
Abstract
This study was to evaluate the response of macrophages to culture with dicalcium silicate nanoparticles (nC2S) and the effect of the nC2S/macrophage-regulated immune environment on osteogenesis directly. Polarity and cytokine of macrophage expression were evaluated. Variations in intracellular homeostasis were assessed by measuring the production of ROS and autophagy. The effect of nC2S on osteogenesis was evaluated by micro-CT, bone immunofluorescence labeling, and immunohistochemical analyses in vivo. The effect of the nC2S/macrophage-regulated immune environment on osteogenesis was evaluated by ALP staining, Alizarin Red solution staining, osteogenesis-related protein expression assessment. The detection results showed that nC2S polarized macrophages toward the M1 phenotype that promoted degradation of P62 and increased ROS production. Besides, nC2S exhibited a good ability to repair bone defects in vivo. The nC2S/macrophage-regulated immune environment promoted osteogenesis. These findings support that nC2S have good biocompatibility and the nC2S/macrophage-regulated environment enhance the osteogenic ability of bone mesenchymal stem cells (BMSCs).
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
β-Dicalcium silicate (β-Ca2 SiO4, β-C2S) is one of the main constituents of Portland cement. The bioactivity and biocompatibility of dicalcium silicate have been reported. In vitro, a bone-like apatite layer covered the β-C2S cement surface after soaking in simulated body fluid (SBF), and its compressive strength was similar to that of human trabecular bone [1]. Dicalcium silicate (C2S) cement promotes bone regeneration in vivo [2]. Some studies have attempted to further modify dicalcium silicate to promote its biological activity. For example, β-dicalcium silicate cement was modified with a silane coupling agent to contain vaterite, which enhanced its biological activity [3]. α′(H))-Dicalcium silicate bone cement doped with tricalcium phosphate supported human adipose-derived stem cell attachment and extension. These novel cements have better properties that are relevant to biological materials, such as improved setting times and degradation rates [4]. α-Tricalcium phosphate cements modified with β-dicalcium silicate exhibited better properties, making them suitable candidates for use as bone substitutes [5]. These novel ceramics improve the proliferation and induce the osteoblastic differentiation of undifferentiated ahMSCs [6]. Dicalcium silicate cement can be used as a root-end filling material and root sealer because the associated cell attachment, cytotoxicity and antibacterial properties were comparable to those of ProRoot WMTA [7]. The cytotoxic effects of Portland cement on cultured human periodontal ligament fibroblasts is the lowest when compared to those exerted by MTA Fillapex and MTA [8]. In vivo, dicalcium silicate cement exhibits a greater ability to promote bone regeneration than WMTA. These results suggest the potential applications of radiopaque dicalcium silicate cement as a substitute for WMTA in endodontic field [9]. The subcutaneous tissue reactions to the implantation of white Portland cement and white mineral trioxide aggregate (MTA) were also investigated. Both white Portland cement and MTA are well tolerated by tissues. White MTA and white Portland cement seem to have comparable biocompatibility [10]. To evaluate the biocompatibility of dicalcium silicate better, the cytotoxic effects of dicalcium silicate particles on immune cells were examined. The toxic effects of a new endodontic cement, which was based on dicalcium and tricalcium silicate and contained hydroxyapatite, on human lymphocytes indicated low genotoxic risk [11]. In our previous research, we assessed the cell toxicity and proinflammatory impacts of dicalcium silicate particles and coatings on immune cells. The results indicated that dicalcium silicate particles and coating have no obvious cytotoxicity on THP-1 and RAW264.7 cells [12, 13]. These materials exert proinflammatory effects. Furthermore, dicalcium silicate induced proinflammatory responses through the TLR2-mediated NF-κB and JNK signaling pathways in RAW264.7 cells [14]. These studies focused on the biocompatibility of dicalcium silicate cements or ion extracts of dicalcium silicate coatings. Few studies have been focused on effects of dicalcium silicate nanoparticles (nC2S). A route for the synthesis of dicalcium silicate nanoparticles was reported [15]. Dong et al. reported that electrospun poly(l-lactic acid) (PLLA) scaffolds containing dicalcium silicate (C2S) nanoparticles (approximately 300 nm) promoted cell proliferation and osteogenic differentiation [16]. The response of immune cells to dicalcium silicate nanoparticles in the smaller size have not been reported. Nanoparticles can interact with the immune system, which may cause immunotoxicity or change the immune microenvironment [17]. It has also been reported that nanoparticles strongly promote the osteogenic differentiation of bone mesenchymal stem cells (BMSCs), and the underlying mechanism is the modulation of macrophages and the related osteoimmune environment by the nanoparticles [18]. The process of bone healing is characterized by three partially overlapping stages: the inflammatory stage, repair and remodeling stage [19]. The initial inflammatory stage is highly crucial to the process of eventual healing. M1 macrophages are predominant at the initial stages of acute inflammation that release proinflammatory cytokines to promote osteogenesis [20]. It has been revealed that M1 macrophages promote bone formation by receptor activation of NF-кB ligand (RANKL) [21]. Therefore, M1 polarization of macrophages is indispensable in the process of bone healing. The osteogenic effects of dicalcium silicate nanoparticles, their safety with respect to macrophages and their effect on the immune microenvironment are still not clearly understood. In the present study, we focused on the dicalcium silicate nanoparticles which the particle size is about 100 nm. The cytotoxic and osteogenic effects of nanoparticles and the macrophage response to dicalcium silicate nanoparticles were researched. Additionally, the effects of the nanoparticle/macrophage-regulated microenvironment on osteogenesis were also evaluated.
Results
Characterization of nC2S
The characteristics of nC2S were described in our previous studies. The XRD results indicated that the spectrum of the sample corresponded to Ca2SiO4. The corresponding peaks were consistent with the standard peaks. Using a nanoparticle size analyser, the particle size of nC2S was 100 nm approximately in diameter (d50 values, previously published). The SEM and EDS are showed in supplement Fig. 1. The endotoxin level of nC2S was below 0.05 EU/mL. The solubility of nC2S in extracellular fluid have been assessed in our previous study. After incubation for 24 h, nC2S gradually dissolved and the dissolution was elevated with increased concentration [22].

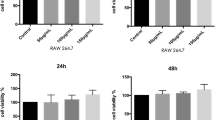
Proliferation and apoptosis of RAW 264.7 cells. (K) Graphical representation of normally cultured RAW264.7 cells (A) and positive control group which cultured with 0.64% phenol solution (B) or 10 μg/mL, 50 μg/mL, or 100 μg/mL nC2S (C, D, and E, respectively) for 24 h. (L) Graphical representations of normally cultured cells (F) and positive control group which cultured with 0.64% phenol solution (G) or 10 μg/mL, 50 μg/mL, or 100 μg/mL nC2S (H, I, and J, respectively) for 48 h. (M) and (N) show the proliferation rates of RAW264.7 cells after incubation for 24 h and 48 h, respectively. All the values are presented as the mean ± SD of triplicate experiments.
Cytotoxicity of nC2S
The cell proliferation rate was more than 100% 24 h after exposure. A more than 95% cell proliferation rate was observed 48 h after exposure (Fig. 1). In the apoptosis analysis, incubation with 10 μg/mL, 50 μg/mL, and 100 μg/mL nC2S increased the apoptosis rate at 24 h and 48 h, as determined by flow cytometry, but this difference had no statistical significance. The apoptosis rate ascended with the increasing concentrations of nC2S and reached the highest rate in 100 μg/mL. The apoptosis rate did not change obviously over time to 48 h compared with positive group (Fig. 1).
Polarization and inflammatory response of macrophages
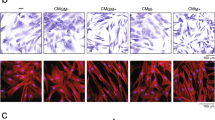
SEM and fluorescence microscopy analysis results indicated that a high concentration of nC2S changed the morphology of cells. Treatment with 100 μg/mL nC2S caused small pseudopodia extruding from the edges of cells. The cells in the control group were round, and the cells in the positive control group branched off and formed many pseudopods. The cells remained round at 10 μg/mL and 50 μg/mL groups, but the edges of the cells spread slightly (Figs. 2 and 3). The expression levels of the cell specific markers CD86 and CCR7 were increased after treatment with 50 μg/mL and 100 μg/mL nC2S. Treatment with 10 μg/mL nC2S did not change CD86 expression but increased CCR7 expression. The expression of the surface marker CD206 was decreased in the 10 μg/mL and 100 μg/mL nC2S groups, but its expression was not significantly changed in the 50 μg/mL nC2S group. CD163 expression was not significantly changed in any of the groups (Fig. 4). Thus, nC2S stimulate RAW264.7 cell polarization toward the M1 phenotype. The expression of TNF-α significantly increased after treatment with 50 and 100 μg/mL nC2S, while IL-1β expression was not significantly changed. The expression of the osteoclastic activity-related gene MCSF was obviously increased after exposure to 100 μg/mL nC2S. High concentrations of nC2S induced proinflammatory cytokine expression. Treatment with 50 and 100 μg/mL nC2S also significantly promoted the expression of the osteogenic factor TGF-β3 (Fig. 4).

SEM observation. (A) In the control group, the cells were round. (B) Positive control group, the cells treated with LPS. The cells developed pseudopods. (C) In the 10 μg/mL nC2S group, morphology of the cells was still round. (D) and (E) Indicated that when treated with 50 and 100 μg/mL nC2S, the cells protrude small pseudopods.

Immunofluorescence staining of RAW264.7 cells cultured with nC2S. (A) Control group. (B) Positive control group. Treatment with LPS caused the cells to swell and spread obviously. (C) The 10 μg/mL nC2S group, the cell was round. (D) and (E) Reveal treatment with 50 and 100 μg/mL nC2S caused the cells to protrude small pseudopods. Take pictures at × 40 magnification (scale bar = 75 μm).

mRNA expression of cell surface markers and cytokines. CD86 and CCR7 expression was increased in the 50 μg/mL and 100 μg/mL nC2S groups. CD206 expression was decreased in the 10 μg/mL and 100 μg/mL nC2S groups. CD163 expression was not significantly changed in any of the groups. TNF-α expression was significantly increased in the 50 and 100 μg/mL nC2S groups. IL-1 β expression was not significantly changed. TGF-β3 expression was significantly increased in the 50 and 100 μg/mL nC2S groups. MCSF expression was obviously increased only in the 100 μg/mL nC2S group. The data are shown as the mean ± SD. *P < 0.05, **P < 0.01 and ***P < 0.001.
Changes in intracellular homeostasis
The expression of the LC3I/II and beclin-1 was not significantly changed after 6 h of incubation with nC2S, which are autophagy-related proteins. P62 expression was significantly decreased in all the experimental groups. The SOD activity increased with increasing concentrations of nC2S at 3 h. nC2S promoted autophagy substrate degradation, and a high concentration of nC2S resulted in oxidative stress (Fig. 8).
Osteogenic effects of nC2S
The BMSC proliferation rate was not affected by nC2S treatment for 24 h (Supplemental Fig. 2). Micro-CT was performed at 4 W and 8 W. The results showed that nC2S promoted osteogenesis at 4 W. The degree of osteogenesis resembled that showed in the autologous bone group. Comparing with the BV/TV value of the control group, that of the nC2S group was obviously increased. The difference between the autologous group and the nC2S group was not statistically significant. At 8 W, more new bone formed in the defect regions in the nC2S group and autologous group. The BV/TV value between two groups had no significant difference (Fig. 6).
The HE staining results also showed little bone formation at the edge of the defect region at 4 W in the autologous bone group and nC2S group. More bone formation occurred from the edge of the defect region into the center of the defect area at 8 W. There was little bone formation at the margin of the defect regions in the control group. According to the results described above, nC2S can promote bone formation similar to autologous bone.
Effects of the nC2S/macrophage-regulated environment on osteogenic differentiation
nC2S increased ALP activity, while conditioned medium (CM group) did not affect ALP activity. When nC2S were combined with CM, ALP activity was obviously increased at 3 days. Treatment with 10 μg/mL and 50 μg/mL nC2S promote mineralization module formation. The degree of mineralization in the CM group was less than that in the nC2S group. More mineralized nodules were formed in the nC2S combined with CM group than in the nC2S group.
BMP-2 and RUNX-2 protein expression was measured by Western blotting at 3 days and 7 days. Runx-2 expression was not significantly changed at 3 days, but its expression was significantly increased at 7 days. Treatment with 10 μg/mL or 50 μg/mL nC2S significantly upregulated Runx-2 expression, but the rate of increase was lower than that after treatment with 10 CM and 50 CM. There was no obvious change in the CM group at 7 days.
BMP-2 expression was distinctly increased in the 10 CM and 50 CM groups compared with the control group at 3 days, but there was no obvious change in the 10 μg/mL, 50 μg/mL and CM groups. The expression level in the 50 CM group was distinctly higher than that in the 50 μg/mL group. At 7 days, BMP-2 expression was significantly increased. The rates of increase in the 10 μg/mL and 50 μg/mL groups were lower than those in the 10 CM and 50 CM groups (Fig. 9).
Discussion
Macrophages have an important regulatory effect on osteogenesis for their plasticity and diversity. RAW264.7 is a murine macrophage cells line originated from Abelson leukemia virus transformed cell linage derived from BALB/c mice. It is recognized as a proper model of macrophages and had a fast growth and less variability than human macrophages [23, 24]. Therefore, we used RAW264.7 as the experimental cells. Nanoparticles play an important role in bone formation and repair because of their specific nanostructure [25, 26]. Nanoparticle size adjust cell behaviors such as cell adhesion, migration and osteogenic differentiation. Researchers have found that Au nanoparticles were likely to be uptaken by MSCs, which caused mechanical signals through Yes-associated protein (YAP) activation and therefore promote osteogenic gene expression [27]. However, the direct effect of nC2S on macrophages and the immunoregulatory effect of bone on stem cells are not clear. The effects of nC2S on immune cells was investigate firstly in this study. Macrophages are the main cells that react to foreign bodies, such as nanoparticles [28]. Determining the cytotoxic effects of nC2S on macrophages helps to further explore their biological effects. In our study, the cytotoxicity of nC2S was evaluated by assessing their effects on the proliferation, apoptosis, and intracellular homeostasis of RAW264.7 cells. Our study indicated that nC2S promoted RAW264.7 cells proliferation and did not cause obvious apoptosis after 24 h and 48 h of incubation. Reports about the effects of calcium silicate materials on immune cell proliferation have been published. In our previous study, the dicalcium silicate coating had no effect on decreasing the proliferation of macrophages compared with the 45S5 coating [12]. Westhauser et al. found that mesoporous bioactive glass nanoparticles (120 ± 10 nm) at concentrations between 0.1 and 0.4 mg/mL promoted the proliferation of BMSCs [29]. To evaluate the effects of nC2S on macrophage intracellular homeostasis, autophagy-related protein expression and oxidative stress levels were investigated. LC3-I, LC3-II, and beclin-1 are closely associated with the induction of autophagy. P62 is an autophagy substrate whose expression level increases when autophagy is suppressed [30, 31]. The results showed that the LC3 I, LC3 II, and beclin-1 expression levels did not change, while P62 expression was significantly decreased, which suggested that nC2S did not promote autophagy in initial stage but promoted the degradation of autolysosomes via the autophagic process (Fig. 5). ROS is a by-product of oxidative stress and responds to exogenous signals or endogenous signals. Nanoparticles can induce ROS production in macrophages. ROS production results in lipid peroxidation, protein denaturation and oxidative DNA damage [32]. In this study, an ROS test kit was used to measure the amount of ROS. Starting with 50 μg/mL, we found that ROS levels increased with increasing concentrations of nC2S, particularly at 150 μg/mL. Based on this result, high concentrations of nC2S induced oxidative stress and disrupted the intracellular homeostasis of RAW264.7 cells. Therefore, we presumed that nC2S might stimulate oxidative stress while the further mechanism did not explore in this study. The nC2S exerted no obvious cytotoxic effects on RAW264.7 cells at concentrations of 10 μg/mL and 50 μg/mL. ROS are indispensable for the induction and maintenance of the M1-type polarization of macrophages. ROS promote proinflammatory cytokine production through activation of the NF-κB and p38 MAPK signalling pathways in macrophages [33]. In this way, nC2S might change the polarization of macrophages. To investigate how nC2S affected the polarization of macrophages, we assessed macrophage polarization by evaluating their morphological changes, phenotypic protein expression, and cytokine production. LPS was classical inflammatory inducer and always used as M1 activator in polarization of macrophages [34, 35]. The polarization of macrophages is heterogenetic, and their functional profiles can be polarized toward proinflammatory (M1) and anti-inflammatory (M2) phenotypes in response to different microenvironments. In the present study, SEM was used to observe the surface morphology of RAW264.7 cells, and cytoskeleton staining was used to observe intracellular structure changes. Many studies have used SEM and cytoskeleton staining to observe the surface morphology and intracellular structural changes of macrophages to assess their polarization [36,37,38]. According to SEM, M2 macrophages exhibited a flattened, expanded morphology, while M1 macrophages were smaller and irregularly shaped [39]. We found that when RAW264.7 cells were incubated with 50 μg/mL and 100 μg/mL nC2S, they gradually changed their functional phenotype and extended tiny pseudopods (Figs. 2, 3). The cytoskeleton is reorganized when macrophages are reprogrammed [40]. According to morphological analysis, RAW264.7 cells polarized toward the M1 phenotype. The expression of proteins associated with the classic phenotypes of macrophages was also investigated in this study. Our results showed that the expression of CCR7 and CD86 was upregulated with increasing concentrations of nC2S. In contrast, CD163 expression did not obviously change, and CD206 expression was suppressed in the nC2S groups compared to the control group. The expression levels of CCR7 and CD86 were investigated as M1 surface marker proteins, while CD206 and CD163 expression was investigated as M2 surface marker proteins [41]. According to the expression patterns of these specific proteins, the phenotypes of RAW264.7 cells were transformed into the M1 after being cultured with nC2S. Macrophages secrete cytokines to perform their functions after polarization. M1 macrophages mainly function in bone resorption by secreting proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β. In contrast, M2 macrophages promote tissue repair by secreting cytokines, such as IL-4 and IL-10 [42]. Here, nC2S with a size of 100 nm were chosen to explore their proinflammatory effects on macrophages. We found that the production of proinflammatory cytokines (TNF-α and MCSF) and TGF-β3 was significantly increased in RAW264.7 cells treated with 100 μg/mL nC2S. In our previous study, the C2S coating exerted fewer inflammatory effects on macrophages than hydroxyapatite (HA) and induced the expression of inflammatory cytokines, such as TNF-α, IL-6, and IL-1β [13]. RAW264.7 cells were polarized toward the M1 phenotype and secreted proinflammatory cytokines when incubated with increasing concentrations of nC2S. Different sizes of biomaterials have different effects on the polarization of macrophages. The size of particles is one of the factors affecting the polarization of macrophages. It is reported that particle size affects the polarization state of cells. Nanoparticles are more likely to polarize macrophages towards M1. Studies have found that 100 μg/mL nano-sized particles were transformed macrophages into the M1 type [43].

Expression of autophagy-related proteins and measurement of ROS levels. (A) and (B) LC3I/II and beclin-1 expression were not significantly changed. P62 expression was significantly decreased in all the experimental groups. (C) The SOD activity increased with increasing concentrations of nC2S.
To verify the direct osteogenic effects of nC2S, we performed micro-CT, bone immunofluorescence labelling, and HE staining. The results showed that nC2S exerted good osteogenic effects, similar to autologous bone (Figs. 6, 7, Supplementary Fig. 3). The osteogenic effect of nC2S on stem cells has been previously reported. In our previous study, nC2S promoted bone formation by mediating autophagy in BMSCs, which was consistent with the present results [22]. Biomaterials are able to regulate the immune microenvironment and, in this way, regulate stem cell bone-forming ability [44]. nC2S is a kind of biological glass (BGs). BGs can regulate the behavior of macrophages and alter local immune microenvironment. It is reported that macro-meso-micro hierarchically porous BGs scaffolds polarized macrophages towards M1 at the early stage (0–3 days) and secreted cytokines IL-1β, which can helpful to bone repair [45]. However, the effects of nC2S on osteogenesis through stimulation of immune regulatory cells, such as macrophages, is not clear yet. Do nC2S inhibit or promote the osteogenesis of BMSCs by polarizing macrophages toward the M1 phenotype? Supernatants from pre-stimulated immune-modulatory cells could be used to stimulate stem cells to determine the effect of these immune-modulatory cells on osteogenesis [46]. It is a strategy to use conditioned medium as the approach to study the interplay between macrophages and skeletal cells [47]. It has been found that supernatants from prestimulated RAW264.7 cells on graphene oxide promoted the osteogenic differentiation of BMSCs [48]. In our study, TGF-β3 was significantly increased in the supernatants from prestimulated RAW264.7 cells on nC2S. TGF-β3 promoted osteogenesis by upregulation of endogenous BMP-2 [49]. Thus, the supernatants from pre-stimulated RAW264.7 cells may have better effects for inducting the osteogenic process of BMSCs than nC2S alone. In this study, the supernatants of RAW264.7 cells after treatment with nC2S were collected and added to BMSC cultures. Combined with the effects of nC2S on macrophages, 50 μg/mL was chosen as the proper concentration of nC2S to generate CM. We used ALP activity staining and Alizarin Red S staining to verify the osteogenic properties of BMSCs. In the present study, our results showed increased ALP activity and mineralization nodule formation by BMSCs in the 10 CM and 50 CM groups compared with the 10 μg/mL and 50 μg/mL nC2S groups. The CM group did not exhibit obvious changes in ALP activity or degrees of mineralization (Fig. 8). The expression levels of both osteogenic proteins (BMP-2) and genes (RUNX-2) were also examined. Interestingly, we found that the expression levels of both RUNX2 and BMP-2 in the 10 CM and 50 CM groups were higher than those in the 10 μg/mL and 50 μg/mL nC2S groups (Fig. 9 ). Based on these findings, BMSCs exhibited better osteogenic effects when cultured with CM. Thus, supernatants from prestimulated RAW264.7 cells exerted better effects for inducting the osteogenic process of BMSCs than nC2S alone. It has been reported that interaction between immunocytes and skeleton system exerts temporal effect on post bone injury [50, 51]. Polarization of macrophages was a dynamic changing process and phenotypes can be reversed after stimulating by microenvironment. M1 macrophages mainly participate in acute inflammation and angiogenesis at the early stage of bone tissue repair while M2 macrophages at a late stage. Some nanomaterials promote osteogenesis by polarizing macrophages toward the M1 phenotype. Copper-doped mesoporous silica nanospheres promoted bone formation by BMSCs by polarizing macrophages toward the M1 phenotype, and these macrophages secreted IL6, IL1β, TNFα, and IFNγ [52]. By polarizing toward the M1 or M2, macrophages can modulate different physiological activities by secreting different cytokines. Generally, M1 macrophages secrete proinflammatory cytokines, and M2 macrophages secrete anti-inflammatory cytokines. Here, we found that M1 macrophages promoted osteogenic ability of BMSCs in vitro by secreting low concentrations of TNF-α after pre-stimulation with 50 μg/mL nC2S. Similarly, a previous study found that M1 could secrete the cytokines TNF-α that reshaped the tumor microenvironment [53]. Inflammation had complicated effects on osteogenesis for its regulatory effect could be favorable or decisive to osteogenesis. There are four stages of bone fracture healing: inflammation, cartilage callus formation, bony callus formation, and bone remodeling. Early inflammation generally occurs in the first one to three days of the bone healing process. Thus, early inflammation is an indispensable stage in bone-induced bone healing [54, 55]. We collected supernatants from RAW264.7 cells treated with nC2S for 24 h and added these supernatants to BMSC cultures. In this way, we established a microenvironment that mimicked the early acute inflammation that occurs in bone fracture. It is possible that this environment regulated the induction of bone formation by BMSCs. In the past, TNF-α was considered a cytokine that negatively regulated the differentiation of osteoblasts. Whereas, evidences are mounting that the regulatory function of TNF-α depends on time and concentration [56, 57]. Many studies have reported that low concentrations of TNF-α exert a positive effect on bone tissue repair. For example, Wang et al. found that TNF-α at concentrations of 1 ng/mL promoted the osteogenic differentiation of mouse bone marrow-derived mesenchymal stem cells [58]. Other researchers revealed the particular signalling pathways by which these cytokines promote osteogenesis. Daniele et al. found that TNF-α was able to promote the osteogenic pro-differentiating effects elicited by adenosine A2B receptor stimulation by suppressing the G protein-coupled receptor desensitization process in BMSCs. This finding revealed the potential function of TNF-α in osteogenesis [59].

Micro-CT evaluation of skull defect repair 4 W and 8 W after implantation. The micro-CT images showed more bone tissue repair in the autologous bone group and nC2S group. The bone volume/total volume (BV/TV) ratio in the new bone formation area was analyzed. The BV/TV values of the autologous bone group and nC2S group were higher than that of the control group. However, there was no significant difference between the autologous bone group and the nC2S group. *P < 0.05.

HE staining of histological sections at 4 W and 8 W. There was little bone formation at the edges of the defect regions at 4 W in the autologous bone group and the nC2S group. Bilateral defect margins were observed at 10x. More bone formation occurred from the edge of the defect region toward the center of the defect areas at 8 W in the autologous bone group and the nC2S group. There was no significant difference between the two groups. *Residual graft in bone defect regions.

ALP activity and Alizarin Red S staining. ALP activity was measured at 3 d and 7 d. Treatment with 10 μg/mL and 50 μg/mL nC2S increased ALP activity. The CM group did not show obvious changes in ALP activity. The 10 CM and 50 CM groups showed obvious increases in ALP activity at 3 d. The Alizarin Red S staining results showed that 10 μg/mL and 50 μg/mL nC2S promoted mineralization module formation compared with the control group. The CM group did not exhibit conspicuous changes in degrees of mineralization. The 10 CM and 50 CM groups showed increased mineralization module formation compared with the nC2S group.

The protein expression of RUNX2 and BMP-2. Runx-2 expression was not significantly changed at 3 d. Treatment with 10 μg/mL and 50 μg/mL nC2S significantly upregulated Runx-2 expression at 7 d, but the rate of increase was lower than those observed after treatment with 10 CM and 50 CM. There was no obvious change in the CM group. BMP-2 expression was significantly increased in the 10 CM and 50 CM groups at 3 d. There was no obvious change in the 10 μg/mL and 50 μg/mL nC2S groups or the CM group. At 7 d, BMP-2 expression was significantly increased. The rates of increase in the 10 μg/mL and 50 μg/mL nC2S groups were lower than 10 CM and 50 CM groups significantly. *P < 0.05 compared to the control group. **P < 0.01 and ***P < 0.001. #P < 0.05 compared to each ether. ##P < 0.01 and ###P < 0.001.
Conclusion
In this study, nC2S exerted no obvious cytotoxic effects on RAW264.7 cells at a certain concentration. When RAW264.7 cells were cultured with nC2S, autophagy substrate P62 was degradated and oxidative stress was induced with increasing concentrations of nC2S. Furthermore, nC2S changed the morphology and cytoskeletal structure of RAW264.7 cells and polarized them toward the M1 phenotype. The expression of the proinflammatory cytokines TNF-α increased, while the expression of IL-1β did not change. After treatment with nC2S, RAW264.7 cells established a slightly proinflammatory microenvironment for BMSCs and promoted osteogenesis. Thus, nC2S exhibit great biocompatibility with RAW264.7 cells and may promote the osteogenesis of stem cells by regulating the immune microenvironment.
Materials and methods
Material preparation and characterization
nC2S particles (100 nm) were synthesized in a laboratory at the Shanghai Institute of Ceramics of the Chinese Academy of Sciences by the sol–gel process. Scanning electron microscopy (SEM; Nova Nano SEM 430, FEI, Finland) combined with energy-dispersive X-ray spectroscopy (EDS; DX-4 system, EDAX, USA) were used to observe the morphologies of the particles and analyze the elements in the particles. The crystal structures were determined by X-ray diffraction (XRD; Geigerflex, Rigaku Co, Japan). A nanoparticle size analyser was used to evaluate the nanoparticle sizes. E-Toxate Kit (Sigma, St. Louis, USA) was performed to measure endotoxin levels to avoid particles contamination. The nanoparticles were dispersed in endotoxin-free water and cultured with Limulus polyphemus in sterilized glass tubes at 37 °C for 1 h.
Proliferation and apoptosis of cells
According to the instructions of American Type Culture Collection (ATCC), macrophages-like cell RAW 264.7 (American Type Culture Collection, TIB71, MD, USA) were cultured. RAW264.7 were cultured in an incubator at 37 °C under 5% CO2/95% air atmosphere. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) added with 10% fetal calf serum (Life Technologies, USA), glutamine (2 mM), penicillin (5000 U/mL) and streptomycin (25 μg/mL) was used to develop RAW264.7.
Balb/c bone marrow stem cells (BMSCs, MUCMX-01001, Cyagen Biosciences Inc., USA) were cultured according to manufacture instructions. In addition, 50 μg/mL, 100 μg/mL and 150 μg/mL nC2S were cultured with RAW264.7 and BMSCs for 24 h separately. After treated with nC2S, cell proliferation was quantified by Cell Counting Kit-8 assay (CCK-8).
CCK-8 was performed to measure the proliferation rates. The absorbance values were measured at 450 nm with a microplate reader (Bio–Rad, California, USA) according to the operating instructions. The percentage of viable cells was calculated based on the OD values obtained in the CCK-8 assay. A FITC Annexin V Apoptosis Detection Kit (Dojindo, Japan) was used to measure the apoptosis levels after the cells were treated for 24 h and 48 h.
Macrophage polarization and inflammatory response
Lipopolysaccharide (LPS, 1 μg/mL, Sigma, St. Louis, USA) was performed as the positive control to observe cell morphological changes. nC2S (10 μg/mL, 50 μg/mL, and 100 μg/mL) and control group were detected after 24 h. 4% glutaraldehyde was used to fix cells for 15 min. Next, gradient dehydrated cells by alcohol. After natural air-drying, the changes of cell surface morphology were observed by SEM (scanning electron microscopy).
Cell shape changes in macrophage phenotype were dependent on contractility within the actin cytoskeleton and therefore change in cytoskeleton represented change of macrophages phenotype [28]. Immunofluorescence staining was used to detect change of cytoskeleton. The fixed cells were permeabilized with 0.1% Triton after fixed by 4% glutaraldehyde. Alexa Fluor® 568-conjugated phalloidin (1:250 dilutions) was used to dye the cytoskeleton. Then, the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI), and images were captured using a confocal microscope (TCS SP5, Leica Microsystem, Solms, Germany) in the dark.
RT–PCR was used to measure the expression of macrophage surface markers and cytokines. The expression of cytokines in culture medium was tested by ELISA. The cells were cultured with 10 μg/mL, 50 μg/mL, and 100 μg/mL C2S nanoparticles for 1 day. The cells of positive control group were cultured with 1 µg/mL LPS. The M1 macrophage surface markers CD86 and CCR7 were measured. The M2 macrophage surface markers CD206 and CD163 were measured. The expression levels of TNF-α, IL-1 β and IL-10 were also measured by RT–PCR (TB green ™ Premi Ex Taq ™ II, Takara Biotechnology, Japan). The expression of the osteogenic factor TGF-β3 and the osteoclastic activity-related gene MCSF in treated RAW264.7 cells were measured by RT–PCR.
Changes in intracellular homeostasis
The expression levels of the autophagy-related proteins LC3I/II, P62 and Beclin-1 (Abcam, USA) and GAPDH were measured by Western blotting at 6 h. As the classic marker, LC3-I/II turnover and P62 were usually evaluated to reflect changes of autophagic flux [60]. P62 was selective autophagy receptor for degradation of ubiqutinated substrates [61]. Beclin-1 was a crucial molecule in autophagic process involved in autophagic pathways, autophagosome formation and maturation [62]. Based on manufacturer’s instruction, the protein of RAW264.7 was extracted. BCA kit (Beyotime, China) was used to quantified concentration of protein. Equivalent amounts of protein were loaded onto 10% SDSPAGE gels, dispersed by electrophoresis and then transferred to PVDF membranes. The PVDF were incubated with anti-GAPDH (1:1000), anti-LC3 (1:1000; Abcam USA), anti-beclin-1 (1:2000 Abcam, USA) and anti-P62 (1:2000; Abcam, USA) as primary antibodies at 4 °C overnight. After that, the membranes were washed and then incubated with a goat anti-rabbit IgG HRP-linked antibody (Beyotime, China) for 1 h, developed and revealed. To analyze the results, the relative ratio was assessed using ImageJ software.
Superoxide Dismutase (SOD) is an antioxidant metal enzyme existing in organisms. Superoxide dismutase (SOD) activity was measured using reactive oxygen species (ROS) test kit (Nanjing Jian Cheng Biology Engineering Institute, Jiangsu, China). RAW264.7 cells were washed with PBS after incubation for 3 h. The plate was incubated with a DCFDA solution for 45 min at 37 °C in the dark according to the instructions. ROS levels was measured using Flow cytometry.
Osteogenic effects of nC2S
Male SD rats (6–8 weeks old) weighing 250–280 g were purchased from the Animal Center of Southern Medical University (Guangzhou, China). The rats were housed under controlled environmental conditions before the experiment. The environmental conditions include room temperature (23 ± 1 °C), relative humidity (60 ± 10%) and light/dark cycle (12 h/12 h). Surgery of 5-mm-diameter calvarial defect was performed in each SD rat. The surgical area was shaved, scrubbed with 70% ethanol. Under aseptic condition, a cranial skin incision was made by sterile instruments along the middle line. The calvarium was exposed by separating the subcutaneous tissue, musculature and periosteum. 5.0 mm outer diameter trephine was used to generate bilateral full-thickness defects. The defect regions were rinsed with sterile PBS to avoid excessive heating of bone edges. The rats were randomly assigned to three groups: the control group, autologous bone group and nC2S group. No graft was received in the control group. The autologous bone group was received autogenous bone graft while the nC2S group were treated with nC2S. The removed calvarial bone of rat after surgery was used as the graft in autologous bone group. The study was affirmed by the animal experiment ethical review board at Southern Medical University. The laboratory ethics review number is L2018033. Skeleton parameters of the groups mentioned above were analyzed by micro-CT and Bone immunofluorescence labelling.
Microcomputed tomography (m-CT) measurement
Micro-CT (mCT-80, Scanco Medical, Bruttisellen, Switzerland) was used to detect the amount of new bone formation in the defect area. The specimens were scanned using a thickness of 0.020 mm per slice in medium-resolution mode, with a 1,024 reconstruction matrix and 200 ms integration time. MIMICS (NRecon, USA; CTvox, USA) was used to evaluate a fixed global threshold of 20% and 3D Creator software was performed to reconstruct tomograms. After three-dimensional reconstruction, the bone volume fraction (BV/TV) was automatically determined. The BV/TV values in the bone defect region represented the amount of new bone formation.
Bone immunofluorescence labelling
One week before sampling, tetracycline (TE, 25 mg/kg of body weight) and calcein (CA, 20 mg/kg of body weight) were intraperitoneally injected into the rats. The calvariae was fixed using buffered glutaraldehyde (4%)-glutaraldehyde (0.25%) fixative (pH 7.4) at 4 °C for 3 days. After sampling, fixation, embedding in polymethylmethacrylate (PMMA) and undecalcified bone slicing were performed. The longitudinal sections of the crania were cut into 150–200 μm thick slices by microtome (Leica Microsystems, Wetzlar, Germany), glued to a plastic support and polished. Finally, approximately 50 μm thickness slices were geted, Fluorescence microscopy was employed to observe mineralized tissue. The blue staining in the images represents TE, and the green staining in the images represents CA, which indicates bone formation and mineralization. The samples were harvested and analyzed at 4 and 8 weeks after the operation, respectively.
Effect of nC2S/macrophage-modulated environment on osteogenic differentiation
Osteogenic components (OM, 2 mM β-glycerrophosphate, 100 mM L-ascorbic acid 2-phosphate, 10 nM dexamethasone (Sigma, Aldrich, USA) were added to culture substrate. Collecting the medium of RAW264.7 cells as basal medium. The medium of RAW264.7 cells treated with 50 μg/mL nC2S for 24 h was also collected which considered as conditioned medium (CM). The negative control group was cultured with medium of RAW264.7 cells. The cells in the CM group were cultured with this CM. The materials groups were cultured with 10 μg/mL or 50 μg/mL nC2S, respectively, with medium of RAW264.7 cells (10 μg/mL group, 50 μg/mL group). The BMSCs were also treated with CM supplemented with 10 μg/mL or 50 μg/mL nC2S (10 CM group or 50 CM group, respectively).
After 3 days and 7 days of cultivation, the activity of alkaline phosphatase (ALP) was analyzed by alkaline phosphatase staining. Cells were washed with PBS for three times and then fixed with 4% cis-butenal for 12 min. BCIP/NBT alkaline phosphatase staining kit (Beyotime, China) was used to staining. After rinsing, images were taken by a stereomicroscope (Leica EZ4HD, Germany).
The amount of mineralization modules was analyzed using alizarin Red S staining (ARS) over 7 days, 14 days and 21 days. The ARS groups were defined as those mentioned in ALP staining. The cells were rinsed with PBS for three times and settled with 4% cis-butenal for 15 min. After that, Samples were invubated with 0.1% salicylin red S (pH 4.1 Solarbio, China) at room temperature for 3 min. After rinsing with ddH2O, samples were photographed using the stereomicroscope (Leica EZ4HD, Germany).
BMP-2 and RUNX-2 protein expression were measured by Western blotting at 3 days and 7 days. BMSCs were treated with 0 μg/mL nC2S, 10 μg/mL nC2S, 50 μg/mL nC2S, CM, 10 CM, 50 CM for 3 days and 7 days. Specimen protein was tested with the following antibodies: anti-GAPDH (1:2000), anti-BMP2 (1:1000; Ab Cam, USA); anti-OPN (1:2000), and anti-RUNX2 (1:2000; Ab Cam, USA). The results were analyzed using ImageJ software.
Statistical analysis
The mean and standard deviation (SD) of triplicate experiments were showed by bar graphs. The data were normalized and passed the normality test by one-way ANOVA with the least significant difference (LSD) test. All analyses were conducted using SPSS 19.0 software (SPSS Inc., Chicago, IL, USA). P values < 0 05 were considered statistically significant.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to the author copyright security issues but are available from the corresponding author on reasonable request.
References
D. Correa, A. Almirall, R. García-Carrodeguas et al., β-Dicalcium silicate-based cement: synthesis, characterization and in vitro bioactivity and biocompatibility studies. J. Biomed. Mater. Res. A 102(10), 3693–3703 (2014)
F. Zuleta, A. Murciano, S.A. Gehrke et al., A new biphasic dicalcium silicate bone cement implant. Materials (Basel) 10(7), 758 (2017)
Y. Zhang, J. Tang, M. Li et al., Enhancing the biological activity of vaterite-containing β-dicalcium silicate cement by silane coupling agent for biomaterials. Mater. Sci. Eng. C 92, 1–10 (2018)
P.N. De Aza, F. Zuleta, P. Velasquez et al., (α′(H))-Dicalcium silicate bone cement doped with tricalcium phosphate: characterization, bioactivity and biocompatibility. J. Mater. Sci. Mater. Med. 25(2), 445–452 (2014)
D. Correa, A. Almirall, R.G. Carrodeguas et al., α-Tricalcium phosphate cements modified with β-dicalcium silicate and tricalcium aluminate: physicochemical characterization, in vitro bioactivity and cytotoxicity. J. Biomed. Mater. Res. B 103(1), 72–83 (2015)
P.N. De Aza, D. García-Bernal, F. Cragnolini et al., The effects of Ca2SiO4-Ca3(PO4)2 ceramics on adult human mesenchymal stem cell viability, adhesion, proliferation, differentiation and function. Mater. Sci. Eng. C 33(7), 4009–4020 (2013)
B.C. Wu, C.K. Wei, N.S. Hsueh et al., Comparative cell attachment, cytotoxicity and antibacterial activity of radiopaque dicalcium silicate cement and white-coloured mineral trioxide aggregate. Int. Endod. J. 48(3), 268–276 (2015)
P. Yoshino, C.K. Nishiyama, K.C. Modena et al., In vitro cytotoxicity of white MTA, MTA Fillapex® and Portland cement on human periodontal ligament fibroblasts. Braz. Dent. J. 24(2), 111–116 (2013)
B.C. Wu, S.C. Huang, S.J. Ding, Comparative osteogenesis of radiopaque dicalcium silicate cement and white-colored mineral trioxide aggregate in a rabbit femur model. Materials (Basel) 6(12), 5675–5689 (2013)
P. Karanth, M.K. Manjunath, Roshni et al., Reaction of rat subcutaneous tissue to mineral trioxide aggregate and Portland cement: a secondary level biocompatibility test. J. Indian Soc. Pedod. Prev. Dent. 31(2), 74–81 (2013)
V. Opačić-Galić, V. Petrović, S. Zivković et al., New nanostructural biomaterials based on active silicate systems and hydroxyapatite: characterization and genotoxicity in human peripheral blood lymphocytes. Int. Endod. J. 46(6), 506–516 (2013)
L. Chen, Y. Zhang, J. Liu et al., Exposure of the murine RAW 2647 macrophage cell line to dicalcium silicate coating: assessment of cytotoxicity and pro-inflammatory effects. J. Mater. Sci. Mater. Med. 27(3), 59 (2016)
L. Chen, P. Zhu, R. Liu et al., Potential proinflammatory and osteogenic effects of dicalcium silicate particles in vitro. J. Mech. Behav. Biomed. Mater. 44, 10–22 (2015)
S. Lai, L. Chen, W. Cao et al., Dicalcium silicate induced proinflammatory responses through TLR2-mediated NF-κB and JNK pathways in the murine RAW 264.7 macrophage cell line. Mediat. Inflamm. 2018, 8167932 (2018)
W. Booncharoen, A. Jaroenworaluck, R. Stevens, A synthesis route to nanoparticle dicalcium silicate for biomaterials research. J. Biomed. Mater. Res. B 99(2), 230–238 (2011)
S. Dong, J. Sun, Y. Li et al., Electrospun nanofibrous scaffolds of poly (l-lactic acid)-dicalcium silicate composite via ultrasonic-aging technique for bone regeneration. Mater. Sci. Eng. C 35, 426–433 (2014)
L. Ou, B. Song, H. Liang et al., Toxicity of graphene-family nanoparticles: a general review of the origins and mechanisms. Part. Fibre Toxicol. 13(1), 57 (2016)
Z. Chen, A. Bachhuka, S. Han et al., Tuning chemistry and topography of nanoengineered surfaces to manipulate immune response for bone regeneration applications. ACS Nano 11(5), 4494–4506 (2017)
L. Claes, S. Recknagel, A. Ignatius, Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 8(3), 133–143 (2012)
H. Lei, M. He, X. He et al., METTL3 induces bone marrow mesenchymal stem cells osteogenic differentiation and migration through facilitating M1 macrophage differentiation. Am. J. Transl. Res. 13(5), 4376–4388 (2021)
R. Huang, X. Wang, Y. Zhou et al., RANKL-induced M1 macrophages are involved in bone formation. Bone Res. 5, 17019 (2017)
R. Wang, L. Chen, L. Shao, The mTOR/ULK1 signaling pathway mediates the autophagy-promoting and osteogenic effects of dicalcium silicate nanoparticles. J. Nanobiotechnol. 18(1), 119 (2020)
S.Y. Choi, J.Y. Choi, J.M. Lee et al., Tartary buckwheat on nitric oxide-induced inflammation in RAW264.7 macrophage cells. Food Funct. 6(8), 2664–2670 (2015)
S. Khatua, J. Simal-Gandara, K. Acharya, Understanding immune-modulatory efficacy in vitro. Chem. Biol. Interact. 352, 109776 (2022)
B. Kupikowska-Stobba, M. Kasprzak, Fabrication of nanoparticles for bone regeneration: new insight into applications of nanoemulsion technology. J. Mater. Chem. 99(26), 5221–5244 (2021)
R. Roy, S. Kumar, A. Tripathi et al., Interactive threats of nanoparticles to the biological system. Immunol. Lett. 158(1–2), 79–87 (2014)
J. Li, J.J. Li, J. Zhang et al., Gold nanoparticle size and shape influence on osteogenesis of mesenchymal stem cells. Nanoscale 8(15), 7992–8007 (2016)
F.Y. McWhorter, T. Wang, P. Nguyen et al., Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. U S A 110(43), 17253–17258 (2013)
F. Westhauser, S. Wilkesmann, Q. Nawaz et al., Effect of manganese, zinc, and copper on the biological and osteogenic properties of mesoporous bioactive glass nanoparticles. J. Biomed. Mater. Res. A 109(8), 1457–1467 (2021)
S.T. Shibutani, T. Saitoh, H. Nowag et al., Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 16(10), 1014–1024 (2015)
N. Mizushima, M. Komatsu, Autophagy renovation of cells and tissues. Cells 147(4), 728–741 (2011)
A. Kermanizadeh, C. Chauché, D.M. Brown et al., The role of intracellular redox imbalance in nanomaterial induced cellular damage and genotoxicity: a review. Environ. Mol. Mutagen 56(2), 111–124 (2015)
E. Rendra, V. Riabov, D.M. Mossel et al., Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 224(2), 242–253 (2019)
L. Liu, H. Guo, A. Song et al., Progranulin inhibits LPS-induced macrophage M1 polarization via NF-кB and MAPK pathways. BMC Immunol. 21(1), 32 (2020)
M. Orecchioni, Y. Ghosheh, A.B. Pramod et al., Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS-) vs. alternatively activated macrophages. Front. Immunol. 10, 1084 (2019)
V. Malheiro, F. Lehner, V. Dinca et al., Convex and concave micro-structured silicone controls the shape, but not the polarization state of human macrophages. Biomater. Sci. 4(11), 1562–1573 (2016)
S. Cui, N. Wienhoefer, U. Bilitewski, Genistein induces morphology change and G2/M cell cycle arrest by inducing p38 MAPK activation in macrophages. Int. Immunopharmacol. 18(1), 142–150 (2014)
M.Z. Guo, M. Meng, S.Q. Duan et al., Structure characterization, physicochemical property and immunomodulatory activity on RAW2647 cells of a novel triple-helix polysaccharide from craterellus cornucopioides. Int. J. Biol. Macromol. 126, 796–804 (2019)
L. Chen, P. Qiao, H. Liu et al., Amorphous calcium phosphate NPs mediate the macrophage response and modulate BMSC osteogenesis. Inflammation 44(1), 278–296 (2021)
D.Y. Vogel, P.D. Heijnen, M. Breur et al., Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. J. Neuroinflamm. 11, 23 (2014)
A. Viola, F. Munari, R. Sánchez-Rodríguez et al., The metabolic signature of macrophage responses. Front. Immunol. 10, 1462 (2019)
D. Yang, Y. Wan, Molecular determinants for the polarization of macrophage and osteoclast. Semin. Immunopathol. 41(5), 551–563 (2019)
W. Xie, X. Fu, F. Tang et al., Dose-dependent modulation effects of bioactive glass particles on macrophages and diabetic wound healing. J. Mater. Chem. B 7(6), 940–952 (2019)
J. Ping, C. Zhou, Y. Dong et al., Modulating immune microenvironment during bone repair using biomaterials: focusing on the role of macrophages. Mol. Immunol. 138, 110–120 (2021)
Y. Liu, L.Y. Cao, S. Zhang et al., Effect of hierarchical porous scaffold on osteoimmunomodulation and bone formation. Appl. Mater. Tdoday 20, 2352–9407 (2020)
G. Valles, F. Bensiamar, L. Maestro-Paramio et al., Influence of inflammatory conditions provided by macrophages on osteogenic ability of mesenchymal stem cells. Stem Cell Res. Ther. 11(1), 57 (2020)
G. Mestres, S.D. Carter, N.P. Hailer et al., A practical guide for evaluating the osteoimmunomodulatory properties of biomaterials. Acta Biomater. 130, 115–137 (2021)
D. Xue, E. Chen, H. Zhong et al., Immunomodulatory properties of graphene oxide for osteogenesis and angiogenesis. Int. J. Nanomed. 13, 5799–5810 (2018)
R.M. Klar, R. Duarte, T. Dix-Peek et al., The induction of bone formation by the recombinant human transforming growth factor-β3. Biomaterials 35(9), 2773–2788 (2014)
D.C. Lacey, P.J. Simmons, S.E. Graves et al., Proinflammatory cytokines inhibit osteogenic differentiation from stem cells: implications for bone repair during inflammation. Osteoarthr. Cartil. 17(6), 735–742 (2009)
M.M. Martino, K. Maruyama, G.A. Kuhn et al., Inhibition of IL-1R1/MyD88 signalling promotes mesenchymal stem cell-driven tissue regeneration. Nat. Commun. 7, 11051 (2016)
M. Shi, Z. Chen, S. Farnaghi et al., Copper-doped mesoporous silica nanospheres, a promising immunomodulatory agent for inducing osteogenesis. Acta Biomater. 30, 334–344 (2016)
M. Song, Z.H. Li, H.S. Gu et al., Ganoderma lucidum spore polysaccharide inhibits the growth of hepatocellular carcinoma cells by altering macrophage polarity and induction of apoptosis. J Immunol. Res. 2021, 6696606 (2021)
F. Loi, L.A. Córdova, J. Pajarinen et al., Inflammation, fracture and bone repair. Bone 86, 119–130 (2016)
J.M. Karnes, S.D. Daffner, C.M. Watkins, Multiple roles of tumor necrosis factor-alpha in fracture healing. Bone 78, 87–93 (2015)
B. Osta, G. Benedetti, P. Miossec, Classical and paradoxical effects of TNF-alpha on bone homeostasis. Front. Immunol. 5, 48 (2014)
B. Zhao, TNF and bone remodeling. Curr. Osteoporos. Rep. 15(3), 126–134 (2017)
L. Wang, J. Zhang, C. Wang et al., Low concentrations of TNF-α promote osteogenic differentiation via activation of the ephrinB2-EphB4 signalling pathway. Cell Prolif. 50(1), e12311 (2017)
S. Daniele, L. Natali, C. Giacomelli et al., Osteogenesis Is improved by low tumor necrosis factor alpha concentration through the modulation of Gs-coupled receptor signals. Mol. Cell Biol. 37(8), e00442-e516 (2017)
D.J. Klionsky, A.K. Abdel-Aziz, S. Abdelfatah et al., Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 17(1), 1–382 (2021)
Y. Katsuragi, Y. Ichimura, M. Komatsu, p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282(24), 4672–4678 (2015)
R. Kang, H.J. Zeh, M.T. Lotze et al., The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18(4), 571–580 (2011)
Funding
This work was supported financially by the National Natural Science Fund (81600904) and the Open Laboratory Project for College Students of Guangzhou Medical University (No. 20205028).
Author information
Authors and Affiliations
Contributions
CL designed the research. ZX was responsible for characterizing nC2S and detecting macrophage polarization and cytokine expression. ROS, autophagy and osteogenic proteins expression were evaluated by ZX. Skull defects repair was performed by CL and ZX. MM generated the figures and drafted the manuscript, which was critically revised by CL. All authors read, corrected and approved the manuscript. CL and ZX contributed equally to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chen, L., Zhou, X. & Mo, M. The response of RAW264.7 cells to dicalcium silicate nanoparticles and the effect of the nanoparticle-regulated immune environment on osteogenesis. Journal of Materials Research 37, 4268–4283 (2022). https://doi.org/10.1557/s43578-022-00793-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/s43578-022-00793-x