
Abstract
Background
The Amyloid theory of Alzheimer’s disease (AD) suggests that the deposition of Amyloid β (Aβ) in the brain triggers a chain of events, involving the deposition of phosphorylated Tau and other misfolded proteins, leading to neurodegeneration via neuroinflammation, oxidative stress, and neurovascular factors. The infectious theory linked various infectious agents with the development of AD, raising the possibility that they serve as etiological causes of the disease. Are these theories mutually exclusive, or do they coincide?
Main body
In this review, we will discuss how the two theories converge. We present a model by which (1) the systemic infectious burden accelerates the development of AD brain pathology via bacterial Amyloids and other pathogen-associated molecular patterns (PAMPs), and (2) the developing AD brain pathology increases its susceptibility to the neurotoxicity of infectious agents -derived PAMPs, which drive neurodegeneration via activated microglia.
Conclusions
The reciprocal effects of amyloid deposition and systemic infectious burden may lead to a vicious cycle fueling Alzheimer’s disease pathogenesis.
Similar content being viewed by others

Background
The neurodegenerative process in Alzheimer’s disease (AD) is considered the consequence of the deposition of misfolded amyloid-β (Aβ) and hyperphosphorylated tau (p-tau) proteins, with histopathological hallmarks that include Aβ-rich extracellular plaques, p-tau-rich neurofibrillary tangles, microgliosis, astrogliosis, and neuronal loss. Aβ is a peptide consisting of about 40 amino acids, formed by sequential cleavages of amyloid β precursor protein (APP) by β-secretase and γ-secretase. In normal subjects, Aβ is released outside the cell, where it is rapidly degraded or removed. However, in aged subjects or under pathological conditions, the metabolic ability to degrade Aβ is decreased, and Aβ peptides may accumulate [1]. The deposition of Aβ peptides is probably one of the earliest pathological events in AD pathogenesis [2]. However, there are still broad discussions on downstream events, triggered by Aβ deposition, which lead to neurodegeneration. Along with the deposition of misfolded protein in the brain, multiple systemic risk factors have been shown to contribute to disease pathogenesis. Among these are infectious agents, which significantly increase the risk of AD. We suggest here a model by which systemic and bacterial amyloids and other Pathogen-associated molecular patterns (PAMPs) accelerate AD brain pathology. While Aβ induces CNS neuroinflammation which is insufficient in itself to cause neurodegeneration, it results in brain visibility to the systemic milieu and increased vulnerability to microbial PAMPs-induced neurotoxicity, leading to neurodegeneration.
Main text
Amyloid deposition induces chronic neuroinflammation: a critical, but insufficient driver of disease
Aβ does not directly cause neurodegeneration
The amyloid theory, which has been the mainstream explanation of AD pathogenesis, proposed originally that amyloid plaques and their major constituents, the Aβ fibrils, are the direct cause of progressive neurodegeneration in AD. However, multiple studies have raised important issues that undermine the amyloid theory, including the large temporal gap and the lack of good anatomic correlation between accumulation of amyloid deposits, clinical deterioration, and neuronal loss. Pathological studies were unable to prove a direct correlation or causality between Aβ deposition, clinical dementia, and neuronal loss [3]. In close agreement, transgenic mouse models of AD that carry mutated human genes associated with excessive Aβ deposition and familial AD, are characterized by heavy amyloid deposition, but exhibit no- to only mild- loss of cortical neurons, starting at an advanced age [4]. These suggest that Amyloid pathology may be necessary, but insufficient to cause neurodegeneration. Indeed, different neuronal-injury biomarkers were found to be independent of Aβ [5]. Amyloid imaging studies have shown that Aβ starts to accumulate in the brain approximately two decades prior to clinical dementia and reaches saturated levels several years before the clinical presentation of early dementia [6,7,8]. This provides a wide gap, during which other pathogenic factors may come into effect and cause neurodegeneration. Further studies found that neurodegeneration in AD was better correlated to local deposition of other misfolded proteins, such as Tau in its highly phosphorylated form, and TDP43 [9, 10], rather than with Aβ. It is thought that Aβ promotes the deposition and dissemination of phosphorylated Tau in the affected brain, leading to neurodegeneration [11,12,13] via several mechanisms, such as a neuroinflammatory process, oxidative stress, and neurovascular factors. However, we suggest here that in addition to promoting Tau pathology, the deposition of Amyloid causes also brain susceptibility to the neurotoxic effect of external (systemic) insults, and in particular to infectious agents – neurotoxicity.
Aβ pathology induces a brain immune response
Multiple studies have shown that Aβ activates the brain’s innate immune system. Pathological Aβ deposits are associated with surrounding (plaque-associated) activated microglia [14, 15]. Monomeric and fibrillar Aβ activates microglia directly via the TLR2 receptor [16]. The Amyloid-burdened brain displays activated glial cells, lymphocytes, and macrophages which release large amounts of inflammatory mediators escalating the inflammatory state and exacerbating other AD pathologies [17].
CNS microglia serve as resident phagocytes that dynamically survey the environment, playing crucial roles in CNS tissue maintenance, injury response, and pathogen defense [18, 19]. Microgliosis, described first by Alois Alzheimer himself, was considered initially the consequence of AD pathology rather than a cause [20]. However, accumulating data have proven that neuroinflammation contributes both to disease initiation and progression. The crucial role of microglia in AD pathogenesis was demonstrated by genome-wide association studies (GWAS) that identified genetic loci which are associated with an increased risk of late-onset AD [21]. These studies have shown that the majority of the loci relate to neuroinflammation and are preferentially or exclusively expressed in microglia. It has become clear that microglia are important players in AD pathogenesis, although it is still highly debated whether the microglial function in AD is beneficial, deleterious, or both. The multiple influences of microglia on AD pathogenesis can be explained by the highly complex nature of these cells, which can polarize into a wide spectrum of phenotypes and activation states, some of them have detrimental effects while others are crucial for disease attenuation and neuroprotection. Among these, several studies have identified the disease-associated microglia (DAM) population, which is increased in both transgenic mouse models of AD and human AD postmortem brains. This sub-population of microglia, which highly express Trem2, is associated with phagocytic activity and Aβ plaque clearance, and is beneficial for AD [22,23,24]. On the other hand, multiple studies suggest that neurotoxic microglia mediate neuronal death in AD [25,26,27]. Microglial toxic activation has harmful effects both through a loss of beneficial functions and through a gain of deleterious functions such as the production of pro-inflammatory cytokines and oxidative stress.
Thus, Aβ deposition induces already from early stages the development of an inflammatory CNS environment, manifesting with marked microglial activation. However, the apparent neuroinflammatory process is insufficient to cause neurodegeneration. This raises the possibility that additional factors may drive activated microglia to become fully neurotoxic. Here we discuss the notion that in addition to endogenous CNS misfolded proteins’ -induced neurodegeneration, there are exogenous insults, and in particular infectious agents that promote neurodegeneration in brains that are inflicted with AD pathology.
CNS visibility and vulnerability to systemic insults in AD
Whether Amyloid or other misfolded proteins drive neurodegeneration, these concepts rely on the traditional thought that Alzheimer’s disease pathogenesis is confined to the nervous system, independent of systemic factors. However, increasing evidence suggests strong bilateral interactions between the brain and the systemic environment that are fundamental to disease pathogenesis.
Systemic factors exacerbate AD
First, multiple systemic risk factors such as diabetes mellitus, midlife hypertension, hypercholesterolemia, smoking, and cardiovascular disease, are associated with a significant increase in developing Alzheimer’s dementia and account for up to 50% of the morbidity. Multiple experimental models showed that the increased risk is not merely by co-morbidity, but rather the exacerbation of AD brain pathology by these risk factors. Exposure of transgenic mice models that carry human genes associated with familial AD to systemic risk factors, resulted in the acceleration of the specific AD pathology [28,29,30]. Moreover, systemic risk factors may affect CNS visibility to the systemic milieu. Specifically, increased plasma glucose levels in diabetes mellitus have been associated with altered blood–brain barrier (BBB) transport functions and oxidative stress in CNS micro-circulation. These changes not only lead to local CNS inflammation but are associated also with upregulation and activation of the receptor for advanced glycation end products (RAGE), which transports Aβ from the blood into the brain across the BBB [31], and therefore may increase Aβ deposition in the brain [32, 33].
Second, different studies have indicated that peripheral immune cells, belonging to either the innate or the adaptive immune system, play an important role in AD pathogenesis. It was shown that circulating myeloid cells mitigate the neuroinflammatory response in AD models and that CNS-infiltrating monocyte-derived macrophages facilitate Aβ plaque removal [34, 35]. Furthermore, systemic regulatory T cells (Tregs) may play a role in disease progression. Some studies suggest that depletion of Tregs accelerated the onset of cognitive decline in mouse AD models, while Tregs administration had neuroprotective effects [36, 37]. Contrarily, others have indicated that pharmacological inhibition of Foxp3 + Tregs is followed by Aβ plaque clearance, mitigation of the neuroinflammation response, and reversal of cognitive decline [38].
BBB integrity is breached in AD
The BBB is formed by a tightly sealed monolayer of brain endothelial cells, which keeps neurotoxic plasma-derived components, RBCs, leukocytes, and pathogens out of the CNS [39]. It is widely agreed that cerebrovascular dysfunction and vascular pathology contribute to cognitive decline and neuronal loss in AD [40, 41]. However, a major issue of discussion is whether this vascular dysregulation is an early pathologic event responsible for disease development, or a late by-product of the toxic brain environment. There are multiple indications for BBB disruption very early in the course of human AD, as shown by using various imaging biomarkers of BBB integrity, cerebrovascular reactivity, resting CBF, increased cerebrovascular resistance, and accumulation of brain microbleeds, indicating cerebral amyloid angiopathy [42, 43]. Recent neuroimaging studies in individuals with mild cognitive impairment (MCI) and early AD have shown BBB breakdown in the hippocampus and in several grey and white matter regions [44,45,46], occurring before brain atrophy or dementia. Moreover, in preclinical AD, changes in vascular biomarkers occur before a detectable increase in standard AD biomarkers, including amyloid deposition, decreased cerebrospinal fluid (CSF) levels of Aβ42 (the most amyloidogenic form of Aβ), and increased CSF levels of tau and phosphorylated tau [47]. In close agreement, BBB integrity is compromised in transgenic AD mice from an early stage, even prior to amyloid deposition [39, 48, 49].
This early BBB breakdown suggests that cerebrovascular changes may be a major driver of the disease pathogenesis, and not just an ‘innocent bystander’ occurring as a result of the dysregulated inflammatory brain environment. The causal role of vascular dysregulation on AD pathogenesis has been suggested as the two-hit vascular hypothesis, where damage to blood vessels is the initial insult, causing BBB dysfunction that eventually leads to neuronal injury and Aβ accumulation [50].
Early BBB breakdown in AD creates CNS visibility to systemic insults, particularly to infectious agents and their products. Blood-borne infections of the CNS in immune-competent subjects with a fully functional BBB are the exception. Indeed, the BBB and CSF barriers prevent the unselective diffusion of vascular and cellular components [51]. In agreement, in healthy mouse models, low and medium doses of endotoxin administered peripherally only minimally entered the brain [3]. However, when the BBB is compromised, various pathogens and pathogens-induced molecules can enter the brain through the bloodstream. It was shown that transgenic AD mice exhibited increased susceptibility to BBB disruption following induction of peripheral inflammatory states [48, 53], providing additional explanation to the observation that AD patients are more vulnerable to the effects of peripheral infection than their age-matched, healthy counterparts [54].
We suggest that early cerebrovascular dysregulation in AD may render the CNS visible to systemic infectious agents, which contribute to disease pathogenesis. Specifically, we will discuss how bacterial Amyloids and microbial PAMPs accelerate AD pathology and cause a direct neurotoxic effect.
The infectious etiology of AD
Among the various systemic factors fueling AD, accumulating evidence imply an association between infections and AD. Systemic infections are associated with long-lasting cognitive decline in patients with pre-existing AD [55, 56]. This has traditionally been viewed as the human analog of sickness behavior, induced in animal models by inflammatory mediators, including pro-inflammatory cytokines and PAMPs, such as endotoxin, and being pronounced in demented patients due to compromised cognitive reserves. However, mounting evidence infers also an association between systemic and CNS infections to the development of AD [57, 58]. Do infectious agents serve merely as risk factors for AD by unknown mechanisms, or do they cause AD pathology directly? We suggest that both the systemic burden of various PAMPs including bacterial amyloids, as well as neuro-invasion of infectious agents may directly accelerate AD brain pathology.
Systemic infections and their products are associated with the development of AD
A well-established infectious-related cause of AD is periodontal disease (periodontitis). Various periodontal pathogens, mainly Treponema denticola, and Porphyromonas gingivalis have been described as potential contributors to AD pathogenesis. Prospective studies indicate that periodontitis is associated with an increased pro-inflammatory state and cognitive decline in AD, independent of baseline cognitive state [59]. The chronic peripheral periodontal infection may elicit a central inflammatory response by two mechanisms. First, periodontal bacteria cause local production of inflammatory molecules, capable of reaching the CNS via systemic circulation [60] and penetrating the dysfunctional BBB. Second, it has been suggested that stimulation of the trigeminal nerve by periodontal disease in the oral cavity, may be transmitted to induce the production of cytokines in the CNS. These cytokines may have a synergic effect with Aβ on activated microglia, causing an amplified reaction favoring AD progression [61].
The huge mass of microbial organisms in the gut, containing more microorganisms than the entire cell population in the brain [62], makes the brain-gut-microbiota axis another important infectious factor in AD pathogenesis. The seemingly silent gut microbiome may produce important effects on the host body (and its brain) during healthy homeostasis and disease [63]. Studies have shown alterations in the gut microbiome in AD patients, with decreased microbial diversity and distinct composition in comparison to age- and sex-matched individuals. In addition, recent studies in transgenic mouse models of AD have demonstrated that manipulation of gut microbiota can influence cerebral amyloid deposition and attenuate neuroinflammation [64, 65], supporting the notion that the resident microbial flora may affect the pathogenesis of AD brain pathology.
Microbial PAMPs contribute to AD pathogenesis
Lipopolysaccharide (LPS) bacterial endotoxins are a major component of the outer membrane of gram-negative bacteria, and an important group of PAMPs. Soluble endotoxin is released when bacteria are destroyed but is also released physiologically as outer membrane vesicles [66]. Therefore, a high load of gram-negative bacteria carrying endotoxins in the microbiome is associated with increased levels of endotoxin in the systemic circulation [67]. When released, endotoxin causes inflammatory activation mainly via activating TLR4 on the cell surface of innate immune cells, including microglia. Animal studies have shown that systemic bacterial endotoxins can induce brain inflammation with accompanying inflammatory-cytokine -induced sickness behavior and cognitive dysfunction [68,69,70]. Furthermore, endotoxin has been shown to exacerbate brain pathology in animal models, specifically Aβ production and aggregation [71] and Tau hyperphosphorylation [71, 72]. These findings are of clinical relevance, as studies found a threefold increase in mean blood endotoxin levels, a 2–threefold increase in brain endotoxin levels in AD patients, and up to a 26-fold increase in hippocampal tissue [68]. Endotoxin is also found in amyloid plaques [73, 74]. Indeed, people with chronic gingival disease (periodontitis) have elevated blood endotoxin [75], a higher risk of AD [76], and a faster rate of cognitive decline [59, 75, 77].
Another major group of PAMPs is TLR2 agonists, derived from gram-positive bacteria and yeasts. For example, Zymosan is a β-Glucan polysaccharide TLR2 agonist derived from the yeast Saccharomyces cerevisiae, and Lipoteichoic acid (LTA), is a TLR2 agonist that is a major constituent of the bacterial wall in Staphylococcus Aureus and other gram-positive bacteria. Thus, TLR2 agonists are produced by multiple systemic infectious agents affecting patients, including chronic gingivitis [78], skin pathogens [79], and gut microbiome [80]. TLR2 agonists are of particular interest since TLR2 serves as a receptor for Aβ-induced microglial activation [81]. TLR2 mediates Aβ ingestion by microglia and its blockage results in extracellular amyloid accumulation [82]. Neurotoxic activation of microglia by TLR2 agonists may be important in AD pathogenesis and neurodegeneration, as disruption of downstream TLR2 signaling prevented the progression of AD pathology and loss of cortical neurons in AD transgenic mice [83].
Invasion of infectious agents to the AD brain
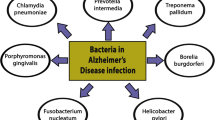
Pathogens invading the brain have been widely studied and suggested also as key causative factors in AD development. Among these are viral pathogens, including Herpes simplex virus (HSV1), Cytomegalovirus (CMV), fungi and bacteria including Chlamydophila pneumoniae, Helicobacter pylori, Borrelia burgdorferi and various periodontal pathogens [84,85,86,87,88,89,90,91].
Porphyromonas gingivalis (P. gingivalis) is a keystone pathogen in the development of chronic periodontitis. In transgenic mice overexpressing mutated human amyloid precursor protein, oral infection with P. gingivalis impaired cognitive function and increased the deposition of AD-like plaques [92]. Furthermore, P. gingivalis LPS has been detected in human AD brains, and P. gingivalis DNA was present in the CSF of clinical AD patients [93, 94]. The brain load of Gingipains, major virulence factors of P. gingivalis, was significantly higher in AD brains compared to non-demented control brains. Moreover, gingipains were shown to colocalize with intraneuronal Aβ and tau tangles. Oral administration of small-molecule gingipain inhibitors significantly reduced P. gingivalis load in mouse brain, decreased the host amyloid response to P. gingivalis brain infection, and successfully blocked gingipain-induced neurodegeneration [93].
One of the most studied viral pathogens in the context of AD is HSV1. HSV1 DNA was found to be present in the brain of AD patients at significantly higher levels compared to age-matched healthy individuals [95]. Viral DNA is found within senile plaques [96], Aβ deposition and tau abnormalities typical of AD are observed after infection with HSV1 and are diminished following antiviral treatments [96,97,98]. In agreement, epidemiological cohort studies showed that HSV1 reactivation, indicated by the presence of both anti-HSV IgM and IgG antibodies, almost doubled the risk for AD in comparison to the presence of anti-HSV IgG alone [99].
Another clue for the importance of HSV1 in AD pathogenesis is indicated by the predilection of the virus to the entorhinal cortex and Temporal lobe, co-localizing with areas presenting early AD pathological changes. This overlap was long described and implies a possible causative role for HSV1 infection in early disease stages [100]. The development of initial AD pathology in the olfactory and entorhinal cortices, and the identification of olfactory dysfunction as one of the earliest clinical symptoms of AD [101, 102], laid the basis for the olfactory hypothesis of AD. This hypothesis suggests that foreign agents are transmitted from the nasal cavity into the brain by the olfactory nerve, as a putative mechanism promoting AD pathogenesis. Studies have suggested that the olfactory system serves as a route of HSV1 entry to the brain [103, 104], and identified HSV1 in the olfactory bulb in post-mortem samples from humans [105]. Furthermore, studies have found Chlamydophila pneumoniae present in the olfactory bulb of AD patients, as well as in the entorhinal cortex, hippocampus, and temporal cortex [106]. Another viral agent that was shown to enter the brain through the olfactory nerve and potentially accelerate cognitive deterioration is SARS-Cov-2 (COVID-19 virus) [107].
Although early involvement of pathogens, penetrating the olfactory and entorhinal cortices is widely agreed, it is yet to be determined whether they are the primary cause of AD, initiating amyloid deposition and BBB disruption, or whether prior AD changes starting in these areas enable their penetration.
An additional unique mechanism by which invading pathogens, and other exogenous insults, including environmental pollutants, may contribute to AD pathogenesis is the activation of retrotransposons and silent human endogenous retroviruses (HERVs) [108]. Transposable elements (TE) dysregulation and HERVs activation have been associated to neurodegenerative processes. TE dysregulation may contribute to neuronal death in tauopathies, a significant increase in HERVs transcripts was found in AD [109], and differential expression of several retrotransposons was observed in association with burden of neurofibrillary tangle in human AD brains [110]. It has been suggested that ERV activation may stimulate continuously inflammatory responses, perpetuating the chronic inflammatory environment in AD brains.
Infectious agents induce amyloid deposition
How do bacterial and viral agents induce AD pathology? First, the increased systemic and CNS burden of microbial PAMPs may increase neuroinflammation in AD. However, there is increasing evidence suggesting that infectious agents may also directly accelerate Aβ deposition. Gut gram-negative bacteria secrete the amyloid protein Curli, which has marked structural similarity to pathological Aβ [111]. Curli is the major constituent of enteric biofilms, inducing both cell–cell and cell-extracellular matrix attachment [112]. Curli creates potent immunogenic complexes that strongly activate immune cells and induce an antibacterial response [113]. Bacterial amyloids are recognized by innate immune cells as a PAMP, leading to their activation via toll-like receptor 2 (TLR2), and CD14 [114]. While this alone can promote neuroinflammation, it has been suggested that microbial components may also accelerate Aβ deposition in the brain [115]. The inoculation of transgenic 5xFAD mice brains with Salmonella Typhimurium bacteria resulted in rapid seeding and accelerated Aβ deposition, in closely anatomic localization with the invading bacteria [116]. Given the structural similarity of Aβ and bacterial Curli, and robust Aβ deposition in response to infection, it has been proposed that Aβ is an anti-microbial peptide, and that pathologic Aβ deposition in the AD brain may be a defensive, anti-bacterial response by the brain’s innate immune system [116]. Brain-derived Aβ entraps and neutralizes invading pathogens, and its oligomerization, a critical step in it becoming pathogenic in AD, may also promote its antimicrobial activities [117].
This association may explain also the robust Aβ deposition in response to HSV-1 infection similar to the brain response to bacterial Curli: Aβ oligomers bind HSV-1 envelope glycoproteins and accelerated β-amyloid deposition was observed in response to herpes virus infection of 5xFAD mice or 3D human neural cell cultures [118].
Aβ is produced in peripheral tissues, including platelets [119], skin fibroblasts, skeletal muscles, and cerebrovascular smooth muscle cells [120,121,122]. Peripherally produced Aβ is secreted into the blood circulation and is able of crossing the blood–brain barrier [123]. It was shown in a parabiosis model that circulating (human) Aβ invaded and accumulated in the brains of wild-type mice (wt), in the form of cerebral amyloid angiopathy and Aβ plaques. Moreover, these led also to neuroinflammation, tau hyperphosphorylation, and neurodegeneration, comprising the full spectrum of AD pathology [124]. Also, transgenic mice expressing human Aβ only in the liver developed pathological features of neurodegenerative disease [125]. Furthermore, the influx of Aβ from the systemic circulation into the brain was enhanced by P. gingivalis infection [126]. Chronic systemic P. gingivalis infection-induced Aβ accumulation in inflammatory monocytes/macrophages, and in the brain of middle-aged mice [127]. Importantly, systemic bacterial amyloids can invade the brain, and cross seed with neuronal amyloid [128], suggesting that there is probably no pre-requisite for the entire pathogen to invade the CNS. Thus, both systemic and CNS infections induce an increase in Aβ deposition and AD pathology.
Molecular mechanism of infection-driven neurodegeneration
Microbial PAMPs kill cortical neurons
The deposition of Aβ as a defensive anti-bacterial response that creates a highly inflammatory CNS environment may underlie the close association between various infections and the development of AD. Although most research on the infectious etiology of AD has focused on individual pathogens, a growing body of evidence supports the hypothesis of polymicrobial causality. Consequently, the cumulative exposure to multiple pathogens may cause an “infectious burden” that contributes to the development of the disease. We further suggest that the presence of AD pathology causes CNS hyper-vulnerability to the neurotoxicity of microbial PAMPs, a process that is made possible by the combination of chronic Aβ-induced neuroinflammation and impaired BBB integrity. We have shown that microbial TLR2- and TLR4-agonists kill cortical neurons and that brains inflicted with AD pathology are significantly more vulnerable to their neurotoxicity by two mechanisms. First, in transgenic AD mouse models, the compromised BBB enabled penetration of systemically administered microbial PAMP to the CNS [129]. Consequently, we demonstrated that systemically administered PAMPs induce neurodegeneration in 5xFAD mice, but not in wt mice [129, 130]. These findings may indicate that the increased visibility of AD brains to systemic infectious agents and PAMPs may contribute to their increased vulnerability to neurotoxic effects of systemic PAMPs, resulting in increased death of cortical neurons. Second, we showed that direct delivery of microbial TLR2- and TLR4- agonists cause cortical neuronal death in a dose-dependent manner and that brains inflicted with AD pathology exhibit a marked increase in cortical neuron death, as compared to wt brains [130]. Thus, microbial PAMPs both penetrate and exhibit increased toxicity to the AD brain than to the normal, wt brain.
Microglia – a key player in inflammation-induced neurodegeneration
How do microbial PAMPS cause neurodegeneration? Our studies suggest that PAMP-induced neurodegeneration is mediated by brain microglia. First, we and others showed a marked increase in TLR2 + and TLR4 + microglia in human AD and murine AD models [129, 131,132,133,134]. Second, we showed that either depletion of microglia by direct Intracerebroventricular (ICV) delivery of Minocycline [129] or modulation of microglial neurotoxic phenotype by direct ICV delivery of a retinoic acid receptor α agonist [130] prevents microbial PAMP-induced neurodegeneration. Finally, we showed that PAMP-induced loss of neurons occurs in the microglia-rich frontal cortex, but not in the microglia-poor CA1 and CA3 regions of the hippocampus [130]. Interestingly, PAMP exposure results in acute death of cortical neurons, rather than inducing a chronic neurodegenerative process [129]. These findings support the notion that recurrent and chronic sub-clinical PAMP exposures may result in a cumulative “infectious burden and cause accelerated neuronal loss. Indeed, we showed that 5xFAD mice housed in a natural environment exhibited accelerated neurodegeneration in comparison to 5xFAD mice housed in a specific-pathogen-free (SPF) facility [130], suggesting that these findings are relevant to the natural infectious milieu. The findings of infectious agents- and microbial PAMPs -induced accelerated neurodegeneration in the 5xFAD mouse model raises the question of its relevance to late-onset AD. Importantly, we studied 7-month-old 5xFAD mice, a time point of heavy amyloid burden and AD pathology, but prior to neurodegeneration. While the 5xFAD model has obvious limitations, such as the lack of deposition of other misfolded proteins, this model, and the choice of mouse age in our experiments, may represent a relatively early stage of late-onset AD, with amyloid accumulation and gliosis. Our findings are compatible with the literature on the early involvement of infections in AD pathogenesis, which may affect the aging population who display Aβ deposition at the pre-clinical phase. Importantly, testing this concept in human patients is possible, for example, by identifying patients who display AD brain pathology at the pre-clinical stage, either by Amyloid-PET imaging [135, 136], or by testing AD-specific biomarkers (eg. Aβ and p-tau) in the blood and CSF [137, 138], and examine prospectively whether a high infectious load is associated with brain atrophy.
Conclusion
Within the multitude of systemic drivers and risk factors for AD, and potential interactions between them, we highlight the role of infectious agents and their products in AD pathogenesis, and suggest the convergence of the amyloid and the infectious hypotheses in AD development. We suggest that systemic infectious agents and pathogen-associated molecules can penetrate the AD brain through the leaky BBB (or via olfactory pathways), accelerate Aβ deposition, and act on local microglia, which are already activated and in increased density. These infectious insults further induce neurotoxic activation of microglia, resulting in neurodegeneration. We propose a model (Fig. 1) by which systemic infectious agents induce neurodegeneration, occurring exclusively in vulnerable brain areas with underlying AD pathology. The Amyloid deposition may not cause neurodegeneration by itself, but rather result in brain susceptibility to the neurotoxic effect of infectious agents. This suggests a “hit and run” mechanism, where the infectious agent-derived PAMPs -induced neurodegeneration may be masqueraded, as it occurs in brain areas already displaying marked microgliosis and AD pathology, invisible to the examining pathologist.

Convergence of the amyloid and infectious theories of Alzheimer's Disease. We suggest that deposition of amyloid β causes increased visibility and susceptibility of the CNS to systemic infectious agents and their components coming from the systemic environment. In turn, infectious agents and PAMPs increase AD pathology and cortical neuron death. These result in a vicious cycle that accelerates neurodegeneration
While additional studies are necessary to determine which is the initial event in AD pathogenesis, we suggest in our model that viral infections, bacterial amyloids, and other PAMPs, accelerate Aβ deposition, which in turn increases the vulnerability of the brain to their neurotoxic effects. This may create a vicious cycle fueling the disease process. Importantly, the PAMP-induced neurodegeneration is mediated by neurotoxic microglial activation, and is reversible through microglial modulation, thus highlighting their potential role as a therapeutic target.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- PAMP:
-
Pathogen associated molecular patterns
- Aβ:
-
Amyloid beta
- APP:
-
Amyloid beta precursor protein
- GWAS:
-
Genome-wide association studies
- DAM:
-
Disease-associated microglia
- RBC:
-
Red blood cells
- MCI:
-
Mild cognitive impairment
- CSF:
-
Cerebrospinal fluid
- TLR:
-
Toll-like receptor
- HSV1:
-
Herpes simplex virus
- LPS:
-
Lipopolysaccharide
- LTA:
-
Lipoteichoic acid
- SPF:
-
Specific pathogen free
- WT:
-
Wild type
- ICV:
-
Intracerebroventricular
- TE:
-
Transposable elements
- HERVs:
-
Human endogenous retroviruses
References
Fuyuki K, Hasegawa H. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease. Front Neurosci. 2018;12(25). https://doi.org/10.3389/fnins.2018.00025.
Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacologica Sinica. 2017;38(9):1205–35.
Murphy MP, Levine H. Alzheimer’s Disease and the β-Amyloid Peptide. J Alzheimers Dis. 2010;19(1):311.
Fainstein N, Dan-Goor N, Ben-Hur T. Resident brain neural precursor cells develop age-dependent loss of therapeutic functions in alzheimer’s mice. Neurobiol Aging. 2018;1(72):40–52.
Chételat G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nature Reviews Neurology. 2013;9(3):123.
Villemagne VL, Pike KE, Chételat G, Ellis KA, Mulligan RS, Bourgeat P, et al. Longitudinal assessment of Aβ and cognition in aging and alzheimer disease. Ann Neurol. 2011;69(1):181–92.
Jack CR, Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, et al. Brain β-amyloid load approaches a plateau. Neurology. 2013;80(10):890–6.
Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67.
Spires-Jones TL, Stoothoff WH, de Calignon A, Jones PB, Hyman BT. Tau pathophysiology in neurodegeneration: a tangled issue. Trends Neurosci. 2009;32(3):150–9.
Brettschneider J, Arai K, del Tredici K, Toledo JB, Robinson JL, Lee EB, et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014;3:423–37.
Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, et al. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2003;100(17):10032–7.
Blurton-Jones M, LaFerla F. Pathways by Which Aβ Facilitates Tau Pathology. Curr Alzheimer Res. 2006;3(5):437–48.
Ballatore C, Lee VMY, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–72.
Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia. 2008;56(13):1438–47.
Yin Z, Raj D, Saiepour N, van Dam D, Brouwer N, Holtman IR, et al. Immune hyperreactivity of Aβ plaque-associated microglia in alzheimer’s disease. Neurobiol Aging. 2017;1(55):115–22.
Jana M, Palencia CA, Pahan K. Fibrillar Amyloid-β peptides activate microglia via TLR2: implications for alzheimer’s disease. J Immunol. 2008;181(10):7254–62.
Ashraf GM, Tarasov VV, Makhmutova A, Chubarev VN, Avila-Rodriguez M, Bachurin SO, et al. The Possibility of an Infectious Etiology of Alzheimer Disease. Mol Neurobiol. 2018;56(6):4479–91.
Nayak D, Roth TL, McGavern DB. Microglia Development and Function*. http://dx.doi.org/10.1146/annurev-immunol-032713-120240 [Internet]. 2014 Mar 21 [cited 2022 Jan 18];32:367–402. Available from: https://www.annualreviews.org/doi/abs/101146/annurev-immunol-032713-120240
Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu Rev Immunol. 2017;35:441.
Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–72.
Hemonnot AL, Hua J, Ulmann L, Hirbec H. Microglia in Alzheimer disease: well-known targets and new opportunities. Front Cell Infection Microbiology. 2019;9(JUL):233.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of alzheimer’s disease. Cell. 2017;169(7):1276-1290.e17.
Deczkowska A, Keren-Shaul H, Weiner A, Colonna M, Schwartz M, Amit I. Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 2018;173(5):1073–81.
Song WM, Colonna M. The identity and function of microglia in neurodegeneration. Nat Immunol. 2018;19(10):1048–58.
Polazzi E, Contestabile A. Reciprocal interactions between microglia and neurons: From survival to neuropathology. Rev Neurosci. 2002;13(3):221–42.
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69.
Lull ME, Block ML. Microglial activation and chronic neurodegeneration. Neurotherapeutics. 2010;7(4):354–65.
Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107(15):7036–41.
Refolo LM, Pappolla MA, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, et al. Hypercholesterolemia accelerates the alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000;7(4):321–31.
Snyder HM, Corriveau RA, Craft S, Faber JE, Greenberg SM, Knopman D, et al. Vascular contributions to cognitive impairment and dementia including alzheimer’s disease. Alzheimer’s Dement. 2015;11(6):710–7.
Deane R, du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–13.
Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate alzheimer disease pathology? Nat Rev Neurol. 2010;6(10):551–9.
Prasad S, Sajja RK, Naik P, Cucullo L. Diabetes mellitus and blood-brain barrier dysfunction: an overview. J Pharmacovigil. 2014;2(2):125.
Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of alzheimer’s disease. J Neurosci. 2011;31(31):11159–71.
Basset NEWSANDVIEWS;, Crone G, Saumon C, Matthay G, Folkesson MA, Clerici HG. Immune cells may fend off Alzheimer disease. Nat Med 2007;13(4):408–9. Available from: https://www.nature.com/articles/nm0407-408
Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, et al. Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain. 2016;139(4):1237–51 https://academic.oup.com/brain/article/139/4/1237/2464189.
Baek H, Ye M, Kang GH, Lee C, Lee G, Choi DB, et al. Neuroprotective effects of CD4+CD25+Foxp3+ regulatory T cells in a 3xTg-AD Alzheimer’s disease model. Oncotarget. 2016;7(43):69347 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5342482/
Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun. 2015;6(1):1–12. Available from: https://www.nature.com/articles/ncomms8967.
Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J Exp Med. 2017;214(11):3151 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5679168/
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–38. Available from: https://www.nature.com/articles/nrn3114.
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and Dysfunction of the Blood-Brain Barrier. Cell. 2015;163(5):1064–78.
Nation DA, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2)270–6. https://doi.org/10.1038/s41591-018-0297-y.
Sweeney MD, et al. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nature reviews. Neurology. 2018;14(3):133–50. https://doi.org/10.1038/nrneurol.2017.188.
van de Haar, Harm J, et al. Subtle blood-brain barrier leakage rate and spatial extent: Considerations for dynamic contrast-enhanced MRI. Medical physics. 2017;44(8):4112–25. https://doi.org/10.1002/mp.12328.
van de Haar, Harm J, et al. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology. 2016;281(2):527–35. https://doi.org/10.1148/radiol.2016152244.
Sweeney MD, et al. Vascular dysfunction-The disregarded partner of Alzheimer's disease. Alzheimer's & dementia: the journal of the Alzheimer's Association. 2019;15(1):158–67. https://doi.org/10.1016/j.jalz.2018.07.222.
Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Weiner MW, et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun [Internet]. 2016 Jun 21 [cited 2021 Dec 24];7. Available from: https://pubmed.ncbi.nlm.nih.gov/27327500/
Takeda S, et al. Increased blood-brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol Aging. 2013;34(8):2064–70. https://doi.org/10.1016/j.neurobiolaging.2013.02.010.
Ujiie M, et al. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation (New York, N.Y. : 1994). 2003;10(6):463–70. https://doi.org/10.1038/sj.mn.7800212.
Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862(5):887–900.
De Chiara G, et al. Infectious agents and neurodegeneration. Mol Neurobiol. 2012;46(3):614–38. https://doi.org/10.1007/s12035-012-8320-7.
Banks WA, Robinson SM. Minimal Penetration of Lipopolysaccharide Across the Murine Blood-brain Barrier. Brain Behav Immun. 2010;24(1):102 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2789209/
Barton SM, Janve VA, McClure R, Anderson A, Matsubara JA, Gore JC, et al. Lipopolysaccharide Induced Opening of the Blood Brain Barrier on Aging 5XFAD Mouse Model. J Alzheimers Dis. 2019;67(2):503–13. Available from: https://pubmed.ncbi.nlm.nih.gov/30584141/.
McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer’s Research and Therapy [Internet]. 2017 Mar 4 [cited 2021 Dec 15];9(1):1–7. Available from: https://alzres.biomedcentral.com/articles/https://doi.org/10.1186/s13195-017-0241-2
Holmes C, et al. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003;74(6):788–9. https://doi.org/10.1136/jnnp.74.6.788.
Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7(2):161–7. Available from: https://www.nature.com/articles/nri2015.
Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7(2):161–7. Available from: https://www.nature.com/articles/nri2015.
Honjo K, van Reekum R, Verhoeff NPLG. Alzheimer’s disease and infection: do infectious agents contribute to progression of alzheimer’s disease? Alzheimer’s Dement. 2009;5(4):348–60.
Ide M, et al. Periodontitis and Cognitive Decline in Alzheimer's Disease. PloS one. 2016;11(3):e0151081. https://doi.org/10.1371/journal.pone.0151081.
Watts A, Crimmins EM, Gatz M. Inflammation as a potential mediator for the association between periodontal disease and Alzheimer’s disease. Neuropsychiatr Dis Treat. 2008;4(5):865. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2626915/.
Dioguardi M, Crincoli V, Laino L, Alovisi M, Sovereto D, Mastrangelo F, et al. The Role of Periodontitis and Periodontal Bacteria in the Onset and Progression of Alzheimer’s Disease: A Systematic Review. J Clin Med. 2020;9(2):495. Available from: https://www.mdpi.com/2077-0383/9/2/495/html.
Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. 2017;474(11):1823 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5433529/.
Jiang C, Li G, Huang P, Liu Z, Zhao B. The Gut Microbiota and Alzheimer’s Disease. J Alzheimers Dis. 2017;58(1):1–15. Available from: https://pubmed.ncbi.nlm.nih.gov/28372330/.
Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer’s disease. Scientific Reports. 2016;6(1):1–12. Available from: https://www.nature.com/articles/srep30028.
Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in Alzheimer’s disease. Scientific Reports. 2017;7(1):1–11. Available from: https://www.nature.com/articles/s41598-017-13601-y.
Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes & development. 2005;19(22):2645–55. https://doi.org/10.1101/gad.1299905.
Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-Gut Microbiota Metabolic Interactions. Science (1979) [Internet]. 2012 Jun 8 [cited 2022 Jan 19];336(6086):1262–7. Available from: https://doi.org/10.1126/science.1223813
Zhang R, Miller RG, Gascon R, Champion S, Katz JS, Lancero M, et al. Circulating endotoxin and systemic immune activation in sporadic amyotrophic lateral sclerosis (sALS). J Neuroimmunol. 2009;206:121–4.
Zhao J, Bi W, Xiao S, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9:5790. https://doi.org/10.1038/s41598-019-42286-8.
Lasselin J, et al. Editorial: Clinical Relevance of the Immune-to-Brain and Brain-to-Immune Communications. Front Behav Neurosci. 2019;12:336. https://doi.org/10.3389/fnbeh.2018.00336.
Asti A, Gioglio L. Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation?. J Alzheimers Dis. 2014;39(1):169–79. https://doi.org/10.3233/JAD-131394.
Lee JW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. https://doi.org/10.1186/1742-2094-5-37.
Yuhai Z, et al. Secretory Products of the Human GI Tract Microbiome and Their Potential Impact on Alzheimer's Disease (AD): Detection of Lipopolysaccharide (LPS) in AD Hippocampus. Front Cell Infect Microbiol. 2017;7:318. https://doi.org/10.3389/fcimb.2017.00318.
Zhan X. Author response: gram-negative bacterial molecules associate with alzheimer disease pathology. Neurology. 2017;88(24):2338.
Shaddox LM, et al. Local inflammatory markers and systemic endotoxin in aggressive periodontitis. J Dent Res. 2011;90(9):1140–4. https://doi.org/10.1177/0022034511413928.
Sadrameli M, et al. Linking mechanisms of periodontitis to Alzheimer's disease. Current opinion in neurology. 2020;33(2):230–8. https://doi.org/10.1097/WCO.0000000000000797.
Wahaidi, Vivian Y, et al. Endotoxemia and the host systemic response during experimental gingivitis. J Clin Periodontol. 2011;38(5):412–7. https://doi.org/10.1111/j.1600-051X.2011.01710.x.
Jain S, Coats SR, Chang AM, Darveau RP. A novel class of lipoprotein lipase-sensitive molecules mediates toll-like receptor 2 activation by porphyromonas gingivalis. Infect Immun. 2013;81(4):1277–86.
Strunk T, Coombs MRP, Currie AJ, Richmond P, Golenbock DT, Stoler-Barak L, et al. TLR2 mediates recognition of live Staphylococcus epidermidis and clearance of bacteremia. PLoS One [Internet]. 2010 [cited 2022 Jan 24];5(4). Available from: https://pubmed.ncbi.nlm.nih.gov/20404927/.
Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil ageing is regulated by the microbiome. Nat. 2015;525(7570):528 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4712631/.
Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J Immunol. 2012;188(3):1098–107. Available from: https://pubmed.ncbi.nlm.nih.gov/22198949/.
McDonald CL, Hennessy E, Rubio-Araiz A, Keogh B, McCormack W, McGuirk P, et al. Inhibiting TLR2 activation attenuates amyloid accumulation and glial activation in a mouse model of Alzheimer’s disease. Brain Behav Immun. 2016;58:191–200. Available from: https://pubmed.ncbi.nlm.nih.gov/27422717/.
Rangasamy SB, Jana M, Roy A, Corbett GT, Kundu M, Chandra S, et al. Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J Clin Invest. 2018;128(10):4297–312. Available from: https://pubmed.ncbi.nlm.nih.gov/29990310/.
Abbayya K, Puthanakar NY, Naduwinmani S, Chidambar YS. Association between Periodontitis and Alzheimer’s Disease. North American Journal of Medical Sciences. 2015;7(6):241. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4488989/.
Olsen I, Singhrao SK. Can oral infection be a risk factor for Alzheimer’s disease? http://dx.doi.org/https://doi.org/10.3402/jom.v729143 [Internet]. 2015 [cited 2022 Jan 19];7(1). Available from: https://www.tandfonline.com/doi/abs/https://doi.org/10.3402/jom.v7.29143
Lövheim H, Olsson J, Weidung B, Johansson A, Eriksson S, Hallmans G, et al. Interaction between cytomegalovirus and herpes simplex virus type 1 associated with the risk of alzheimer’s disease development. J Alzheimer’s Disease. 2018;61(3):939–45.
Barnes LL, Capuano AW, Aiello AE, Turner AD, Yolken RH, Torrey EF, et al. Cytomegalovirus Infection and Risk of Alzheimer Disease in Older Black and White Individuals. The Journal of Infectious Diseases [Internet]. 2015 Jan 15 [cited 2022 Jul 15];211(2):230–7. Available from: https://academic.oup.com/jid/article/211/2/230/2910562.
Doulberis M, Kotronis G, Thomann R, Polyzos SA, Boziki M, Gialamprinou D, et al. Review: Impact of Helicobacter pylori on Alzheimer’s disease: What do we know so far? Helicobacter [Internet]. 2018 Feb 1 [cited 2022 Aug 3];23(1). Available from: https://pubmed.ncbi.nlm.nih.gov/29181894/.
Gérard HC, Dreses-Werringloer U, Wildt KS, Deka S, Oszust C, Balin BJ, et al. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunology & Medical Microbiology [Internet]. 2006 Dec 1 [cited 2022 Aug 3];48(3):355–66. Available from: https://academic.oup.com/femspd/article/48/3/355/506227.
Pisa D, Alonso R, Rábano A, Rodal I, Carrasco L. Different Brain Regions are Infected with Fungi in Alzheimer’s Disease. Scientific Reports 2015 5:1 [Internet]. 2015 Oct 15 [cited 2022 Aug 3];5(1):1–13. Available from: https://www.nature.com/articles/srep15015.
Miklossy J. Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation [Internet]. 2011 Aug 4 [cited 2022 Aug 3];8. Available from: https://pubmed.ncbi.nlm.nih.gov/21816039/.
Ishida N, Ishihara Y, Ishida K, Tada H, Funaki-Kato Y, Hagiwara M, et al. Periodontitis induced by bacterial infection exacerbates features of Alzheimer’s disease in transgenic mice. npj Aging and Mechanisms of Disease 2017 3:1 [Internet]. 2017 Nov 6 [cited 2022 Jul 15];3(1):1–7. Available from: https://www.nature.com/articles/s41514-017-0015-x.
Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Science Advances [Internet]. 2019 Jan 23 [cited 2022 Jul 15];5(1). Available from: /pmc/articles/PMC6357742/
Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean SJ. Determining the presence of periodontopathic virulence factors in short-term postmortem alzheimer’s disease brain tissue. J Alzheimer’s Disease. 2013;36(4):665–77.
Itzhaki RF. Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus. Front Aging Neurosci [Internet]. 2014 [cited 2022 Jul 9];6(AUG). Available from: https://pubmed.ncbi.nlm.nih.gov/25157230/.
Wozniak M, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol [Internet]. 2009 Jan [cited 2022 Jan 19];217(1):131–8. Available from: https://pubmed.ncbi.nlm.nih.gov/18973185/.
Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;429(2–3):95–100.
Wozniak MA, Frost AL, Itzhaki RF. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimer’s Disease. 2009;16(2):341–50.
Lövheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement [Internet]. 2015 Jun 1 [cited 2022 Jan 19];11(6):593–9. Available from: https://pubmed.ncbi.nlm.nih.gov/25043910/
Ball M, Ball MJ. “Limbic Predilection in Alzheimer Dementia: Is Reactivated Herpesvirus Involved?” Canadian Journal of Neurological Sciences [Internet]. 1982 [cited 2022 Jul 15];9(3):303–6. Available from: https://www.cambridge.org/core/journals/canadian-journal-of-neurological-sciences/article/limbic-predilection-in-alzheimer-dementia-is-reactivated-herpesvirus-involved/C00C8CCF59A0268FD7461BC0257215D5.
Devanand DP, Michaels-Marston KS, Liu X, Pelton GH, Padilla M, Marder K, et al. Olfactory deficits in patients with mild cognitive impairment predict Alzheimer’s disease at follow-up. American Journal of Psychiatry [Internet]. 2000 Sep 1 [cited 2022 Jul 13];157(9):1399–405. Available from: https://ajp.psychiatryonline.org/doi/https://doi.org/10.1176/appi.ajp.157.9.1399.
Zou YM, Lu D, Liu LP, Zhang HH, Zhou YY. Olfactory dysfunction in Alzheimer’s disease. Neuropsychiatric Disease and Treatment [Internet]. 2016 Apr 15 [cited 2022 Jul 13];12:869. Available from: /pmc/articles/PMC4841431/
Tomlinson AH, Esiri MM. Herpes simplex encephalitis: immunohistological demonstration of spread of virus via olfactory pathways in mice. J Neurol Sci. 1983;60(3):473–84.
Mori I, Nishiyama Y, Yokochi T, Kimura Y. Olfactory transmission of neurotropic viruses. Journal of NeuroVirology 2005 11:2 [Internet]. 2005 Apr [cited 2022 Jul 13];11(2):129–37. Available from: https://springerlink.bibliotecabuap.elogim.com/article/.
Lin WR, Casas I, Wilcock GK, Itzhaki RF. Neurotropic viruses and Alzheimer’s disease: a search for varicella zoster virus DNA by the polymerase chain reaction. Journal of Neurology, Neurosurgery, and Psychiatry [Internet]. 1997 [cited 2022 Jul 10];62(6):586. Available from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1074141/?report=abstract.
Balin BJ, Gérard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, et al. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Medical Microbiology and Immunology 1998 187:1 [Internet]. 1998 [cited 2022 Aug 3];187(1):23–42. Available from: https://springerlink.bibliotecabuap.elogim.com/article/https://springerlink.bibliotecabuap.elogim.com/article/
Serrano-Castro PJ, Estivill-Torrús G, Cabezudo-García P, Reyes-Bueno JA, Ciano Petersen N, Aguilar-Castillo MJ, et al. Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: a delayed pandemic? Neurología (English Edition). 2020;35(4):245–51.
Licastro F, Porcellini E. Molecular Sciences Activation of Endogenous Retrovirus, Brain Infections and Environmental Insults in Neurodegeneration and Alzheimer’s Disease 1. Retrotransposons and Human Endogenous Retrovirus in Neurodegenerative Diseases. J Mol Sci. 2021;22:7263.
Sun W, Samimi H, Gamez M, Zare H, Frost B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nature Neuroscience 2018 21:8 [Internet]. 2018 Jul 23 [cited 2022 Jul 13];21(8):1038–48. Available from: https://www.nature.com/articles/s41593-018-0194-1.
Guo C, Jeong HH, Hsieh YC, Klein HU, Bennett DA, de Jager PL, et al. Tau Activates Transposable Elements in Alzheimer’s Disease. Cell Rep [Internet]. 2018 Jun 5 [cited 2022 Jul 13];23(10):2874–80. Available from: https://pubmed.ncbi.nlm.nih.gov/29874575/.
Perov S, Lidor O, Salinas N, Golan N, Tayeb-Fligelman E, Deshmukh M, et al. Structural Insights into Curli CsgA Cross-β Fibril Architecture Inspire Repurposing of Anti-amyloid Compounds as Anti-biofilm Agents. PLOS Pathogens [Internet]. 2019 [cited 2022 Apr 1];15(8):e1007978. Available from: https://journals.plos.org/plospathogens/article?id=https://doi.org/10.1371/journal.ppat.1007978.
Miller AL, Bessho S, Grando K, Tükel Ç. Microbiome or infections: amyloid-containing biofilms as a trigger for complex human diseases. Front Immunol. 2021;26(12):514.
Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity. 2015;42(6):1171–84.
Friedland RP. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J Alzheimers Dis [Internet]. 2015 [cited 2022 Apr 1];45(2):349–62. Available from: https://pubmed.ncbi.nlm.nih.gov/25589730/.
Friedland RP, McMillan JD, Kurlawala Z. What Are the Molecular Mechanisms by Which Functional Bacterial Amyloids Influence Amyloid Beta Deposition and Neuroinflammation in Neurodegenerative Disorders? Int J Mol Sci [Internet]. 2020 Mar 1 [cited 2022 Apr 4];21(5). Available from: https://pubmed.ncbi.nlm.nih.gov/32121263/.
Kumar DKV, Choi HS, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Science Translational Medicine [Internet]. 2016 May 25 [cited 2022 Apr 1];8(340). Available from: https://www.science.org/doi/abs/https://doi.org/10.1126/scitranslmed.aaf1059.
Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of alzheimer’s disease. Alzheimer’s Dementia. 2018;14(12):1602–14.
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s disease-associated β-Amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. 2018;99(1):56-63.e3.
Li QX, Evin G, Small DH, Multhaup G, Beyreuther K, Masters CL. Proteolytic Processing of Alzheimer’s Disease βA4 Amyloid Precursor Protein in Human Platelets ∗. Journal of Biological Chemistry [Internet]. 1995 Jun 9 [cited 2022 Apr 1];270(23):14140–7. Available from: http://www.jbc.org/article/S0021925818922118/fulltext.
Citron M, Vigo-Pelfrey C, Teplow DB, Miller C, Schenk D, Johnston J, et al. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc Natl Acad Sci U S A [Internet]. 1994 Dec 6 [cited 2022 Apr 1];91(25):11993–7. Available from: https://pubmed.ncbi.nlm.nih.gov/7991571/.
Kuo YM, Kokjohn TA, Watson MD, Woods AS, Cotter RJ, Sue LI, et al. Elevated Aβ42 in skeletal muscle of alzheimer disease patients suggests peripheral alterations of AβPP metabolism. Am J Pathol. 2000;156(3):797–805.
van Nostrand WE, Melchor JP. Disruption of pathologic amyloid beta-protein fibril assembly on the surface of cultured human cerebrovascular smooth muscle cells. Amyloid [Internet]. 2001 [cited 2022 Apr 1];8 Suppl 1(SUPPL. 1):20–7. Available from: https://pubmed.ncbi.nlm.nih.gov/11676286/.
Zlokovic BV, Martel CL, Mackic JB, Matsubara E, Wisniewski T, Mccomb JG, et al. Brain Uptake of Circulating Apolipoproteins J and E Complexed to Alzheimer′s Amyloid β. Biochem Biophys Res Commun. 1994;205(2):1431–7.
Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL, et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Molecular Psychiatry 2018 23:9 [Internet]. 2017 Oct 31 [cited 2022 Apr 1];23(9):1948–56. Available from: https://www.nature.com/articles/mp2017204.
Lam V, Takechi R, Hackett MJ, Francis R, Bynevelt M, Celliers LM, et al. Synthesis of human amyloid restricted to liver results in an Alzheimer disease–like neurodegenerative phenotype. PLOS Biology [Internet]. 2021 Sep 1 [cited 2022 Apr 1];19(9):e3001358. Available from: https://journals.plos.org/plosbiology/article?id=https://doi.org/10.1371/journal.pbio.3001358.
Zeng F, Liu Y, Huang W, Qing H, Kadowaki T, Kashiwazaki H, et al. Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after Porphyromonas gingivalis infection. Journal of Neurochemistry [Internet]. 2021 Aug 1 [cited 2022 Apr 1];158(3):724–36. Available from: https://onlinelibrary.wiley.com/doi/full/https://doi.org/10.1111/jnc.15096
Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas gingivalis infection induces Amyloid-β accumulation in monocytes/macrophages. J Alzheimer’s Disease. 2019;72(2):479–94.
Friedland RP, McMillan JD, Kurlawala Z. What Are the Molecular Mechanisms by Which Functional Bacterial Amyloids Influence Amyloid Beta Deposition and Neuroinflammation in Neurodegenerative Disorders? International Journal of Molecular Sciences 2020, Vol 21, Page 1652 [Internet]. 2020 Feb 28 [cited 2022 Apr 1];21(5):1652. Available from: https://www.mdpi.com/1422-0067/21/5/1652/htm.
Lax N, Fainstein N, Nishri Y, et al. Systemic microbial TLR2 agonists induce neurodegeneration in Alzheimer’s disease mice. J Neuroinflammation. 2020;17:55. https://doi.org/10.1186/s12974-020-01738-z.
Ganz T, Fainstein N, Elad A, Lachish M, Goldfarb S, Einstein O, et al. Microbial pathogens induce neurodegeneration in Alzheimer’s disease mice: protection by microglial regulation. Journal of Neuroinflammation 2022 19:1 [Internet]. 2022 Jan 6 [cited 2022 Jan 19];19(1):1–15. Available from: https://springerlink.bibliotecabuap.elogim.com/articles/https://doi.org/10.1186/s12974-021-02369-8.
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the Toll-Like Receptor 4 in Neuroinflammation in Alzheimer’s Disease. Cellular Physiology and Biochemistry [Internet]. 2007 [cited 2022 Mar 21];20(6):947–56. Available from: https://www.karger.com/Article/FullText/110455
Frank S, Copanaki E, Burbach GJ, Müller UC, Deller T. Differential regulation of toll-like receptor mRNAs in amyloid plaque-associated brain tissue of aged APP23 transgenic mice. Neurosci Lett. 2009;453(1):41–4.
Letiembre M, Liu Y, Walter S, Hao W, Pfander T, Wrede A, et al. Screening of innate immune receptors in neurodegenerative diseases: a similar pattern. Neurobiol Aging. 2009;30(5):759–68.
Fiebich BL, Batista CRA, Saliba SW, Yousif NM, de Oliveira ACP. Role of microglia TLRs in neurodegeneration. Front Cell Neurosci. 2018;2(12):329.
Teipel S, Drzezga A, Grothe MJ, Barthel H, Chételat G, Schuff N, et al. Multimodal imaging in alzheimer’s disease: validity and usefulness for early detection. Lancet Neurol. 2015;14(10):1037–53.
Chételat G, Arbizu J, Barthel H, Garibotto V, Law I, Morbelli S, et al. Amyloid-PET and 18F-FDG-PET in the diagnostic investigation of alzheimer’s disease and other dementias. Lancet Neurol. 2020;19(11):951–62.
Blennow K, Zetterberg H. Biomarkers for Alzheimer’s disease: current status and prospects for the future. Journal of Internal Medicine [Internet]. 2018 Dec 1 [cited 2022 Jul 15];284(6):643–63. Available from: https://onlinelibrary.wiley.com/doi/full/https://doi.org/10.1111/joim.12816.
Teunissen CE, Verberk IMW, Thijssen EH, Vermunt L, Hansson O, Zetterberg H, et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. The Lancet Neurology [Internet]. 2022 Jan 1 [cited 2022 Jul 15];21(1):66–77. Available from: http://www.thelancet.com/article/S1474442221003616/fulltext.
Acknowledgements
Not applicable.
Funding
This work was supported by a gift from Mrs. Rosa Rasiel.
Author information
Authors and Affiliations
Contributions
T.G. and T.B.H. conceptualized and wrote the manuscript, N.F critical revision of the manuscript. All author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ganz, T., Fainstein, N. & Ben-Hur, T. When the infectious environment meets the AD brain. Mol Neurodegeneration 17, 53 (2022). https://doi.org/10.1186/s13024-022-00559-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-022-00559-3