 4 and 8 weeks after conversion to the tablet formulation of sirolimus (from Kelly et al.,[35] with permission).
A nested group of 24 de novo kidney transplant patients had full pharmacokinetic profiles on sirolimus oral solution (n = 13) or tablet form (n = 11), which showed similar C0, AUC and Cmax but significantly longer tmax in patients taking the tablet form (5.8 ± 4.1 vs 2.0 ± 1.2h; p < 0.05).[34] Again, there was lower interpatient variability in patients taking the tablet form. Another recent multicentre, randomised study in de novo kidney transplant recipients confirmed the results of the previous study, with tmax being the only pharmacokinetic parameter significantly different between patients on the oral solution versus the tablet form (2.12 ± 0.84 vs 3.46 ± 2.40h; p = 0.054).[36] In 22 healthy adult volunteers, a high fat meal decreased sirolimus AUC by 35%.[28] It is recommended that patients take the drug consistently with or without food to reduce fluctuations in drug exposure. Furthermore, concomitant administration with the microemulsion but not the oil-based formulation of cyclosporin promotes sirolimus absorption.[37]
In similar fashion to cyclosporin, overall sirolimus absorption is probably affected by the activity of P-glycoprotein as well as intestinal cytochrome P450 (CYP) activity.[38] The 2 compounds are likely to cross-inhibit these activities, possibly accounting for the pharmacokinetic interaction as well as the interpatient variability at the absorptive interface.
3.2 Distribution
In human whole blood, sirolimus is distributed among red blood cells (94.5%), plasma (3.1%), lymphocytes (1.01%) and granulocytes (1.0%). In contrast to tacrolimus and cyclosporin, the distribution is not temperature- or concentration-dependent.[13] The sequestration of sirolimus, like tacrolimus, in red blood cells is believed to be at least partially due to their abundant content of immunophilins.[39] The mean blood-to-plasma ratio in 36 stable renal transplant recipients treated with a single oral dose of sirolimus was 34.5: 1,[31] with significant interindividual variability (CV% 52.3, range 10 to 70). In the plasma compartment, a smaller proportion of sirolimus (compared with cyclosporin) is bound to lipoproteins (40%), although this ratio increases with increasing sirolimus concentrations. The distribution of sirolimus among lipoprotein fractions has not yet been reported, although it is known that the metabolic effects of sirolimus increase the content of high, low, intermediate and very low density lipoproteins.[40] Among the 60% in the free fraction, only a small proportion (about 4%) is bound to soluble plasma proteins.[13] Thus, whole blood is considered the appropriate matrix for therapeutic drug monitoring.
Because of its hydrophobic nature, sirolimus is widely distributed in lipid membranes of body tissues as well as erythrocytes, showing a large apparent volume of distribution (5.6 to 16.7 L/kg).[12,32] Data from rats support this observation. Following 14 days of intravenous or oral administration, spleen, kidney, liver, intestine and heart tissue-to-blood partition coefficients were as high as 40.[41] Because the tissue distribution of sirolimus has not formally been studied in humans, the possible relationship of these concentrations to the efficacy and toxicity of the drug is currently unknown.
3.3 Metabolism and Elimination
The large interindividual variability in cyclosporin biotransformation is related to 8.5- and 9.4-fold variability in enterocyte P-glycoprotein and CYP3A4 content, respectively,[42,43] and similar interindividual variability in the actions of these systems on sirolimus biotransformation[44] and efflux can be expected.[43,45] Other CYP family members potentially play a role in sirolimus metabolism; however, there are no studies documenting their participation.
In 16 stable renal transplant recipients receiving cyclosporin and prednisone at the time of administration of a single sirolimus dose, there was an 8-fold difference in apparent clearance (0.042 to 0.339 L/h/kg) and a range of terminal half-lives of 43.8 to 86.5 hours.[27] Among 40 stable renal transplant recipients who received a 14-day course of sirolimus in addition to cyclosporin and prednisone, there was 4.5-fold variation in apparent clearance (0.090 to 0.416 L/h/kg, mean 0.210 L/h/kg) and a terminal half-life of 62 ± 12 hours.[31] The long terminal half-life of the drug necessitates a loading dose (2.56 ± 0.70 × maintenance dose) to achieve steady-state concentrations rapidly,[31] and also allows a once daily administration schedule, which is more convenient for the patient than the twice daily regimen generally used for calcineurin inhibitors; this advantage may improve compliance with the therapeutic regimen. The tablet form of sirolimus appears to have similar apparent oral clearance to the oral solution.[34,36]
Variations in drug clearance and absorption rates result in a wide range of sirolimus Cmin ss values among patients receiving the same dose. In a pivotal phase III study (Global 302) among 212 patients receiving 2 mg/day, the Cmin ss over the 6-month period was 2.34 to 31.30 µg/L; the values for patients on 5 mg/day were 5.95 to 50.90 µg/L.[46] Presumably, the primary clearance route for sirolimus metabolites is biliary: 91% of the transformation products after administration to healthy volunteers of a single dose of radioactive sirolimus were found in faeces, in contrast to 2.2% excreted in urine.[47] As with cyclosporin and tacrolimus, hepatic dysfunction results in increased concentrations of sirolimus and its metabolites.
More than 16 metabolites have been identified in the bile of rats receiving sirolimus.[48] In humans, trough whole blood samples contain 56 ± 9% of drug-derived compounds as metabolites, particularly the 12-hydroxy-, 16-O-demethyl-, 39-O-demethyl-, 27,39-O-di-demethyl- and dihydroxy-sirolimus (fig. 4), which form 71.8 ± 68%, 35 ± 42%, 20 ± 20% and 20 ± 31% of total metabolites, respectively.[14] Although sirolimus metabolites seem to display less than 10% of the activity of the parent compound,[11,49,50] it is possible that detailed characterisation of the toxic versus immunosuppressive effects of sirolimus metabolites may provide insights for therapeutic drug monitoring.

Chemical structures of sirolimus metabolites.
3.4 Drug Interactions
Drugs affecting CYP3A4 or P-glycoprotein levels or activities would be expected to alter sirolimus pharmacokinetics. Diltiazem and ketoconazole increased sirolimus AUC by 60 and 990%, respectively, whereas rifamycin reduced AUC by 82%.[46] Simultaneous administration of cyclosporin in the microemulsion, but not the oil-based, formulation increased sirolimus AUC by 50%, but sirolimus did not alter cyclosporin exposure.[37] In rats, concomitant oral administration of cyclosporin and sirolimus increased both the blood and the tissue concentrations of both drugs,[51] and, in phase III studies, patients receiving sirolimus required lower cyclosporin dosages to achieve similar target cyclosporin concentrations.[52] The nature, extent and clinical implications of the pharmacokinetic interactions between sirolimus and cyclosporin require further investigation.
Administration of a 2-week course of 1 to 13 mg/m2 sirolimus to 40 stable renal transplant patients resulted in a significant increase in prednisolone AUC (mean ratio 1.27; p = 0.0001).[53] The clinical significance of this finding needs further study.
4. Therapeutic Drug Monitoring
Both experimental animal and clinical data suggest that the immunosuppressive efficacy and the occurrence and severity of adverse effects of sirolimus correlate with blood concentrations.[8,54] In a phase I/II study of 40 de novo mismatched living donor kidney transplant recipients, Cmin ss values >15 µg/L (LC/UV assay) were associated with adverse reactions of increased triglyceride levels or reduced haemoglobin, leucocyte and particularly platelet counts (fig. 5).[55] In a retrospective single-centre review of 150 kidney transplant recipients treated de novo with a sirolimus/cyclosporin combination, sirolimus Cmin ss values >15 µg/L (LC/UV assay) had a positive predictive value for hypertriglyceridaemia (>300 mg/dl), thrombocytopenia (<100 000/mm3) and leucopenia (<4000/mm3).[20] Furthermore, dose-corrected Cmin ss values <1.7 µg/L/mg were associated with a 3.6-fold increase in, and observed Cmin ss values >5 µg/L had an 89.5% negative predictive value for, the occurrence of acute rejection episodes (fig. 6). Thus it appears that, when sirolimus is used in addition to cyclosporin and prednisone, a therapeutic concentration range of 5 to 15 µg/L is associated with protection from acute rejection episodes and adverse effects. Sirolimus allows a 50% reduction in cyclosporin exposure,[56] a property that can be especially useful in patients who receive cadaveric renal grafts, which tend to show slow recovery of renal function.

Receiver operating characteristic functions to predict the correlation between trough steady-state concentrations (Cmin ss) and sirolimus-induced adverse drug reactions. (a) Thrombocytopenia (<100 000/mm3); (b) leucopenia (<4000/mm3); (c) hypertriglyceridaemia (>400 mg/dl); (d) hypercholesterolaemia (>240 mg/dl) [from Kahan et al.,[20] with permission].

Receiver operating characteristic functions to predict the occurrence of an acute rejection episode. (a) Use of dose-corrected trough steady-state concentration (Cmin ss/mg); (b) use of dose-corrected area under the concentration-time curve (AUC/ mg) [from Kahan et al.,[20] with permission].
The appropriate sirolimus Cmin ss for acute rejection episode prophylaxis is a function of the concomitant immunosuppressive regimen (table III). When used as base therapy with azathioprine and prednisone, a regimen stipulating initial Cmin ss values equal to 30 µg/L during the first 2 months, and 15 µg/L (LC/UV assay) thereafter, led to a 41% rate of acute rejection episodes among 41 cadaveric kidney transplant recipients.[57] When combined with mycophenolate mofetil and prednisone, this sirolimus regimen was associated with a 27.5% rate of acute rejection episodes among 40 cadaveric renal transplant recipients.[58] Indeed, the combination of sirolimus (Cmin ss of 10 to 20 µg/L; LC/UV assay) and basiliximab with late introduction of low dosage cyclosporin has provided excellent prophylaxis of acute rejection episodes and renal function for primary, non-African-American recipients of cadaveric kidney transplants that displayed delayed graft function.[59,60] In purely Caucasian low-risk liver and kidney-pancreas transplant recipients, Cmin ss of 6 to 12 µg/L (IMx™ assay) in combination with low dosage tacrolimus has been reported to yield low rates of acute rejection episodes and toxicity.[10]

Sirolimus concentrations and acute rejection prophylaxis in combination with other immunosuppressive agents
The appropriate regimen for long term drug monitoring has not been established. Because of the long half-life and extensive tissue distribution of the drug, steady-state concentrations are not reached before day 6 after initiation of therapy or after a dosage change. Thus, daily concentration monitoring is not necessary; the first sirolimus measurements should not be obtained before day 4 after inception of, or change in, therapy. Thereafter, we recommend monitoring Cmin ss weekly for the first month and bi-weekly for the next month, targeting a 5 to 15 µg/L range if cyclosporin is being used concomitantly at Cmin ss concentrations of 75 to 150 µg/L. If the patient fails to attain these values despite a dosage of 20 mg/day, a full pharmacokinetic study should be performed to assess whether the defect is due to limited absorption or rapid clearance rates. After the second month, Cmin ss estimates are necessary only if warranted clinically: namely, upon changes in or addition to the therapeutic regimen of a drug that affects CYP3A4 biotransformation or because of suspicion of unusual circumstances or noncompliance, gastrointestinal disturbances, or exaggerated toxicity.
The solid formulation has been approved by the US Food and Drug Administration (FDA) for use in kidney transplant recipients. The solid form showed similar pharmacokinetic behaviour, except for lower Cmax, to the liquid preparation in stable renal transplant recipients (section 3.1). Conversion from one formulation to the other was not associated with any significant adverse effects or with any acute rejection episodes.[35]
In a nonblind, multicentre, randomised trial, 477 de novo kidney transplant recipients receiving 2 mg/day of either oral solution or tablet form of sirolimus in addition to cyclosporin and corticosteroids showed similar efficacy and adverse effect profiles.[33] The incidences of acute rejection (21 vs 19%), graft survival (94 vs 91%) and patient survival (97 vs 98%) were similar between the 2 groups. The main adverse effects were anaemia, decrease in platelet count and increase in triglyceride and cholesterol levels, which occurred with similar frequency in both groups.
5. Conclusion
Although structurally similar to tacrolimus, sirolimus has a novel mechanism of action, which leads to synergy with cyclosporin. The long half-life of the drug necessitates a loading dose to achieve therapeutic concentrations quickly, and also allows for once daily administration. Highly variable absorption and metabolism of the drug result in large differences in blood concentrations among patients receiving the same dose. Efficacy for the prevention of acute rejection episodes, and the rate of common adverse effects (thrombocytopenia, leucopenia and hypertriglyceridaemia), are concentration-dependent. Thus, despite the FDA-mandated pivotal blinded trials using fixed dosages of sirolimus in conjunction with full exposure to cyclosporin and prednisone, it is now recognised that therapy in renal transplant recipients is optimised by marked reductions in the dosages of immunosuppressive agents, a strategy that demands therapeutic drug monitoring.[30] The excellent correlation between Cmin and AUC makes the former a simple and reliable index for monitoring sirolimus exposure. However, automated assays to measure whole blood concentrations are not available. Although the addition of sirolimus to the current armamentarium of immunosuppressive drugs has resulted in novel approaches to reduce or avoid the nephrotoxicity caused by calcineurin inhibitors, the effect of these regimens or of pharmacokinetic parameters on long term graft survival, as evidenced by the incidence of chronic rejection, is not yet known. The pharmacokinetic behaviour and therapeutic drug monitoring guidelines for sirolimus in other solid organ transplant recipients await further investigation.
References
Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic: II. Fermentation, isolation and characterization. J Antibiot (Tokyo) 1975; 28: 727–32
Calne RY. The rejection of renal homograft inhibition in dogs by 6-mercaptopurine. Lancet 1960; I: 417–8
Kahan BD, Chang JY, Sehgal SN. Preclinical evaluation of a new potent immunosuppressive agent, rapamycin. Transplantation 1991; 52: 185–91
Kahan BD, Gibbons S, Tejpal N, et al. Synergistic interactions of cyclosporine and rapamycin to inhibit immune performances of normal human peripheral blood lymphocytes in vitro. Transplantation 1991; 51: 232–9
Stepkowski SM, Napoli KL, Wang ME, et al. Effects of the pharmacokinetic interaction between orally administered sirolimus and cyclosporine on the synergistic prolongation ofheart allograft survival in rats. Transplantation 1996; 62: 986–94
Dumont FJ, Staruch MJ, Koprak SL, et al. Distinct mechanisms of suppression of murine T cell activation by the related mac-rolides FK-506 and rapamycin. J Immunol 1990; 144: 251–8
Salm P, Tresillian MJ, Taylor PJ, et al. Stability of sirolimus (rapamycin) in whole blood. Ther Drug Monit 2000; 22: 423–6
Granger DK, Cromwell JW, Chen SC, et al. Prolongation of renal allograft survival in a large animal model by oral rapamycin monotherapy. Transplantation 1995; 59: 183–6
Kahan BD. Established immunosuppressive drugs: clinical and toxic effects. In: Kahan BD, Ponticelli C, editors. Principles and practice of renal transplantation. London: Dunitz, 2000: 349–414
McAlister VC, Gao Z, Peltekian K, et al. Sirolimus-tacrolimus combination immunosuppression [letter]. Lancet 2000; 355: 376–7
Christians U, Sattler M, Schiebel HM, et al. Isolation of two immunosuppressive metabolites after in vitro metabolism of rapamycin. Drug Metab Dispos 1992; 20: 186–91
Yatscoff RW, Wang P, Chan K, et al. Rapamycin: distribution, pharmacokinetics, and therapeutic range investigations. Ther Drug Monit 1995; 17: 666–71
Yatscoff R, LeGatt D, Keenan R, et al. Blood distribution of rapamycin. Transplantation 1993; 56: 1202–6
Streit F, Christians U, Schiebel HM, et al. Sensitive and specific quantification of sirolimus (rapamycin) and its metabolites in blood of kidney graft recipients by HPLC/electrospray-mass spectrometry. Clin Chem 1996; 42: 1417–25
Taylor PJ, Johnson AG. Quantitative analysis of sirolimus (Rapamycin) in blood by high-performance liquid chromatography-electrospray tandem mass spectrometry. J Chro- matogr B Biomed Sci Appl 1998; 718: 251–7
Napoli KL, Kahan BD. Sample clean-up and high-performance liquid chromatographic techniques for measurement of whole blood rapamycin concentrations. J Chromatogr B Biomed Appl 1994; 654: 111–20
Davis DL, Murthy JN, Napoli KL, et al. Comparison of steady-state trough sirolimus samples by HPLC and a radioreceptor assay. Clin Biochem 2000; 33: 31–6
Salm P, Taylor PJ, Pillans PI. Analytical performance of microparticle enzyme immunoassay and HPLC-tandem mass spectrometry in the determination of sirolimus in whole blood. Clin Chem 1999; 45: 2278–80
Svensson JO, Brattström C, Säwe J. Determination of rapamycin in whole blood by HPLC. Ther Drug Monit 1997; 19: 112–6
Kahan BD, Napoli KL, Kelly PA, et al. Therapeutic drug monitoring of sirolimus: correlations with efficacy and toxicity. Clin Transplant 2000; 14: 97–109
Holt DW, Lee T, Johnston A. Measurement of sirolimus in whole blood using high-performance liquid chromatography with ultraviolet detection. Clin Ther 2000; 22 Suppl. B: B38–48
Napoli KL, Kahan BD. Routine clinical monitoring of sirolimus (rapamycin) whole-blood concentrations by HPLC with ultraviolet detection. Clin Chem 1996; 42: 1943–8
Goodyear N, Murthy JN, Gallant HL, et al. Comparison of binding characteristics offour rapamycin metabolites to the 14 and 52 kDa immunophilins with their pharmacologic activity measured by the mixed-lymphocyte culture assay. Clin Biochem 1996; 29: 309–13
Davis DL, Murthy JN, Gallant-Haidner H, et al. Minor immunophilin binding of tacrolimus and sirolimus metabolites. Clin Biochem 2000; 33: 1–6
Goodyear N, Napoli KL, Murthy JN, et al. Radioreceptor assay for sirolimus in patients with decreased platelet counts. Clin Biochem 1997; 30: 539–43
Salm P, Taylor PJ, Pillans PI. The quantification ofsirolimus by high-performance liquid chromatography-tandem mass spectrometry and microparticle enzyme immunoassay in renal transplant recipients. Clin Ther 2000; 22 Suppl. B: B71–85
Brattstrom C, Sawe J, Tyden G, et al. Kinetics and dynamics of single oral doses of sirolimus in sixteen renal transplant recipients. Ther Drug Monit 1997; 19: 397–406
Zimmerman JJ, Ferron GM, Lim H-K, et al. The effect of a high-fat meal on the oral bioavailability of the immunosuppressant sirolimus (rapamycin). J Clin Pharmacol 1999; 39: 1155–61
Johnson EM, Zimmerman J, Duderstadt K, et al. A randomized, double-blind, placebo-controlled study of the safety, tolerance, and preliminary pharmacokinetics of ascending single doses of orally administered sirolimus (rapamycin) in stable renal transplant recipients. Transplant Proc 1996; 28: 987
Yatscoff RW. Pharmacokinetics of rapamycin. Transplant Proc 1996; 28: 970–3
Zimmerman J, Kahan BD. Pharmacokinetics of sirolimus in stable renal transplant patients after multiple oral dose administration. J Clin Pharmacol 1997; 37: 405–15
Ferron GM, Mishina EV, Zimmerman JJ, et al. Population pharmacokinetics of sirolimus in kidney transplant patients. Clin Pharmacol Ther 1997; 61: 416–28
Van Buren CT, Rapamune Study Group. Sirolimus oral solution and tablets demonstrate equivalent safety and efficacy in renal allografts [abstract no. 157]. American Society of Transplantation/American Society of Transplant Surgeons Joint Meeting; 2000 May 13-17; Chicago. Transplantation 2000; 69 Suppl.: S153
Kelly P, Napoli KL, Kahan BD. Comparison of the pharmacokinetics of sirolimus (Rapamune) in renal transplant recipients following administration of the liquid or solid table formations [abstract no. 158]. Transplantation 2000; 69: S154
Kelly PA, Napoli KL, Dunne C, et al. Conversion from liquid to solid sirolimus formulations in stable renal allograft transplant recipients. Biopharm Drug Dispos 1999; 20: 249–53
Zimmerman J, Rapamune Study Group. A comparative study of the pharmacokinetic profiles of sirolimus oral solution and tablets in renal allograft patients [abstract no. 159]. Transplantation 2000; 69: S154
Kaplan B, Meier-Kriesche HU, Napoli KL, et al. The effects of relative timing of sirolimus and cyclosporine microemulsion formulation co-administration on the pharmacokinetics of each agent. Clin Pharmacol Ther 1998; 63: 48–53
Yacyshyn BR, Bowen-Yacyshyn MB, Pilarski LM. Inhibition by rapamycin of P-glycoprotein 170-mediated export from normal lymphocytes. Scand J Immunol 1996; 43: 449–55
Kay JE, Sampare-Kwateng E, Geraghty F, et al. Uptake of FK 506 by lymphocytes and erythrocytes. Transplant Proc 1991; 23: 2760–2
Hoogeveen RC, Ballantyne CM, Pownall HJ, et al. Effect of sirolimus on the metabolism of ApoB-100 containing lipoproteins in renal transplant patients. Transplantation. In press
Napoli KL, Wang ME, Stepkowski SM, et al. Distribution of sirolimus in rat tissue. Clin Biochem 1997; 30: 135–42
Lown KS, Mayo RR, Leichtman AB, et al. Role of intestinal P-glycoprotein (mdr1) in interpatient variation in the oral bioavailability of cyclosporine. Clin Pharmacol Ther 1997; 62: 248–60
Lampen A, Zhang Y, Hackbarth I, et al. Metabolism and transport of the macrolide immunosuppressant sirolimus in the small intestine. J Pharmacol Exp Ther 1998; 285: 1104–12
Sattler M, Guengerich FP, Yun CH, et al. Cytochrome P-450 3A enzymes are responsible for biotransformation of FK506 and rapamycin in man and rat. Drug Metab Dispos 1992; 20: 753–61
Crowe A, Lemaire M. In vitro and in situ absorption of SDZ-RAD using a human intestinal cell line (Caco-2) and a single pass perfusion model in rats: comparison with rapamycin. Pharm Res 1998; 15: 1666–72
MacDonald A, Scarola J, Burke JT, et al. Clinical pharmacokinetics and therapeutic drug monitoring of sirolimus. Clin Ther 2000; 22 Suppl. B: B101–21
Wyeth Laboratories. Sirolimus (Rapamune®): prescribing in formation. Philadelphia (PA): Wyeth-Ayerst Pharmaceuticals Inc., 2001
Trepanier DJ, Gallant H, Legatt DF, et al. Rapamycin: distribution, pharmacokinetics and therapeutic range investigations: an update. Clin Biochem 1998; 31: 345–51
Wang CP, Chan KW, Schiksnis RA, et al. High performance liquid chromatographic isolation, spectroscopic characterization and immunosuppressive activities of two rapamycin degradation products. J Liq Chromatogr 1994; 17: 3383–92
Wang CP, Lim HK, Chan KW, et al. High performance liquid chromatographic isolation and spectroscopic characterization of three major metabolites from the plasma of rats receiving rapamycin (sirolimus) orally. J Liq Chromatogr 1995; 18: 2259
Napoli KL, Wang ME, Stepkowski SM, et al. Relative tissue distributions of cyclosporine and sirolimus after concomitant peroral administration to the rat: evidence for pharmacokinetic interactions. Ther Drug Monit 1998; 20: 123–33
Kahan BD, Rapamune U.S. Study Group. Sirolimus (Rapamune, rapamycin) is more effective than azathioprine to reduce the incidence of acute renal allograft rejection episodes when used in combination with cyclosporine and prednisone: a phase III U.S. multicenter trial. Lancet 2000; 356: 194–202
Jusko WJ, Ferron GM, Mis SM, et al. Pharmacokinetics of prednisolone during administration of sirolimus in patients with renal transplants. J Clin Pharmacol 1996; 36: 1100–6
Fryer J, Yatscoff RW, Pascoe EA, et al. The relationship of blood concentrations of rapamycin and cyclosporine to suppression of allograft rejection in a rabbit heterotopic heart transplant model. Transplantation 1993; 55: 340–5
Kahan BD, Podbielski J, Napoli KL, et al. Immunosuppressive effects and safety of a sirolimus/cyclosporine combination regimen for renal transplantation. Transplantation 1998; 66:1040–6
Kahan BD, Julian BA, Pescovitz MD, et al. Sirolimus reduces the incidence of acute rejection episodes despite lower cyclosporine doses in Caucasian recipients of mismatched primary renal allografts: a phase II trial. Transplantation 1999; 68: 1526–32
Groth CG, Backman L, Morales JM, et al., Sirolimus European Renal Transplant Study Group. Sirolimus (rapamycin)-based therapy in human renal transplantation: similar efficacy and different toxicity compared with cyclosporine. Transplantation 1999; 67: 1036–42
Kreis H, Cisterne JM, Land W, et al. Sirolimus in association with mycophenolate mofetil induction for the prevention of acute graft rejection in renal allograft recipients. Transplantation 2000; 69: 1252–60
Hong JC, Kahan BD. Use of anti-CD25 monoclonal antibody in combination with rapamycin to eliminate cyclosporine treatment during the induction phase of immunosuppression [brief communication]. Transplantation 1999; 68: 701–4
Hong JC, Kahan BD. A calcineurin-free strategy for induction immunosuppression for delayed graft function in cadaveric kidney transplantation. Transplant Proc 2001; 33: 1271–2
Acknowledgements
This work was supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK 38016-14).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mahalati, K., Kahan, B.D. Clinical Pharmacokinetics of Sirolimus. Clin Pharmacokinet 40, 573–585 (2001). https://doi.org/10.2165/00003088-200140080-00002
Published:
Issue Date:
DOI: https://doi.org/10.2165/00003088-200140080-00002