
Abstract
Background
In Antarctic P. syringae RNase R play an essential role in the processing of 16S and 5S rRNA, thereby playing an important role in cold-adapted growth of the bacterium. This study is focused on deciphering the in vivo functional activity of mesophilic exoribonuclease R and its catalytic domain (RNB) in an evolutionary distant psychrophilic bacterium Pseudomonas syringae Lz4W.
Results
Our results confirm that E. coli RNase R complemented the physiological functions of the psychrophilic bacterium P. syringae RNase R and rescued the cold-sensitive phenotype of Pseudomonas syringae ∆rnr mutant. More importantly, the catalytic domain (RNB) of the E. coli RNase R is also capable of alleviating the cold-sensitive growth defects of ∆rnr mutant as seen with the catalytic domain (RNB) of the P. syringae enzyme. The Catalytic domain of E. coli RNase R was less efficient than the Catalytic domain of P. syringae RNase R in rescuing the cold-sensitive growth of ∆rnr mutant at 4°C, as the ∆rnr expressing the RNBEc (catalytic domain of E. coli RNase R) displayed longer lag phase than the RNBPs (Catalytic domain of P. syringae RNase R) complemented ∆rnr mutant at 4°C. Altogether it appears that the E. coli RNase R and P. syringae RNase R are functionally exchangeable for the growth requirements of P. syringae at low temperature (4°C). Our results also confirm that in P. syringae the requirement of RNase R for supporting the growth at 4°C is independent of the degradosomal complex.
Conclusion
E. coli RNase R (RNase REc) rescues the cold-sensitive phenotype of the P. syringae Δrnr mutant. Similarly, the catalytic domain of E. coli RNase R (RNBEc) is also capable of supporting the growth of Δrnr mutant at low temperatures. These findings have a vast scope in the design and development of low-temperature-based expression systems.
Similar content being viewed by others


Background
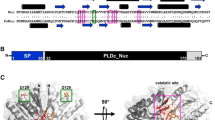
RNase R is a conserved hydrolytic ribonuclease (3' to 5') that belongs to RNase II family of exoribonucleases. This processive enzyme is capable of degrading the RNA molecules through their complex secondary structures, unlike the exoribonuclease RNase II, which can degrade only single-stranded RNAs [1,2,3,4]. The domain arrangement of RNase R is similar to that of RNase II and Rrp44, with the CSD1, CSD2, and S1 domains capping the top of the RNB domain [1, 5] (Fig. 1). The crystal structure of E. coli RNase R revealed a unique feature that differs from the crystal structures of other reported exoribonucleases is the presence of two open channels that act as important RNA binding sites, i.e., a top channel between the S1 and CSD1 domains and a side channel between the RNB and CSD1 domains [1, 6,7,8]. Mutational analysis of the enzyme has established that the aspartic acid residues D272, D278, and D280 in the catalytic pocket of the E. coli RNase R (RNase REc) are important for the ribonuclease activity, and D280 is directly involved in the catalysis during the hydrolytic cleavage of terminal nucleotides from the RNA chain. Further, the residue Y324 plays a key role in deciding the final length of end-products. The substrate binding domains (CSD1, CSD2, and S1) bind only to single-stranded 3′-overhang ends of RNA molecules, thereby screening the substrates and regulating the entry of RNA into the catalytic pocket of the enzyme [9]. Structural comparison of E. coli RNase R with other RNase II family proteins revealed two open RNA-binding channels in RNase R and suggested a tri-helix ‘wedge’ region in the RNB domain that may induce RNA unwinding. Construction of two tri-helix wedge mutants revealed that mutants, indeed lost their RNA unwinding but not RNA binding or degrading activities. Structural studies revealed that duplex RNA with an overhang is bound in the two RNA-binding channels in RNase R. The 3′ overhang is channeled into the active site and duplex RNA is unwound upon reaching the wedge region during RNA degradation [1].

Domain organization of RNase R. Schematic representing the domain organization in E.coli and P. syringae RNase R. Individual domains are shaded in separate colors and numbers represent the amino acids in the primary sequence of protein
Thus, the RNA binding domains CSD1, CSD2, and S1 play multiple roles in substrate recognition and sensing of 3′ overhangs of RNA molecules, whereas RNB domain plays a critical role in unwinding and processing/degradation of double-stranded RNA molecules [1, 2, 10].
Physiologically, RNase R plays a key role in the RNA metabolism of cell via degradation of polyadenylated RNAs, degradation of mRNA transcripts containing REP (repetitive extragenic palindromes) sequences [3], degradation of defective and non-functional tRNAs, trans-translation and quality control of ribosomes [11], turnover of ribosomal RNAs (e.g., 16S and 23S rRNAs) [8, 12,13,14,15] and growth-phase specific (e.g., stationary phase) removal of ompA mRNA in E. coli [16]. Depletion of RNase R affects tmRNA (transfer-messenger RNA) metabolism as evidenced by the accumulation of tmRNA precursors and tmRNA degradation intermediates [17]. The tmRNA-dependent trans-translation pathway is important for releasing the stalled ribosomes from truncated or defective mRNAs, tagging the proteins and peptides produced from the truncated mRNAs and facilitating the degradation of both defective RNAs and proteins. Thus, RNase R also plays a role in the maintenance of protein quality in the cells [18].
RNase R is a stress-induced protein that shows increased expression at low temperatures and stationary phase [16, 18]. In E. coli RNase R is encoded by rnr gene, and its levels in the cell are regulated by RNase G, RNase E, and SmpB [19, 20]. In Streptococcus pneumoniae, Aeromonas hydrophila, pathogenic E. coli, and Shigella flexneri, disruption of rnr gene leads to a decrease in virulence [21,22,23]. RNase R is also required for the low-temperature growth of Legionella pneumophila and A. hydrophila [24]. In Mycoplasma genitalium bearing the smallest bacterial genome, RNase R is the only exoribonuclease that plays a crucial role in all RNA metabolic processes including the processing and degradation of different types of RNA molecules [25,26,27]. Recent studies have illustrated that methylated ribose in the ribonucleotides acts as stop signals for RNase R-mediated RNA degradation [26]. This indicates that ribose methylation status of RNA might be used as a signaling mechanism by bacteria for the screening of RNA molecules to be sent for degradation or processing and maturation.
In our laboratory, RNase R was found to be a component of the novel RNA degradosomal complex of P. syringae Lz4W, in which RNase R is associated with the endoribonuclease RNase E and RNA helicase RhlE [28, 29]. The disruption of rnr gene led to a cold-sensitive phenotype of the Antarctic bacterium. Further biochemical investigations revealed that cold-sensitive ∆rnr is defective in the 3′-end processing of 16S and 5S rRNAs, as a result of which rnr mutants accumulated unprocessed 16S rRNA and 5S rRNA precursor molecules in the cells [30]. Collectively, these studies have established three important things: first, psychrophilic bacteria like P. syringae Lz4W possess novel degradosome that has replaced the ss-RNA degrading exoribonuclease PNPase with the ds-RNA degrading enzyme RNase R in the protein complex; second, RNase R plays a crucial role in cold adaptation, and third, remarkably, the exoribonuclease enzyme plays a role in the 3′-end processing of 16S rRNAs which was hitherto unknown, but predicted to be an endoribonuclease mediated process [30].
RNase R is essential for growth of P. syringae at low temperature. P. syringae Δrnr mutants display cold sensitive phenotype when grown at 4°C, whereas E. coli Δrnr mutants do not display any low temperature associated growth defect. At molecular level, ∆rnr mutants of P. syringae and E. coli accumulate rRNA degradation intermediates suggesting their role in rRNA degradation or quality control [28, 31]. However, ∆rnr mutant of P. syringae accumulates 3′-end unprocessed 16S and 5S rRNA precursors at low temperature, while E. coli mutant does not. The P. syringae Δrnr mutant is also defective in tmRNA degradation and processing. Thus, there were distinct similarities and differences in the activities and requirements of exoribonucleases (RNase RPs and RNase REc) during growth of the psychrophilic and mesophilic bacteria. Therefore, it was important to know whether E. coli RNase R would be able to complement the cold-sensitive growth defect of Δrnr mutant of the Antarctic P. syringae.
The objectives of this study are (i) Will E. coli RNase R (RNase REc) complement the cold-sensitive phenotype of P. syringae Δrnr mutant? (ii) Will the catalytic domain (RNBEc) of E. coli RNase R be able to carry out similar in vivo functions like the catalytic domain (RNBPs) of P. syringae RNase R despite their differences in in vitro activities?
Our study clearly demonstrates that the E. coli-specific exoribonuclease R (RNase REc) has retained all the functions that are necessary to support the growth of psychrophilic P. syringae at 4 °C. This is remarkable as E. coli itself does not grow at 4°C, and the enzymes in spite of their evolutionary divergence and alteration in the biochemical properties, have maintained the common essential activities. The individual catalytic domains (RNBPs and RNBEc) are also functionally similar, as both of them are capable of supporting the growth of Δrnr mutant at 4°C.
Methods
Growth media and bacterial cultures
The Antarctic P. syringae Lz4W was routinely grown at 22°C or 4°C (for optimum and low temperatures respectively) in Antarctic bacterial medium (ABM) composed of 5 g/l peptone and 2.0 g/l yeast extract, as described earlier [32, 33]. E. coli strains were cultured at 37°C in Luria–Bertani medium, which contained 10 g/l tryptone, 5 g/l yeast extract, and 10 g/l NaCl [34]. For solid media, 15 g/l bacto-agar (Hi Media) was added to ABM or LB. Both ABM and LB media were supplemented with ampicillin (100 μg/ml), kanamycin (50 μg/ml), and tetracycline (10 μg/ml) as per requirement.
Fresh ABM broth was inoculated with 1% of primary culture and incubated at 22°C or 4°C with constant shaking. The optical density of bacterial culture was measured after different time intervals at 600 nm [OD600] and plotted against time.
Molecular biology methods
General molecular biology techniques including isolation of genomic DNA, polymerase chain reactions (PCR), restriction enzyme digestion and ligation, transformation, etc. were performed as described [32]. All restriction enzymes, T4 DNA ligase, and other enzymes used in this study were purchased from New England Biolabs (NEB). Plasmids were prepared by using a plasmid isolation kit (Qiagen). Oligonucleotides were purchased from a commercial source (Bioserve Biotechnology, India). Gene amplifications were carried out using high-fidelity pfx DNA polymerase (Invitrogen). The conjugal transfer of recombinant plasmids into P. syringae was carried out by a biparental mating method using the donor E. coli strain S17-1, as described earlier [35].
Construction of plasmids for expression and complementation studies
All gene cloning experiments were performed in E. coli DH5α cells. The detailed methodology has been reported earlier [34, 36, 37]. All plasmids used for protein expression and genetic complementation are listed in Table 1.
Cloning and expression of E. coli RNase R
The amplification and cloning of the RNase R encoding gene (rnr) of P. syringae has been reported earlier [30]. The E. coli rnr gene was amplified by using a set of gene-specific Forward and Reverse primers with incorporated NdeI and SalI sites, and cloned into pET28a expression vector (Table S 1). The plasmid (pET28arnrEc) was transformed into E. coli BL-21 (DE3) strain, and expression of RNase REc was observed under IPTG induction at various time intervals (Data not shown) [40]. The His-tagged E. coli rnr gene along with vector-specific RBS (Ribosome binding site) was released from pET28arnrEc plasmid by XbaI and SacI digestion and subcloned into broad host range vector pGL10 [38]. The construct (pGLrnrEc) was transformed into E. coli S-17 strain and mobilized into P. syringae ∆rnr mutant by biparental mating as described earlier [35, 41]. Expression of the RNase REc in ∆rnr strain was confirmed by Western analysis using anti-His antibodies (Fig. 2).

Expression of E.coli RNase R. Expression of RNase RPs and RNase REc in ∆rnr strain was analyzed by western blotting where cell lysate from ∆rnr strains expressing RNase RPs and RNase REc were transferred to a nylon membrane and probed by anti-His antibodies
Cloning and expression of RNB domain of RNase REc
The truncated gene fragment (1239 base pairs) encoding catalytic (RNB) domain (413 amino acids) of E. coli RNase R was amplified using gene-specific forward and reverse primers with incorporated NdeI and BamHI sites respectively, and cloned into pET28a expression vector. (Table S 1). The resultant plasmid pET28aRNBEc was transformed into E. coli BL-21(DE3) strain and expression of RNBEc domain under IPTG induction was confirmed by SDS-PAGE analysis (Data not shown). The fragment encoding the His-tagged RNBEc domain was released from pET28aRNBEc along with vector-specific RBS (Ribosome binding site) by XbaI and SacI digestion and subcloned into broad host range plasmid pGL10 [38]. The construct (pGLRNBEc) was transformed into E. coli S-17 strain and mobilized into P. syringae ∆rnr mutant by bi-parental mating as reported earlier [35, 41]. Expression of the RNBEc in ∆rnr mutant was confirmed by Western analysis using anti-RNase R antibodies (Fig. 3).

Expression of RNB (Catalytic) domain of E. coli RNase R. (a) Color-coded schematic diagram showing different domains (color shaded) and position of primers employed for amplification of catalytic RNB domain. The numbers represent the nucleotide base pairs in rnr gene. (b) Expression of RNB domain in ∆rnr strain complemented by pGLRNBEc was analyzed by western blotting. Cell lysate from wild-type cells (Lane 2), ∆rnrpGLRNBEc strain (Lane 1) were loaded on polyacrylamide gel, transferred to a nylon membrane, and probed with anti RNase RPs antibodies
Functional complementation studies
Broad host range vectors pGL10rnrEc and pGL10rnbEc were mobilized into cold-sensitive ∆rnr strain, and growth pattern of complemented strains was analyzed at both optimal and low temperatures (22 °C and 4 °C). All bacterial strains used for genetic complementation studies are listed in Table 2.
Results
Bioinformatic analysis of P. syringae rnr gene
Analysis of rnr gene (Gene encoding RNase R) sequences from different bacteria revealed that rnr locus is highly conserved among the Pseudomonas. sp. The operon consists of rnr (RNase R encoding) and trmH gene (encoding putative tmRNA or rRNA methyl transferase). Up-stream of the Bi-cistronic operon are two genes that code for tRNA-leucine, whereas downstream of the rnr operon is a highly conserved gene (rpsF) that codes for S6 ribosomal protein [39].
Sequence alignment studies (T-COFFEE, EMBL-EBI) of rnr gene from Antarctic P. syringae, P. aeruginosa, P. fluorescens, and mesophilic representative strains (E. coli and B. subtilis) revealed that P. syringae Lz4W displayed maximum similarity with P. fluorescens (88.91%), followed by P. aeruginosa (74.97%) (Fig. 4a). The Antarctic bacterium shows only a modest sequence similarity (53.17%) with E. coli and even lesser similarity with B. subtilis (37.52%). Sequence alignment results illustrate that there is high similarity among the representative strains from Pseudomonas sp., as compared to their sequence similarity with representative strains from mesophiles (E. coli and B. subtilis).

Multiple sequence alignment [T-coffee, www.ebi.ac.uk] of the amino acid sequence of the RNA helicases. Accordingly, B. subtilis has been indicated as B whereas E. coli has been abbreviated as E. Similarly P. syringae Lz4W, P. aeruginosa, and P. florescens have been indicated as Lz4W, P_1, and P_2 respectively. The alignment results also illustrate the identity among amino acid residues in different [N and C] regions of the protein
A gene sequence-based phylogeny of exoribonuclease (rnr) genes from Pseudomonas. sp., and mesophilic E. coli, B. subtilis is shown in Fig. 4b. The analysis indicates that the rnr genes belonging to different strains have been clustered into three distinct groups representing, Pseudomonas. sp. (P. syringae Lz4W, P. aeruginosa, and P. fluorescens), E. coli, and B. subtilis. The representative strains from Pseudomonas group display sufficient homology among each other to be clustered as a group and provide a possible explanation for a convergent evolution among the representative strains in pseudomonas group or divergence from the mesophilic representatives which have been clustered in two separate groups.
Growth analysis of ∆rnr strain complemented with RNase REc
To confirm whether the E. coli RNase REc is capable of complementing the cold-sensitive growth defect of P. syringae ∆rnr mutant, the complemented ∆rnr mutant (∆rnr/pGLrnrEc) expressing the E. coli RNase R was monitored for growth, and the growth profiles were compared with the wild-type and ∆rnr mutant. Experimentally, all bacterial strains were grown at optimal (22°C) and low (4°C) temperatures, and OD600 values of the cultures were recorded at regular intervals and plotted against time. All strains displayed optimal growth pattern at 22°C (Fig. 5a). As expected, the cold-sensitive ∆rnr mutant did not display any measurable growth at 4°C; however, the cold sensitive phenotype of P. syringae ∆rnr mutant was rescued by E. coli-specific RNase REc in the complemented mutant (∆rnr/pGLrnrEc) (Fig. 5b). The results confirmed that the E. coli specific RNase REc is active in P. syringae, and the enzyme is capable of complementing the functions of P. syringae RNase RPs at low temperature. The activity of the RNase REc was comparable to the endogenous RNase RPs, as the growth profiles of the two complemented mutants (∆rnr/pGLrnrEc and ∆rnr/pGLrnrPs) were similar.

Mesophilic RNase R complements cold-sensitive phenotype of ∆rnr. (a) Growth profile of wild-type, ∆rnr, ∆rnrpGLrnrPs, and ∆rnrpGLrnrEc strains at 22°C and (b) at 4°C confirmed over-expression of RNase REc from broad host range plasmid (pGL10) complements cold-sensitive phenotype of Pseudomonas syringae ∆rnr strain. For measurement of growth, samples were collected from each culture at regular intervals, OD at 600 nm was recorded and plotted against time. Each growth curve was performed at least three times
Complementation of cold-sensitive phenotype of ∆rnr mutant by catalytic domain (RNBEc)
To assess the biological activity of the catalytic domain (RNBEc) of E. coli-specific RNase REc by complementation analysis, we expressed the RNBEc domain in P. syringae ∆rnr mutant from a broad host range plasmid pGL10. For biological activity, growth profiles of ∆rnr strain (∆rnr/pGLRNBEc) expressing the RNBEc of E. coli RNase R were compared with ∆rnr expressing P. syringae RNB (∆rnr/pGLRNBPs) and the wild-type. The above strains were grown at 22°C and 4°C, and optical densities of the cultures were measured at OD600 at regular intervals and plotted against time. All strains displayed normal growth at 22°C (Fig. 6a). At 4°C, ∆rnr mutant displayed a cold-sensitive phenotype, whereas the complemented mutants ∆rnr/pGLRNBPs and ∆rnr/pGLRNBEc displayed measurable growth to stationary phase but with a long lag time (Fig. 6b). The lag time was longer in ∆rnr/pGLRNBEC compared to ∆rnr/pGLRNBPS strain. The results confirmed that catalytic domains of both the mesophilic and psychrophilic exoribonuclease R (RNase R) are physiologically active and sufficient for rescuing the cold-sensitive phenotype of P. syringae ∆rnr mutant. At low temperatures (4°C), the longer growth lag exhibited by ∆rnr mutant complemented by RNBEc, compared to ∆rnr mutant expressing the RNBPs might be related to the physiological efficiency of RNBPs over the heterologous E. coli specific RNBEc.

Complementation of ∆rnr strain by catalytic domain (RNB) of E. coli RNase R. (a) Growth analysis of P. syringae wild type, ∆rnr, ∆rnrpGLRNBPs, and ∆rnrpGLRNBEc strains at 22°C and (b) at 4°C established that complementation of cold-sensitive ∆rnr strain by RNBEc alleviates the cold-sensitive phenotype of mutant strain but with a long Lag phase even longer than with RNBPs. For measurement of growth in cell cultures at 22°C or 4°C, samples were collected from each culture at regular intervals, and their OD at 600 nm was recorded and plotted against time. Each growth curve was repeated at least three times
Discussion
The major focus of the current study was to examine the activity of E. coli RNase R in the cold-adapted Antarctic P. syringae, as RNase R plays an important role in the growth and viability of the P. syringae, especially at low temperatures (4°C). RNase R functions inside the cells either as an integral component of a multi-enzyme complex or as a freely soluble enzyme. This is important, as bacteria possess a huge RNA degrading multi-enzyme complex (degradosome) for efficient processing and degradation of different RNA substrates with variable complexity. The RNA degradosome is generally composed of Endo-ribonuclease E (RNase E), that acts as a scaffold for the assembly of other components like exoribonucleases (e.g., PNPase and RNase R), RNA helicases (e.g., RhlB, RhlE, and Rho factors), regulatory proteins (e.g., metabolic enzymes enolase and aconitase) and many transient proteins (e.g., DnaK, GroEL, GroES, Hfq, poly(A) polymerase [42] and polyphosphate kinase, etc.) that functionally interact with each other for the efficient processing and degradation of the substrates. The degradosome is a highly dynamic structure which undergoes changes in composition under different growth conditions within the cells of a species and in different bacterial species with diverged adaptability to different environmental stresses. This has been achieved by the binding activity of highly variable and intrinsically disordered regions of the C-terminal domain of RNase E. In E. coli degradosome, C-terminal region of RNase E acts not only as a scaffold for the assembly of exoribonuclease PNPase, RNA helicase RhlB, and the glycolytic enzyme enolase [43, 44] but also helps in localizing the complex to plasma membrane using a membrane targeting sequence (MTS) motif on this domain [45]. Since Antarctic P. syringae is adapted to grow at low temperatures, the degradosomal components include RNase E that provides a scaffold for the assembly of the ds-RNA degrading exoribonuclease RNase R, and RNA helicase [46]. Disruption of rnr gene leads to defects in the processing of rRNA (16S rRNA and 5S rRNA) and consequent cold-sensitive phenotype accompanied by cell death [30]. On the other hand, the disruption of rnr gene in E. coli has no deleterious effect on the processing of RNA or growth, although double mutant of rnr and pnp is not viable [13]. This study has convincingly proved that in P. syringae, the important functions of exoribonuclease R in rRNA processing and maintenance of cellular physiology are independent of the degradosomal complex.
Maintaining the rate of enzyme-catalyzed reactions at an acceptable limit to sustain growth by synthesizing cold-active and thermo-labile enzymes in cold environments is the most important adaptation of psychrophiles [46,47,48,49,50,51]. Lack of strong selective pressure for structural stability in cold environments probably helped in the evolution of cold-active enzymes with increased destabilization and flexibility of active site or whole protein [52]. Reactions catalyzed by cold-active enzymes progress with decreased ∆G and ∆H reflecting that these enzymes are more efficient with high specific activity and are less temperature dependent [53,54,55,56]. The active site of these enzymes is less stable and heat-labile [57, 58] whereas these enzymes unfold at relatively lower temperatures than their mesophilic homologs [59]. It has been observed that the majority of cold-adapted enzymes have a half-life of less than 12 min at 50 °C [49, 60, 61]. Cold-adapted enzymes are prone to increased error in folding [62] and cold denaturation, most probably caused by the hydration of polar and non-polar groups [59, 63, 64]. Psychrophilic enzymes are also accompanied by structural changes outside the active site that modulate the activity of critical residues at freezing temperatures and enhance the flexibility of loops around the active site. Cold active enzymes have more accessible and large-sized active sites [65, 66], better channeling of the substrate to the active site, enhanced electrostatic potential, and better release of products [53, 65,66,67,68,69].
Recent studies have provided valuable insights into the structure–function relationship of various enzymes including the ribonucleases. These studies have provided the role of structural flexibility, side chain flexibility, and the role of hydrophobic amino acids in the functioning of the protein. Our results have provided an important input that, despite having variable structural adaptability and substrate specificity, conservation of function allowed the mesophilic enzyme to perform its function in an evolutionary distinct cold-adapted species of bacteria. The role of different polar/non-polar amino acids, amino acid side chains, and structural flexibility may be more related to protein thermostability, thermolability, substrate specificity, and catalysis. Our study has comprehensively proved that exoribonuclease R has a flexible structure that allows it to interact even with less specific substrates and perform its function at a physiologically acceptable rate.
In the light of the differences between two types of degradosomal assembly exemplified by the E. coli and P. syringae, variations in structure–function relationship among cold-adapted and mesophilic enzymes, differences in biochemical properties and divergent in vivo functions of two exoribonucleases, two questions were raised: (i) will mesophilic E. coli RNase R (RNase REc) be able to complement the cold-sensitive growth defect of P. syringae Δrnr mutant? (ii) Despite bearing differences in in vitro activities, will the catalytic domain (RNBEc) of E. coli RNase R be able to carry out similar in vivo functions like the catalytic domain (RNBPs) of P. syringae RNase R?
Our results illustrate that E. coli RNase R (RNase REc) is capable of rescuing the cold-sensitive growth defect of P. syringae Δrnr mutant. The findings of the current study also illustrate that the catalytic domains of two exoribonucleases from E. coli and P. syringae (RNBEc and RNBPs) are also capable of complementing the growth defects of P. syringae Δrnr mutant at low temperature (4°C). However, P. syringae Δrnr mutant expressing the catalytic domain derived from E. coli RNase R (RNBEc) displays a longer lag time (~ 240 hours) as compared to the lag time (~ 120 hours) displayed by the P. syringae Δrnr mutant complemented by catalytic domain derived from the P. syringae RNase R (RNBPs). The difference in the catalytic efficiencies of RNBPs and RNBEc at low temperatures is probably related to the substrate specificity, substrate binding, and structural flexibility associated with the two catalytic domains.
Altogether, the results presented here suggest that E. coli RNase R could alleviate the cold-sensitive phenotype of P. syringae ∆rnr mutant at 4°C. The activities of the conserved catalytic domains (RNBPs and RNBEc) are largely intact in the two diverged bacteria (P. syringae and E. coli) adapted to grow in different temperature ranges. These results also provide valuable insights into the flexibility of protein structure, structure–function relationship, and conservation of function among the exoribonucleases. Since cold-sensitive ∆rnr mutant accumulates unprocessed 5S and 16S rRNA at low temperatures, the rescue of cold sensitivity in ∆rnr mutant by RNase REc is indicative of an essential role being played by E. coli RNase R in 16S and 5S rRNA processing [30, 31].
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- RNase R:
-
Exoribonuclease R
- RNase REc :
-
E. coli-Specific RNase R
- RNase RPs :
-
P. syringae-Specific RNase R
- RNBPs :
-
Catalytic domain of RNase R [P. syringae-Specific]
- RNBEc :
-
Catalytic domain of RNase R [ E. coli specific]
- tmRNA:
-
Transfer-messenger RNA
References
Chu L-Y et al (2017) Structural insights into RNA unwinding and degradation by RNase R. Nucleic Acids Res 45(20):12015–12024
Hossain ST, Malhotra A, Deutscher MP (2016) How RNase R Degrades structured RNA: Role Of The Helicase Activity And The S1 Domain. J Biol Chem 291(15):7877–7887
Cheng ZF, Deutscher MP (2005) An important role for RNase R in mRNA decay. Mol Cell 17(2):313–318
Suzuki H et al (2006) Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res 34(8):e63
Matos RG et al (2011) Swapping the domains of exoribonucleases RNase II and RNase R: conferring upon RNase II the ability to degrade ds RNA. Proteins 79(6):1853–1867
Cairrao F, Arraiano CM (2006) The role of endoribonucleases in the regulation of RNase R. Biochem Biophys Res Commun 343(3):731–737
Cheng ZF, Deutscher MP (2002) Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J Biol Chem 277(24):21624–9
Vincent HA, Deutscher MP (2006) Substrate recognition and catalysis by the exoribonuclease RNase R*. J Biol Chem 281(40):29769–29775
Matos RG, Barbas A, Arraiano CM (2009) RNase R mutants elucidate the catalysis of structured RNA: RNA-binding domains select the RNAs targeted for degradation. Biochem J 423(2):291–301
Vincent HA, Deutscher MP (2009) The roles of individual domains of RNase R in substrate binding and exoribonuclease activity. The nuclease domain is sufficient for digestion of structured RNA. J Biol Chem 284(1):486–494
Domingues S et al (2015) The role of RNase R in trans-translation and ribosomal quality control. Biochimie 114:113–118
Awano N et al (2010) Escherichia coli RNase R Has Dual Activities, Helicase and RNase. J Bacteriol 192:1344–1352
Cheng ZF, Deutscher MP (2003) Quality control of ribosomal RNA mediated by polynucleotide phosphorylase and RNase R. Proc Natl Acad Sci U S A 100(11):6388–6393
Sulthana S, Deutscher MP (2013) Multiple exoribonucleases catalyze maturation of the 3’ terminus of 16S ribosomal RNA (rRNA). J Biol Chem 288(18):12574–12579
Tejada-Arranz A et al (2021) RNase R is associated in a functional complex with the RhpA DEAD-box RNA helicase in Helicobacter pylori. Nucleic Acids Res 49(9):5249–5264
Andrade JM, Cairrao F, Arraiano CM (2006) RNase R affects gene expression in stationary phase: regulation of ompA. Mol Microbiol 60(1):219–228
Venkataraman K, Zafar H, Karzai AW (2014) Distinct tmRNA sequence elements facilitate RNase R engagement on rescued ribosomes for selective nonstop mRNA decay. Nucleic Acids Res 42(17):11192–11202
Cairrao F et al (2003) Cold shock induction of RNase R and its role in the maturation of the quality control mediator SsrA/tmRNA. Mol Microbiol 50(4):1349–1360
Liang W, Deutscher MP (2010) A novel mechanism for ribonuclease regulation: transfer-messenger RNA (tmRNA) and its associated protein SmpB regulate the stability of RNase R. J Biol Chem 285(38):29054–29058
Moreira RN et al (2012) Synergies between RNA degradation and trans-translation in Streptococcus pneumoniae: cross regulation and co-transcription of RNase R and SmpB. BMC Microbiol 12:268
Erova TE et al (2008) Cold shock exoribonuclease R (VacB) is involved in Aeromonas hydrophila pathogenesis. J Bacteriol 190(10):3467–3474
Tobe T et al (1992) vacB, a novel chromosomal gene required for expression of virulence genes on the large plasmid of Shigella flexneri. J Bacteriol 174(20):6359–6367
Bárria C et al (2022) RNase R, a new virulence determinant of Streptococcus pneumoniae. Microorganisms 10(2):317
Charpentier X et al (2008) Loss of RNase R induces competence development in Legionella pneumophila. J Bacteriol 190(24):8126–8136
Abula A et al (2021) Molecular mechanism of RNase R substrate sensitivity for RNA ribose methylation. Nucleic Acids Res 49(8):4738–4749
Lalonde MS et al (2007) Exoribonuclease R in Mycoplasma genitalium can carry out both RNA processing and degradative functions and is sensitive to RNA ribose methylation. RNA 13(11):1957–1968
Hutchison CA et al (1999) Global transposon mutagenesis and a minimal Mycoplasma genome. Science 286(5447):2165–2169
Purusharth RI et al (2005) Exoribonuclease R interacts with endoribonuclease E and an RNA helicase in the psychrotrophic bacterium Pseudomonas syringae Lz4W. J Biol Chem 280(15):14572–14578
Shivaji S, et al (1989) Isolation and identification of Pseudomonas spp. from Schirmacher Oasis, Antarctica. Appl Environ Microbiol 55(3):767–70
Purusharth RI, Madhuri B, Ray MK (2007) Exoribonuclease R in Pseudomonas syringae is essential for growth at low temperature and plays a novel role in the 3’ end processing of 16 and 5 S ribosomal RNA. J Biol Chem 282(22):16267–16277
Sulthana S, Basturea GN, Deutscher MP (2016) Elucidation of pathways of ribosomal RNA degradation: an essential role for RNase E. RNA 22(8):1163–1171
Janiyani KL, Ray MK (2002) Cloning, sequencing, and expression of the cold-inducible hutU gene from the antarctic psychrotrophic bacterium Pseudomonas syringae. Appl Environ Microbiol 68(1):1–10
Regha K, Satapathy AK, Ray MK (2005) RecD plays an essential function during growth at low temperature in the antarctic bacterium Pseudomonas syringae Lz4W. Genetics 170(4):1473–1484
Malke H, Sambrock J, Fritsch EF, Maniatis T (1989) Molecular Cloning, A Laboratory Manual (Second Edition), Volumes 1, 2 and 3. 1625 S., zahlreiche Abb. und Tab. Cold Spring Harbor: Cold Spring Harbor Laboratory Press. $ 115.00. ISBN: 0–87969–309–6. 1990. 30(8): p. 623–623
Strand TA et al (2014) A new and improved host-independent plasmid system for RK2-based conjugal transfer. PLoS ONE 9(3):e90372
Liu J et al (2018) An improved method of preparing high efficiency transformation Escherichia coli with both plasmids and larger DNA fragments. Indian J Microbiol 58(4):448–456
Froger A, Hall JE (2007) Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp 6:253
Bidle KA, Bartlett DH (1999) RecD function is required for high-pressure growth of a deep-sea bacterium. J Bacteriol 181(8):2330–2337
Sulthana S et al (2011) rnr gene from the antarctic bacterium Pseudomonas syringae Lz4W, encoding a psychrophilic RNase R. Appl Environ Microbiol 77(22):7896–7904
Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189(1):113–130
Sinha AK et al (2013) Replication arrest is a major threat to growth at low temperature in Antarctic Pseudomonas syringae Lz4W. Mol Microbiol 89(4):792–810
Braman J, Papworth C, Greener A (1996) Site-directed mutagenesis using double-stranded plasmid DNA templates. Methods Mol Biol 57:31–44
Carpousis AJ (2002) The Escherichia coli RNA degradosome: structure, function and relationship in other ribonucleolytic multienzyme complexes. Biochem Soc Trans 30(2):150–155
Worrall JA et al (2008) Reconstitution and analysis of the multienzyme Escherichia coli RNA degradosome. J Mol Biol 382(4):870–883
Khemici V et al (2008) The RNase E of Escherichia coli is a membrane-binding protein. Mol Microbiol 70(4):799–813
Ponnada PSk, et al (2011) Cold active enzymes from the marine psychrophiles: biotechnological perspective. Adv Biotech 10:16–20
Åqvist J, Isaksen GV, Brandsdal BO (2017) Computation of enzyme cold adaptation. Nat Rev Chem 1(7):0051
Gerday C (2014) Fundamentals of cold-active enzymes, in cold-adapted yeasts: biodiversity, adaptation strategies and biotechnological significance. Buzzini P, Margesin R, Editors. Springer Berlin Heidelberg, Berlin, 325–350
Siddiqui KS, Cavicchioli R (2006) Cold-adapted enzymes. Annu Rev Biochem 75:403–433
Lonhienne T, Gerday C, Feller G (2000) Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility. Biochim Biophys Acta 1543(1):1–10
Rishi N et al (2022) Enzymatic Behaviour of Cold Adapted Microbes. In: Goel R et al (eds) Survival Strategies in Cold-adapted Microorganisms. Springer Singapore, Singapore, pp 113–131
Gerday C et al (2000) Cold-adapted enzymes: from fundamentals to biotechnology. Trends Biotechnol 18(3):103–107
Collins T, et al (2008) Fundamentals of Cold-Adapted Enzymes, in Psychrophiles: from Biodiversity to Biotechnology, R. Margesin, et al., Editors. Springer Berlin Heidelberg, Berlin, p. 211–227
Deniz AA (2018) Enzymes can adapt to cold by wiggling regions far from their active site. Nature 558(7709):195–196
Peterson ME et al (2007) The dependence of enzyme activity on temperature: determination and validation of parameters. Biochem J 402(2):331–337
Daniel RM et al (2008) The effect of temperature on enzyme activity: new insights and their implications. Extremophiles 12(1):51–59
Marx JC et al (2007) Cold-adapted enzymes from marine Antarctic microorganisms. Mar Biotechnol (NY) 9(3):293–304
Sočan J, Purg M, Åqvist J (2020) Computer simulations explain the anomalous temperature optimum in a cold-adapted enzyme. Nat Commun 11(1):2644
D’Amico S et al (2003) Activity-stability relationships in extremophilic enzymes. J Biol Chem 278(10):7891–7896
Chen Y, Tian Q, Wang H, Ma R, Han R, Wang Y et al (2022) A manganese-based metal-organic framework as a cold-adapted nanozyme. Adv Mater e2206421. https://doi.org/10.1002/adma.202206421
Georlette D et al (2004) Some like it cold: biocatalysis at low temperatures. FEMS Microbiol Rev 28(1):25–42
D’Amico S, Gerday C, Feller G (2001) Structural determinants of cold adaptation and stability in a large protein. J Biol Chem 276(28):25791–25796
Makhatadze GI, Privalov PL (1995) Energetics of protein structure. Adv Protein Chem 47:307–425
Aurilia V, et al (2009) Structure and dynamics of cold-adapted enzymes as investigated by FT-IR spectroscopy and MD. The Case of an Esterase from Pseudoalteromonas haloplanktis. J Phys Chem B 113(22):7753–7761
Aghajari N et al (2003) Crystal structures of a psychrophilic metalloprotease reveal new insights into catalysis by cold-adapted proteases. Proteins 50(4):636–647
Russell RJ et al (1998) Structural adaptations of the cold-active citrate synthase from an Antarctic bacterium. Structure 6(3):351–361
Khan S, Farooq U, Kurnikova M (2016) Exploring protein stability by comparative molecular dynamics simulations of homologous hyperthermophilic, mesophilic, and psychrophilic proteins. J Chem Inf Model 56(11):2129–2139
Kim SY, et al (1999) Structural basis for cold adaptation. Sequence, biochemical properties, and crystal structure of malate dehydrogenase from a psychrophile Aquaspirillium arcticum. J Biol Chem 274(17):11761–7
Smalås AO et al (2000) Cold adapted enzymes. Biotechnol Annu Rev 6:1–57
Acknowledgements
The authors acknowledge the Council of Scientific & Industrial Research, India and Indian Council of Medical Research (ICMR), India for financial support in the form of a fellowship to AH during this study.
Funding
No external funding was received during this study.
Author information
Authors and Affiliations
Contributions
Conception or design of the work: Ashaq Hussain and Malay Kumar Ray. Acquisition and interpretation of data: Ashaq Hussain. Drafted and reviewed the manuscript: Ashaq Hussain. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Functional activity of E. coli RNase R in the Antarctic Pseudomonas syringae Lz4W.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hussain, A., Ray, M.K. Functional activity of E. coli RNase R in the Antarctic Pseudomonas syringae Lz4W. J Genet Eng Biotechnol 21, 101 (2023). https://doi.org/10.1186/s43141-023-00553-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43141-023-00553-2