
Abstract
The poorly water-soluble chemotherapeutic agents, paclitaxel (PTX), exhibit serious clinical side effects upon oral administration due to poor aqueous solubility and a high degree of toxic effects due to non-specific distribution to healthy tissues. In our efforts, we formulated biocompatible dietary lipid-based nanostructured lipidic carriers (NLCs) to enhance the oral bioavailability of PTX for treatment of the liver cancer. A three-factor, three-level Box–Behnken design was employed for formulation and optimization of PTX-loaded NLC formulations. PTX-loaded NLC formulation prepared by melt-emulsification in which glyceryl monostearate (GMS) was used as solid lipid and soybean oil as liquid lipid, while poloxamer 188 and Tween 80 (1:1) incorporated as a surfactant. In vitro drug release investigation was executed by dialysis bag approach, which indicated initial burst effect with > 60% drug release within a 4-h time period. Moreover, PTX-NLCs indicated high entrapment (86.48%) and drug loading efficiency (16.54%). In vitro cytotoxicity study of PTX-NLCs performed on HepG2 cell line by MTT assay indicated that PTX-NLCs exhibited comparatively higher cytotoxicity than commercial formulation (Intaxel®). IC50 values of PTX-NLCs and Intaxel® after 24-h exposure were found to be 4.19 μM and 11.2 μM. In vivo pharmacokinetic study in Wistar rats also indicated nearly 6.8-fold improvement in AUC and Cmax of the drug from the PTX-NLCs over the PTX suspension. In a nutshell, the observed results construed significant enhancement in the biopharmaceutical attributes of PTX-NLCs as a potential therapy for the management of human liver carcinoma.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The worldwide incidence of hepatocellular carcinoma (HCC) is very high and accounts for the second most leading cause of death due to liver carcinoma (1). The pathogenesis of liver carcinoma is considered to be highly complicated based on its progression, which depends on several factors such as environment, genetics, and lifestyle. Moreover, a variety of risk factors and toxicants tends to influence the occurrence of liver cancer such as aflatoxin, ethanol, sex steroid, hepatitis B and C virus infection, liver cirrhosis, unclean water, nitrosamine, and trace elements. Hence, pathogenesis and progression of liver cancer is considered to be highly complicated, which makes diagnosis and treatment of liver cancer become highly challenging in biomedical research (2).
The oral route of drug administration is the most chosen and handy route but hydrophobic drugs lack high systemic exposure because of its poor water solubility. Not only the solubility as well as other intrinsic qualities of the drug, the physiological milieu in the gastrointestinal tract (GIT) which includes buffer species, pH values, and bile salts, do hamper the absolute absorption (3,4).
Paclitaxel (PTX) is a semi-synthetic cyclodecane derivative of taxol, obtained from the Western yew plant Taxus brevifolia exhibit powerful antineoplastic activity and primarily indicated for the prevention and treatment of diverse varieties of carcinomas. It interferes with the normal function of microtubules in the process of cell division through binding with tubulin, acts as a mitotic poison, and inhibits the progression of cell division. PTX is the first-line drug of choice for several types of cancers like breast, ovary, and non-small cell lung cancer. It has also been highly useful in modulating the incidence and progression of HCC. However, PTX drug therapy is challenging in clinical use owing to poor water solubility, high lipophilicity (log P 3.96) and P-gp efflux, thus showing slow dissolution rate and poor oral bioavailability (< 8%), along with non-specific distribution related toxicity to the healthy tissues (2,5,6,7).
In this regard, lipidic nanoparticles, especially nanostructured lipid carriers (NLCs), which are considered as second-generation solid lipid nanoparticles (SLNs), are highly popular in drug delivery due to their potential to enhance the oral bioavailability of hydrophobic drugs. NLCs deliver the drug without affecting the physicochemical characteristics of the drugs. These biocompatible lipidic nanoparticles are made up of a blend of dimensionally dissimilar lipid molecules in which solid lipid is mixed with liquid lipid to circumvent the challenges of SLNs. The use of a combination of solid and liquid lipids accommodates the drugs in molecular form in their spatial structure or in amorphous clusters. This allows significantly higher encapsulation of both hydrophilic and lipophilic therapeutic agents, reduction of the expulsion of the drug upon storage, and regulation of drug release profile by altering lipid matrix and significantly improving stability in different pH of GIT (8,9,10,11,12). NLCs on account of their hydrophobic nature and particle size in nanometric range (< 200 nm) are extensively explored as oral drug delivery system for a large number of chemotherapeutic drugs and showed excellent therapeutic efficacy in the studies performed in animals. NLCs can facilitate the distribution of chemotherapeutic drugs and enhances the retention of the drugs at tumor site through enhanced permeation and retention (EPR) effect. Furthermore, it was envisaged that cholesterol-rich NLCs may efficiently accumulate in liver carcinomas marked with over expression of low-density lipoprotein (LDL) receptors (3,13,14,15,16).
Systematic optimization of nanopharmaceutical products using quality by design (QbD) principles is a customary practice nowadays (17,18). This involves application of suitable experimental designs for optimization of the highly influential formulation and process parameters (19). Besides, QbD facilitates a thorough understanding of the system and helps in reducing developmental cost, time, efforts, and failures at the end stage of product development life cycle (20,21,22). Several literature reports have demonstrated the application of QbD principles in the development of nanopharmaceutical formulations (17,18).
Therefore, the endeavor of the contemporary research was to develop the biocompatible NLCs of PTX using dietary lipids for improving the oral bioavailability and anticancer activity. NLCs were prepared using soybean oil as liquid lipid and GMS as solid lipid, as well as poloxamer 188 as a surfactant. Box–Behnken design (BBD) was employed for systematic optimization of the NLCs and evaluated for different response variables. The optimized PTX-loaded NLCs were further screened through in vitro, ex vivo, and in vivo investigations.
MATERIALS AND METHOD
Materials
PTX was procured from Fresenius Kabi Oncology Limited, Gurugram, India. Intaxel®, a commercial formulation (Fresenius Kabi Oncology Limited, Gurugram, India) of PTX containing Cremophor EL/ethanol was procured from the market. Compritol 888 ATO, GMS, stearic acid, oleic acid, Gelucire44/44, and Labrafil M 1944 CS were purchased from Gattefosse, CEDEX, France. Olive oil, soybean oil, canola oil, Tween 80 and 20, and methyl alcohol (AR grade) were procured from Merck, India, while Poloxamer 188 was procured from BASF, Mumbai, India. Human hepatocellular carcinoma (HepG2) cell line was procured from the National Centre for Cell Science (NCCS), Pune, India.
Methods
Screening of Formulation Excipients
An excess amount of PTX was dissolved in 1 mL of different liquid lipids using 2-mL glass vials to determine the saturation solubility. The glass vials were tightly closed and kept on constant stirring using a mechanical water bath shaker maintained at 37 ± 2°C for 72 h. The excipient mixtures were centrifuged at 10,000 rpm for 10 min by the use of a high-speed centrifuge (Thermo Fisher Scientific, India). The separated supernatant was further dissolved in acetonitrile to determine the saturation solubility of the drug using RP-HPLC technique at 227 nm. For solid lipid, precisely weighed quantity (1 g) of the solid lipids were taken in the beaker followed by constant stirring (200 rpm) and heating at just above its melting point by the use of magnetic stirrer with heating facility (Remi Instrument Ltd., India). In the above completely melted solid lipid, PTX was dissolved in increases of 1 mg until PTX was fully dissolved and the quantity of solid lipids needed to dissolve PTX was determined in triplicate and presented as average value (mg/g) ± SD (23). Binary lipid phase was selected on account of good solubility of solid and liquid lipid in the fixed ratio. For this, selected solid and liquid lipid was intermixed in the ratio ranging from 90:10 to 10:90 to calculate the equilibrium solubility. Lipid blends were put on the magnetic stirrer with a hot plate and stirred continuously at 200 rpm for 1 h at 80 ± 2°C. The samples were assessed visually for turbidity and phase separation. Surfactant concentration was also chosen on the basis of aqueous titration study. Primary emulsions were prepared by melting and agitating the lipid mixture using a magnetic stirrer, where the different ratio of surfactants was added followed by dropwise addition of double-distilled water. The developed preparation was analyzed through naked eye for phase clarity and homogeneity (24).
Development of PTX-NLCs
PTX-NLCs were developed by melt-emulsification ultrasonication process as per the reported procedure with suitable modification (23). Accurately weighed amount of PTX and solid lipid (GMS) were placed a magnetic stirrer with heating facility to melt at 70 ± 2°C to obtain a homogeneous mixture and transparent oil phase. The liquid lipid (soybean oil) and cholesterol were mixed in a separate beaker with required quantity of ethanol to obtain a clear solution. In the meantime, 10 mL of the aqueous phase was separately prepared in a beaker by dispersing surfactant/co-surfactant (Tween 80 and poloxamer 188 at 1:1 ratio) in purified water and kept on the hot plate to heat (70 ± 2°C). After that, the liquid lipid and aqueous phase was put into the molten solid lipid phase drop by drop followed by stirring at 70 ± 2°C and allowed to stand for 30 min. Finally, the resultant emulsion was put on Ultrasonicator (Hielscher, Ultrasound UP-50H, Darmstadt, Germany) to sonicate for 3–5 min to obtain the desired NLCs.
Optimization of PTX-NLCs by BBD
A 3-factor, 3-level containing Box–Behnken design was used to optimize the PTX-NLCs using Design-Expert® software ver. 9.0.1 (State-Ease Inc., Minneapolis, USA). The concentration of lipid (X1), surfactant (X2), and sonication time (X3), were chosen as the factors or independent variables at low (− 1), medium (0), and high (+1) levels, respectively. A total of 17 experimental trials were generated and were evaluated for particle size (Y1), % entrapment efficiency (Y2), and % drug loading (Y3) as the dependent or response variables. Table I enlists the design matrix with experimental trials of NLCs containing different combination of the factors and obtained values of the responses. Mathematical modeling was carried out using the quadratic polynomial model to establish the cause-and-effect relationship between the independent and dependent variables. Finally, the best possible optimum formulation was identified by mathematical optimization using desirability function and graphical optimization by overlay plot.
Characterization of the Optimized PTX-NLCs
Particle Size, Polydispersity Index, and Zeta Potential Analysis
The particle size, polydispersity index (PDI), and zeta potential of PTX-NLCs were determined after 100-fold dilution in double-distilled water followed by dynamic light scattering (DLS) approach employing Malvern Zetasizer (Malvern Instruments Ltd., Worcestershire, UK).
Determination of Drug Entrapment and Loading Efficiency
The drug entrapment and loading efficiency of PTX-NLCs were analyzed using the reported procedure (23). The PTX-NLCs dispersion was subjected to high-speed centrifugation (15,000 rpm for 15 min). Subsequently, the supernatant fraction was collected and filtered by Millipore VR membrane (0.2 μm), suitably diluted with acetonitrile and analyzed by RP-HPLC technique at 227 nm to quantify the amount of unencapsulated drug. The trial was run in triplicate and outcomes were presented as average value ± SD. Consequently, the drug entrapment efficiency as well as drug loading capacity was calculated by the use of Eqs. 1 and 2 as follows.
where, Wtotal is the sum total of PTX, Wfree is the quantity of unencapsulated PTX, and Wlipid is the total weight of solid and liquid lipid.
Transmission Electron Microscopy Analysis
The optimized PTX-NLCs were subjected for transmission electron microscopy (TEM) analysis to find out the surface morphology and particle size. The optimized PTX-NLC was diluted 50-folds with double-distilled water and stained negatively by 1% phosphotungstic acid and then dried on the carbon-coated grid. Finally, it was subjected to observation using TEM (Morgagni 268D, Fei Electron Optics, Netherlands) at 100 kV.
In vitro Drug Release Study
The in vitro drug release analysis was executed employing a dialysis bag approach using 0.25 L of simulated intestinal juice (pH 6.8) maintaining at 37 ± 0.5°C for 24 h at 100 rpm. The dialysis membrane with molecular weight cutoff 12 kDa (Merck, India) was used (25,26). Correctly weighed quantity (1 mL) of the optimized PTX-NLCs (equivalent to 20 mg of the PTX) was kept in the dialysis bag and put into the dissolution medium. An aliquot (2 mL) of the sample was taken at a series of time points (0.5, 1, 2, 4, 6, 8, 12, and 24 h), followed by replenishment with an equal volume of fresh dissolution medium to maintain the sink conditions. The release of PTX from optimized NLCs was evaluated in contrast to the PTX suspension release data. The previously filtered sample (passed through 0.22-μm filtering unit) was subjected for drug content analysis employing RP-HPLC technique (21). The analysis was performed in triplicate and outcomes were shown as average value ± SD. Further, the obtained drug release data was subjected to the evaluation of release kinetic using various mathematical models (zero-order, first-order, Higuchi and Korsmeyer-Peppas).
In vitro Cell Line Study
Cell Culture and Growth Condition
The HepG2 cancer cell line was cultivated in Eagle’s minimum essential medium (MEM) with 2.0 mM L-glutamine, 1.5 g/L sodium bicarbonate, 100 U/mL penicillin and 100 μg/mL streptomycin, and boosted with 10% (v/v) fetal bovine serum. Cells were developed at 37°C and 5% CO2 humidified ambience in an incubator (Remi Instruments, Mumbai, India).
MTT Cell Viability Assay
MTT assay was employed to identify the cell viability of optimized PTX-NLCs and commercial formulation (Intaxel®) as per the previously reported procedure (27). About 1 × 104cells/well of HepG2 were seeded in 100 μL absolute culture medium in 96-well culture plate and incubated for the whole night in humidified air. The stock of optimized PTX-NLCs and commercial formulation (Intaxel®) were made in phosphate buffer saline (PBS) and diluted into culture medium to the preferred concentrations (2, 5, 10, and 20 μM) and transferred to the culture wells. Subsequent to 24 h of the incubation phase, 10 μL of MTT (5 mg/mL in PBS) was supplemented and re-incubated at 37°C until purple formation of formazan crystals takes place. The formazan blue crystals were mixed in 100 μL of dimethyl sulfoxide (DMSO) and analyzed at 540 nm by the use of microplate ELISA reader (BIORAD 680, USA). The plot of percentage cell viability against PTX concentrations was considered to determine the IC50. The alterations in cellular morphology were examined using inverted phase contrast microscopy (Nikon ECLIPSE Ti-S, Nikon Instruments, Co., Ltd., China).
Intracellular ROS Assay
The intracellular reactive oxygen species (ROS) production was examined by fluorescence microscopic imaging method as per the earlier protocol (28). The cells (1 × 104 per well) were treated with two different concentrations 2 μM and 5 μM of optimized PTX-NLC for 12 h. Afterward, the cells were incubated with dichloro-dihydro-fluorescein diacetate (10 mM) at 37°C for 30 min and cleansed with PBS. Intracellular fluorescence intensity of cells was observed with an inverted fluorescent microscope (Nikon ECLIPSE Ti-S, Nikon Instruments, Co., Ltd., China). In fluorometric examination, the cells (1 × 104 per well) were seeded and exposed with optimized PTX-NLC in 96-well (8 × 12 matrix) black bottom culture plate. Fluorescence intensity was recorded with multiwell microplate photometers (Synergy H1 Hybrid Multi-Mode Reader, BioTek, Mumbai, India) at an excitation wavelength of 485 nm and an emission wavelength of 528 nm. Results were presented as percentage of fluorescence intensity compared to the control wells.
Cellular Apoptosis Study
Fluorescent nuclear dye DAPI was utilized to investigate the apoptotic potential of optimized PTX-NLC as per earlier procedure (28). HepG2 cells (1 × 105 cells per well) were seeded in 24-well culture plate for the night and exposed with optimized PTX-NLC for 24 h. After the incubation phase, cells were cleansed and placed in 4% p-formaldehyde for 15 min. Then, permeabilization was done with permeabilizing buffer (3% p-formaldehyde and 0.5% Triton X-100) for 10 min. Subsequent to staining with DAPI dye (50 μg/mL), photographs of condensed nuclei facing apoptosis were taken with the help of an inverted fluorescent microscope (Nikon ECLIPSE Ti-S, Nikon Instruments, Co., Ltd., China). Apoptosis was semi-quantified by morphological alterations of nuclei employing approximately 500 cells/well denoting one sample.
Animal Study
All the animal experimental protocol was executed successfully as per prior approval by the Animal Ethics Committee of Integral University, Lucknow, U.P, India. Albino Wistar rats (200–250 g each) were chosen and acclimatized separately in a suitable cages with adequate food (ad libitum) and water.
Ex Vivo Intestinal Permeation Assessment
The ex vivo intestinal permeation examination was performed to compare optimized PTX-NLCs with the PTX-suspension as per the previously reported procedure (29). The animals were subjected to overnight fasting and sacrificed by cervical dislocation. The intestine was obtained by surgical removal followed by flushing with saline and jejunum portion was harvested. The PTX (5 mg) suspension was prepared using 0.2% w/v carboxy-methylcellulose in 5-mL water and then 2 mL of sample was put into the mucosal side and firmly tied both the ends of the intestinal sac. The sac filled with PTX suspension was dipped in a jacketed glass fabrication accommodated with 100 mL of Kreb’s ringer solution pre-warmed at 37 ± 2°C by an outer water jacketed tube. Then, 2 mL of sample was taken at different time hiatuses from serosal medium and refilled with freshly prepared Kreb’s ringer solution. The withdrawn samples were passed through a 0.22-μm membrane filter and drug content was quantified through HPLC. A similar procedure was used for evaluating the permeability of optimized PTX-NLCs. The investigation was done in triplicate and outcomes were shown as mean value ± SD. The apparent permeability coefficient (Papp) of PTX suspension and optimized PTX-NLCs was determined by the use of Eq. 3.
where F is the membrane permeation flux, A is the barrier membrane surface area, and C0 is the preliminary concentration of the drug in the mucosal medium. Membrane permeation flux (F) was determined by drawing the slope of the linear portion of the graph between cumulative amount of drug permeated (μg) from the intestinal sac against time (min).
In Vivo Pharmacokinetic Study
A unit dose paralleled design pharmacokinetic examination was executed in rats. The rats were randomly divided into two groups (n = 6 in each group) and subjected to overnight fasting condition with free access to water. PTX suspension and optimized PTX-NLCs containing PTX equivalent to animal dose were administered orally to the animals using a cannula. Samples (~ 0.5 mL blood) were collected from the retro-orbital plexus from time to time in heparinized micro-centrifuge tubes. Plasma was centrifuged at 10,000 rpm for 10 min and the PTX was separated by the use of acetonitrile as an extracting solvent. All the samples were passed through 0.22-μm membrane filter and subjected for drug content analysis by the use of previously reported and validated bioanalytical RP-HPLC technique (29). Pharmacokinetic modeling and data analysis were performed using Win-Nonlin software version 5.0 (M/s Pharsight, CA, USA). One-compartment open body model with zero lag time using modified Wagner-Nelson method was used for computing various pharmacokinetic parameters.
Statistical Analysis
All the obtained data in different studies were articulated as mean ± SD and statistically evaluated by one-way analysis of variance (ANOVA) followed by Tukey’s test at 5% level of statistical significance.
RESULTS AND DISCUSSION
Screening of Excipients
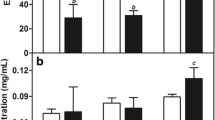
As indicated by the results of solubility studies in different liquid lipids (Fig. 1a), PTX showed maximum solubility in soybean oil (78.42 ± 7.78 mg/mL), whereas screening of different solid lipids (Fig. 1b) showed maximum solubility in GMS (67.76 ± 6.52 mg/g). Thus, soybean oil and GMS were chosen as liquid and solid lipids, respectively. Using the selected lipids in the ratio 7:3 w/w, the aqueous titrations were performed for screening of surfactant. The visual observation for turbidity and phase separation was taken as the end point. On the basis of phase clarity and stability of primary emulsion formed by aqueous titration, polysorbate 80 and poloxamer 188 in the ratio of 1:1 was selected as the surfactant for the development of NLCs (30).

Solubility of PTX in different a liquid lipids and b solid lipids; data expressed as mean ± 1 SD (n = 3)
Preparation and Optimization of PTX-loaded NLC
The optimization data analysis was performed by multiple linear regression analysis, where the experimental data are fitted to the second-order (quadratic) mathematical model with interaction terms, as shown in Eq. 4. Based on the selected model, the recorded values of the responses were subjected for comparison with predicted values, as enlisted in Table II. The selected mathematical model, which indicated goodness of fit of the experimental data, as is evident from Table III indicating quadratic model fitness and statistical parameters for all the studied response variables.
where Y is the response related to every factor level amalgamation; β0 is the intercept; β1 to β9 are the coefficients of different model terms; X1, X2, and X3 are the linear model terms; X1X2, X2X3, and X1X3 are the linear interaction terms; while X12, X22, and X33 are the quadratic model terms.
The consequences of regression analysis indicated that the quadratic nature of the relationship between the factors is best suitable. The studied independent variable viz. the concentration of total lipids (GMS and soybean oil) have a positive effect on the response Y1 (particle size), Y2 (entrapment efficiency), and Y3 (drug loading). The concentration of surfactants (poloxamer 188) exhibited positive effect on response Y2 (entrapment efficiency) and Y3 (drug loading) whereas the concentration of surfactant also revealed an inverse effect on response Y1 (particle size). On the other hand, sonication time revealed an inverse effect on all the responses, i.e., Y1 (particle size), Y2 (entrapment efficiency), and Y3 (drug loading).
Effect of Independent Variables on Particle Size (Y 1)
The polynomial equation indicating the relationship among the factors on particle size (Y1) is given in Eq. 5 as follows:
As per the above polynomial equation, the concentration of total lipids (A) was found to be directly dependent on particles size (Y1). Figure 2a illustrates the 3D-response surface plot for particle size indicating the relationship among the factors. Small particle size is desired for efficient oral delivery of NLCs. It was noticed that particle size of PTX-NLCs increases with the increment made in the concentration of total lipid (GMS and soybean oil). Increase in surfactant concentration (B) and sonication time (C), however, resulted in a decrease in the particle size. This drop in size at higher concentration of surface-active agent may be attributed to the reduced interfacial tension between two phases (aqueous and lipid phases), which causes the production of emulsion droplets of smaller size (31). Higher concentration of surfactants efficiently stabilizes the nanoparticles by developing a steric barrier on the surface of nanoparticles and thereby prevents aggregation of nanoparticles. Another probable elucidation for such reduction in the particle size (Y1) may happen from the duration of sonication time (C), involved in the last stage of development of NLCs (32,33,34).

Three-dimensional (3D) response surface plots showing simultaneous influence of independent variables on response parameters: a particle size, b entrapment efficiency, and c drug loading of PTX-loaded NLC formulated as per Box-Behnken experimental design
Effect of Independent Variables on Entrapment Efficiency (Y 2)
The polynomial equation indicating the relationship among the factors on entrapment efficiency (Y2) is given in Eq. 6 as follows:
As per the above polynomial equation, the concentration of total lipids (A) was directly dependent on entrapment efficiency. Figure 2b illustrates the 3D-response surface plot for entrapment efficiency indicating the relationship among the factors. Higher entrapment efficiency is desirable for NLCs to retain the maximum amount of drug in it. From the response graph, it was revealed that increasing the amount of lipid (GMS and soybean oil) from 2 to 4% resulted in a gradual increment in entrapment efficiency. This may be due to the hydrophobic nature of GMS and soybean oil that increased the encapsulation of drug in the lipid as the higher concentration of lipid is available to encapsulate the drug. Increment in the final concentration of surfactant (B) and sonication time (C) leads the reduced entrapment efficiency of NLCs. Greater concentration of surfactant in the continuous phase may enhance the partition of the drug from dispersed to the continuous phase of the medium. Enhancement in the partition of the drug can be attributed to the enhanced solubilization of the drug in the continuous aqueous phase which facilitates easy dispersion in the lipid matrix. On the contrary, increase in sonication time leads reduced entrapment efficiency because the decrease in particle size results in poor entrapment of drug (32,33,34).
Effect of Independent Variables on Drug Loading (Y 3)
The polynomial equation indicating the relationship among the factors for drug loading (Y3) is given in Eq. 7 as follows:
As per the above polynomial equation, the concentration of total lipids (A) was directly dependent on drug loading. Figure 2c illustrates the 3D-response surface plot indicating the effect of studied factors on the response drug loading. Higher drug loading is desirable for NLCs so that the maximum amount of drug can be stacked, and a higher amount can reach the target site. In our study, it was noticed that the increase in total lipid concentration (A) causes an increase in drug loading (Y3). Increase in the lipid concentration also reduces the efflux of drug from the lipid matrix to the external phase (35). An enhancement in drug loading was seen with the increment in the quantity of liquid lipid. Surfactant concentration (B) and sonication time (C) showed a contrary effect on drug loading. Our results are in consonance with earlier findings (33,36). Such an effect of independent variables on drug loading (Y3) is represented in a three-dimensional (3D) response surface plot.
Selection of Optimum PTX-loaded NLC
The optimized PTX-NLCs was chosen based on the criterion of acquiring the highest value of entrapment efficiency and drug loading while reducing the particle size by mathematical optimization technique of Design Expert® software. Upon “trading off” different response variables using numerical desirability function, the formulation with total lipid (3%), surfactant (2%), and sonication (6 min) was found to fulfill prerequisites of an optimized formulation. The optimized PTX-NLCs (NLC3) exhibited the particle size of 153.82 ± 5.58 nm, entrapment efficiency of 86.48 ± 6.46%, and drug loading of 16.54 ± 2.92%, respectively, which was further demarcated in the optimal design space region depicted in Fig. 3.

Overlay plot acquired from graphical optimization, indicating yellow region as design space and flagged point as the optimized PTX-loaded nanolipid carrier
Characterization of the Optimized PTX-Loaded NLC
Determination of Particle Size, PDI, and Zeta Potential
The particle size and PDI of the optimized PTX-NLCs were found to be 153.82 ± 5.58 nm and 0.221 ± 0.026, respectively (Fig. 4a). Zeta potential of the optimized PTX-NLCs was found to be − 22.12 ± 2.48 mV (Fig. 4b). Zeta potential signifies the stability of the particles in the continuous medium. Greater value of zeta potential exhibits high repulsion between the particles and minimizes the chances of particle aggregation (23). A negative value of the zeta potential demonstrated that the optimized PTX-NLCs had attained better stability and dispersion feature.

Images showing a particle size distribution, b zeta potential distribution profiles, and c TEM image of optimized PTX-loaded NLC formulation
Entrapment Efficiency and Drug Loading
The recorded entrapment efficiency and drug loading capacity of optimized PTX-NLCs was 86.48 ± 6.46% and 16.54 ± 2.92%. The improvement in entrapment efficiency and drug loading capacity are possible because of the distinctive binary lipid matrix of NLCs.
Transmission Electron Microscopy Analysis
The surface morphology and average diameter of optimized PTX-NLCs were analyzed under TEM. The TEM photomicrographs illustrated that more or less spherical surface of the optimized PTX-NLCs with uniform size distribution (< 200 nm) and it validated the particle size analysis by dynamic light scattering. The obtained results point out that the particles were evenly distributed and alienated from each other. The TEM images of optimized PTX-NLCs are shown in Fig. 4c.
In Vitro Drug Release and Release Kinetics
The % cumulative PTX released from optimized PTX-NLCs and PTX suspension in simulated intestinal fluid pH 6.8 was represented as a function of time in Fig. 5. The in vitro release study of the optimized PTX-NLCs exhibited biphasic performance with initial burst release for 4 h subsequently a sustained release pattern up to 24 h. The initial burst release effect was considered as the highest drug release (%) initially for 4 h (> 70%) and after 24 h % cumulative release was calculated as > 90%. The same procedure was followed for in vitro release study of the free PTX suspension, and after 24 h of the study, 24.65 ± 4.56% release of PTX was found. There are numerous factors that may directly influence the drug release from the NLCs like drug solubility in the lipid, type of lipid matrix, and its concentration, partition coefficient, as well as particle size (23).

In vitro release profile of the drug from PTX suspension and optimized PTX-NLC formulation in simulated intestinal fluid; data expressed as mean ± SD (n = 3)
In order to evaluate the release kinetics of the optimized PTX-NLCs, the in vitro release data was fitted to different release kinetic models. The values of correlation coefficients (R2) for various kinetic models of optimized PTX-NLCs are shown in Table IV. The model with the maximum R2 value was regarded as the best-suited model for drug release mechanism. On the basis of obtained results, the Korsmeyer–Peppas model was chosen as the best fit model for optimized PTX-NLCs, as it showed the highest R2 value, i.e., 0.948.
In Vitro Cytotoxicity Assay
As shown in Figs. 6a and 7a, the control cells (unexposed to treatment) experienced no toxicity and remained healthy. The cells exposed to optimized PTX-NLCs experienced morphological alterations as revealed by the obtained photomicrograph. Also, the cells exhibited shrinkage and fragmentation in contrast to the unexposed cells. The above finding indicated apoptotic nature of the cells (27). The data obtained on the cytotoxic effect further indicated that 1 μM of optimized PTX-NLCs decreased the cell viability to ~ 84.68% (p < 0.05) in comparison to the control. Apart from this, the cell viability was severely reduced to about 55.34, 25.24, 14.47, and 11.3% (p < 0.05) at concentrations 2, 5, 10, and 20 μM of PTX-NLC, respectively (Fig. 6b), and IC50 of PTX-NLC on HepG2 cells was found to be 4.19 μM. Furthermore, the commercial formulation (Intaxel®) reduced the viability of cells to 96.28, 89.4, 64.01, 45.05, and 26.87% (p < 0.05), at concentrations 1, 2, 5, 10, and 20 μM, respectively (Fig. 7)b. Besides, IC50 of Intaxel® on HepG2 cells was found to be 11.2 μM. The present results suggested that treatment of PTX-NLC significantly (p < 0.001) minimizes the cell viability in a dose-dependent manner.

In vitro cytotoxicity test of PTX-NLC formulation against human hepatoma HepG2 cells. a Photomicrograph of HepG2 cell line treated with 1- to 20-μM concentration of HepG2. Photomicrographs were taken with an inverted phase contrast microscope. Scale bar = 100 μm. b Dose-response effects of PTX-NLC formulation on cytotoxicity against HepG2 cell line at different concentrations for 24 h. Values are expressed as mean ± SEM of at least three independent experiments. *p < 0.05 as compared with their respective control

In vitro cytotoxicity test of commercial formulation (Intaxel®) against human hepatoma HepG2 cells. a Photomicrograph of HepG2 cell line treated with 1- to 20-μM concentration of HepG2. Photomicrographs were taken with an inverted-phase contrast microscope. Scale bar = 100 μm. b Dose-response effects of commercial formulation (Intaxel®) on cytotoxicity against HepG2 cell line at different concentrations for 24 h. Values are expressed as mean ± SEM of at least three independent experiments.*p < 0.05 as compared with their respective control
Intracellular ROS Assay
ROS generation plays an important role in the apoptosis induction of cell death and therefore, dichloro-dihydro-fluorescein diacetate (DCFHDA) staining was performed to measure the oxidative stress in HepG2 cells. Figure 8a depicts data on HepG2 cells exposed to the optimized PTX-NLCs formulation, which exhibited a significant enhancement in ROS intensity in a dose-dependent manner with respect to the untreated cells. The quantitative measurement of ROS expressed from 2-μM concentration of optimized PTX-NLCs showed 157.29% (p < 0.05) improvement in ROS production in comparison to control. Furthermore, ROS production was increased by 195.32% (p < 0.05) at 5-μM concentration of optimized PTX-NLCs in contrast to untreated cells (Fig. 8b). ROS are more reactive than molecular oxygen due to activation of signal molecules and destabilization of mitochondrial membrane triggering the discharge of apoptotic agents leads enhanced cytotoxic effect on cancerous cells (27,28). In our study, the obtained results clearly confirmed that optimized PTX-NLCs prompted the cells death by triggering the ROS production.

Photomicrographs showing intracellular ROS generation in HepG2 cells induced by PTX-NLC formulation. a Photomicrographs showing intracellular ROS generation induced at 2 and 5 μM of PTX-NLC formulation after 12-h incubation. Photomicrographs were taken with a florescence microscope. Scale bar = 100 μm. b Graph showing extent of ROS generation with respect to the control. Values are expressed as the percentage of fluorescence intensity relative to the control. Values are expressed as mean ± SEM of at least three independent experiments, *p < 0.05 as compared with their respective control
Further, the effect of optimized PTX-NLCs induced nuclear condensation was evaluated by fluorescence photomicrograph. As per the photomicrograph depicted in Fig. 9a, HepG2 cells exposed to increasing concentrations of optimized PTX-NLCs prompted the chromatin condensation in contrast to control cells. The concentrations 2 and 5 μM of optimized PTX-NLCs showed the highest condensation as seen under an inverted fluorescence microscope. Moreover, at 2 and 5 μM of optimized PTX-NLCs, about 23% and 46.6% of apoptotic cells were present (Fig. 9b). The presence of condensed and fragmented nuclei in the images revealed that optimized PTX-NLCs triggered the cell death through apoptosis mechanism in an effective manner.

Chromatin condensation of HepG2 cells treated with PTX-NLC formulation after 24 h. a Cells were treated with 2- and 5-μM of PTX-NLC formulation, Photomicrographs were taken with fluorescence microscope that showed fragmented and condensed nuclei as indicated by arrows. Scale bar = 100 μm. b Numerical data were expressed as percentage apoptotic cells respective to their control. Values are expressed as mean ± SEM of at least three independent experiments. *p < 0.05 as compared with their respective control
Ex Vivo Permeation Studies
Ex-vivo gut permeation studies suggested that the NLCs formulation composed of lipids (GMS and Soybean oil) and surfactants (poloxamer 188) have better permeation than API suspension, analyzed at the same time with a similar experimental protocol. A graph was plotted between the cumulative amount of PTX released (μg) vs. time (min) to get permeation flux (F) from the linear portion of the graph illustrated in Fig. 10a. Moreover, the cumulative drug transported (μg/cm2) was plotted against time as demonstrated in Fig. 10b clearly denoted that higher quantity of the PTX was transported through the intestinal sac from the optimized PTX-NLCs in comparison to PTX suspension. Further, an apparent permeability coefficient (APC; Papp) for the transport of PTX suspension was compared with that for optimized PTX-NLCs in ileum as shown in Fig. 10c. The Papp for the optimized PTX-NLCs and PTX-suspension was found to be 4.84 × 10−4 and 0.82 × 10−4 cm/s respectively. The result indicated that the cumulative drug transport (μg/cm−2) and Papp were observed in the order optimized PTX-NLCs > PTX suspension. Optimized PTX-NLCs comprised of lipids and surfactants with the concurrent cutback in particle size had proved to enhance the permeability of the PTX from the intestine. Surfactant (poloxamer 188) which is present in the developed PTX-NLCs not only offers stability to the preparation but also alleviate intestinal efflux by the preventing the P-glycoprotein efflux pumps that are available in the villus tip of enterocytes in the GIT (29).

Ex vivo gut permeation study showing a cumulative amount of drug permeated (μg) versus time, b cumulative drug transported (μg cm−2) versus time, and c Papp (APC) for PTX suspension and PTX-NLC formulation
In Vivo Pharmacokinetic Study
The proportional pharmacokinetic profiles of optimized PTX-NLCs and PTX-suspension, after oral administration, are presented in Fig. 11 and the pharmacokinetic factors are given in Table V. At all time points, the drug plasma concentrations in optimized PTX-NLC-treated animals were significantly higher (p < 0.001) than PTX suspension. Cmax of optimized PTX-NLC was found to be 6.8-fold higher (p < 0.001) compared to PTX suspension, respectively. Statistically, the difference in AUC0 → 12 and Cmax of the optimized PTX-NLCs was highly significant (p < 0.001) in comparison to the PTX suspension. The AUC0 → 12 of optimized PTX-NLCs was found to be 1852.56 ± 42.56 ng h/mL, which is highly significant (p < 0.05) compared to AUC0 → 12 of PTX suspension (291.025 ± 13.94 ng h/mL). Nearly 6.3-fold improvement in AUC0 → 12 was observed from optimized PTX-NLCs as compared to the PTX suspension. High differences in Cmax and AUC0 → 12 of a drug from NLCs might be attributed to enhanced drug absorption by nanosized structure, lipophilic nature, inhibition of P-gp efflux, etc. Thus, the results corroborated improved oral drug absorption profile of PTX when formulated in NLCs.

Plasma concentration–time profiles of drug in rats after oral administration of PTX-NLC formulation and PTX suspension; data expressed as mean ± SD (n = 6)
CONCLUSION
The PTX-loaded NLCs were of successfully prepared with the help of systematic optimization approach using melt emulsification ultrasonication technique. The conclusion made from the current study, the developed NLCs could be an efficient nanoparticulate target enabled carrier system for the delivery of PTX. In the current investigation, we have presented a novel approach for the fabrication of lipid-based nanocarrier system and introduced physico-chemical heterogeneity in the nanocarrier design while optimizing the nanoparticle formulation approach to a clinically certified set of nanocarrier. Due to its excellent nanoscale reservoir structures for better encapsulation of drugs, greater tumor targetability, and bio-safety. NLC-based preparations can be utilized as a potential drug delivery carrier for other hydrophobic chemotherapeutic agents, accelerating the effective cancer therapy whereas plummeting nonspecific side effects. Moreover, the in vitro, ex vivo, and in vivo characterization studies are corroborated a remarkable improvement in the oral biopharmaceutical performance and anticancer activity of PTX-NLC over the PTX-suspension and marketed formulation. The research outcomes from the current studies effectively endorsed the high degree of suitability of the NLC containing dietary lipids as one of the versatile systems for enhancing the oral bioavailability of PTX having low aqueous solubility along with hepatic first-pass metabolism and restricted intestinal permeability.
References
Best J, Schotten C, Theysohn JM, Wetter A, Müller S, Radünz S, et al. Novel implications in the treatment of hepatocellular carcinoma. Ann Gastroenterol. 2017;30(1):23–32.
Li S, Zheng L. Effect of combined treatment using wilfortrine and paclitaxel in liver cancer and related mechanism. Med Sci Monit. 2016;22:1109–14.
Soni K, Rizwanullah M, Kohli K. Development and optimization of sulforaphane-loaded nanostructured lipid carriers by the Box-Behnken design for improved oral efficacy against cancer: in vitro, ex vivo and in vivo assessments. Artif Cells Nanomed Biotechnol. 2017:1–17. https://doi.org/10.1080/21691401.2017.1408124.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–48.
Yang XY, Li YX, Li M, Zhang L, Feng LX, Zhang N. Hyaluronic acid coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013;334(2):338–45.
Sandhu PS, Beg S, Mehta F, Singh B, Trivedi P. Novel dietary lipid-based selfnanoemulsifying drug delivery systems of paclitaxel with p-gp inhibitor: implications on cytotoxicity and biopharmaceutical performance. Expert Opin Drug Deliv. 2013;12(11):1809–22.
Rivkin I, Cohen K, Koffler J, Melikhov D, Peer D, Margalit R. Paclitaxel clusters coated with hyaluronan as selective tumor-targeted nanovectors. Biomaterials. 2010;31(27):7106–14.
Akhter MH, Rizwanullah M, Ahmad J, Ahsan MJ, Mujtaba MA, Amin S. Nanocarriers in advanced drug targeting: setting novel paradigm in cancer therapeutics. Artif Cells Nanomed Biotechnol. 2018;46(5):873–84.
Ahmad J, Akhter S, Rizwanullah M, Amin S, Rahman M, Ahmad MZ, et al. Nanotechnology-based inhalation treatments for lung cancer: state of the art. Nanotechnol Sci Appl. 2015;8:55–66.
Beg S, Saini S, Bandopadhyay S, Katare OP, Singh B. QbD-driven development and evaluation of nanostructured lipid carriers (NLCs) of olmesartan medoxomil employing multivariate statistical techniques. Drug Dev Ind Pharm. 2018;44(3):407–20.
Barkat MA, Harshita, Ahmad J, Khan MA, Beg S, Ahmad FJ. Insights into the targeting potential of Thymoquinone for therapeutic intervention against triple-negative. Breast Cancer Curr Drug Targets 2018;19(1):70–80.
Hussain A, Wais M, Singh SK, Samad A, Nazish I, Beg S. Nanostructured lipidic carriers for oral bioavailability enhancement. Recent Pat Nanomed. 2015;5(2):78–86.
Granja A, Vieira AC, Chaves LL, Nunes C, Neves AR, Pinheiro M, et al. Folate-targeted nanostructured lipid carriers for enhanced oral delivery of epigallocatechin-3-gallate. Food Chem. 2017;237:803–10.
Gao X, Zhang J, Xu Q, Huang Z, Wang Y, Shen Q. Hyaluronic acid-coated cationic nanostructured lipid carriers for oral vincristine sulfate delivery. Drug Dev Ind Pharm. 2017;43(4):661–7.
Sun B, Luo C, Li L, Wang M, Du Y, Di D, et al. Core-matched encapsulation of an oleate prodrug into nanostructured lipid carriers with high drug loading capability to facilitate the oral delivery of docetaxel. Colloids Surf B Biointerfaces. 2016;143:47–55.
Aditya NP, Shim M, Lee I, Lee Y, Im MH, Ko S. Curcumin and genistein coloaded nanostructured lipid carriers: in vitro digestion and antiprostate cancer activity. J Agric Food Chem. 2013;61(8):1878–83.
Beg S, Rahman M, Kohli K. Quality-by-design approach as a systematic tool for the development of nanopharmaceutical products. Drug Discov Today. 2018.
Beg S, Rahman M, Rahman Z, Akhter S. Perspectives of quality by design approach in nanomedicines development. Curr Nanomed. 2017;7:191–7.
Singh B, Raza K, Beg S. Developing “optimized” drug products employing “designed” experiments. Chem Ind Digest. 2013;23:70–6.
Singh B, Beg S. Attaining product development excellence and federal compliance employing quality by design (QbD) paradigms. Pharma Rev. 2015;13(9):35–44.
Singh B, Beg S. Product development excellence and federal compliance via QbD. Chronicle Pharmabiz. 2014;15(10):30–5.
Singh B, Beg S. Quality by design in product development life cycle. Chronicle Pharmabiz. 2013;22:72–9.
Rizwanullah M, Amin S, Ahmad J. Improved pharmacokinetics and antihyperlipidemic efficacy of rosuvastatin-loaded nanostructured lipid carriers. J Drug Target. 2017;25(1):58–74.
Qidwai A, Khan S, Md S, Fazil M, Baboota S, Narang JK, et al. Nanostructured lipid carrier in photodynamic therapy for the treatment of basal-cell carcinoma. Drug Deliv. 2016;23(4):1476–85.
Xu W, Lee MK. Development and evaluation of lipid nanoparticles for paclitaxel delivery: a comparison between solid lipid nanoparticles and nanostructured lipid carriers. J Pharm Investig. 2015;45(7):675–80.
Barkat MA, Harshita AI, Ali R, Singh SP, Pottoo FH, et al. Nanosuspension-based aloe vera gel of silver sulfadiazine with improved wound healing activity. AAPS PharmSciTech. 2017;18(8):3274–85.
Ahamad MS, Siddiqui S, Jafri A, Ahmad S, Afzal M, Arshad M. Induction of apoptosis and antiproliferative activity of naringenin in human epidermoid carcinoma cell through ROS generation and cell cycle arrest. PLoS One. 2014;9(10):e110003.
Siddiqui S, Ahmad E, Gupta M, Rawat V, Shivnath N, Banerjee M, et al. Cissus quadrangularis Linn exerts dose-dependent biphasic effects: osteogenic and anti-proliferative, through modulating ROS, cell cycle and Runx2 gene expression in primary rat osteoblasts. Cell Prolif. 2015;48:443–54.
Ahmad J, Mir SR, Kohli K, Chuttani K, Mishra AK, Panda AK, et al. Solid-nanoemulsion preconcentrate for oral delivery of paclitaxel: formulation design, biodistribution, and γ-scintigraphy imaging. Biomed Res Int. 2014;2014:984756.
Tan SW, Billa N, Roberts CR, Burley JC. Surfactant effects on the physical characteristics of amphotericin-B containing nanostructured lipid carriers. Colloids Surf A Physicochem Eng Asp. 2010;372(1–3):73–9.
Liu J, Gong T, Wang C, Zhong Z, Zhang Z. Solid lipid nanoparticles loaded with insulin by sodium cholate-phosphatidylcholine-based mixed micelles: preparation and characterization. Int J Pharm. 2007;340(1–2):153–62.
Rahman Z, Zidan AS, Khan MA. Non-destructive methods of characterization of risperidone solid lipid nanoparticles. Eur J Pharm Biopharm. 2010;76(1):127–37.
Mishra A, Imam SS, Aqil M, Ahad A, Sultana Y, Ameeduzzafar, et al. Carvedilolnano lipid carriers: formulation, characterization and in-vivo evaluation. Drug Deliv. 2016;23(4):1486–94.
Moghddam SM, Ahad A, Aqil M, Imam SS, Sultana Y. Optimization of nanostructured lipid carriers for topical delivery of nimesulide using Box-Behnken design approach. Artif Cells Nanomed Biotechnol. 2017;45(3):617–24.
Alam S, Aslam M, Khan A, Imam SS, Aqil M, Sultana Y, et al. Nanostructured lipid carriers of pioglitazone for transdermal application: from experimental design to bioactivity detail. Drug Deliv. 2016;23(2):601–9.
Khan A, Imam SS, Aqil M, Ahad A, Sultana Y, Ali A, et al. Brain targeting of temozolomide via the intranasal route using lipid-based nanoparticles: brain pharmacokinetic and scintigraphic analyses. Mol Pharm. 2016;13(11):3773–82.
Acknowledgments
The authors are grateful to Gattefosse for providing gift samples of oils and co-surfactants, and Fresenius Kabi India Pvt. Ltd. for PTX. The authors also acknowledge the kind help from Dr. M. Arshad, Deptt. Zoology, Lucknow University, India, for in vitro cell culture studies.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interests.
Additional information
Guest Editor: Sanyog Jain
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Harshita, Barkat, M.A., Rizwanullah, M. et al. Paclitaxel-loaded Nanolipidic Carriers with Improved Oral Bioavailability and Anticancer Activity against Human Liver Carcinoma. AAPS PharmSciTech 20, 87 (2019). https://doi.org/10.1208/s12249-019-1304-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-019-1304-4