
Abstract
Purpose of Review
Herein, we provide a critical review of the clinical and translational research examining the relationship between viral and bacterial pathogens and Alzheimer’s disease. In addition, we provide an overview of the biological pathways through which chronic infection may contribute to Alzheimer’s disease.
Recent Findings
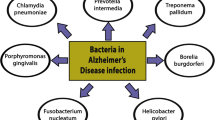
Dementia due to Alzheimer’s disease is a leading cause of disability among older adults in developed countries, yet knowledge of the causative factors that promote Alzheimer’s disease pathogenesis remains incomplete. Over the past several decades, numerous studies have demonstrated an association of chronic viral and bacterial infection with Alzheimer’s disease. Implicated infectious agents include numerous herpesviruses (HSV-1, HHV-6, HHV-7) and various gastric, enteric, and oral bacterial species, as well as Chlamydia pneumonia and multiple spirochetes.
Summary
Evidence supports the association between multiple pathogens and Alzheimer’s disease risk. Whether these pathogens play a causal role in Alzheimer’s pathophysiology remains an open question. We propose that the host immune response to active or latent infection in the periphery or in the brain triggers or accelerates the Alzheimer’s disease processes, including the accumulation of amyloid-ß and pathogenic tau, and neuroinflammation. While recent research suggests that such theories are plausible, additional longitudinal studies linking microorganisms to Aß and phospho-tau development, neuroinflammation, and clinically defined Alzheimer’s dementia are needed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Dementia is a leading cause of disability and death within the USA. Alzheimer’s disease (AD), which accounts for approximately two-thirds of all dementia cases, was estimated to effect 5.8 million Americans in the year 2020, including 10% of individuals over age 65 [1]. AD is a neurodegenerative condition that is characterized clinically by a gradual and progressive cognitive decline (particularly memory decline) and loss of functional capacity. Pathologically, AD is defined by cerebral accumulation of two insoluble proteinaceous aggregates: amyloid-ß (Aß) in the form of extracellular neuritic plaques (ß-pleated sheets surrounded by swollen nerve terminals) and tau in the form of intracytoplasmic neurofibrillary tangles (NFTs) in the proximal dendrites and distal axons of the neuron. It is widely accepted that both Aß and tau NFTs are necessary for the development of pathologically defined AD.
Despite enormous efforts focused on developing treatments for AD, currently, there are no approved therapies that modify the disease course. The bulk of drug development research to date has focused on targeting brain Aß deposits, either by inhibiting upstream enzymes (e.g., ß-secretase) or through stimulating the immune response to remove Aß (i.e., active or passive immunization). The recent string of phase 3 clinical trial failures of drugs targeting Aß has led many to begin to seriously consider alternative biological pathways as drivers of AD progression [2]. The theory that infectious agents may play a role in initiating or accelerating AD has received increasing attention in recent years, especially in light of several recent studies linking specific infections to AD risk. However, this is not a new idea.
While Dr. Alois Alzheimer is reported to initially have postulated a connection between infection and AD in 1901, primary evidence for the role of chronic infection in AD was not established until decades later [3]. The relationship between AD occurrence and presence of viral genome in the brain was first established in 1991 by Jamieson and colleagues, who found herpes simplex virus type 1 (HSV-1) genetic material in autopsy brain samples [4]. In 2006, the connection between infectious agents and AD risk was expanded beyond viral infection to include infection with various bacterial species. Among autopsied AD patient brains, Chlamydophila (Chlamydia) pneumoniae-infected neural cells, including astrocytes, microglia, and neurons, were found in close proximity to senile plaques and neurofibrillary tangles [5, 6]. More recent studies have provided additional support for the connection between bacterial infection and AD incidence. For example, micro colonies of spirochetes, such as Borrelia burgdorferi, have been found to play a role in AD plaque formation [7]. Additionally, constituents of the oral microbiome, including Porphyromonas gingivalis [8], have been associated with AD risk. Such findings suggest a potential connection between chronic infection and AD risk; however, the field of AD research is still far from reaching a consensus on whether chronic infections may actually influence AD pathogenesis. This review will provide a critical examination of the evidence for the role chronic infection in AD and highlight the biological pathways through which chronic infection may influence brain function and contribute to development of AD.
Alzheimer’s Disease Pathophysiology
Over the past 3 decades, the amyloid hypothesis has provided perhaps the most widely accepted conceptualization for AD pathogenesis. Central to this hypothesis is the idea that the accumulation of amyloid within the CNS and its deposition into plaques throughout the cortex leads to a cascade of downstream events, including tau formation, synaptic dysfunction, and neurodegeneration [9]. This cascade of events is believed to be initiated by the aggregation of Aß1-40 and Aß1-42 peptides, which are cleaved from the amyloid precursor protein (APP) by ß- and γ-secretases. Soluble Aß peptides then form Aß oligomers before assembling into ß-pleated sheets, protofilaments, and fibrils, and eventually aggregating to form insoluble amyloid plaques. In individuals who develop AD, amyloid plaques begin accumulating as early as 2 decades before the onset of significant clinical symptoms [10]. Though the original amyloid hypothesis posits that amyloid plaques promote downstream neurodegenerative changes, this hypothesis has been revised in more recent years to suggest that Aß oligomers and fibrils (rather than amyloid plaques) are responsible for the proteotoxicity that initiates and perpetuates AD.
Central to the amyloid hypothesis are the downstream pathological events resulting from Aß accumulation, most notably the formation of pathological tau. The tau protein is a microtubule-binding protein that plays a key role in assembling and stabilizing tubulin polymers localized within the axon of neuronal cells. Because microtubules play a central role in axonal transport, formation of pathological tau causes a loss of microtubule binding and impaired transport of organelles to and from the neuronal synapse, resulting ultimately in cellular dysregulation and synaptic dysfunction and loss [11]. The accumulation of Aß oligomers has been proposed to cause abnormal phosphorylation (hyperphosphorylation) of tau and the assembly of hyperphosphorylated tau filaments into insoluble aggregates called neurofibrillary tangles (NFTs). Tau NFTs, compared to cortical Aß, tend to occur later in the disease course and show a stronger association with cognitive decline and dementia risk [10]. Ultimately, synaptic dysfunction, neuronal loss, and cognitive decline are believed to occur as a result of the aggregation of phospho-tau and/or tau NFTs. Given this hypothesized link between tau and neurocognitive dysfunction, a tau hypothesis has been put forth wherein tau, rather than amyloid, triggers AD pathogenesis.
With the exception of several anti-amyloid drugs, such as aducanumab, donanemab, and BAN2401, which have shown some efficacy in recent trials, the overwhelming majority of therapies targeting Aß and tau in humans with AD or mild cognitive impairment (MCI) have been unsuccessful [2]. Although there are multiple explanations for the lack of efficacy, this pattern of findings underscores the need to, at the very least, refine the amyloid and tau hypotheses. Alternatively, hypotheses that do not consider amyloid or tau as the biological linchpins for AD should also be considered. The blueprint of AD biology provided by genome-wide association studies (GWAS) suggests that a range of non-amyloid/tau pathways are etiologically relevant to AD. For example, a GWAS of sporadic AD found nearly 30 genetic variants that influence AD risk, the majority of which were on or near genes known to be involved in immune function, lipid and cholesterol metabolism, and endocytosis [12]. Involvement of the innate immune response in AD pathogenesis may be one of the most consistent findings from genetic and genomic AD research, supporting the idea that immune function or dysfunction is an important determinant of who ultimately develops Alzheimer’s dementia [13,14,15,16].
Microglia, the brain’s resident immune cell, have been shown to influence AD pathophysiology by regulating disease progression through both phagocytic and neuroinflammatory processes [17]. In fact, genes coding for myeloid receptors prominently expressed by microglia, such as triggering receptor expressed on myeloid cells 2 (TREM2) and CD33, are included among the major AD risk variants, further supporting the role of innate immune activation in AD pathogenesis. In light of this evidence, a framework described as the amyloid cascade-neuroinflammation hypothesis has been proposed [18]. Though several variations have been put forth, at its core, the amyloid cascade-neuroinflammation hypothesis posits that the accumulation of Aß triggers a neuroimmune response, i.e., activation of microglia, early in the disease course. In this setting, a proportion of microglia transition from a homeostatic to an active phenotype, initiating an immune response designed to remove toxic forms of Aß. Though microglial activation and removal of Aß at this stage may initially be protective, microglial phagocytosis is believed to be unable to keep up with the rapid rate of Aß production, ultimately causing the microglial response to become anergic and pro-inflammatory [17]. This pro-inflammatory neuroimmune activation has been shown to promote the hyperphosphorylation and spreading of tau and downstream neurodegeneration [19]. Importantly, there is an alternative conceptualization of the amyloid cascade-neuroinflammation hypothesis that proposes an initial disease aggravating low-grade neuroinflammatory response to Aß oligomers that is followed by a secondary Aß plaque-associated neuroimmune response characterized by microglial activation, phagocytosis, and CNS inflammation [20].
Alzheimer’s Disease and the Infectious Hypothesis
The amyloid/tau hypothesis of AD pathogenesis is not fully at odds with theories that propose a causal role for chronic infection. Although both theories generally propose a functional role for soluble Aß and Aß plaques, infectious theories also posit that a pathogen, or the immune response to a pathogen, can trigger or perpetuate the Alzheimer’s disease process. While there is still no consensus regarding the role of infection in AD, studies and leading theories have identified at least three distinct frameworks that may ultimately characterize the role of chronic infection in AD [21].
The first possibility is that infectious agents may actually trigger or initiate AD pathogenesis (Fig. 1). This has been described as the “infectious origin” hypothesis. The second possibility is that rather than trigger AD, infectious processes may instead accelerate a disease process that has already been set in motion. This theory has not been formally named but can be described as the “infectious accelerant” hypothesis. The third possibility is that there is no causal relationship between chronic infection and AD. This theory further postulates that previous associations described in the literature may be accounted for by reverse causation (i.e., AD increasing rate of infection), residual confounding (i.e., factors that jointly influence risk of infection and risk of AD), or other methodological or technical confounds. In the sections below, we use this framework to review the body of literature that has sought to understand whether chronic infection plays a mechanistic role in AD. Evidence supporting the role for chronic infection in AD has emerged from studies of viral, bacterial, parasitic, and fungal pathogens. Given the broad nature of this topic, this review will focus primarily on the putative role of chronic viral and chronic bacterial infections in AD, as research in this area has been most extensive. We refer the reader elsewhere for a comprehensive review of the evidence for the role of parasitic [22, 23] and fungal [24,25,26,27] pathogens in AD.

There are at least three hypotheses which characterize the potential relationship between chronic infection and Alzheimer’s disease (AD). Hypothesis 1, also described as the “infectious origin” hypothesis, posits that infectious agents can trigger AD pathogenesis. Hypothesis 2, also described as the “infectious accelerant” hypothesis, posits that infectious agents are not necessary, but can accelerate the development and progression of AD pathology. Hypothesis 3, which posits that there is no causal relationship between chronic infection and AD, suggests further that infection may be only an associative feature of AD. Figure created with Biorender.com
Viral Infection in Alzheimer’s Disease
Over the years, numerous viral pathogens have been proposed to play a role in initiating or promoting AD. However, evidence for a causal link between chronic viral infection and AD has been mixed. A large number of pathogens have been implicated, including human immunodeficiency virus (HIV), influenza A virus (H3N2 and H1N1 strains), and numerous herpesviruses. Although HIV has been associated with cognitive deficits, specifically in association with a syndrome called HIV-associated neurocognitive disorder (HAND), epidemiological studies have yet to demonstrate a clear association between HIV infection and greater risk for AD. HIV has been associated with increased Aß plaque presence [28] and greater levels of brain p-tau at autopsy, compared to uninfected individuals, though studies examining AD CSF biomarkers have yielded inconsistent results [29, 30]. Studies examining influenza infection have failed to demonstrate a consistent relationship with AD risk. One recent study found evidence supporting the protective effects of the influenza vaccine against AD [31, 32]. However, a large case-control study of nearly 40,000 individuals that compared people with past influenza infection to those without previous influenza infection found no evidence of increased AD risk among those previously infected [33].
Other candidate pathogens include numerous herpesviruses: herpes simplex virus 1 (HSV-1) or oral herpes, herpes simplex virus 2 (HSV-2) or genital herpes, human herpesvirus 3 (HHV-3) or varicella zoster virus (VZV), human herpesvirus 4 (HHV-4) or Epstein-Barr virus (EBV), human herpesvirus 5 (HHV-5) or cytomegalovirus (CMV), human herpesvirus 6 (HHV-6), human herpesvirus 7 (HHV-7), and human herpesvirus 8 (HHV-8). Herpes simplex virus 1 (HSV-1) is most commonly known for causing sores around the mouth but is also responsible for herpes simplex encephalitis (HSE), a rare and severe neurological disorder that affects limbic brain regions, the same regions affected by AD [34]. HSV-1 was among the first herpesviruses established to play a potential role in AD, as numerous early studies found evidence for detectable HSV-1 DNA in a high proportion of the brains of AD patients, particularly in temporal brain regions [4, 35,36,37]. However, these same studies demonstrated that HSV-1 DNA can also be found at high rates among older adults without pathologically defined AD. A small study which examined six AD brains found that 90% of the Aß plaques contained HSV-1 DNA and that 72% of the HSV-1 DNA was colocalized with Aß plaques. In contrast, among older adults without AD, 80% of the Aß plaques contained HSV-1 DNA, and only 24% of the HSV-1 DNA was found within Aß plaques [38]. It was postulated that reactivated HSV-1 present in trigeminal ganglia may cause the neuronal lesions seen in AD as well as the normal neuronal degeneration seen in aged brain tissue [34]. However, whether HSV-1 has a causal role in AD and through which biological pathways HSV-1 may exert direct pathogenic effect on the brain has not been established.
Although observational studies do not show a clear increase of HSV-1 in AD brains, translational research suggests HSV-1 may initiate or accelerate AD pathogenesis. For example, in a study which examined Alzheimer’s disease (5XFAD) transgenic mice with HSV-1 injected into the hippocampal region of each hemisphere, HSV-1 promoted the seeding of Aß plaques and accelerated amyloidosis [39]. Though the authors found that Aß protected against development of HSV-1 encephalitis, they concluded that this antiviral effect of Aß accumulation may also promote neurodegeneration. Additional research indicates that both acute and chronic HSV-1 infections promote neuroinflammation and oxidative damage in mouse brains in a process likely mediated by microglial activation [40, 41]. Considering the possible contribution of HSV-1 infection to neuroinflammation and the central role of microglial activation in AD pathogenesis [17], it is possible that HSV-1 infection accelerates AD onset through microglial-mediated processes.
While epidemiological studies do not suggest a consistent association between HSV-1 and AD risk in the overall population, there does appear to be a relationship between HSV-1 infection and possession of the APOE genotype linked to AD risk. The APOE gene codes apolipoprotein E, a lipid carrier of the central and peripheral nervous systems. Individuals who carry one or two copies of the ε4 allele on APOE are at increased risk for developing AD. Ruth Itzhaki found that the APOEε4 genotype was more common among AD patients who had HSV-1 in their brains at autopsy than among AD patients who were negative for brain HSV-1 [42]. While this study was limited by small sample size (n = 84), these findings suggest that either (a) APOEε4 allele possession increases the risk of HSV-1 brain infection or that (b) HSV-1 infection increases risk of AD but only in the context of APOEε4 allele possession. This finding was subsequently replicated, including by a study which found higher rates of APOEε4 genotype among individuals with a history of oral herpetic lesions, a clinical manifestation of HSV-1 infection [43]. APOEε4 is thought to contribute to AD pathogenesis via its effects on lipid metabolism and neuronal repair [44]. Given that both apolipoprotein E and HSV-1 bind at the heparin sulfate proteoglycan receptor on the cell surface and use this as an entry into cells, it is possible that more HSV-1 is let into neural cells of individuals with the weaker isoform of apolipoprotein E (ε4 isoform) [45, 46]. While the evidence presented is not sufficient to suggest a causative role of HSV-1 in AD, it remains plausible that HSV-1 increases risk for AD among individuals who carry the APOEε4 variant.
Human herpesviruses 4 and 5, more commonly referred to as Epstein-Barr virus (EBV) and cytomegalovirus (CMV), respectively, have also been implicated in AD pathogenesis. For example, EBV seropositivity has been associated with increased risk of developing AD [47]. While this study points to EBV as a potential risk factor for AD development, further evidence is needed to confirm such a relationship and to shed light on a biological mechanism. In a sample of 1204 participants, the presence of CMV antigen was associated with a greater rate of cognitive decline over a 4-year period [48]. CMV presence in the blood was also associated with reduced learning, recall, and coding speed in children aged 6–16 years from the National Health and Nutrition Examination Survey (NHANES) III, suggesting a connection between CMV infection and early cognition [49]. In contrast, a study of persons aged 80 and older found CMV infection was unrelated to functional and cognitive impairment [50]. Such contradictory findings indicate the need for continued investigation of the roles of CMV and EBV in AD.
Other herpes viruses proposed to play a role in AD pathogenesis include HSV-2, HHV-3 (or VZV), HHV-6, and HHV-7. HSV-2 is commonly known as the cause of genital herpes and has been investigated for its role in AD. As early as 1987, southern blot hybridization data from 18 AD and 5 control brains found no relationship between HSV-2 viral presence and AD occurrence [51]. In 2002, another study used polymerase chain reaction (PCR) to determine the presence of HSV-2 in AD and non-AD brains and found no difference in viral presence [52]. A 2019 meta-analysis which examined the rate of HSV-2 in the brains of AD patients versus a non-AD control group found no evidence for an increased prevalence of HSV-2 in AD brains [53••]. While APOE genotype has been associated with HSV-1 infection, no link between HSV-2 infection and APOE genotype was found in a study of 200 adults [43]. Together, these results do not support the role of HSV-2 in AD pathogenesis.
Recent evidence suggests that HHV-6 and HHV-7, both betaherpesviruses associated with the febrile and rash-causing illness called roseola or sixth disease [54], may play a role in AD pathogenesis. A 2002 autopsy study of 85 older adult brains found HHV-6 to be present in AD brains at a higher rate (60%) than in age-matched control brains without AD pathology (40%) [52]. More recently, an autopsy study by Readhead and colleagues found viral involvement in the regulation of AD-associated brain gene expression networks. Prompted by these findings, an examination of the AD-associated virome in postmortem brain tissue revealed viral DNA from HHV-6A and HHV-7 at higher rates in AD brains across three independent cohorts. Supporting these findings, expression of certain HHV-6A and HHV-7 genes in brain tissue was associated with AD traits, including symptom severity, amyloid plaque density, and progression of tau NFTs. Additionally, genetic loci coding brain HHV-6A were found to overlap with AD risk genes [55]. Though these findings provide compelling evidence for the role of HHV-6A and HHV-7 in AD pathogenesis, a recent analysis which performed RNA-seq on over 1200 autopsied brains across three cohorts failed to replicate the finding of increased HHV-6 in AD [56••]. At present, there is no consistent evidence for increased HHV-6A and HHV-7 infection among individuals with AD. However, the genetic overlap found between brain HHV-6A, AD-associated gene expression networks, and AD risk variants provides compelling evidence for the role of HHV-6A in AD.
Less evidence is available for other herpesviruses, including HHV-3 and HHV-8. HHV-3, also known as Varicella zoster virus (VZV), is the cause of chickenpox in its acute form and shingles upon reactivation from latency [54]. Homozygosity for the APOEe4 allele confers greater risk of shingles development following previous VZV infection [57], suggesting a connection between HHV-3 and AD. However, the nature of such a connection remains unknown. Although HHV-8 has been associated with such neurological conditions as AIDS-dementia, amyotrophic lateral sclerosis (ALS), and primary CNS lymphoma, complications of the CNS in HHV-8 infection are rare, and evidence for a role of HHV-8 in AD is lacking [58]. Overall, the role of herpesviruses in AD remains unclear. Though mounting evidence suggests a relationship between HSV-1, HSV-6A, and AD, further research is required to elucidate the biological mechanisms that may underlie the connection between each of these herpesviruses and AD pathogenesis.
Evidence for the Role of Specific Bacterial Infections in Alzheimer’s Disease
Bacterial species have also been suggested to play a role in AD pathogenesis. In particular, the bacterial species Chlamydophila pneumoniae, alternatively called Chlamydia pneumoniae or C. pneumoniae, has been repeatedly implicated in AD pathogenesis [59]. C. pneumoniae most commonly infects the epithelial lining of the respiratory tract, is thought to play a role in both bronchial asthma and chronic obstructive pulmonary disease (COPD), and is responsible for about 20% of lower respiratory tract infections. [60]. Research has additionally pointed to C. pneumoniae as a potential factor in the neuropathogenesis of AD [6, 61]. PCR assay for bacterium DNA sequences found C. pneumoniae presence in 17/19 AD brain samples, compared to 1/19 in control brain samples. Culture studies were positive for C. pneumoniae in a subset of AD brain samples when compared to brains of controls, suggesting that C. pneumoniae was not only present, but alive and active in these brain samples at the time of death [62]. A 2006 study found greater C. pneumoniae presence in AD brains (20 of 27) compared to control brains (3 of 27) when PCR was run targeting two key C. pneumoniae genes. Assessment of chlamydial RNA transcript in these samples indicated that the bacteria were viable and metabolically active. Immunohistochemistry confirmed bacterial presence in a variety of neural cells, including astrocytes, microglia, and neurons, and found that infected cells were often in close proximity to both senile plaques and neurofibrillary tangles. These findings support the hypothesis that extra-respiratory infection with C. pneumoniae may play a role in AD pathogenesis [5]. Like other pathogens, C. pneumoniae may interact with the major AD risk gene, APOE. In situ hybridization analyses revealed that the number of C. pneumoniae-infected cells of the AD brain was greater in AD patients also displaying the APOEε4 genotype. qRT-PCR confirmed that ε4-bearing AD patients had a significantly higher bacterial load than congruent samples from AD patients without the allele [63]. Follow-up study from the same team indicated that apoE4 protein, as compared to apoE2 and apoE3 proteins, seems to enhance attachment of C. pneumoniae in its infectious elementary body (EB) form to astrocytes and microglia in vitro [6, 64].
Spiral-shaped bacterial spirochetes have also been implicated as pathogens in the infectious AD hypothesis [65]. Bacterial species of the order spirochetes include Borrelia burgdorferi (B. burgdorferi) and Treponema pallidum (T. pallidum). Infection with B. burgdorferi, the tick-borne etiologic agent of Lyme disease, induces pro-inflammatory cytokines and chemokines and causes systemic symptoms in some cases [66]. A small 2019 case-control study found greater prevalence of B. burgdorferi IgG in AD (29%) and MCI (23%) patients, compared to controls (10%) [67]. Contrasting these findings, a 2014 study which examined whether the incidence of death due to AD was higher in regions with the highest rates of Lyme disease found no correlation between Lyme disease incidence and AD deaths [68]. T. pallidum, the syphilis spirochete, has earned the epithet “stealth pathogen” because of its notable ability to evade the host immune system [69]. Three decades of historic evidence suggest that T. pallidum infection can cause cortical atrophy, amyloid deposition, and dementia [70]. Accumulation of spirochetes in the gray matter of the brain, a result of chronic syphilis infection, can cause syphilitic dementia. The morphology of the spirochetal “plaques” cannot be distinguished from the senile plaque characteristic of AD [27]. Though it is possible that systemic and chronic syphilitic infection, possibly in tandem with viral or bacterial co-infectors, may promote AD-related brain changes, additional research is required to confirm such a relationship [71].
Oral, gastric, and enteric bacterial species have also been previously implicated in AD. The gut-brain axis (GBA), defined as the two-way crosstalk between the central and enteric nervous systems, has become an area of increasing research interest [72]. The enteric microbiome, which plays a pivotal role in regulating the GBA [73], has been linked to AD risk. For example, a study which examined the gut microbiome using fecal samples from 25 individuals with dementia and 25 age-matched controls found that AD participants had overall decreased gut microbiome diversity, including decreased Firmicutes and Bifidobacterium, as well as increased Bacteroidetes [74]. Bacteroidetes makes up the largest phylum of Gram-negative bacteria found in the human GI tract and is known to promote secretion of a variety of pro-inflammatory neurotoxins, including surface lipopolysaccharides (LPSs). Bacteroides fragilis (B. fragilis), one of the most abundant Bacteroidetes species found in the gut, secretes LPSs known to contribute to systemic inflammation when present in blood. Both peripherally and in the CNS, LPSs given off by Bacteroidetes can promote pro-inflammatory transcription via NF-kB (p50/p65) complex activation [75]. Beyond bacteria of the phylum Bacteroidetes, other symbiotic and opportunistically pathogenic bacteria of the gut are postulated to promote AD, including Enterococcus faecalis (E. faecalis) and Salmonella typhimurium (S. typhimurium) [3, 76]. However, additional research is needed to confirm or refute the connection between each of these microbes and AD pathogenesis.
In addition to bacteria of the human gut, numerous mutualistic and pathogenic bacteria of the oral cavity have been implicated in AD. The human oral microbiome, the personalized microscopic ecosystem inhabiting the lips, tongue, tonsils, palate, and gums, has historically been the most studied subset of human microflora [77]. Among aged persons, oral pathogens cause periodontal disease, edentulism, and dental caries [78]. In recent years, alterations in oral health status and oral microbiota have also been associated with declining cognition, dementia, and AD [79, 80]. While it is established that AD, due to resulting difficulty performing activities of daily living, can cause a reduction in oral health maintenance [81], it has also been speculated that poor oral health, namely periodontal disease, can promote AD. Chronic periodontitis (CP) is an oral infection caused by anaerobic bacteria of the subgingival biofilm (e.g., Porphyromonas gingivalis) [82]. CP has been studied in relation to systemic disorders for several decades [83], and multiple studies published within the last 2 decades have demonstrated a relationship between periodontitis and AD, all-cause dementia, and MCI [78, 81, 84,85,86,87, 88••]. Among the largest of these was a 2017 retrospective matched cohort study using 9291 newly diagnosed individuals with CP matched to 18,672 individuals without CP. Those with a 10-year history of CP exhibited a 1.7-fold increase in AD risk [87]. While these studies may indicate that periodontal disease plays a role in AD pathogenesis, there remains possibility of reverse causation due to changes in oral care ability in those with Alzheimer’s dementia, prodromal AD, or even preclinical AD.
Bacterial species known to colonize parts of the digestive system other than the gut and mouth include Helicobacter pylori or H. pylori, a gram-negative spiral bacterium colonizing the human stomach. While full-scale, randomized controlled trials are lacking, results from epidemiologic and translational research suggest that H. pylori may play a role in AD [89, 90]. Infection with H. pylori affects between 85 and 95% of people in developing countries and between 30 and 50% in developed countries as of 2019, and it is known to play a role in gastric pathologies such as gastroduodenal ulcer disease as well as gastric carcinoma [91, 92]. Potential mechanisms by which H. pylori may play a role in neurological disease include bacterial infection of neuronal tissue directly via the oral-nasal-olfactory pathway or indirectly via H. pylori-infected monocytes that fail to auto-destruct [90, 93,94,95]. H. pylori infection can cause leukocyte infiltration into the gastric mucosa and subsequent neutrophil activation, which creates a massive infiltration of reactive oxygen species (ROS) and resulting tissue damage [96]. Such oxidative stress has been implicated in blood-brain barrier (BBB) disruption and subsequent infiltration of inflammatory mediators into the brain, particularly in relation to multiple sclerosis (MS), an autoimmune condition of the nervous system [97]. This BBB breakdown and concomitant inflammation may also contribute to AD pathogenesis [98].
A 2004 study which examined H. pylori antibodies (HP-IgG and HP-IgA) in blood found greater HP-IgG among vascular disease (VaD) patients compared to AD patients and controls and greater HP-IgA presence in both VaD and AD patients compared to controls [99]. Additionally, a small case-control study of AD patients and controls screened for current H. pylori in a histologic analysis of gastric mucosa biopsy found a greater presence of H. pylori in AD patients (88%) compared to controls (47%) upon upper GI endoscopy [100]. A relationship between H. pylori infection and AD severity has also been demonstrated. For example, H. pylori antibody presence in the serum was associated with decreased Mini-Mental State Examination (MMSE) scores and increased CSF tau and phospho-tau concentrations in patients with AD [101]. These studies present evidence for a connection between H. pylori and AD. However, further work is needed to determine whether this pathogen functions as an AD trigger, an accelerant, or merely an associative feature of AD.
Chronic Infection and Alzheimer’s Disease Pathways
The findings reviewed above provide support for a potential role of multiple infectious agents in AD. However, the contribution of chronic infection to AD may not be organism specific. Instead, a stereotyped host immune response common to invading pathogens may constitute a unifying disease pathway. Although the antiviral/antimicrobial host immune response is considered protective in many cases, infections that cannot be completely cleared may trigger low-grade inflammation and, in some cases, chronic inflammation [102,103,104]. The development of what is often referred to as chronic low-grade inflammation may act as a final common pathway through which both CNS and non-CNS infectious processes contribute to AD and other neurodegenerative conditions (Fig. 2). This framework, which has been referred to previously as the excessive inflammation theory, aligns with both the infectious origin and infectious accelerant hypotheses.

a Viral and bacterial products are recognized by macrophages, neutrophils, and dendritic cells in peripheral tissues. Viral products in infected cells can be recognized by Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and retinoid acid-inducible gene-I (RIG-I)-like helicases (RLHs). Bacteria are recognized by TLRs, scavenger receptors, C-type lectins, and integrins. This recognition of pathogens by innate immune cells causes an inflammatory response characterized by the release of cytokines and chemokines, small proteins which interact with other immune cells to coordinate the inflammatory response. b Cytokines and chemokine released from peripheral immune cells can trigger neuroimmune activation by interacting with microglia. c Inflammatory cytokines can alter the microglial phenotype, promoting a shift from a homeostatic or protective/disease resolving phenotype to a cytotoxic pro-inflammatory phenotype. d Viral or bacterial pathogens that infiltrate the central nervous system can also be recognized by microglia. These pathogens can shift microglia toward a cytotoxic pro-inflammatory phenotype. e In the context of Alzheimer’s disease pathogenesis, amyloid-ß oligomers interact with TLRs and scavenger receptors on microglial cells. While microglia may initially clear brain amyloid-ß via phagocytosis, excessive amyloid-ß accumulation may promote a pro-inflammatory cytotoxic microglial response, causing tau hyperphosphorylation and neuronal synaptic dysfunction. f Through activation of the host immune response, chronic infection can promote atherosclerosis and endothelial dysfunction, ultimately increasing risk for cerebrovascular dysfunction, specifically thrombotic stroke. Figure created with Biorender.com
Just as chronic infections can occur both outside and within the CNS, so too can the resulting host immune and inflammatory response. Inflammation that takes place outside of the CNS, often referred to as systemic inflammation, has been considered by some a key feature of AD pathogenesis [105]. Basic and translational research, as well as large-scale epidemiological and pharmacoepidemiologic studies, have all provided support for the role of systemic inflammation in AD. For example, dozens of studies have demonstrated that individuals with MCI and AD (particularly early AD) tend to have higher levels of inflammatory cytokines, chemokines, and acute phase proteins in their blood. A recent meta-analysis found consistent support for elevations in IL-1ß, IL-6, and IL-18 among older adults with AD [106]. An open question, however, is whether systemic inflammation represents a mechanistic driver of AD or is simply a byproduct of the AD pathogenesis. Supporting the mechanistic role of systemic inflammation, experimental animal studies suggest that peripheral inflammatory challenges can initiate or exacerbate Aß and tau pathology [107, 108]. Several human studies suggest that elevated inflammatory proteins in blood during middle adulthood, well before the typical onset of dementia, are associated with subsequent cognitive decline, dementia risk, and late-life brain abnormalities [109,110,111,112]. Moreover, individuals who use anti-inflammatory medication over an extended period during middle adulthood have been found to be at reduced risk of developing AD in subsequent years [113]. Together, these findings suggest that infections, particularly if present during middle adulthood, may increase AD risk by promoting systemic inflammation.
There is a high degree of crosstalk between the peripheral immune system and the brain through what is often labeled the neuroimmune axis. Accordingly, systemic inflammation caused by chronic infection can have immediate and consequential effects on neuronal function. A potent example of systemic infection causing neuronal dysfunction is provided in the case of infectious delirium, a condition in which an acute infection restricted to one part of the body (e.g., urinary tract infection) can cause transient alterations in cognition and behavior. In this case, an inflammatory response caused by an infection outside of the CNS triggers a pro-inflammatory immune response within the CNS, affecting brain function. Systemic inflammation can cause a neuroinflammatory response through multiple routes, including (1) neural signaling via the vagus nerve which innervates multiple organ systems; (2) flow of cytokines and chemokines through gaps in the blood-brain barrier called circumventricular organs; (3) active transport of cytokines and chemokines through the blood-brain barrier; and (4) indirect signaling through endothelial cells of the cerebrovascular system [114,115,116]. Brain changes caused by acute systemic inflammation are likely to be driven, at least in part, by pro-inflammatory activation of microglia [117]. However, it is not clear whether the peripheral immune response generated by chronic infection initiates or perpetuates neuroinflammation in humans. However, factors such as increased age, microglia senescence, and the presence of proteinaceous aggregates in the brain can prime microglia, making an aberrant response to chronic infection more likely [118].
Chronic infection may also contribute to neurodegeneration by promoting cardiovascular and cerebrovascular disease, both of which are well known risk factors for AD and vascular dementia. For example, H. pylori, C. pneumoniae, P. gingivalis, and HIV are each believed to promote atherosclerosis via activation of TLR2- and TLR4-dependent pathways [119]. C. pneumoniae [120] and H. pylori [121], in particular, have been associated with increased risk for stroke (particularly stroke caused by atherosclerotic lesions), so too has total infectious burden [122]. Acute infections, such as pneumonia and septicemia, have also been associated with an elevated risk for stroke [123], suggesting that there is a set of common biological processes that occur with infection and promote vascular disease. Infectious agents have, for example, been shown to promote inflammation of vascular endothelial cells both in the periphery and in the brain, a process closely tied to atherogenesis [124].
As described above, viral and bacterial pathogens have been found in brain tissue. These infectious processes can enter the CNS through neural routes (e.g., trigeminal ganglia) and by crossing the blood-brain barrier, at which point they can exert direct effects on neural function. Infectious agents within the brain can trigger a neuroinflammatory response via signaling of pattern recognition receptors on the surface of microglial cells, a process that can initiate a reactive microglial response characterized by the release of potentially cytotoxic inflammatory mediators known as reactive oxygen species. Persistent inflammatory signaling as a result of chronic CNS infection may have adverse consequences on microglial and neuronal function (e.g., synaptic loss) and influence the rate of amyloid accumulation or tau phosphorylation, thus perpetuating or accelerating the development of AD [105]. Consistent with this theory, murine models of CNS infection (e.g., HSV-1, CMV) show microglial activation, evidence of neuroinflammation, and the infiltration of peripheral immune cells into the brain [125,126,127].
There is mounting evidence that Aß, a central component of AD pathogenesis, may play a compensatory role in infected brains as an antimicrobial peptide (AMP) [39, 128]. AMPs are a component of the innate immune system that protects organisms from a broad spectrum of pathogens [39, 128, 129]. Chemical and physiological similarities between Aß peptides and previously identified AMPs have been described, particularly LL-37, an AMP widely expressed in humans and rodents which is found in the CNS [130]. This AMP hypothesis gained support after Socia and colleagues demonstrated that Aß42 peptides inhibit the overnight growth of bacteria and fungi in vitro [129]. Further support for these findings comes from studies which demonstrate that Aß and the over-expression of Aß in mice and nematodes was associated with increased survival after exposure to bacterial and yeast pathogens [128, 131]. Although Aß AMP may be effective in protecting the brain from infection, continual AMP expression may lead to excessive neuroinflammation and therefore perpetuation of AD pathogenesis. Thus, the antimicrobial protection hypothesis of Aß suggests that infection, at least for a subset of patients, can be an initiating factor.
Evidence From Pharmacoepidemiological Studies and Clinical Trials
If viral or bacterial infections do have a mechanistic role in AD pathogenesis, then treatment of such infections with antivirals or antibiotics would be expected to yield protective effects. Observational cohort studies, pharmacoepidemiologic research, and randomized clinical trials have each been informative in this regard. Some of the most compelling data for the role of viral infection comes from a series of studies conducted using the Taiwan National Health Insurance Research Database. These investigators found that, although VZV was associated with a small increase in dementia risk overall, individuals taking antiherpetic medication for VZV infection had a comparatively lower risk for dementia compared to participants with VZV infection not taking antiherpetic medication [132]. A second study using the same database found similar results for HSV infection, i.e., that antiherpetic treatment reduced the risk associated with infection status [133]. Recently, a study which examined VZV infection in a South Korean population provided further support for a reduction in dementia risk associated with antiherpetic treatment [134]. A study that examined data from multiple European population-based healthcare databases amounting to 2.5 million participants found additional evidence supporting the link between herpesvirus infection and dementia. Among infected individuals, use of antiviral medication was associated with a lower risk for incident dementia. Notably, the findings did not differ based on type of virus or antiviral drug, or dementia etiology (AD and vascular dementia) [135].
Findings demonstrating reduced dementia risk with antiviral medication and in vitro evidence showing that antiviral and antibiotic medication can slow or halt aspects of AD pathology have prompted the initiation of multiple clinical trials [136,137,138]. Currently, the antiherpetic agent valacyclovir (Valtrex) is being examined in a 78-week randomized, double-blind, placebo-controlled phase 2 trial of 130 adults with mild AD who are positive for HSV-1 or HSV-2 antibodies [139]. A smaller open-label phase 2 pilot trial is also examining the efficacy of valacyclovir for treatment of AD or MCI due to AD in 36 individuals with HSV IgG titers (ClinicalTrials.gov Identifier: NCT02997982). This study was completed in 2020, but the results were not published at the time this review was drafted. Other antivirals, such as Efavirenz (NCT03706885) and Emtriva (NCT04500847), drugs used to treat HIV, are also being examined for their ability to slow the progression of AD. However, these drugs are being examined in non-infected individuals based on evidence that they may reduce AD risk through their effects on brain cholesterol metabolism and inflammation. Trials to examine the efficacy of antibiotic therapies for reduction of AD risk have also been conducted, but with limited success thus far. An early randomized triple-blind, placebo-controlled trial of a 3-month doxycycline and rifampin intervention targeting C. pneumoniae in AD patients found evidence for symptom reduction at 6 months, but not at 12 months [140]. A recent randomized double-blind, placebo-controlled trial of a 2-year treatment with another antibiotic, minocycline, found no evidence for slowing of cognitive or functional decline [141]. Currently, there are multiple ongoing or a recently completed phase 2 trials examining the effects of other antibiotics, such as rifaximin (NCT03856359), on cognitive decline in individuals with AD.
Conclusion
The past decade has seen increasing recognition of the role of immune function in the pathophysiology of AD and many other neurodegenerative conditions. Though the infectious hypothesis for AD was initially proposed some time ago, the idea that pathogens may contribute to AD pathogenesis has been brought back to the forefront. As such, there has been a steady increase in research focused on testing both new and old theories relevant to understanding the role that chronic infection may play in AD. There is still much to be done, however. At present, there is little consensus within the field about whether chronic infection may actually promote AD development. We have put forth a framework that outlines three possible relationships between chronic infection and AD. Though the clinical and translational research described in the sections above has already provided valuable insight into the relationship between infection and AD, additional work is needed to address a number of remaining research gaps.
From an epidemiological perspective, it will be essential in the coming years to better understand the timing of infection (or reactivation of infection) in relation to various components of AD pathophysiology. Given that AD pathogenesis takes place over 2 or more decades, primary infection that occurs only after the appearance of clinical symptoms in the late phase of the disease is less likely to be causally relevant and certainly would not be solely responsible for AD. For example, it is possible that the immunologic changes known to occur in the context of Alzheimer’s dementia may increase the risk for viral or bacterial infection, particularly of the CNS. Both increased age and AD have been associated with an exhaustion of adaptive immune response (e.g., reduced T-cell repertoire), a change that may make it easier for microorganisms to infiltrate host defenses. Understanding the temporal relationship between chronic infection, Aß deposition, neuroimmune activation, pathogenic tau formation, and longitudinal cognitive decline will therefore be essential for gaining a more complete understanding of the relationship between pathogens and AD risk and addressing legitimate concerns about reverse causation. Moving forward, the field will also need to carefully parse out the potential contribution of systemic infection versus CNS infection. CNS penetrance of certain viral or bacterial products may not be necessary to influence AD pathogenesis, but this link has only been hinted at using data from observational studies. The key question remains of whether chronic infection in the periphery is able to trigger a chronic inflammatory response powerful enough to alter neuroimmune (microglial) function. To our knowledge, this link has not been clearly demonstrated in humans or in animal models.
We anticipate that these and other questions will be addressed within the next decade with the improvement in research tools, the increased availability of clinical and multi-omic data to scientists around the world, and the willingness of major funding agencies, including the National Institutes of Health and the American Society for Infectious Disease, to support research examining the role that pathogens may play in development of AD. The evidence linking a range of microorganisms to Aß development, clinically defined Alzheimer’s dementia, and pathologically defined AD certainly warrants continued research efforts, especially given the repeated failures of drugs targeting Aß.
Data Availability
N/A.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
2020 Alzheimer’s disease facts and figures. Alzheimer’s & Dementia, 2020. 16(3): p. 391-460
Vaz M, Silvestre S. Alzheimer’s disease: recent treatment strategies. Eur J Pharmacol. 2020;887:173554.
Fulop T, et al. Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front Aging Neurosci. 2018;10:224.
Jamieson GA, et al. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol. 1991;33(4):224–7.
Gérard HC, et al. Chlamydophila (Chlamydia) pneumoniae in the Alzheimer’s brain. FEMS Immunol Med Microbiol. 2006;48(3):355–66.
Balin BJ, et al. Chlamydia pneumoniae: an etiologic agent for late-onset dementia. Front Aging Neurosci. 2018;10:302.
Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimers Dis. 2016;53(4):1459–73.
Dominy SS, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595–608.
Villemagne VL, et al. Amyloid ß deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67.
Guo T, et al. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener. 2020;15(1):40.
Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease Pathogenesis. Biol Psychiatry. 2015;77(1):43–51.
Kunkle BW, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aß, tau, immunity and lipid processing. Nat Genet. 2019;51(3):414–30.
Fan Z, et al. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain. 2017;140(3):792–803.
Craft JM, Watterson DM, Van Eldik LJ. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia. 2006;53(5):484–90.
Sims R, et al. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet. 2017;49(9):1373–84.
Heneka MT, et al. Neuroinflammation in Alzheimer’s disease. The Lancet Neurology. 2015;14(4):388–405.
Van Eldik LJ, et al. The roles of inflammation and immune mechanisms in Alzheimer’s disease. Alzheimer's & dementia (New York, N Y). 2016;2(2):99–109.
Ising C, et al. NLRP3 inflammasome activation drives tau pathology. Nature. 2019;575(7784):669–73.
Cuello AC. Early and late CNS inflammation in Alzheimer’s disease: two extremes of a continuum? Trends Pharmacol Sci. 2017;38(11):956–66.
Itzhaki RF, et al. Do infections have a role in the pathogenesis of Alzheimer disease? Nat Rev Neurol. 2020;16(4):193–7.
Torres L, et al. Toxoplasma gondii alters NMDAR signaling and induces signs of Alzheimer’s disease in wild-type, C57BL/6 mice. J Neuroinflammation. 2018;15(1):57–7.
Kusbeci OY, et al. Could Toxoplasma gondii have any role in Alzheimer disease? Alzheimer Dis Assoc Disord. 2011;25(1):1–3.
Parady B. Innate immune and fungal model of Alzheimer’s disease. Journal of Alzheimer's disease reports. 2018;2(1):139–52.
Pisa D, et al. Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimers Dis. 2015;43(2):613–24.
Alonso R, et al. Identification of fungal species in brain tissue from Alzheimer’s disease by next-generation sequencing. J Alzheimers Dis. 2017;58(1):55–67.
Fülöp T, et al. Role of microbes in the development of Alzheimer’s disease: state of the art - an International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front Genet. 2018;9:362.
Fulop T, et al. Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J Neuro-Oncol. 2019;25(5):634–47.
Canet G, et al. HIV neuroinfection and Alzheimer’s disease: similarities and potential links? Front Cell Neurosci. 2018;12:307–7.
Rubin LH, Sundermann EE, Moore DJ. The current understanding of overlap between characteristics of HIV-associated neurocognitive disorders and Alzheimer's disease. J Neuro-Oncol. 2019;25(5):661–72.
Amran A, et al. Influenza vaccination is associated with a reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2020;16(S10):e041693.
Verreault R, et al. Past exposure to vaccines and subsequent risk of Alzheimer’s disease. CMAJ : Canadian Medical Association journal = journal de l’Association medicale canadienne. 2001;165(11):1495–8.
Imfeld P, et al. Influenza infections and risk of Alzheimer’s disease. Brain Behav Immun. 2016;57:187–92.
Ball MJ. Limbic predilection in Alzheimer dementia: is reactivated herpesvirus involved? Can J Neurol Sci. 1982;9(3):303–6.
Sequiera LW, et al. Detection of herpes-simplex viral genome in brain tissue. Lancet. 1979;2(8143):609–12.
Jamieson GA, et al. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol. 1992;167(4):365–8.
Wozniak MA, et al. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer’s disease patients. J Med Virol. 2005;75(2):300–6.
Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol. 2009;217(1):131–8.
Eimer WA, et al. Alzheimer’s disease-associated ß-amyloid is rapidly seeded by Herpesviridae to protect against brain infection. Neuron. 2018;99(1):56–63.e3.
Valyi-Nagy T, et al. Herpes simplex virus type 1 latency in the murine nervous system is associated with oxidative damage to neurons. Virology. 2000;278(2):309–21.
Schachtele SJ, et al. Herpes simplex virus induces neural oxidative damage via microglial cell Toll-like receptor-2. J Neuroinflammation. 2010;7:35.
Itzhaki RF, et al. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349(9047):241–4.
Koelle DM, et al. APOE genotype is associated with oral herpetic lesions but not genital or oral herpes simplex virus shedding. Sex Transm Infect. 2010;86(3):202–6.
Zhao N, et al. Apolipoprotein E, receptors, and modulation of Alzheimer’s disease. Biol Psychiatry. 2018;83(4):347–57.
Honjo K, van Reekum R, Verhoeff NP. Alzheimer’s disease and infection: do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement. 2009;5(4):348–60.
Itzhaki RF, Wozniak MA. Herpes simplex virus type 1, apolipoprotein E, and cholesterol: a dangerous liaison in Alzheimer’s disease and other disorders. Prog Lipid Res. 2006;45(1):73–90.
Carbone I, et al. Herpes virus in Alzheimer’s disease: relation to progression of the disease. Neurobiol Aging. 2014;35(1):122–9.
Aiello AE, et al. The influence of latent viral infection on rate of cognitive decline over 4 years. J Am Geriatr Soc. 2006;54(7):1046–54.
Tarter KD, et al. Persistent viral pathogens and cognitive impairment across the life course in the third national health and nutrition examination survey. J Infect Dis. 2014;209(6):837–44.
Matheï C, et al. Associations between cytomegalovirus infection and functional impairment and frailty in the BELFRAIL Cohort. J Am Geriatr Soc. 2011;59(12):2201–8.
Pogo BG, Casals J, Elizan TS. A study of viral genomes and antigens in brains of patients with Alzheimer’s disease. Brain. 1987;110(Pt 4):907–15.
Lin WR, et al. Herpesviruses in brain and Alzheimer’s disease. J Pathol. 2002;197(3):395–402.
•• Warren-Gash C, et al. Human herpesvirus infections and dementia or mild cognitive impairment: a systematic review and meta-analysis. Sci Rep. 2019;9(1):4743–3 This meta-analysis of 57 human studies across various geographic settings examined the pooled association of human herpesviruses (HSV-1/2, HHV6, VZV, EBV) with risk for dementia and mild cognitive impairment (MCI).
Payne S. Chapter 34 - family Herpesviridae. In: Viruses SP, editor. : Academic Press; 2017. p. 269–78.
Readhead B, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99(1):64–82.e7.
•• Allnutt MA, et al. Human herpesvirus 6 detection in Alzheimer’s disease cases and controls across multiple cohorts. Neuron. 2020;105(6):1027–1035.e2 This study examined HHV-6 presence in three independent cohorts using RNA sequencing and DNA samples derived from the autopsied brains of individuals with and without Alzheimer’s disease. The authors did not find a strong association between HHV-6 and Alzheimer’s disease.
Wozniak MA, et al. Does apolipoprotein E determine outcome of infection by varicella zoster virus and by Epstein Barr virus? Eur J Hum Genet. 2007;15(6):672–8.
Volpi A. Epstein-Barr virus and human herpesvirus type 8 infections of the central nervous system. Herpes. 2004;11(Suppl 2):120a–7a.
Itzhaki RF, et al. Microbes and Alzheimer’s disease. Journal of Alzheimer’s disease : JAD. 2016;51(4):979–84.
Choroszy-Król I, et al. Infections caused by Chlamydophila pneumoniae. Adv Clin Exp Med. 2014;23(1):123–6.
Shima K, Kuhlenbäumer G, Rupp J. Chlamydia pneumoniae infection and Alzheimer’s disease: a connection to remember? Med Microbiol Immunol. 2010;199(4):283–9.
Balin BJ, et al. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;187(1):23–42.
Gérard HC, et al. The load of Chlamydia pneumoniae in the Alzheimer’s brain varies with APOE genotype. Microb Pathog. 2005;39(1):19–26.
Gérard HC, et al. Apolipoprotein E4 enhances attachment of Chlamydophila (Chlamydia) pneumoniae elementary bodies to host cells. Microb Pathog. 2008;44(4):279–85.
Holmes C, Cotterell D. Role of infection in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs. 2009;23(12):993–1002.
Petzke M, Schwartz I. Borrelia burgdorferi pathogenesis and the immune response. Clin Lab Med. 2015;35(4):745–64.
Herrera-Landero A, et al. Borrelia burgdorferi as a risk factor for Alzheimer’s dementia and mild cognitive impairment. European Geriatric Medicine. 2019;10(3):493–500.
O’Day DH, Catalano A. A lack of correlation between the incidence of Lyme disease and deaths due to Alzheimer’s disease. J Alzheimers Dis. 2014;42:115–8.
Radolf JD, et al. Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat Rev Microbiol. 2016;14(12):744–59.
Miklossy J, Biology and neuropathology of dementia in Syphilis and Lyme Disease, in Handbook of Clinical Neurology. 2008, Elsevier. p. 825-844
Miklossy J. Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci. 2015;7:46.
Carabotti M, et al. The gut-brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28(2):203–9.
Wang H-X, Wang Y-P. Gut microbiota-brain axis. Chin Med J. 2016;129(19):2373–80.
Vogt NM, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537–7.
Lukiw WJ. Bacteroides fragilis Lipopolysaccharide and inflammatory signaling in Alzheimer’s disease. Front Microbiol. 2016;7:1544.
Li H, et al. Amyloid, tau, pathogen infection and antimicrobial protection in Alzheimer’s disease –conformist, nonconformist, and realistic prospects for AD pathogenesis. Translational Neurodegeneration. 2018;7(1):34.
Chen T, et al., The human oral microbiome database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database : the journal of biological databases and curation, 2010. 2010: p. baq013-baq013
Sureda A, et al. Oral microbiota and Alzheimer’s disease: do all roads lead to Rome? Pharmacol Res. 2020;151:104582.
Orr ME, et al. Can oral health and oral-derived biospecimens predict progression of dementia? Oral Dis. 2020;26(2):249–58.
Aguayo S, et al. Association between Alzheimer’s disease and oral and gut microbiota: are pore forming proteins the missing link? J Alzheimers Dis. 2018;65(1):29–46.
Aragón F, et al. Oral health in Alzheimer’s disease: a multicenter case-control study. Clin Oral Investig. 2018;22(9):3061–70.
How KY, Song KP, Chan KG. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front Microbiol. 2016;7:53–3.
Tamse A, Schwartz Y. Unusual findings in heart and dental pulp in systemic primary amyloidosis. J Oral Med. 1981;36(1):16–7.
Kim J-M, et al. Dental health, nutritional status and recent-onset dementia in a Korean community population. International Journal of Geriatric Psychiatry. 2007;22(9):850–5.
Martande SS, et al. Periodontal health condition in patients with Alzheimer’s disease. Am J Alzheimers Dis Other Dement. 2014;29(6):498–502.
Sochocka M, et al. Association between periodontal health status and cognitive abilities. The role of cytokine profile and systemic inflammation. Curr Alzheimer Res. 2017;14(9):978–90.
Chen C-K, Wu Y-T, Chang Y-C. Association between chronic periodontitis and the risk of Alzheimer’s disease: a retrospective, population-based, matched-cohort study. Alzheimers Res Ther. 2017;9(1):56.
. Demmer RT, et al. Periodontal disease and incident dementia: the atherosclerosis Risk in communities study (ARIC). Neurology. 2020;95(12):e1660–71 Using a community-based cohort of 8,275 participants, the authors demonstrated that midlife periodontal disease is associated with a modest increase in risk for dementia or mild cognitive impairment later in life.
Cuomo P, et al. An in vitro model to investigate the role of Helicobacter pylori in type 2 diabetes, obesity, Alzheimer’s disease and cardiometabolic disease. Int J Mol Sci. 2020;21:21.
Doulberis M, et al. Review: impact of Helicobacter pylori on Alzheimer’s disease: what do we know so far? Helicobacter. 2018;23:1.
Fischbach W, Malfertheiner P. Helicobacter pylori infection. Dtsch Arztebl Int. 2018;115(25):429–36.
Khoder G, et al. Prevalence of Helicobacter pylori and its associated factors among healthy asymptomatic residents in the United Arab Emirates. Pathogens (Basel, Switzerland). 2019;8(2):44.
Kovács T, Cairns NJ, Lantos PL. Olfactory centres in Alzheimer’s disease: olfactory bulb is involved in early Braak’s stages. NeuroReport. 2001;12:2.
Kountouras J, et al. A proposed role of human defensins in Helicobacter pylori-related neurodegenerative disorders. Med Hypotheses. 2014;82(3):368–73.
Figura N, et al. Extragastric manifestations of Helicobacter pylori infection. Helicobacter. 2010;15(s1):60–8.
Kountouras J, Chatzopoulos D, Zavos C. Reactive oxygen metabolites and upper gastrointestinal diseases. Hepatogastroenterology. 2001;48(39):743–51.
Kountouras J, et al. Potential implications of Helicobacter pylori-related neutrophil-activating protein. World J Gastroenterol. 2012;18(5):489–90.
Kountouras J, et al. Impact of reactive oxygen species generation on Helicobacter pylori-related extragastric diseases: a hypothesis. Free Radic Res. 2017;51(1):73–9.
Malaguarnera M, et al. Helicobacter pylori and Alzheimer’s disease: a possible link. European Journal of Internal Medicine. 2004;15(6):381–6.
Kountouras J, et al. Relationship between Helicobacter pylori infection and Alzheimer disease. Neurology. 2006;66(6):938.
Roubaud-Baudron C, et al. Impact of chronic Helicobacter pylori infection on Alzheimer’s disease: preliminary results. Neurobiol Aging. 2012;33(5):1009.e11–9.
Moutsopoulos NM, Madianos PN. Low-grade inflammation in chronic infectious diseases: paradigm of periodontal infections. Ann N Y Acad Sci. 2006;1088:251–64.
Oshima T, et al. Association of Helicobacter pylori infection with systemic inflammation and endothelial dysfunction in healthy male subjects. J Am Coll Cardiol. 2005;45(8):1219–22.
Jackson L, et al. A population-based epidemiologic study of Helicobacter pylori infection and its association with systemic inflammation. Helicobacter. 2009;14(5):108–13.
Newcombe EA, et al. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation. 2018;15(1):276.
Lai KSP, et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry. 2017;88(10):876–82.
Kahn MS, et al. Prolonged elevation in hippocampal Aß and cognitive deficits following repeated endotoxin exposure in the mouse. Behav Brain Res. 2012;229(1):176–84.
Liu Y, et al. Peripheral inflammation promotes brain tau transmission via disrupting blood-brain barrier. Biosci Rep. 2020;40:2.
Schmidt R, et al. Early inflammation and dementia: a 25-year follow-up of the Honolulu-Asia Aging Study. Ann Neurol. 2002;52(2):168–74.
Walker KA, et al. Systemic inflammation during midlife and cognitive change over 20 years: The ARIC Study. Neurology. 2019;92(11):e1256–67.
Walker KA, et al. The association of mid-to late-life systemic inflammation with white matter structure in older adults: The Atherosclerosis Risk in Communities Study. Neurobiol Aging. 2018;68:26–33.
Tao Q, et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1(6):e183597.
McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and Alzheimer’s disease: the last 22 years. J Alzheimers Dis. 2016;54(3):853–7.
Thayer JF, Sternberg EM. Neural aspects of immunomodulation: focus on the vagus nerve. Brain Behav Immun. 2010;24(8):1223–8.
Banks WA. Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Curr Pharm Des. 2005;11(8):973–84.
Quan N. Immune-to-brain signaling: how important are the blood-brain barrier-independent pathways? Mol Neurobiol. 2008;37(2-3):142–52.
Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013;61(1):71–90.
Niraula A, Sheridan JF, Godbout JP. Microglia priming with aging and stress. Neuropsychopharmacology. 2017;42(1):318–33.
Li B, Xia Y, Hu B. Infection and atherosclerosis: TLR-dependent pathways. Cell Mol Life Sci. 2020;77(14):2751–69.
Chen J, et al. Chlamydia pneumoniae infection and cerebrovascular disease: a systematic review and meta-analysis. BMC Neurol. 2013;13:183.
Wang ZW, et al. Helicobacter pylori infection contributes to high risk of ischemic stroke: evidence from a meta-analysis. J Neurol. 2012;259(12):2527–37.
Elkind MS, et al. Infectious burden and risk of stroke: the northern Manhattan study. Arch Neurol. 2010;67(1):33–8.
Elkind Mitchell SV, et al. Infection as a stroke risk factor and determinant of outcome after stroke. Stroke. 2020;51(10):3156–68.
Shah PK. Inflammation, infection and atherosclerosis. Trends Cardiovasc Med. 2019;29(8):468–72.
Bortolotti D, et al. HHV-6A infection induces amyloid-beta expression and activation of microglial cells. Alzheimers Res Ther. 2019;11(1):104.
Kaushik DK, Gupta M, Basu A. Microglial response to viral challenges: every silver lining comes with a cloud. Front Biosci (Landmark Ed). 2011;16:2187–205.
Marques CP, et al. Microglial cells initiate vigorous yet non-protective immune responses during HSV-1 brain infection. Virus Res. 2006;121(1):1–10.
Kumar DK, et al. Amyloid-ß peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72.
Soscia SJ, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505.
Bergman P, et al. Induction of the antimicrobial peptide CRAMP in the blood-brain barrier and meninges after meningococcal infection. Infect Immun. 2006;74(12):6982–91.
Bourgade K, et al. ß-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology. 2015;16(1):85–98.
Chen VC, et al. Herpes zoster and dementia: a nationwide population-based cohort study. J Clin Psychiatry. 2018;79:1.
Tzeng NS, et al. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections-a nationwide, population-based cohort study in Taiwan. Neurotherapeutics. 2018;15(2):417–29.
Bae S, et al., Association of herpes zoster with dementia and effect of antiviral therapy on dementia: a population-based cohort study. Eur Arch Psychiatry Clin Neurosci, 2020.
Schnier C, et al., Antiherpetic medication and incident dementia: observational cohort studies in four countries. medRxiv, 2020: p. 2020.12.03.20241497
Forloni G, et al. Anti-amyloidogenic activity of tetracyclines: studies in vitro. FEBS Lett. 2001;487(3):404–7.
Tomiyama T, et al. Inhibition of amyloid beta protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. J Biol Chem. 1996;271(12):6839–44.
Familian A, et al. Inhibitory effect of minocycline on amyloid beta fibril formation and human microglial activation. Glia. 2006;53(3):233–40.
Devanand DP, et al. Antiviral therapy: valacyclovir treatment of Alzheimer’s disease (VALAD) trial: protocol for a randomised, double-blind, placebo-controlled, treatment trial. BMJ Open. 2020;10(2):e032112.
Loeb MB, et al. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J Am Geriatr Soc. 2004;52(3):381–7.
Howard R, et al. Minocycline at 2 different dosages vs placebo for patients with mild Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2020;77(2):164–74.
Code Availability
N/A.
Funding
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging. This study was also supported by contracts K23 AG064122 (Dr. Walker). Compliance with the National Institutes of Health (NIH) Public Access Policy requires proper submission of this work to PubMed Central (PMC).
Author information
Authors and Affiliations
Contributions
LB and KW conceptualized, drafted, and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Ethics Approval and Consent to Participate
N/A.
Consent for Publication
Obtained.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Infectious Involvement in Neurological Disease
Rights and permissions
About this article

Cite this article
Butler, L., Walker, K.A. The Role of Chronic Infection in Alzheimer’s Disease: Instigators, Co-conspirators, or Bystanders?. Curr Clin Micro Rpt 8, 199–212 (2021). https://doi.org/10.1007/s40588-021-00168-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40588-021-00168-6