
Abstract
Human safety issues arise throughout the life cycle of pharmaceutical products and relevant information comes from a multitude of sources. Assessment and management of risks to humans requires a problem-based analysis to bring together relevant information regardless of source. The Safety Evaluation Plan (SEP) is a tool to support problem-oriented safety analysis. Safety issues are specified and the evaluation and management of each problem is based on a status summary that integrates the most current information from all relevant sources. The status summary is updated regularly during the course of clinical development to reflect the results of new studies and new clinical trials. In the postmarketing period, relevant postmarketing data is incorporated. Recent regulatory initiatives emphasise early identification of product safety risks so that appropriate risk-management measures can be instituted at the time of approval. A problem-oriented approach supports growing regulatory expectations regarding risk assessment and risk management. The problem-oriented approach facilitates early identification of safety issues and an evidence-based approach to their evaluation. Proactive management of safety problems leads to prompt assessment of risks and timely and appropriate steps aimed at risk reduction. The SEP provides a single global assessment for each safety issue. Regulatory submissions for pharmaceutical and biological products are organised by type of information. International Conference of Harmonisation documents covering clinical safety issues structure and analyse information separately by type, for example, adverse events, serious adverse events, laboratory data, vital signs, etc. A problem-oriented analysis would need to find a place in the regulatory process. A problem-oriented approach to safety cuts across typical structures in the pharmaceutical industry where different groups handle preclinical, clinical and postmarketing safety information. The SEP can improve communication within the company and externally. Nonetheless, supporting structures need to be adapted to support such an interdisciplinary process. Overall, the problem-oriented approach, supported by a SEP, contributes to realistic expectations and sustained credibility when dealing with safety issues.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1. A Problem-Oriented Approach to Human Safety Issues
Human safety issues arise throughout the life cycle of pharmaceutical products, and information relevant to understanding these issues comes from a multitude of sources. The assessment and management of risks to humans requires a problem-based analysis to bring together relevant information regardless of source.
Unfortunately, neither the organisation of product development within most pharmaceutical or biotechnology companies nor the structure of key regulatory documents supports a problem-based approach to safety. The purpose of this paper is to highlight the need for such an approach. A tool for use by sponsors that encourages such thinking during the prolonged and complex process of developing and marketing a therapeutic product is suggested.
How can such problem-oriented safety analysis be incorporated in marketing authorisation applications and related documents? With increased awareness of the social and public health costs of adverse events related to medical treatment, risk assessment and risk management plans (RMPs) are at the forefront of regulatory initiatives.[1] This is a timely moment to incorporate a problem-oriented approach to support these efforts and thus encourage effective collaboration between sponsors and regulatory agencies.
2. The Safety Evaluation Plan (SEP): A Tool to Support Problem-Oriented Analysis
A safety evaluation plan (SEP) is a tool designed to delineate the human safety issues under evaluation and brings together material from various sources to define specific safety issues. The SEP as described here is a document maintained by the sponsor to provide a summary and overview of the most important safety issues that have emerged in a clinical development project. It serves as an indexed executive summary of safety concerns under surveillance. This article also discusses the interface between the SEP and other important documents, including the investigator brochure (IB), core safety information and sections of the registration dossier.
3. Safety Information Comes from Many Sources
A New Drug Application (NDA) may consist of as many as 100 000 pages, with millions of datapoints, collected over the course of a number of years. Scores of people contributing different expertise and working in different departments, buildings, companies, cities and countries ultimately produce an application for a licence to market a medicinal product.Footnote 1 After submission, regulatory reviewers, again representing many different disciplines, are assigned to focus on subsets of the massive document.
Major components of the submission dealing with quality, preclinical and efficacy data (which includes clinical safety) are compiled separately. To render the mass of data digestible, integrated summaries of clinical efficacy and safety are prepared. This critical process is one of the last in the preparation of the dossier since it can be finished only when the last clinical study report is complete. Thus, the full safety profile of the product materialises at the last stages of the filing process. Ideally, the reverse should be true: information from successive studies should confirm and enhance what is already known or suspected. For this to happen, new processes need to be introduced to track safety problems throughout drug development.
Recent US FDA efforts emphasise early clarification of product safety risks[2] so that appropriate risk management measures can be instituted at the time of approval.[3] This places the onus on the industry sponsors to better characterise safety issues during development.[4,5] Novel approaches to monitoring safety issues are needed to address these expectations.
4. The Weed Method
Interestingly, clinical medicine offers an analogous problem to that encountered in drug development and suggests a parallel solution. Care of chronically ill patients generates numerous data, collected by many individuals over years or even decades. The medical records of a patient may be divided among different institutions. Even in one institution, it may run to hundreds of pages, representing encounters in various wards and outpatient departments. Data are collected and structured by discipline (e.g. doctor’s progress notes, nurse’s notes) or information type (e.g. clinical notes, laboratory data, medication records, x-ray reports) and are generally ordered temporally. Different individuals representing different disciplines enter information into the record. Add chronically high personnel turnover to the mix, and you end up with the well known ‘discontinuity of care’, with important information buried and forgotten.
In 1970, Lawrence Weed developed an integrated approach to patient records: a problem-oriented, rather than information-oriented, approach to the medical record, now known as the Weed Method.[6,7] Weed recommended a fundamentally different organisation of physician’s admission and progress notes.
A standard medical note lists the chief complaint, the patient’s report of symptoms and findings on physical examination, followed by a summary of pertinent laboratory results, radiological studies and other tests. The medical note then concludes with diagnostic impressions and treatment recommendations. Weed instead recommends that physicians structure their progress notes not by information type but rather by medical problem, with the relevant information from each source arrayed under the corresponding problem. Each problem would include subjective symptoms and objective information from the physical examination and from tests. The assessment, and the plan for managing that problem, follows. The acronym applied to the process was ‘SOAPing’ the medical record (subjective, objective, assessment, plan).
For example, a patient who was admitted for pneumonia might also have diabetes mellitus, glaucoma and a urinary tract infection. Using Weed’s approach, the patient’s respiratory symptoms, chest auscultatory findings, chest x-rays and sputum bacteriology, and response to prescribed antibiotic would be ordered under the problem ‘pneumonia’. Under ‘diabetes’, the medical note would list appropriate symptoms along with laboratory tests, such as blood and urine glucose measurements and response to treatment. Glaucoma and urinary tract infection would be handled in a similar fashion. The problem list would be carried through to future admissions.
This strategy translates well to a new approach to analysing data collected during the drug development process. Sponsors can identify a safety issue, give it a name, summarise the status, evaluate the problem and propose a plan. Using an approach like Weed’s, they can then organise information by problem rather than exclusively by information type.
5. Structure of the SEP
The SEP requires that problems be specified. Key information is summarised for each problem, an evaluation is made and a corresponding plan of action is defined. By identifying a safety issue, the sponsor commits to collect sufficient information to determine the nature of the risk and to ensure that it is appropriately managed. Safety issues should not be listed lightly, but the process of clarifying an issue demonstrates the sponsor’s exercise of due diligence.
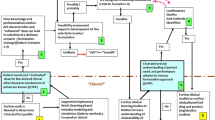
The SEP starts with a ‘problem name’ list. A section on ‘status’ summarises and references key findings, positive or negative, as of that point in development, for each respective problem. A section on evaluation, associated with each problem, includes the sponsor’s assessment of the risk as well as its implications. Finally, a plan to elucidate or manage the problem is presented, which may include additional studies or, if warranted, specific measures to reduce risk. Figure 1 demonstrates the basic structure of the SEP.

Components of the safety evaluation plan.
5.1 Problem Name
Choosing the correct problem name will help delineate the nature and scope of the issue while accurately reflecting the current information status. For example, if animal studies show human ether-a-go-go-related gene (HERG) channel abnormalities, the problem might be named ‘HERG channel abnormalities’, not ‘torsade de pointes’. If phase I studies show QT interval changes, the problem heading might be updated to ‘QT prolongation’.[8]
Potential risks identified at the beginning of clinical development, based on theoretical concerns and those suggested by preclinical studies, also correspond to named safety problems. Thus, the problem list includes findings from toxicology studies and safety pharmacology studies that represent potential risks to humans. A further set of problems may reflect theoretical concerns, for example the possibility of autoimmune disease, perhaps manifesting as vitiligo in cancer vaccines aimed at melanoma. For a biological product such as a monoclonal antibody, the potential risks may derive from the target receptors found on nontarget tissues. Alternatively, problems may reflect adverse events that are considered characteristics of the drug class, for example cough with a new ACE inhibitor.
The sponsor may anticipate other safety issues based on the population to be treated. For example, a large proportion of patients with early multiple sclerosis are women of childbearing age. In such a population, pregnancy-related concerns might be addressed throughout the entire development process, for example in determining what contraceptives can be used safely. Similarly, a potential for drug interactions or synergistic toxicity with commonly used concomitant medications might be designated a potential safety issue. Early identification of such safety issues leads to proactive study, assessment and management. Once there is experience in humans, observations made in clinical studies can identify new safety signals. An example of a problem list is shown in table I.

Hypothetical safety issue list for an angiogenic cytokine at the end of phase I studies[9]a
5.2 Problem Status
For each problem, a status summary reflects available information. The ‘problem status’ section is dynamic, with information added as new studies are completed. This section offers a synopsis of the information on the problem, available up to the current point in product development. The status section should include pertinent positive and negative findings.
For problems based on theoretical concerns or information from other products, a brief (referenced) summary of the current literature or other product information is presented. Information is added as it becomes available from toxicology and safety pharmacology studies. For example, in the list of problems shown in table I the status section for ‘promotion of malignancy’ might point out that this problem is listed based on theoretical concerns and some experimental models using other angiogenic agents. It might point out that toxicology studies with this agent have been negative and that no new malignancies have been reported in human studies to date. Appropriate citations would support these statements.
Clinical studies provide preliminary human data for each problem. Relevant findings from phase I studies are parsed over the existing safety issue list. If, for example, ‘gastroenteritis’ had been identified as an issue based on preclinical data from repeat dose studies of a new chemical entity in dogs, gastrointestinal symptoms reported by human volunteers would be summarised by their nature, severity, outcome and frequency. If 20% of subjects in a phase I repeat dose study of the drug reported flatulence and mild diarrhoea without abdominal pain, the problem status section would note this with a reference to the study report. This might also alert the sponsor to a potential tolerability issue. On the other hand, if no human volunteers reported any gastrointestinal adverse effects, this would also be mentioned in the problem status section.
Alternatively, signals coming out of phase I studies may lead to the addition of completely new safety issues for the problem list. For example, if unexplained ventricular extra-systoles developed in a presumed healthy volunteer, this would be added to the problem list. Phase I studies also provide information on symptoms such as headache or complaints about the bad taste of an oral product. Even though they are not major safety issues, such problems may be relevant to product tolerability and may need to be managed. As phase II studies are completed, the status of each problem is updated to incorporate data on more patients and a range of doses.
When information is available from phase III trials, the problem status for each issue should emphasise the experience most relevant to the formulation and mode of administration of the product to be marketed. By this time, the sponsor should have a good idea of the characteristics (nature, severity, frequency and outcome) of each problem and have formed some conclusions about its implications.
The accrual of information about a product does not end at the time of the first marketing approval. Information will continue to be added as a result of routine postmarketing surveillance, as summarised in periodic safety update reports (PSURs),[10] as well as through any additional postmarketing commitments. Significant new information from the marketed product experience is incorporated under the appropriate problem status section of the SEP.
In summary, the problem status section for each safety issue summarises key positive and negative findings as they accrue throughout the product life cycle. The information in the problem status section may begin with theoretical concerns and preclinical data, but will have human experience from clinical trials and ultimately data from postmarketing experience added. Figure 2 highlights the many sources of problem status information.

The many sources of problem status information in a safety evaluation plan.
5.3 Problem Evaluation
For each problem, an ‘evaluation’ follows the status section. The evaluation has two components: (i) an assessment of how well a risk is understood; and (ii) an analysis of the implications of the risk.
5.3.1 Risk Assessment
Risk assessment considers the amount and quality of the information presented in the status section. Is the purported risk well characterised? How complete is the information from key studies? Are there major sources of bias? Is information from various sources contradictory? Will it be possible to get better information or is the issue inherently difficult to clarify? For example, if there is a theoretical risk of promoting growth of malignant tumours, but no such tumours have yet been seen, what can be concluded? Was follow-up of individual patients long enough? Was there a systematic method of identifying new tumours? Based on this information, how large a risk can be excluded? A critical evaluation of current information clarifies the limits of current understanding and thereby suggests the steps needed to assess the risk adequately.
5.3.2 Risk Implications
Safety issues range from the well understood and readily manageable to the unanticipated and fatal. Steps to assess and manage a given issue need to be commensurate with the magnitude and urgency of the identified risk and should be appropriate to the indication under study.
A safety issue with a potentially life-threatening or fatal outcome urgently demands a full assessment and appropriate action. Still, such a risk may be acceptable in the context of untreatable life-threatening diseases. For example, T-cell leukaemia developed in two children who had received gene therapy using a retrovirus vector for X-linked severe combined immunodeficiency (SCID).[11] Although clinical holds were imposed on these trials after the second case of leukaemia, the studies were ultimately restarted. The conclusion of the FDA Biological Response Modifier Advisory Committee (BRMAC) was that “the benefit of gene therapy over existing treatments for children with X-linked SCID who do not have an HLA-identical bone marrow donor is significant, and provides an impetus for allowing retroviral vector mediated clinical trials in subjects with SCID to proceed”.[12] In the example given above, there was strong evidence of benefit, albeit in the preapproval setting. A more difficult problem emerges when the risks are evident before there is empirical evidence of a benefit. In the above example, had the leukaemia cases occurred in the first SCID patients to be treated, before there was strong evidence of efficacy, the decision to proceed with the studies would have been more difficult.
At the other end of the spectrum are safety issues that may not constitute major risks to individual patients but that are important to the clinical development of a product. Flu-like symptoms occur in most patients treated with interferons. These symptoms can affect patient compliance and they make it impossible to blind a study adequately. Thus, severity is not the only consideration determining the implications of a risk. The risk implication section of the problem evaluation might consider whether a risk, if confirmed, is acceptable for the indication under development. Emerging risks may make the benefit-risk ratio unacceptable. Alternatively, they may be unacceptable within a specific population. For example, relapsing-remitting multiple sclerosis often afflicts young women, who in the early stages of the disease are living normal lives with normal expectations. Significant cosmetic problems such as severe acne or skin discolouration might well be unacceptable to this group of patients, particularly if treatment is administered to slow disease progression. The same issues may be of less concern for patients with secondary progressive multiple sclerosis who are already experiencing considerable disability and a diminished quality of life. Early recognition of such issues and clear definitions of decision points are essential for both clinical safety and decision making regarding product development.
The risk implication section may conclude that simple labelling will be sufficient to manage the risk, particularly for safety issues that are anticipated consequences of a product’s mode of action (e.g. hypoglycaemia with insulin) or those that are characteristic of a product class, such as cough for ACE inhibitors. The evaluation of such issues may well conclude that they can be managed through appropriate labelling.
However, writing off anticipated safety issues without sufficient evidence may be risky. Even if a safety issue is characteristic of a drug class, it may be atypically severe or frequent in a given product. Cerivastatin, like other HMG CoA reductase inhibitors, was labelled for rhabdomyolysis. What turned out to be different was the frequency and severity: fatal rhabdomyolysis occurred 16–80 times more often with cerivastatin than with other drugs in this class.[13]
The reverse may also be the case. Greater tolerability or a lower frequency of an adverse effect that is typical of a drug class may be an important part of its competitive product profile, either vis-à-vis another product group (e.g. gastrointestinal bleeding with cyclo-oxygenase-2 inhibitors compared with conventional NSAIDs[14]) or within its class (e.g. sexual dysfunction amongst atypical antipsychotics[15]). If such considerations will be important to a product’s acceptance in the market, evaluation of the risk may show that additional data are needed to support claims for a more favourable safety profile.
Once a product is in phase III, there are important decisions regarding what, if any, safety issues are likely to require additional measures, beyond standard postmarketing surveillance once marketed. Based on this assessment, the sponsor will consider developing an RMP with proposals for additional studies in the perimarketing period.
In summary, the thoughtful evaluation of each safety issue starting at an early stage in product development and reconsidered periodically to reflect accruing information is the key to managing product safety during clinical development. This evaluation must take into account the level of knowledge about the risk, as well as appreciation of its implications for patient well-being and the population to be treated.
5.4 Problem Plan
The plan for managing a problem should derive from the evaluation. Urgent issues may be the subject of ‘stopping rules’, particularly in early studies. For example, if a study of cynomolgus monkeys showed a small number of animals with severe neutropenia, the problem plan might anticipate that in early studies absolute neutropenia might require a stopping rule. Such a plan would have substantial implications for study conduct and timelines. The specifics of how such a rule might be implemented would go beyond the scope of the SEP.
Certain issues have such significance that a crisis management plan is necessary. This is especially likely when large numbers of patients are exposed, for example with marketed products or when there is a latent period associated with the risk, such as carcinogenicity concerns. The vast majority of safety issues do not have such urgency; nonetheless, the timeline for resolution of the issue is important and will depend on a plethora of different factors, some clinical, some regulatory and some business related.
Where there is uncertainty about risk, the plan suggests additional steps to be undertaken, for example repetition of a questionable preclinical study or incorporation of specialised clinical assessments such as regular retinal examinations or Holter monitoring into clinical protocols. The plan might also propose an ‘exit strategy’ so that such surveillance measures do not necessarily become a part of the product label.
Should the evaluation section have identified significant potential risks that labelling alone cannot handle, the plan should include a commitment to a specific postmarketing RMP. The major strategic elements of that commitment should be defined in the plan, for example the intention to undertake a large, simple safety study or to institute a pregnancy registry.
The SEP may also highlight issues for which a communication plan is of special importance. A communication plan can ensure that a suitable and consistent message about a safety issue is presented to different regulatory authorities, health professionals and the public.
6. Investigator Brochure
When the IB is written and updated, the information presented should be consistent with the SEP. Entering phase I studies, since there is no human experience with the study treatment, the risks outlined in the IB will be based on theoretical concerns and preclinical information. In subsequent IB updates, the problems listed should be updated with new information corresponding to the SEP. The risk assessment of specific problems will also guide decisions as to which adverse experiences related to treatment can be clearly classified as ‘expected’. As problems go from potential risks to well documented and well characterised adverse drug reactions, this should be reflected in the IB section on expected undesirable effects of the treatment. Changes in the informed consent will also need to be conducted in association with this process.
7. Core Safety Information
The SEP, IB and core safety information are interrelated documents. Key elements of the IB and core safety information evolve directly from the SEP, as does the decision to undertake an RMP (figure 3). Much of the core safety information will also develop as a natural output of the safety issue list and the associated assessments. Decisions can be made prospectively about issues that will need special attention, including those that have the potential to become contraindications, warnings or precautions.

Key safety documents derived from the safety evaluation plan.
8. Managing Safety for Marketed Products
8.1 Risk Assessment for Marketed Products: Pharmacovigilance and Pharmacoepidemiology
Even if the overall benefit-risk ratio is sufficient to support registration, there are invariably open issues that need to be resolved. The FDA describes premarketing risk assessment as “the process of identifying, estimating and evaluating the nature and severity of risk from a product”, with the additional observation that “good risk assessment underlies good risk management and pharmacovigilance”.[16]
The process of producing and updating the SEP defines the status of various ongoing issues at the time the regulatory dossier is ready for submission. For most safety issues, information continues to evolve after registration and routine postmarketing surveillance (i.e. spontaneous reporting plus monitoring of ongoing clinical research and the scientific literature) will be sufficient. Some issues need more proactive steps. For example, suspected rare but serious adverse drug reactions (e.g. organ failure) may need to be monitored through special steps, such as studies of transplant waiting lists. The safety of the product in the very old or the very young may require additional assessment. The impact of concomitant therapies that may have been excluded during clinical development may need to be addressed after marketing. In these settings, problem-oriented risk assessment efforts may continue during the postmarketing period. A targeted approach may be called for to collect specific missing information.[17] The sponsor’s risk assessment strategy should include the use of appropriate tools such as active surveillance, postmarketing clinical trials, epidemiological studies or registries to provide the needed additional information. The plan section of the SEP presents the chosen strategy. A specific tool may address a number of problems. For example, a prospective epidemiological cohort study may provide information relevant to a variety of problems. On the other hand, a case control study of acute hepatic failure focuses on that single safety problem.
9. Risk-Management Plan
An RMP, as defined by the FDA,[18] is a strategic safety effort to reduce risk that sets at least one risk reduction goal and that uses an intervention tool other than the package insert. An RMP can be introduced at any time in the product life cycle.
For issues that have been under review as part of the SEP, the need for an RMP should be no surprise. The sponsor will have explored various options and should be prepared to propose a sound RMP at filing. In addition to the steps recommended to reduce risk, the RMP should incorporate concrete measures to demonstrate the effectiveness of the proposed actions.
Alternatively, if contacts with regulatory authorities have suggested an area of concern, prior planning can help the sponsor present data and analyses that put off the need for a formal RMP. Tracking the issue through the SEP ensures it receives appropriate attention.
9.1 Fostering Communication
Regardless of the stage of drug development, the SEP serves to improve communication within the company and with important external stakeholders. Within the company, it provides a longitudinal record of safety issues associated with a product. This record is particularly valuable when there is a change of personnel and/or responsibilities within the organisation.
Table II lists four elements considered important to constructive interactions between sponsors and the US FDA.[19] Communications based on an up-to-date SEP can keep regulatory agencies abreast of which safety issues the sponsor is tracking. This ensures clarity about which concerns both parties consider important. In the status section, the sponsor summarises what it considers the key relevant information. By making this specific, there can be a discussion of the value and interpretation of specific types of evidence. In the problem evaluation section, the sponsor articulates an assessment of the importance and implications of the issue. If this perspective is different from that of the regulator, those differences will be apparent early on and will stimulate appropriate steps towards a mutually agreed plan to resolve these differences. The sponsor explicitly commits to steps to protect patients in ongoing studies and may describe plans to clarify specific safety issues during subsequent steps in the drug development process.

The safety evaluation plan (SEP) facilitates communication between sponsors and regulatory agencies
Communications based on the SEP facilitate consistent messages on risks to all involved regulatory agencies. This is particularly important in complex organisations with different regional units that provide information to local regulatory agencies. The SEP provides a single global assessment for each safety issue.
9.2 Problem-Oriented Analyses and Key Regulatory Documents
International Conference of Harmonisation (ICH) documents covering clinical safety do not support integrated evaluations of a safety issue since information is primarily presented and analysed by type. Specific summary sections present conclusions and propose actions (i.e. with regard to labelling), but the documents do not ask that the logic behind these critical recommendations be articulated.
9.2.1 International Conference of Harmonisation (ICH) E3: Structure and Content of the Clinical Study Report
The safety evaluation section of the E3, the ICH document on the Clinical Study Report, segregates discussion of different types of safety information, as shown in the following excerpt on safety evaluation:[20] “The more common adverse events, laboratory test changes etc. should be identified, classified in some reasonable way, compared for treatment groups, and analysed, as appropriate, for factors that may affect the frequency of adverse reactions/events, such as time dependence, relation to demographic characteristics, relation to dose or drug concentration etc. Finally, serious adverse events and other significant adverse events should be identified, usually by close examination of patients who left the study prematurely because of an adverse event, whether or not identified as drug related, or who died”.
Although it would seem self-evident that information from adverse events, serious adverse events, vital signs and laboratory tests would be organised somewhere by safety issue, this is not a part of the Clinical Study Report format. Even in the ‘Safety Conclusions’ section, there is no expectation that information be integrated or evaluated by safety issue even though the study findings are to be evaluated to determine their clinical implications.
9.2.2 Common Technical Documents
The Common Technical Documents (CTDs) guide data presentation to aid the submission of the same registration material in each ICH region. The organisation of the CTDs[21–23] is principally by type of information: quality, preclinical, clinical. Clinical safety is discussed within the ICH ME4 Efficacy module. In that document, section 2.5.5., entitled ‘Overview of Safety’, allows for a discussion of information from various sources, but as table III shows, this information is by type and is intended to be the basis for prescribing information. Although the instructions are to provide “concise critical analysis of the safety data, noting how results support and justify proposed prescribing information”,[23] the data are presented by type and the section format is an introduction into the core safety information: there is no place for a problem-oriented discussion in this section.

Examples of the clinical safety information presented in the International Conference of Harmonisation ME4 Efficacy module of the Common Technical Document, section 2.5.5: Overview of Safety
Within the clinical Efficacy module, material from individual clinical study reports is integrated across studies (section 2.7.4), but the structure essentially duplicates that of the Clinical Study Report. Data from the specific sections of several key studies are presented and the findings are compared. Again, there is no place to bring together information on particular safety issues. Although one can use the overall safety summary section of the CTD to list specific problem risk assessments, neither the CTD nor its component documents request that the sponsor provide an integrated presentation of the relevant information that led the assessment.
9.2.3 ICH E2E: Pharmacovigilance Specification and Pharmacovigilance Plan
In Osaka, Japan, the ICH recently brought the E2E document on pharmacovigilance planning to step 2.[17] The E2E document develops the concept of a ‘Pharmacovigilance Specification’ and ‘Pharmacovigilance Plan’ to be submitted at the time of licence application. As stated in this document, “The Pharmacovigilance Specification is a summary of the identified risks of a drug, the potential for important unidentified risks, the populations potentially at-risk and situations that have not been adequately studied”.[17] So described, the Pharmacovigilance Specification would be a logical output of the SEP. The E2E document emphasises the need to include relevant nonclinical and clinical information in the Pharmacovigilance Specification. It also requires assessment and evaluation, with an explicit definition of specific data needs (‘missing data’). It points out that postmarketing risk assessment tools, such as large simple safety studies and epidemiological investigations, may provide data on a variety of safety issues. This developing document provides an opportunity to explicitly further an integrated, problem-oriented analysis and presentation of safety issues. It would be important to add a specific component to this document that permits the sort of problem-oriented analysis that has been the focus of this article.
9.3 Introducing Problem-Oriented Safety Analysis within a Company
9.3.1 Responsibility for the SEP
While the utility of a problem-oriented approach to safety issues is inherently appealing to anyone involved in managing risk, it cuts across typical structures in the pharmaceutical industry where different groups handle preclinical, clinical and postmarketing safety information. Construction and maintenance of the SEP require input and support from all these groups; however, unless responsibility for the SEP is clear it will not be maintained as a viable document.
The introduction of an SEP is likely to lead to a variety of organisational questions. For example, who should be responsible for maintaining the SEP? The answer depends on organisational structures within a company. Pharmacovigilance or risk management groups, if they have responsibility for safety risks from early in the clinical development process, are appropriate owners. Alternatively, it may be the responsibility of the clinical research group until commercialisation and then be assumed by the group responsible for postmarketing safety. The advantage of having the SEP in the hands of the pharmacovigilance organisation is to maintain ownership throughout the life of the product. Furthermore, global organisation is more likely in pharmacovigilance than in other departments.
Since the judgements made in developing the SEP may affect patient safety within clinical development, the person ultimately responsible for the SEP should be medically qualified. However, this person must be trained in the problem-oriented approach, since the SEP requires a rather different slant to information than most current approaches. The SEP requires that preclinical information be assessed to identify human risks. The process of identifying those risks requires input from the toxicology and safety pharmacology groups as well as those dealing with clinical development and risk management. The person responsible for developing the initial SEP will need the support of all those parties. Early identification of risks and evaluation of their long-term implications for product development at this stage can be extremely edifying to all team participants and decision makers.
The very essence of an SEP is the reallocation of relevant safety information from documents based on information type to an integrated problem-oriented structure. The SEP owner must integrate information from various information sources. At the same time, each department contributing information should concur that the information is appropriately interpreted within the SEP. The process of preparing the SEP facilitates interdisciplinary communication.
The sponsorship and support of senior management are critical to the successful introduction of the concept of problem-oriented safety evaluation and the institution of SEPs. Since the creation and maintenance of the SEP require resources as well as cooperation across organisational units within the company, appropriate systems and procedures must be created to support it. Management needs to provide adequate resources to develop and maintain an SEP, at least for designated products.
Most companies have procedures in place for the identification and evaluation of safety signals. Documents created to support the introduction of SEPs should be coordinated with those existing standard operating procedures. In addition, they should be integrated with procedures for creating core safety information, establishing RMPs and implementing crisis management.
Since safety information assessment and the corresponding action plans require the agreement and coordination of activities by a number of disciplines, the updated SEP needs to be a commitment among all concerned parties. Those functions that are responsible for executing the actions defined in the plan must support the conclusions of the SEP. Thus, the relevant product or project team functions may wish to sign off on SEP updates.
9.3.2 Technical Support
The SEP can be maintained as a document but, preferably, it should be supported by an appropriate information system. A database built on specified safety problems that can cross reference specific project documents and that is related to the existing document management system ensures that the SEP is a living document, rather than a bureaucratic encumbrance.
9.3.3 Some Caveats
Although the SEP supports the management of known or potential safety issues, it carries with it some risks. The SEP adds transparency to risk assessment and risk management. Although these evaluations, and the process of evaluation itself, are evidence of due diligence, it is essential that the assessments in the SEP be appropriate to the current information and understanding of the problem. Moreover, the sponsor must take commitments made in the plan seriously. The plan should not commit to actions that will not be possible. It is better to formulate commitments cautiously, including feasibility studies, than to make a commitment that will not be funded or is not possible because of time or population constraints. For example, if an epidemiological study is under consideration to assess a possible risk, the action plan should include an evaluation of the feasibility of such a study. Is there a suitable database? Are there sufficient numbers of patients to give the study sufficient power? The managers of the SEP should not commit to a specific risk assessment or RMP without assurance that the programme itself is realistic and there are resources available to support it.
10. Conclusion
Human safety issues associated with the development of drug therapies should be managed via a problem-oriented approach. This method supports evolving regulatory expectations regarding risk assessment and risk management. The problem-oriented approach facilitates early identification of safety issues and an evidence-based approach to their evaluation. Proactive management of safety problems leads to a prompt assessment of risks and timely and appropriate steps aimed at risk reduction.
The SEP is a tool to support problem-oriented safety analysis. Safety issues are specified and the evaluation and management of each problem is based on a status summary integrating information from all relevant sources. The SEP can improve communication within the company and externally. It supports a consistent message for the sponsor’s communications globally. Overall, the problem-oriented approach, supported by an SEP, contributes to realistic expectations and sustained credibility when dealing with safety issues.
Notes
Although this article focuses on pharmaceutical and biotechnology products, the same considerations apply to medical devices.
References
Kohn L, Corrigan J, Donaldson M, editors. To err is human: building a safer health system. Washington, DC: Committee on Quality of Health Care in America, Institute of Medicine, National Academy Press, 1999
FDA concept paper: premarketing risk assessment [online]. Available from URL: http://www.fda.gov/cder/meeting/riskManageI.htm [Accessed 2003 Mar 3]
FDA concept paper: risk management programs [online]. Available from URL: http://www.fda.gov/cder/meeting/riskManageII.htm [Accessed 2003 Mar 3]
FDA concept paper: risk assessment of observational data: good pharmacovigilance practices and pharmacoepidemiologic assessment [online]. Available from URL: http://www.fda.gov/cder/meeting/riskManageIII.htm [Accessed 2003 Mar 3]
Heads of Agencies Working Group report. Establishing a European risk management strategy: summary report of the Heads of Agencies Ad Hoc Working Group. 2003 Jan
Weed LL. Medical records, medical education, and patient care. Chicago; Year Book Medical Publishers Inc., 1969
Weed LL. Medical records that guide and teach. N Engl J Med 1968; 278(12): 652–7
Roden DM. Drug therapy: drug-induced prolongation of the QT interval. N Engl J Med 2004 Mar; 350(10): 1013–22
Isner JM, Vale PR, Symes JF, et al. Assessment of risks associated with cardiovascular gene therapy in human subjects. Circ Res 2001; 89: 389–400
ICH harmonized tripartite guideline: clinical safety data management. Periodic Safety Update Reports for marketed products E2C. Recommended for adoption at step 4 of the ICH process, 6 Nov, 1996 [online]. Available from URL: http://www.ich.org/MediaServer.jser?.@_ID=477&@_TYPE=MULTIMEDIA&@_TEMPLATE=616&@_MODE=GLB [Accessed 7 May 2004]
McCormack MP, Rabbitts TH. Mechanisms of disease: activation of the T-cell oncogene LMO2 after gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 2004 Feb; 350(9): 913–22
US FDA. BRMAC #34, topic III: briefing document [online]. Available from URL: http://www.fda.gov/ohrms/dockets/ac/03/briefing/3924B2_1.pdf [Accessed 2004 May 3]
Davidson MH. Controversy surrounding the safety of cerivastatin. Expert Opin Drug Saf 2002 Sep; 1(3): 207–12
Silverstein FE, Faich G, Goldstein JL, et al. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study. A randomized controlled trial: Celecoxib Long-term Arthritis Safety Study. JAMA 2000 Sep 13; 284(10): 1247–55
Knegtering R, Castelein S, Bous H, et al. A randomized open-label study of the impact of quetiapine versus risperidone on sexual functioning. J Clin Psychopharmacol 2004 Feb; 24: 56–61
FDA Center for Drug Evaluation and Research. CDER/CBER risk management public workshop [online]. Available from URL: http://www.fda.gov/cder/meeting/riskManagement.htm [Accessed 2004 May 1]
ICH draft consensus guideline: pharmacovigilance planning (PvP) E2E. Released for consultation at step 2 of the ICH process on 11 November 2003 by the ICH Steering Committee [online]. Available from URL: http://www.ich.org/UrlGrp-Server.jser?.@_ID=276&@_TEMPLATE=254 [Accessed 2004 May 7]
Seligman PJ. Safety issues: regulatory update. Temple University School of Pharmacy/Industry Workshop, 2004 April 20 [online]. Available from URL: http://www.fda.gov/cder/Offices/OPaSS/safetyIssues_files/frame.htm [Accessed 2004 May 7]
Vaillancourt JM, Wallman L. Improve agency/industry communication throughout the drug development process. The 5th Joint Project Management Workshop, Drug Information Association (DIA) and FDA; Bethesda (MD); 2004 May 11-13
ICH Harmonized Tripartate Guidelines. Structure and content of clinical study reports (E3). Recommended adoption at step 4 of the ICH process on 30 Nov, 1995 [online]. Available from URL: http://www.ich.org/MediaServer.jser?.@_ID=479&@_MODE=GLB [Accessed 2004 May 7]
ICH harmonized tripartite guideline: the common technical document for the registration of pharmaceuticals for human use. Quality-M4Q. Quality overall summary of module 2, module 3: quality [online]. Available from URL: http://www.ich.org/MediaServer.jser?.@_ID=556&@_MODE=GLB [Accessed 2004 May 7]
ICH harmonized tripartite guideline: the common technical document for the registration of pharmaceuticals for human use. Safety-M4S. Non-clinical overview and nonclinical summaries of module 2, organization of module 4 [online]. Available from URL: http://www.ich.org/MediaServer.jser?.@_ID=559&@_MODE=GLB [Accessed 2004 May 7]
ICH harmonized tripartite guideline: the common technical document for the registration of pharmaceuticals for human use. Efficacy -M4E. Clinical overview and clinical summary of module 2, module 5: clinical study reports [online]. Available from URL: http://www.ich.org/MediaServer.jser?.@_ID=561&@_MODE=GLB [Accessed 2004 May 7]
Acknowledgements
No sources of funding were used to assist in the preparation of this article. The author has no conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haas, J.F. A Problem-Oriented Approach to Safety Issues in Drug Development and Beyond. Drug-Safety 27, 555–567 (2004). https://doi.org/10.2165/00002018-200427080-00007
Published:
Issue Date:
DOI: https://doi.org/10.2165/00002018-200427080-00007