
Abstract
The nonlinear mixed effects models are commonly used modeling techniques in the pharmaceutical research as they enable the characterization of the individual profiles together with the population to which the individuals belong. To ensure a correct use of them is fundamental to provide powerful diagnostic tools that are able to evaluate the predictive performance of the models. The visual predictive check (VPC) is a commonly used tool that helps the user to check by visual inspection if the model is able to reproduce the variability and the main trend of the observed data. However, the simulation from the model is not always trivial, for example, when using models with time-varying input function (IF). In this class of models, there is a potential mismatch between each set of simulated parameters and the associated individual IF which can cause an incorrect profile simulation. We introduce a refinement of the VPC by taking in consideration a correlation term (the Mahalanobis or normalized Euclidean distance) that helps the association of the correct IF with the individual set of simulated parameters. We investigate and compare its performance with the standard VPC in models of the glucose and insulin system applied on real and simulated data and in a simulated pharmacokinetic/pharmacodynamic (PK/PD) example. The newly proposed VPC performance appears to be better with respect to the standard VPC especially for the models with big variability in the IF where the probability of simulating incorrect profiles is higher.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Nonlinear mixed effects models (NLMEM) are commonly used modeling techniques for drug development, pharmacokinetic/pharmacodynamic (PK/PD) analysis, and epidemiological studies because they are able to quantify the individual and population parameters and to identify the biological sources of inter-individual and intra-individual variability. Using NLMEM, clinical trials can be better designed to reduce costs and increase in efficacy, dose regimens can be optimized and, in general, the source of stochastic variability can be minimized. However, as usual in any modeling analysis, before drawing clinical conclusions driven by the model, it is important to use correct diagnostics to evaluate the NLMEM predictive performance. In these last two decades, many sophisticated diagnostic tools were proposed (1) that rely, for example, on numerical assessment (e.g., objective function, standard error of the parameter estimates), on graphical evaluation of model predictions (e.g., individual/population residuals), or on a simulation-based visualization that investigate the model capability of reproducing the observed data (e.g., posterior predictive checks (2) and normalized prediction distribution error (3)). Recently, a simulation-based diagnostic has been particularly popular for its simplicity: the visual predictive check (VPC) (4). The basic idea of VPC is to assess by visual inspection whether the model is able or unable to catch the variability of the observed data. To do so, multiple simulations are performed using the individual parameters that are realizations drawn from the population parameter estimates and keeping the structure of the observed dataset. Then, the prediction interval (PI), typically the 5th and the 95th percentiles of the simulated datasets, is compared to the corresponding percentiles of the observed data. To make the interpretation of the VPC less subjective, the 95% CI of the percentiles of the simulated data are used instead of just the PI (5). Many VPC adaptations have been proposed since then (6–8) but there are still pitfalls that have to be explored, for example, during the simulation step. This step is particularly critical because it is not always trivial to simulate profiles that are consistent with the original dataset. For instance, when models with time-varying known input function (IF) are evaluated, there is a potential mismatch between the set of simulated individual parameters and the associated individual IF which can cause an incorrect profile simulation. This kind of modeling strategy is well-established in the diabetes area where IFs are introduced to partition the glucose-insulin feedback system and to better identify the insulin action and secretion models (9–13). In particular, the glucose and insulin system is decomposed in two subsystems where the glucose signal is modeled and the insulin signal is assumed as an error-free time-varying input function and vice versa depending on which part of the system needs to be described (14). In PKPD modeling, IFs are used when no assumptions are made on the PK model and the PD part needs to be modeled. In these situations, the PD is fitted using the PK data as a known input function. Note that to avoid introducing further misspecification on the PD estimates, the number of samples in the PK curve should be sufficient and with a limited noise level.
This study aims to overcome this VPC limitation in the simulation step by taking into account a term that correlates the set of simulated parameters with the most appropriate IF. This correlation term is the minimum distance calculated for each set of simulated individual parameters with the previously estimated individual parameters on the observed dataset in the estimation step of VPC. We assessed the newly proposed corrected version of VPC, the distance VPC (dVPC), and compared its performance to the standard VPC on four models (the intravenous glucose tolerance test (IVGTT) and the meal/oral glucose tolerance test (meal/OGTT) glucose and C-peptide minimal models (9–12)) and on a typical PKPD example such as the warfarin model (15).
MATERIAL AND METHODS
Population Modeling
Nonlinear mixed effects models (NLMEM) are able to quantify both the population and the individual parameters and identify by a hierarchical approach the biological sources of intra-individual and inter-individual variability. More specifically, in a first step, the observed data are described by:
where y ij is the j th observation of the i th subject at some known time instant X ij . Here, n is the number of individuals and m i is the number of observation of individual i. p ind i is the vector of individual parameters of the i th subject. Note that IF is defined as an error-free input that varies along time and that represents an individual sampled kinetic that is not modeled. The variability due to measurement and model misspecifications, better known as the residual unknown variability (RUV), is explained through ε ij which is assumed to be independently distributed with a zero mean and Gaussian distribution.
In a second step, the individual model parameters are assumed to be a realization of usually a normal or lognormal distribution that has mean (θ, fixed effects, i.e., values that are common to all subjects) and variability (Ω, between subject variability—BSV) form, what will be called in this article population parameters (p pop). In case of a lognormal distribution, the individual realization of a parameter is:
where p ind ki is the k th model parameter of the i th subject, θ k is the typical value of the k th parameter common to the entire population, and η ki is the random effect of the k th model parameter of the i th subject that is assumed to be independently distributed with a zero mean and Gaussian distribution:
VPC
The standard VPC is calculated from 1000 simulations of the original observed dataset. For each simulated dataset and in each bin, the median value together with the 5th and 95th percentiles (the simulated prediction interval—simPI) are calculated. Note that binning in this article is chosen in a way that each bin corresponds to one time point because the sampling time is the same for each subject. From these collections of values, the 95% confidence interval CI are calculated and then compared to the median and observed prediction interval (obsPI) calculated in the original dataset.
dVPC
The standard VPC approach does not take into account the relation between the simulated set of individual parameters and the individual IF associated. The distance VPC (dVPC) proposed in this article, as described in Fig. 1, creates a correlation that drives the simulation, firstly, by calculating for each set of individual simulated parameters a vector of distances between the simulated set and all the previously estimated sets of individual parameters on the observed data and, subsequently, by looking for the minimum distance in each vector. The minimum distance detects the closest set of estimated parameters to the simulated one and consequently associates in the simulation step the IF of the selected estimated parameters to the simulated set of parameters. The distances used are the Mahalanobis distance (MD) and the normalized Euclidean distance (NED) that is a simplified version of the first. The rationale to use one or the other is based on how the covariance matrix (Ω) of the random effects is declared in the model: if it has covariance terms, the Mahalanobis distance is used; otherwise, the Euclidean distance is employed. By definition, the distance represents the distance between two random vectors X = [X 1, …, X K]T and Y = [Y 1, …, Y K]T that belong to the same multivariate distribution with mean μ = [μ 1, …, μ K]T and covariance Σ. Note that K is the number of parameters that have a random effect The covariance Σ matrix is diagonal when the normalized Euclidean distance is calculated, whereas it takes into account the off-diagonal terms when the Mahalanobis distance is applied. In formula, the squared Mahalanobis distance can be described as in Eq. 4:

Schematic representation of the VPC and dVPC diagnostics
Equation 4 can be simplified in the following way (Eq. 5) when the normalized Euclidean distance is applied, since only the diagonal terms are considered:
Note that in our case, if we assume a normal distribution, the mean vector μ is θ and the covariance matrix ∑ is Ω.
Models
The glucose and C-peptide meal/oral glucose tolerance test (MTT/OGTT) and intravenous (IVGTT) models (9–13) are commonly used tools to quantify the glucose-insulin system. All these four models present at least one known input function. The dataset consist of 120 healthy volunteers (16) (71 males and 49 females age 62 ± 17.5 and body weight 79.2 ± 13.5 kg) that underwent both an MTT and IVGTT. The MTT (10 kcal/kg, 45% carbohydrate, 15% protein, and 40% fat) contains 1 ± 0.02 g/kg glucose, and plasma samples are collected at −120, −30, −20, −10, 0, 5 10, 15, 20, 30, 40, 50, 60, 75, 90, 120, 150, 180, 210, 240, 260, 280, 300, 360, and 420 min for the measurement of plasma glucose, insulin, and C-peptide concentrations. The insulin-modified IVGTT consists of an administration of a dose of 330 mg/kg glucose at time 0 and a dose of 0.02 units/kg of insulin at time 20 min. Blood samples are collected at −120, −30, −20, −10, 0, 2, 4, 6, 8, 10, 15, 20, 22, 25, 26, 28, 31, 35, 45, 60, 75, 90, 120, 180, and 240 min for measurement of glucose, insulin, and C-peptide concentrations.
The process used to generate MTT simulated glucose and C-peptide data was the following. First of all, the individual parameter values of each subject were estimated using the NLMEM approach. Once the individual estimates were obtained, they were considered as true and used to simulate new concentration profiles using their corresponding individual IF as input functions and by introducing the previously estimated measurement error.
MTT Glucose and C-Peptide Minimal Models
The MTT glucose minimal model (11,13) has two input functions: insulin and rate of appearance (IF = [I, Ra]) (Eq. 8). The parameters S G (min−1), V (dL kg−1), S I (μU−1 mL min−1), and p 2 (min−1) are declared lognormal with mean equal to the fixed effects θ and variance Ω that is a diagonal matrix:
where Q (mg kg−1) is the glucose mass in plasma; \( {G}_{\mathrm{ss}} \) and \( {G}_{\mathrm{b}} \) (mg dL−1) the steady state and the basal glucose concentration in plasma, respectively; I (μU mL−1) insulin plasma concentration and \( {I}_{\mathrm{b}} \) (μU mL−1) its basal value; X (min−1) insulin action; and Ra (mg kg−1 min−1) the glucose rate of appearance in plasma.
The MTT C-peptide minimal model (12,13) has two input functions: the glucose and the first derivative of glucose (IF = [G, dG]) (Eq. 9). The parameters are β (min−1 mg−1 dL pmol L−1) that represents the static sensitivity, k (min−1) the dynamic sensitivity, α (min−1) a constant rate, and CPb (pmol L−1) the basal C-peptide. They are declared lognormal with mean equal to the fixed effects θ and variance Ω that is a full matrix. Note that k 01, k 12, and k 21 (min−1) are the C-peptide kinetic parameters fixed to standard population values following the method proposed by Van Cauter et al (17).
where CP1 and CP2 (pmol L−1) are C-peptide concentration above basal in the accessible and peripheral compartments, respectively; G (mg dL−1) the glucose concentration; G b its basal value; and SR the pancreatic secretion made up of two components: a static (SRS) and a dynamic (SRd) component controlled by glucose and glucose rate of change, respectively.
IVGTT Glucose and C-Peptide Minimal Models
The IVGTT glucose minimal model (9) (Eq. 6) has one input function insulin (IF = [I]). The estimated parameters are glucose effectiveness (S G—min−1), distribution volume (V—dL kg−1), insulin sensitivity (S I—min−1pmol−1 L), and insulin action (p 2—min−1). They are all declared lognormal with mean equal to the fixed effects θ and variance Ω with only two covariance term between S G and V and S I and p 2 as in Denti et al (18).
where Q is the glucose mass in plasma (mg kg−1); G ss and G b the steady state and basal glucose concentration in plasma (mg dL−1), respectively; I insulin plasma concentration (pmol L−1) and I b its basal value; X the insulin action (min−1); and D the dose (mg kg−1). Note that glucose measurements prior to 8 min are excluded from the parameter estimation, because the 1-compartment minimal model is not designed to account for the fast glucose kinetics after the glucose bolus.
The IVGTT C-peptide minimal model (10) (Eq. 7) has glucose as input function (IF = [G]). The estimated parameters are the first phase secretion index (X 0—pmol L−1), the second phase secretion index (β—pmol L−1 dL mg−1 min−1), the basal C-peptide (CPb—pmol L−1), and the secretion parameters (m—min−1 and α—min−1). They are all declared lognormal with mean equal to the fixed effects θ and variance Ω that is a full matrix. Note that k 01, k 12, and k 21 (min−1) are the C-peptide kinetic parameters fixed to standard population values following the method proposed by Van Cauter et al (17).
where CP1 and CP2 (pmol L−1) are the C-peptide concentration above basal in the accessible and in the peripheral compartments, respectively, G glucose concentration (mg dL−1), whereas X (pmol L−1) and Y (pmol L−1 min−1) are the C-peptide amount and provision in the β cells, respectively.
Warfarin Model
To demonstrate the approach feasibility in a different area, we applied the method on a well-known PKPD example such as the warfarin model (15) where the PK of the drug (C—mg L−1) is assumed to be the known input function and the PD of the effect, the prothrombin (PCA), is the fitted data sampled at time 0, 24, 36, 48, 72, 96, and 120 h. In particular, the dataset is based on a simulation of 128 subjects obtained from the population estimates of 32 healthy subjects that underwent an oral single dose of warfarin (1.5 mg/kg) (19). On these subjects, 250 samples of warfarin concentrations together with 232 samples of prothrombin complex activity (PCA) were measured. The model was implemented as a turnover model as in Mager et al (20) to characterize warfarin delayed effect with an indirect mechanism of action due to the interaction between the drug and the endogenous enzymes (the prothrombin) (20). The estimated parameters p = [E0, Emax, C50, Tover] are declared lognormal with mean equal to the fixed effects θ and variance Ω that is a diagonal matrix. Note that Tover is the turnover half-life and that Rin is set be equal to kout × E0 whereas kout is equal to ln2/Tover.
Analysis of the Results
We investigated the performance of the VPC and dVPC in some simulated examples (MTT glucose and C-peptide, warfarin) and in some real data scenarios (IVGTT glucose and C-peptide). Within the simulated example of the MTT C-peptide model, we investigated the capability of the dVPC to detect misspecifications in the model. In particular, the C-peptide model was simulated with the structure declared in Eq. 7 and then was estimated introducing two misspecifications in the model: the Ω matrix was declared diagonal and the C-peptide kinetics were described by a one-compartment model. The results that were obtained using the standard VPC and the newly proposed dVPC were firstly evaluated by visual inspection of the graphs and, then, compared in a more quantitative way. In particular, the sum of squared residuals (RSS) was calculated among the 5th, 95th percentile and the median values calculated in the observed dataset and the median of the corresponding values calculated in the simulated dataset. Finally, some VPC statistics were calculated: the percentages of false positive (FP) and false negative (FN). Note that an observation is defined as false positive if it lay inside its corresponding simulated PI but outside the VPC PI, whereas it is defined false negative if it lies outside its corresponding simulated PI but inside the VPC PI. All the estimation steps were done using NONMEM 7.2 (21) and the algorithm First-Order Conditional Estimation (FOCE) approximation with eta-epsilon interaction, whereas all the simulation steps were done using Matlab R2010a (22).
RESULTS
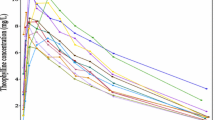
Preliminary, the standard goodness of fit was checked in all models by examination of scatter plots of predicted versus measured data and weighted residuals both at the population and at the individual level. The results (not shown) indicate that all the models well describe the data, and the weighted residuals match the measurement error assumptions. Shrinkage (23) values both in the individual parameter estimates (η-shrinkage) and in the residual error (ε-shrinkage) were below the suggested critical threshold of 30%. In Fig. 2 is presented the motivational example based on simulated MTT glucose data where a standard VPC simulation of unrealistic glucose profiles is shown. Note that there are glucose profiles that are significantly lower than the 5th percentiles and significantly higher than the 95th percentiles calculated on the observed dataset. These profiles are not physiologically plausible for a healthy population.

Spaghetti plots based on simulated MTT glucose data of time versus observed glucose concentration (on the left) and of time versus simulated glucose concentration profiles (on the right) through the standard VPC procedure. The solid line is the median of the observed data and the dashed lines are the 5th and 95th percentile of the observed data
MTT Glucose and C-Peptide Minimal Model
In Fig. 3, we present the standard VPC of the MTT glucose and C-peptide minimal models applied on simulated data. The simulated profiles without the correction were sometimes not physiological and, as a consequence, the CIs of the percentiles calculated on the simulated data do not match the reference percentiles calculated on the observed data. If we take into consideration in the simulation step, respectively, the Mahalanobis distance for the C-peptide model as its Ω matrix is not diagonal and the normalized Euclidean distance for the glucose model as its Ω matrix is diagonal, the VPC performance improves: the CIs of the percentiles calculated on the simulation with the dVPC better match the percentiles of the observed data (Fig. 3). In Table I, the average sum of squared residuals (RSS) show that dVPC performs better than the standard VPC in both models as the RSS are smaller in both examples using the dVPC. Moreover, the percentages of FP and FN that were obtained using the new dVPC (Table II) are smaller than the percentages that were obtained with the standard VPC. In Fig. 4 is shown that the dVPC is able to detect a misspecified model. By looking at the highest percentile in the misspecified case, it is evident that the dVPC is able to detect the misspecification as the CIs of the simulated percentile are not matching perfectly the corresponding observed percentile.

VPC (on the left part of the panel) and dVPC (on the right part of the panel) of the MTT glucose and C-peptide minimal models applied on simulated dataset. The solid line is the median of the observed data and the dashed lines are the 5th and 95th percentile of the observed data. The grey bands correspond to 95% CI of the median and 5th and 95th percentiles in the simulated data

dVPC of a misspecified MTT C-peptide minimal model (on the left) and dVPC of a non-misspecified C-peptide minimal model (on the right) applied on simulated dataset. The solid line is the median of the observed data, and the dashed lines are the 5th and 95th percentile of the observed data. The grey bands correspond to 95% CI of the median and 5th and 95th percentiles in the simulated data
IVGTT Glucose and C-Peptide Minimal Model
In Fig. 5, we present the standard VPC of the IVGTT glucose and C-peptide minimal models. The CIs of the percentiles calculated on the simulated dataset present some mismatch with the corresponding percentile calculated on the observed data. In particular, the VPC of the IVGTT glucose shows a clear mismatch in the 5th percentile, whereas the VPC of the C-peptide shows clear mismatches in the 95th percentile and the median. Note that the VPC of the IVGTT glucose minimal model is presented from min 8 since the model explains the dynamics of glucose from that minute on. In this case, the difference between the simulated profiles and the observed one is less marked than the one obtained in the MTT data. In Fig. 5, the new dVPCs are presented applied to both the IVGTT glucose and C-peptide minimal models. Note that since the Ω matrix has covariance terms in both models, the Mahalanobis distance was calculated. The CIs of the percentiles calculated on the simulated dataset follow better the dynamics of the observed percentiles. In Table I, the RSS for the standard and the new dVPC for both the IVGTT models are presented. The RSS are smaller in both IVGTT models using the new dVPC compared to the standard VPC. In Table II, the percentages of FP and FN are presented: for both the IVGTT models, the percentages again are smaller using the new dVPC approach.

VPC (on the left) and dVPC (on the right) of the IVGTT glucose and C-peptide minimal models applied on real dataset. The solid line is the median of the observed data and the dashed lines are the 5th and 95th percentile of the observed data. The grey bands correspond to 95% CI of the median and 5th and 95th percentiles in the simulated data
Warfarin Model
In Fig. 6, we present the standard VPC of the PD of the warfarin model together with the new dVPC technique of the same data, respectively. The improvement due to the correction obtained using the new technique can be noted from the graphs by looking at the 95th percentile. Note that the distance calculated in this example is the normalized Euclidean distance as its Ω matrix is defined diagonal. In Table I, the average RSS shows that the new VPC is better performing than the standard as the VPC has larger RSS. Moreover, the percentages of FP and FN (Table II) are bigger or comparable using the standard VPC compared to the new dVPC.

VPC (on the left) and dVPC (on the right) of the warfarin model applied on simulated dataset. The solid line is the median of the observed data and the dashed lines are the 5th and 95th percentile of the observed data. The grey bands correspond to 95% CI of the median and 5th and 95th percentiles in the simulated data
DISCUSSION
During model building, it is fundamental to evaluate the model performance with appropriate tools. In the PKPD area, the VPC is a commonly used diagnostic to test whether or not the model is able to reproduce the variability and the main trend of the data. However, this diagnostic tool still presents pitfalls in the simulation step when models are identified with input functions (IF). In fact in this case, there is a lack of correlation between each set of individual simulated parameters and the associated individual IF which can cause an incorrect simulation profile. This problem is illustrated in a plot (Fig. 2): the standard VPC simulation procedure generates some unrealistic simulated glucose profiles that reach, for example, a glucose steady state at 50 mg/dl which is physiologically implausible. Note also that this lack of correlation, due to fact that the system is separated in two subparts, would have not been present if there had been a simultaneous fit of the two signals that would have offered a flexible full parameter framework that, in turn, would have allowed simulation of consistent profiles. This study aims to overcome this VPC limitation by taking into account in the simulation step a correlation term using the Mahalanobis or normalized Euclidean distance (depending on the Ω matrix) that bounds the set of individual simulated parameters with the most appropriate individual input function. We assess the performance of this new diagnostic dVPC on simulated and real metabolic data examples and in a simulated PKPD case.
By looking at the graphs, the standard VPC in the various presented examples shows some mismatch between the CI of the percentiles calculated on the simulated dataset and the percentiles of the observed data (Figs. 3, 4, 5, and 6). This mismatch is more evident in the metabolic examples relative to MTT (Fig. 3) that, by definition, is a less controlled experiment compared to IVGTT (Fig. 5) since it includes additional variability due to the gastrointestinal tract. MTT produces very variable glucose and insulin profiles and, as a consequence, a wrong association of the individual simulated parameters with IF is more likely, which it translates into an incorrect profile simulation. Moreover, there is a clear underperformance of the standard VPC of the MTT glucose minimal model with respect to the MTT C-peptide minimal model (Fig. 3), where we can see in both the 5th and the 95th percentile a mismatch with the CI of percentiles calculated on the simulated dataset. This might be due to the fact that the input functions of the glucose minimal model are two different signals (rate of appearance of glucose and insulin), whereas in the C-peptide minimal model the IFs are the glucose signal and its first derivative. Regarding the standard VPC relative to the IVGTT and the warfarin models (Figs. 5 and 6), they present fewer mismatches in the CIs of the simulated percentiles with the observed percentiles because the profiles measured are less variable among the subjects. Moreover, note that the warfarin model is the only example that presents one parameter with 27% of shrinkage (the rest of the examples have shrinkage lower than 20%) that might introduce some bias in the association procedure at the individual parameter level and contribute to make the impact of the dVPC technique less evident. It is interesting to point out that the glucose and insulin model examples have a close loop mechanism of control in which the data (output) has an effect on the IF (input) and the input has an effect on the output. The warfarin model instead presents an open loop mechanism of control in which only the input has an effect on the output. This might explain why the impact of the dVPC is less evident in the warfarin PKPD example as there is less dependency between the input and output of the system. The new VPC, the dVPC, is a clear improvement compared to the standard VPC, and it is easy to grasp this from visual inspection of the five examples (Figs. 3, 4, 5, and 6) where the CI of the percentiles calculated on the simulated dataset are better matching the corresponding percentiles of the observed data.
In Fig. 4 is shown that the dVPC diagnostic is able to detect misspecifications in a simulated C-peptide example as the 95th observed percentile is not well described by the simulated CI of the corresponding percentile.
As far as the sum of squared residuals is concerned, the underperformance of the standard VPC method with respect to the newly proposed dVPC is evident in all the five examples as the average of RSS is larger (Table I) using the standard VPC. Moreover, the relative largest drops of RSS between VPC and the dVPC are detectable, as expected, in the glucose (47%) and the C-peptide (40%) oral minimal models since the MTT produces more variable signals. Note that this is in agreement with the previously discussed results obtained by visual inspection of the graphs. In the C-peptide and warfarin models, the relative RSS drop is still large, around 32%, for both, whereas in the warfarin example the drop is less significant, around 5%.
Finally, the VPC statistics presented in Table II also yields the same results: dVPC is better performing as the percentages of FP and FN are smaller or comparable to the percentages obtained with the standard VPC. It is interesting to note that looking at the standard VPC results, the oral C-peptide minimal model and the oral glucose minimal model have, respectively, the largest percentage of FP and FN which again confirms the same trend that has been previously discussed in VPC graphs and RSS.
This newly proposed dVPC is an informative tool to evaluate correctly models with IF because it maintains the characteristic of easy visualization and interpretation of standard VPC, while avoiding simulating unrealistic profiles. Moreover, dVPC is appealing because it preserves the dynamics of the profiles in the time course unlike other methods proposed in literature, such as pcVPC (8) that, even if it is still suitable to handle input functions, has both data and predictions corrected or where, instead of the measurements, the percentiles in which the observed data is falling with respect to its simulated distribution are plotted (7) or where the distribution of the observed data around the model predicted median at each observation time is plotted (6). Drawbacks of dVPC are that to have robust results the dataset under analysis needs a relevant number of subjects and a rich sampling of the input function. The first prerequisite guarantees a good characterization of the population and, consequently, a good coverage of the possible combinations of the individual simulated set of parameters with the input functions. This in other words means avoiding associating IF based on too big distances between parameter vectors and avoiding sampling a relevant number of times the same IF. The second prerequisite ensures a reliable IF. This prerequisite is a common feature of all models with known input functions, i.e., IFs rich in sampling and with small measurement error allow not to introduce further bias in the analyzed model. Finally, it is important to underline that since the method relies on an association process at the individual parameter level, the individual estimate precision needs to be satisfactory and the shrinkage level needs to be low in order to guarantee a reliable matching step. Note also that the drawbacks that we discussed, such as small number of subjects and sparse and noisy data, are potential weak points of not only the dVPC per se but also of the standard VPC.
CONCLUSION
This work proposes a refinement of the standard diagnostic VPC, the dVPC, which is built for a particular class of models that present a time-varying known input function that cannot be modeled or it is not relevant to model. Despite the simplicity of the method, the results show both on real and simulated examples that dVPC is a more appropriate diagnostic with respect to the standard VPC. We suggest using the dVPC during model building for a more accurate performance evaluation of this class of models and, as a consequence, to obtain more reliable model-based clinical conclusions.
REFERENCES
Karlsson MO, Savic RM. Diagnosing and model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20.
Yano Y, Beal LS, Sheiner BL. Evaluating pharmacokinetic/pharmacodynamic models using the posterior predictive check. J Pharmacokinet Pharmacodyn. 2001;28(2):171–92.
Brendel K, Comets E, Laffont C, Laveille C, Mentre F. Metrics for external model evaluation with an application to the population pharmacokinetics of Gliclazide. Pharm Res. 2006;23(9):2036–49.
Holford N. The visual predictive check-superiority to standard diagnostic (Rorschach) plots. In PAGE 14; 2005; Abstr 738 [www.page-meeting.org/?abstract=738].
Karlsson MO, Holford N. A tutorial on visual predictive checks. In PAGE 17; 2008; Abstr 1434 [www.page-meeting.org/?abstract=1434].
Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn. 2008;35:185–202.
Wang D D, Zhang S. Standardized visual predictive check—how and when to use it in model evaluation. In PAGE 18; 2009; Abstr 1501 [www.page-meeting.org/?abstract=1501].
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. APPS J. 2011;13(2):143–51.
Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Phy. 1979;236:E667–77.
Toffolo G, De Grandi F, Cobelli C. Estimation of beta-cell sensitivity from IVGTT C-peptide data. Knowledge of the kinetics avoids errors in modeling secretion. Diabetes. 1995;44:845–54.
Dalla Man C, Caumo A, Cobelli C. The oral glucose minimal model: estimation of insulin sensitivity from a meal test. IEEE Trans Biomed Eng. 2002;49:419–29.
Breda E, Cavaghan MK, Toffolo G, Polonsky KS, Cobelli C. Oral glucose tolerance test minimal model indexes of beta cell function and insulin sensitivity. Diabetes. 2001;50(1):150–8.
Cobelli C, Dalla Man C, Toffolo G, Basu R, Vella A, Rizza R. The oral minimal model method. Diabetes. 2014;63:1203–13.
Cobelli C, Dalla Man C, Sparacino G, Magni L, De Nicolao G, Kovatchev BP. Diabetes: models, signals, and control. IEEE Rev Biomed Eng. 2009;2:54–96.
Holford NH. Clinical pharmacokinetics and pharmacodynamics of warfarin. Understanding the dose-effect relationship. Clin Pharmacokin. 1986;11:483–504.
Basu R, Dalla Man C, Campioni M, Basu A, Klee G, Toffolo G, et al. Effects of age and sex on postprandial glucose metabolism: differences in glucose turnover, insulin secretion, insulin action and hepatic extraction. Diabetes. 2006;55:2001–14.
Van Cauter E, Mestrez F, Sturis J, Polonsky KS. Estimation of insulin secretion rates from C-peptide levels. Comparison of individual and standard kinetic parameters for C-peptide clearance. Diabetes. 1992;41:368–77.
Denti P, Bertoldo A, Vicini P, Cobelli C. IVGTT glucose minimal model covariate selection by nonlinear mixed-effects approach. Am J Phy. 2010;298:E950–60.
O’ Reilly RA, Aggeler PM. Studies on coumarin anticoaugulant drugs. Initiation of warfarin therapy without a loading dose. Circulation. 1968;38:169–77.
Mager DE, Wyska E, Jusko WJ. Diversity of mechanism-based pharmacodynamic models. Drug Metab Dispos. 2003;31:510–8.
Beal SL, Sheiner LB, Boeckmann AJ, Nonmem User’s Guides. 1989-2012. Icon Development Solution, Maryland, USA.
Matlab R. 2010a, MathWorks. http://www.mathworks.com/.
Savic RM, Karlsson MO. Importance of shrinkage in empirical bayes estimates for diagnostics: problems and solutions. AAPS J. 2009;11:558–69.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Largajolli, A., Bertoldo, A., Campioni, M. et al. Visual Predictive Check in Models with Time-Varying Input Function. AAPS J 17, 1455–1463 (2015). https://doi.org/10.1208/s12248-015-9808-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9808-7