
Abstract
The influence of phosphorus doping on the properties of \( \alpha^{\prime}_{\text{H}} \)-dicalcium silicate (C2S) bone cement was analyzed, in addition to bioactivity and biocompatibility. All the cements were composed of a solid solution of TCP in C2S (\( \alpha^{\prime}_{\text{H}} \)-C2Sss) as the only phase present. The compressive strength ranged from 3.8–16.3 MPa. Final setting times ranged from 10 to 50 min and were lower for cements with lower L/P content. Calcium silicate hydrate was the principal phase formed during the hydration process of the cements. The cement exhibited a moderate degradation and could induce carbonated hydroxyapatite formation on its surface and into the pores. The cell attachment test showed that the \( \alpha^{\prime}_{\text{H}} \)-Ca2SiO4 solid solution supported human adipose stem cells adhesion and spreading, and the cells established close contacts with the cement after 24 h of culture. The novel \( \alpha^{\prime}_{\text{H}} \)-C2Sss cements might be suitable for potential applications in the biomedical field, preferentially as materials for bone/dental repair.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Traditionally, dicalcium silicate (Ca2SiO4) has been an important constituent in Portland cement, refractories and heat-resistant coatings and can spontaneously develop strength toward water [1–4]. Mineral trioxide aggregate (MTA) powder is basically a mixture of Portland cement and bismuth(III) oxide [5, 6]. At present these materials are proposed for many clinical applications in dentistry, including root-end filling, root-perforation repair, pulp capping, apicogenesis and dentin hypersensitivity reduction [7–10]. The main disadvantage when using MTA as a dental material is primarily due to its long setting time of approximately 2 h [11]. For this purpose, a material should ideally have a relatively short setting time to avoid being washed away by saliva and to reduce the possibility of the unset material irritating oral tissues. Recently, the setting time of Portland cement was successfully reduced by adding calcium chloride (CaCl2) [12].
However, to our knowledge, the preparation and characterization of Ca2SiO4 cements doped with phosphorus have not yet been reported. Among other bioactive components, the incorporation of tricalcium phosphate into calcium silicate bone cement has been attempted in order to achieve biological anchorage of the cement, which would be promoted by the growing of bone into the pores produced by the resorption of the cement particles. Phase relations in the Ca3(PO4)2–Ca2SiO4 system were described in an early paper by Nurse et al. [13] and Fix et al. [14], and later revised by other authors [15]. The diagram shows the possibility of obtaining dicalcium silicate doped with phosphorus. At the temperature of the invariant point (518 ± 6 °C), the maximum solid solution of tricalcium phosphate (TCP) in Ca2SiO4 is about ~21 wt%.
Thus, the objective of this study was to investigate the physicochemical properties and in vitro bioactivity and biocompatibility of novel dicalcium silicate cements doped with tricalcium phosphate with compositions lying in the field of the \( \alpha^{\prime}_{\text{H}} \)–Ca2SiO4 solid solution in the Ca2SiO4-Ca3(PO4)2 system [15], by solid state reaction, at high temperature and slow cooling to room temperature.
2 Materials and methods
2.1 Cements preparation
Dicalcium silicate (C2S) was obtained by solid state reaction-sintering, starting from an appropriate mixture of calcium carbonate (CaCO3 > 99.0 wt% Fluka) with an average particle size of 13 μm, and silicon oxide (SiO2 > 99.7 wt%, Strem Chemicals), with an average particle size <30 μm. The powders were cold isostatically pressed at 200 MPa and heat treated at 1,525 °C for 12 h at a heating rate of 8.3 °C/min followed by cooling rate of 5 °C/min. The powder was then dried and sieved thorough 30 mesh. TCP was synthesized by solid state reaction from a stoichiometric mixture of calcium hydrogen phosphate anhydrous (CaHPO4, Sigma) and calcium carbonate (CaCO3, Sigma) with an average particle size of <30 μm. The CaHPO4 and CaCO3 mixture was heated in a platinum crucible at 1,500 °C for 3 h. Then it was liquid-nitrogen quenched by rapid withdrawal from the furnace. The obtained material was ground and characterized by X-ray diffraction (XRD).
C2S and TCP powders were ground to an average particle size of ~15 μm, and an 85 wt% C2S–15 wt% TCP composition was mixed in a manual agate mortar under acetone. This was then isostatically pressed into bars at 200 MPa and heated up to 1,500°C/3 h, then rapid withdrawal from the furnace and liquid-nitrogen quenched. The bars were ground after heat treatment, then pressed and reheated again. This procedure was repeated three times to homogenize the entire composition. The reaction-sintering temperatures were selected bearing in mind the information provided by the C2S–TCP phase equilibrium diagram [15]. The obtained materials were ground and characterized by XRD.
To prepare the cements (or cement pastes), equal-mass powders (10 g) were mixed with 2 g of KH2PO4 as an accelerator. Then they were mixed with distilled water so that the liquid- to powder ratio (L/P ratio) was 0.35, 0.37, 0.38, 0.41 mL g−1. The mixtures were stirred to form homogeneous pastes, within 1 min by a stainless-steel spoon. The paste was used to fill cylindrical holes in a Teflon mold with a diameter of 26 mm and a height of 10 mm and then stored in a 37 °C, 100 % humidity incubator.
2.2 Setting time
Initial (I) and final (F) setting times were measured for each batch of water according to UNE-EN196-3.The initial setting time I is defined as the time necessary for the needle (300 g, 45 mm length, 1.13 ± 0.05 mm diameter) to plunge into the paste and leave a print of 6 ± 3 mm. The final setting time F is defined as the time necessary so that a cylindrical needle with flat tip area of 19.6 mm2, (A = π r2, r = 2.5 mm) no longer leaves a visible print on the paste surface. Each sample was used for only one penetration/indentation test and then discarded. Approximately fifty samples were used to assess the setting time of each experimental material.
2.3 Characterization of the cements
After the cements were set for given intervals, they were transferred into acetone (100 %) to stop hydration and then dried in an electric furnace at 37 °C. The bulk density and total porosity of the cements were characterized by means of mercury pycnometry as described elsewhere [16, 17]. The phase composition was characterized by X-ray diffraction (Bruker AXS D8-Advance) using λCuKα1 radiation (0.15418 nm) and a secondary curved graphite monochromator. The surface and broken surfaces of the specimens previously tested in compression and kept to this end were observed by scanning electron microscopy (SEM) (Hitachi S-3500 N). The compressive strength was measured on the samples at a loading rate of 100 N seg−1 using a universal testing machine (Instron-1195, USA) according to ASTM D695-91. Five replicates were tested for each group, and the results were expressed as means ± standard deviation.
2.4 In vitro test in SBF
In order to estimate sample bioactivity, we used what Kokubo et al. [18] proposed in 2006, the Tris-buffered Simulated Body Fluid (SBF), with an ion concentration nearly equal to that of human blood plasma. The cements with L/P ratio of 0.38 mL g−1 were stored in an incubator at 100 % relative humidity and 37 °C for 7 days and then immersed in 10 mL SBF at 37 °C and pH 7.30. The solid/liquid weight ratio was equal to 5 × 10−3, close to the value used by other authors [19–21]. The solution in a shaker water bath was refreshed with 25 % of fresh SBF daily. After different soaking periods (up to 7 days) the specimens were removed from the SBF, rinsed with deionized water, and dried at room temperature.
The samples surfaces were examined with SEM–EDS at 20 keV after the exposure to the SBF. Additional changes in ionic concentration of the SBF were examined using inductively couple plasma optical emission spectroscopy (ICP-OES).
For the transmission electron microscopy (TEM) study the samples were prepared by careful removal of the reaction layer from sample surfaces using a razor blade, and dispersing the powder on the surface of ethanol in a Petri dish. After drying the powder specimens were then collected on carbon coated TEM copper grids of 300 mesh. Electron beam transparent particles were chosen for TEM examination by selected area diffraction (SAD) and also EDS. A Jeol Jem 2010 microscope was operated at 200 keV, and an 80 cm camera length condition was applied for SAD patterns.
2.5 In vitro cell test
Human adipose stem cells (hASC) were isolated from subcutaneous adipose tissue of volunteer female donors undergoing elective liposuction procedures, after obtaining their informed consent according to the procedures approved by the ethics committee [22]. Samples were collected from three different patients aged 25–35 years.
The isolation procedure has been previously established [23]. Briefly, the adipose tissue was washed several times with sterile phosphate buffer saline (PBS). Washed aspirates were treated with 0.075 % w/v collagenase in PBS (Sigma-Aldrich, Steinheim, Germany) for 30 min at 37 °C under gentle agitation. The collagenase was inactivated with an equal volume of control culture media composed of Dulbecco’s modified eagle medium (DMEM) supplemented with 10 % fetal bovine serum (FBS) and antibiotics (100 U mL−1 penicillin and 100 μg mL−1 streptomycin). This suspension was quickly centrifuged (172×g for 10 min) and the pellet was resuspended in a control culture medium and filtered through sterile gauze to remove tissue debris. The percentage of living cells was determined by trypan blue staining. Afterwards, cells were seeded in adherent dishes at a density of 12,000 cells/cm2 and incubated in a 5 % CO2 atmosphere at 37 °C. It was important to wash the cells several times with PBS after the 24-hour incubation. This manipulation allowed the elimination of non-adherent cells these being mainly dead cells, red blood cells and adipocytes. The medium was replaced every 3 days and split by trypsin treatment when cultures reached 80–90 % of confluence.
The hASC were grown in the cement with L/P ratio of 0.38 mL g−1 in a number of 700 cells/mm2. The medium was replaced every 2 days during the course of the experiment (1–10 days). Sample analysis results were obtained in triplicate from three separate experiments.
The surface morphology of specimens was analyzed by SEM–EDS in order to evaluate the cell growth and adhesion to the cements surface. After incubation for 1, 3 and 10 days, the samples were removed from the culture well, rinsed with PBS and fixed with 3 % glutaraldehyde in a 0.1 M cacodylate buffer for 1.5 h at 4 °C. Then they were rinsed and post-fixed in osmium tetroxide for 1 h, before being dehydrated through increasing concentrations of ethanol (30, 50, 70, 90 vol%) with final dehydration in absolute alcohol. After this, they were dried by the critical-point method and palladium coated and examined by SEM–EDS.
2.6 Statistical analysis
Statistical analysis was carried out using the Student’s test. In all cases, the results were considered statically significant at P < 0.05.
3 Results
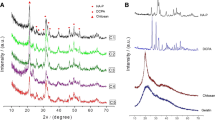
Figure 1 shows the XRD patterns of the as-prepared 85wt%C2S–15wt%TCP powders, and the cement with L/P ratio of 0.35 at 7, 14 and 28 days of setting at 37 °C and 100 % relative humidity as representative of all cements. Before setting, all the peaks correspond with \( \alpha^{\prime}_{\text{H}} \)-C2Sss (JCPDS card no. 083-1494), regardless TCP addition. As the phase is a solid solution, the diffraction peaks are slightly displaced with respect to the corresponding JCPDS card. The displacement range for \( \alpha^{\prime}_{\text{H}} \)-C2Sss phase was 0.1 degrees. After 7 days of setting, there was a remarkable change in XRD patterns, as indicated by the decrease in the \( \alpha^{\prime}_{\text{H}} \)-C2S peaks and the increase of new peaks. This was due to the formation of calcium silicate hydrate (CSH) (JCPDS card no. 29-0374). After 28 days, the intensity of peaks caused by α′-C2Sss was almost negligible, and the intensities at 2θ = 29.042 and 18.504 were enhanced. The XRD patterns of the hydrate resembled that of the CSH reported by Taylor [24, 25].

XRD patterns of (a) as-prepared \( \alpha^{\prime}_{\text{H}} \)-C2Sss powders and the cements with L/P ratio of 0.35 after setting for (b) 7, (c) 14, and (d) 28 days. The peaks intensity of the \( \alpha^{\prime}_{\text{H}} \)-C2Sss (asterisks) decreased gradually with setting time, whereas that of calcium silicate hydrate (black square) increased gradually
The surfaces and broken surfaces of the cements with L/P of 0.35 and 0.41 after setting for 7 days are shown in Fig. 2. The results showed the same features for both cements, which have rough surfaces with tiny agglomerated particles. Moreover, it seemed that cement with L/P of 0.35 had a denser structure than the other cement. On broken surfaces (Figs. 2b, d), it is possible to notice that the pore size was larger than those on the surface and needle-like crystals of aggregated CSH. The cement with a higher L/P showed more porous structures with larger agglomerate grains.

SEM micrographs of the surface and broken surface of the pastes with L/P ratio of 0.35 (a, b) and 0.41 (c, d), respectively after setting for 7 days
Table 1 shows the effect of L/P on setting time, porosity and compressive strength. It is clear to see that the porosity decreased slightly with the prolonged times, and the porosity especially decreased with the decrease of L/P, which was in agreement with the SEM observation showed in Fig. 2. A smaller amount of liquid in the paste led to a large reduction in porosity and increased pressure resistance. Analysis of variance of the compressive strength data showed a significant variation in strength between specimens (P < 0.05).
Table 1 also shows data for the experiments to evaluate the influence of the L/P ratio on cement setting. The results showed that the L/P ratio greatly affected setting times. An increase in setting time was observed with the higher L/P because of lower viscosity in the pastes. Increasing the L/P from 0.35 to 0.41 resulted in a clear increase in I from 6 to 25 min and F from 10 to 50 min.
The essential condition for bioactive material is to establish a permanent attachment with living bone by the formation of a surface carbonate hydroxyapatite (CHA) layer in the body environment. To determine the bioactivity of this material, the cement was soaked in SBF. The SEM micrograph of the samples after soaking in SBF for 1 day is shown in Fig. 3a. It is clear that precipitation of globular particles took place on the cement surface and also into the pores. This morphology does not change further with soaking time although the size of the aggregates increases on average to about 20 μm in diameter and forms a thicker and dense layer. These small spherical particles were determined to be bone-like apatite although from EDS microanalysis the Ca/P ratio was on average 2.3, higher than that in hydroxyapatite. This fact suggests that CHA was forming on the surface according to the results reported by other authors in Si–Ca–P-base materials [26, 27].

a SEM micrograph of the surface of the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement with L/P ratio of 0.35 mL g−1 after soaking in SBF for 1 day, b Changes in the Ca, P and Si concentrations of the SBF solution measured by ICP-OES after soaking \( \alpha^{\prime}_{\text{H}} \)-Ca2SiO4 solid solution cement for various periods
Figure 3b shows the profile of Ca, P and Si ion release from the cement in SBF. There was a burst release of Ca and Si during the dissolution of the specimen in the initial soaking. After that, Ca concentration decreased markedly because of fast consumption of Ca2+ ions for CHA precipitation, which also acted as a “protective” layer on the surface of the matrix, which retarded the dissolution of Si. Unlike the changes in Ca and Si, the P concentration decreased because of the absorption of PO4 3− ions from SBF. After prolonged soaking up to 7 days, the Si concentration continued to increase with a corresponding decrease in the Ca and P concentration indicating, that the ions were used up to form the CHA.
TEM was used to examine the ultrastructure of the surface product formed after the exposure of cement to SBF for 7 days. The characteristic plate-like morphology of the HA-like surface product at low magnification is shown in Fig. 4a. When the specimens were appropriately oriented, the SAD pattern often displayed the (210) ring and (002) arcs corresponding to the 0.308 and 0.344 nm lattice spacing respectively, indicating the preferential orientation of the CHA crystals in the layer (Fig. 4b).

a TEM micrograph of the CHA phase formed on the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement during exposure to SBF after 7 days. b SAD pattern of the phase
Figure 5a shows the hASC adhesion to the cement after the incubation experiment for 1 day. In general, the cells appeared flat and adhered well onto the cement surfaces in all time intervals. The hASC undergo their morphological changes to stabilize the cell-material interface of the hydrated cement. They stabilized close contacts with the cement, and numerous filopodia anchored the cells to the material within 1 day, which indicates that the \( \alpha^{\prime}_{\text{H}} \)-Ca2SiO4 solid solution cement supported hASC adhesion and spreading. The hASC viability assay (Fig. 5b) confirmed the SEM observation, demonstrating the proliferation capability of the cells during the culture study (P < 0.05).

a Morphological aspect of the hASC spreading on \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement for 1 day. b Cell proliferation after culturing in dissolution extracts of the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement for different periods
4 Discussion
Calcium silicate cements, such as ProRootMTA and other Portland-based cements, have been extensively studied in recent years for their mechanical, physical and biological properties [28–30]. Unfortunately, the principal limitation of original calcium silicate MTA cement was related to the slow setting time (approximately more than 170 min) and the difficulty in hand manipulation and clinical use. In this study we have demonstrated that the addition of KH2PO4 to the basic composition of all the experimental dicalcium silicate cements doped with tricalcium phosphate reduced the final setting time to 10–50 min.
Particle size, sintering temperature, liquid phase, and composition of powders, as well as the ratio of liquid to powder, played crucial roles in the setting time of the paste materials [26]. When the powder and liquid phases were mixed in an appropriate ratio, they formed a paste that hardened by entanglement of the crystals precipitated in the paste at both body and room temperature, as observed by SEM (Fig. 2b, d). The entanglement structure, consisting of fine particle agglomerates or needle-like crystals, could be considered a hydration product of a CSH phase that might be responsible for causing the particles to adhere to one another. According to the setting reaction of calcium silicate-based Portland cement [30], it is possible to consider that the product could be related to the hardening mechanism of the dicalcium silicate cements doped with tricalcium phosphate.
The setting property of \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement results from the progressive hydration of the SiO4 4− ions in \( \alpha^{\prime}_{\text{H}} \)-C2Sss. When \( \alpha^{\prime}_{\text{H}} \)-C2Sss reacts with water, a nanoporous, amorphous CSH phase is deposited on the original \( \alpha^{\prime}_{\text{H}} \)-C2Sss, while Ca(OH)2 crystals nucleate and grow in the available capillary pore space in the previously deposited CSH phase (Fig. 1). As time proceeds, the CSH phase polymerizes and hardens. This phenomenon suggests that the setting progress is mainly attributed to the formation of a solid network, which is also associated with the densification and the increase of the mechanical strength. The decrease of the porosity is due to development of microstructure of the cement and is associated with an increase of the strength.
On the other hand, the compressive strength of the calcium phosphate cements with various L/P ratios varied from 3.8 to 16.3 MPa (Table 1). Thus, if the convenient operability and the final mechanical property after injection are taken into account, the pastes with low L/P (0.35–0.37) may be the preferred candidate for surgical applications.
Several in vivo studies have suggested that the formation of the CHA layer is an essential requirement in events leading to subsequent osseointegration of implants [31, 32]. Compared to synthetic hydroxyapatite, the surface CHA layer of silicon- containing bioactive materials is more similar, in terms of crystallinity, to the apatite of bone tissue, and consequently a greater proportion of bone bonding has been reported for silicon-containing bioactive materials than for hydroxyapatite [33, 34]. After immersion in a simulated body fluid for as little as 1 h, the cement specimens induced the precipitation of apatite spherical particles (Fig. 3a), indicating its high bioactivity. Similar apatite deposit has been observed precipitatinf on MTA cement surfaces immersed in SBF after 1 week [35, 36]. The Ca2+ and OH− ions released from MTA, mainly because of calcium hydroxide dissociation, react with phosphorus ions of SBF solution, resulting in the precipitation of carbonated apatite on the MTA surface.
For the in vitro solution-driven paste degradation on the basis of the Si release, it can be seen that the rate of Si release increased rapidly for the first day, and then continued moderately after a prolongation of soaking time. This dissolution is primarily governed by the chemical composition and physical characteristics (porosity, surface area, grain size, etc.) of the hydrated calcium silicate. Moreover, the precipitated CHA also acts as a shielding layer on the surface of the cement which results in a decrease in the rate of Si release. This hydrated cement exhibits moderate degradation with regard to Si release. The combined SEM–EDS, ICP, TEM, and SAD analyses showed that the \( \alpha^{\prime}_{\text{H}} \)-Ca2SiO4 solid solution cement is not only bioactive, but also dissolvable in SBF. However, more in vitro and in vivo degradation studies need to be conducted in order to confirm the degradation properties of the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement.
In vitro cell–material interaction is a useful criterion in the evaluation of new biomaterials. The present study indicates that cement supports hASC cells proliferation. Cells have been found in close contact with cements. This cell behavior suggests that the surfaces of the material are non-irritant and do not affect the structural integrity of the cell. The cells appeared flat and exhibited an intact, well-defined morphology, with cytoplasmic extensions. The preservation of cytoplasmic extensions is important because they allow a vital three-dimensional network within bone.
Our results suggest that the ionic products of the cement dissolution can provide an adequate stimulus for cell proliferation. Previous studies showed that the ionic products of the bioactive ceramic containing calcium, silicate and phosphorous stimulated cell proliferation [37–39]. \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement is a calcium- and silicate-containing material with phosphorous in a solid solution and our results showed that the dissolution extracts of the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement in a certain concentration range also promoted cells growth.
5 Conclusions
In conclusion, the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cements that exhibited shortened setting times were successfully developed. Among the four cements studied, those containing 0.35 and 0.37 L/P ratio cements might prove the most useful for bone/dental repair requiring a setting time of a few minutes. The new \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement could quickly form carbonate hydroxyapatite spherical particles after immersion in a simulated body fluid for 1 day. The hASC cell viability increased on the cement surfaces after 24 h of incubation. This study supports the hypothesis that the \( \alpha^{\prime}_{\text{H}} \)-C2Sss cement obtained displays in vitro bioactivity and biocompatibility, which makes it a potential candidate for surgical applications. However, further in vivo studies are required to evaluate the clinical potential of \( \alpha^{\prime}_{\text{H}} \)-dicalcium silicate cements doped with tricalcium phosphate.
References
Nicoleau L, Nonat A. The di- and tricalcium silicate dissolutions. Cem Concr Res. 2013;47:14–30.
Wesselsky A, Jensen OM. Synthesis of pure Portland cement phases. Cem Concr Res. 2009;39(11):973–80.
Kalousek GL, Nelson EB. Hydrothermal reactions of dicalcium silicate and silica. Cem Concr Res. 1978;8(3):283–9.
Öztürk A, Suyadal Y, Oǧuz H. The formation of belite phase by using phosphogypsum and oil shale. Cem Concr Res. 2000;30(6):967–71.
Gandolfi MG, Perut F, Ciapetti G, Mongiorgi R, Prati C. New Portland cement-based materials for endodontics mixed with articaine solution: a study of cellular response. J Endod. 2008;34:39–44.
Coleman NJ, Nicholson JW, Awosanya K. Preliminary investigation of the in vitro bioactivity of white Portland cement. Cem Concr Res. 2007;37(11):1518–23.
Saunder WP. A prospective clinical study of periradicular surgery using mineral trioxide aggregate as a root-end filling. J Endod. 2008;34:6655–60.
Pace R, Giuliani V, Pagavino G. Mineral trioxide aggregate as repair material for furcal perforation: case series. J Endod. 2008;34:1130–3.
Holden DT, Schwartz SA, Kirkpatrick TC, Schindler WG. Clinical outcomes of artificial root-end barriers with mineral trioxide aggregate in teeth with immature apices. J Endod. 2008;34:812–7.
Gandolfi MG, Farascioni S, Pashley DH, Gasparotto G, Prati C. Calcium silicate coating derived from Portland cement as treatment for hypersensitive dentine. J Dent. 2008;36:565–78.
Torabinejad M, Hong CU, McDonald F, Pitt Ford TR. Physical and chemical properties of a new root-end filling material. J Endod. 1995;2:349–53.
Abdullaha D, Pitt Ford TR, Papaioannouc S, Nicholsond J, McDonald F. An evaluation of accelerated Portland cement as a restorative material. Biomaterials. 2002;23:4001–10.
Nurse RW, Welch JH, Gutt W. High-temperature equilibria in the system dicalcium silicate–tricalcium phosphate. J Chem Soc. 1959;1077–83.
Fix W, Heymann H, Heinke R. Subsolidus relations in the system 2CaO·SiO2–3CaO·P2O5. J Am Ceram Soc. 1969;52(6):346–7.
Rubio V, de la Casa-Lillo MA, de Aza S, de Aza PN. The system Ca3(PO4)2–Ca2SiO4. The sub-system Ca2SiO4–7CaOP2O52SiO2. J Am Ceram Soc. 2011;94(12):4459–62.
Takagi S, Chow LC. Formation of macropores in calcium phosphate cement implants. J Mater Sci Mater Med. 2001;12(2):135–9.
Almirall A, Larrecq G, Delgado JA, Martinez S, Planell JA, Ginebra MP. Fabrication of low temperature macroporous hydroxyapatite scaffolds by foaming and hydrolysis of an alpha-TCP paste. Biomaterials. 2004;25(17):3671–80.
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone activity? Biomaterials. 2006;27:2907–15.
Kokubo T. Novel bioactive materials. An Quim. 1997;93(1):49–55.
Martinez IM, Velasquez P, Meseguer-Olmo L, de Aza PN. Production and study of in vitro behaviour of monolithic α-tricalcium phosphate based ceramics in the system Ca3(PO4)2–Ca2SiO4. Ceram Int. 2011;37:2527–35.
Carrodeguas RG, de Aza AH, Jimenez J, de Aza PN, Pena P, Lopez-Bravo A, de Aza S. Preparation and in vitro characterization of wollastonite doped tricalcium phosphate bioceramics. Key Eng. 2008;361–363:237–40.
Dubois SG, Floyd EZ, Zvonic S, Kilroy G, Wu X, Carling S, Halvorsen YD, Ravussin E, Gimble JM. Isolation of human adipose-derived stem cells from biopsies and liposuction specimens. Methods Mol Biol. 2008;449:69–79.
Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, Benhaim P, Lorenz HP, Hedrick MH. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7:211–26.
Taylor HFW. Hydrated calcium silicates: part I. Compound formation at ordinary temperatures. J Chem Soc. 1950;25:3682–90.
Taylor HFW. Cement chemistry. London: Academic Press; 1990.
Gutierrez GV, Nonell JM, Ojeda LL, de Aza PN, de Aza S. Dental cements from polyacrilic acid and wollastonite. Bol Soc Ceram V. 2005;44(2):89–94.
de Aza PN, Luklinska ZB, Anseau M. Bioactivity of diopside ceramic in human parotid saliva. J Biomed Mater Res B. 2005;73B:54–60.
Bortoluzzi EA, Broon NJ, Durante MAH, de Demarchi ACO, Bramante CM. The use of a setting accelerator and its effect on pH and calcium ion release of mineral trioxide aggregate and white portland cement. J Endod. 2006;32:1194–7.
Bortoluzzi EA, Broon NJ, Bramante CM, Felippe WT. Tanomaru Filho M, Esberard RM. The influence of calcium chloride on the setting time, solubility disintegration, and pH of mineral trioxide aggregate and white portland cement with a radiopacifier. J Endod. 2009;35:550–4.
Older I. Hydration, setting and hardening of Portand cement. In: Hewlett PC, editor. Lea’s chemistry of cement and concrete. 4th ed. Oxford: Butterworth-Heinemann; 2007. p. 241–97.
Minarelli-Gaspar AM, Saska S, Carrodeguas RG, de Aza AH, Pena P, de Aza PN, et al. Biological response to wollastonite doped α-tricalcium phosphate implants in hard and soft tissues in rats. Key Eng Mater. 2009;396–398:7–10.
de Val JE Mate-Sanchez, Calvo-Guirado JL, Delgado-Ruiz RA, Ramirez-Fernandez MP, Martinez IM, Granero-Marin JM, Negri B, Chiva-Garcia F, Martinez-Gonzalez JM, De Aza PN. New block graft of α-TCP with silicon in critical size defects in rabbits: Chemical characterization, histological, histomorphometric and micro-CT study. Ceram Int. 2012;38:1563–70.
Kenny SM, Buggy M. Bone cements and fillers: a review. J Mater Sci Mater Med. 2002;13:119–1206.
Minarelli Gaspar AM, Saska S, da Cunha LR, Bolini PD, Carrodeguas RG, De Aza AH, Pena P, De Aza PN, De Aza S. Comparison of the biological behavior of wollastonite bioceramics prepared from synthetic and natural precursors. Key Eng. 2008;396-363:1083–6.
Tay FR, Pashley DH, Rueggeberg FA, Loushine RJ, Weller RN. Calcium phosphate phase transformation produced by interaction of the Portland cement component of white MTA with a phosphate-containing fluid. J Endod. 2007;33:1347–51.
Oliveira IR, Andrade TL, Jacobovitz M, Pandolfelli VC. Bioactivity of calcium aluminate endodontic cement. J Endod. 2013;39(6):774–8.
Magallanes-Perdomo M, De Aza AH, Mateus AY, Teixeira S, Monteiro FJ, De Aza S, Pena P. In vitro study of the proliferation and growth of human bone marrow cells on apatite–wollastonite-2 M glass ceramics. Acta Biomater. 2010;6(6):2254–63.
Martinez IM, Velasquez PA, De Aza PN. Synthesis and stability of α-tricalcium phosphate doped with dicalcium silicate in the system Ca3(PO4)2–Ca2SiO4. Mater Charact. 2010;61:761–7.
Meseguer-Olmo L, Aznar-Cervantes S, Mazon P, De Aza PN. In vitro behaviour of adult mesenchymal stem cells of human bone marrow origin seeded on a novel bioactive ceramics in the Ca2SiO4–Ca3(PO4)2 system. J Mater Sci Mater Med. 2012;23:3003–14.
Acknowledgments
Part of this work was supported by Generalitat Valenciana ACOM/2009/173.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
de Aza, P.N., Zuleta, F., Velasquez, P. et al. \( {\alpha^{\prime}}_{\text{H}} \)-Dicalcium silicate bone cement doped with tricalcium phosphate: characterization, bioactivity and biocompatibility. J Mater Sci: Mater Med 25, 445–452 (2014). https://doi.org/10.1007/s10856-013-5084-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10856-013-5084-1