
Abstract
Osteoarthritis (OA) is a chronic joint disease characterized by the degeneration, destruction, and excessive ossification of articular cartilage. The prevalence of OA is rising annually, concomitant with the aging global population and increasing rates of obesity. This condition imposes a substantial and escalating burden on individual health, healthcare systems, and broader social and economic frameworks. The etiology of OA is multifaceted and not fully understood. Current research suggests that the death of chondrocytes, encompassing mechanisms such as cellular apoptosis, pyroptosis, autophagy, ferroptosis and cuproptosis, contributes to both the initiation and progression of the disease. These cell death pathways not only diminish the population of chondrocytes but also exacerbate joint damage through the induction of inflammation and other deleterious processes. This paper delineates the morphological characteristics associated with various modes of cell death and summarizes current research results on the molecular mechanisms of different cell death patterns in OA. The objective is to review the advancements in understanding chondrocyte cell death in OA, thereby offering novel insights for potential clinical interventions.
Similar content being viewed by others

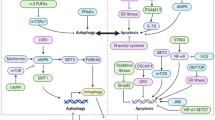
Introduction
OA is a prevalent global health condition [1].Symptoms of OA include pain and temporary morning stiffness. The advanced stage of OA is distinguished by instability and physical impairment, significantly diminishing quality of life. Initially, OA was attributed to the degradation of articular cartilage [2], It is now understood as a complex interplay of local and systemic inflammatory and metabolic fact the extracellular matrix degradation [3]. Research has indicated that the progression of OA is influenced by various mechanisms of cell death [4, 5], Various cell death patterns exhibit distinct mechanisms of action in the pathogenesis of OA. As OA research progresses, advancements in understanding its mechanism of action have been made [6]. However, the field continues to encounter numerous challenges. The complex pathophysiology and risk factors associated with OA contribute to the lack of effective treatment options.
Due to the intricate and demanding characteristics of OA, there is an urgent requirement for the identification of novel therapeutic targets and tools to address the needs of patients with OA. Cell death, serving as a crucial regulatory mechanism, significantly contributes to the maintenance of the body’s internal homeostasis and the equilibrium of cell populations [7]. In recent years, there has been significant progress in the development of pharmaceutical agents that specifically target the cell death pathway, showing promise for the prevention and management of various human diseases, including OA [8]. These medications are formulated to prevent chondrocyte loss, consequently decelerating the advancement of OA. Research has shown advancements in understanding cell death mechanisms and their relation to OA. Numerous studies have concentrated on investigating the impact of various cell death pathways on OA and have conducted extensive research in this area. Nevertheless, there remain numerous unexplored facets concerning the interplay and consequences of cell death pathways on OA that necessitate further comprehensive investigation.
This paper aims to examine the recent advancements in OA research, specifically exploring the correlation between regulators of chondrocyte death, various forms of cell death, and targeted therapeutic interventions in OA. The objective is to investigate the association between patterns of cell death and OA pathogenesis. By examining various mechanisms of cell death and their interplay, a more profound understanding of the etiology of OA can be attained, providing a theoretical basis for the prevention and treatment of this medical condition.
Apoptosis and OA
Apoptosis is a programmed cell death process that occurs in response to specific physiological or pathological stimuli, regulated by endogenous genetic pathways [9], which is a fundamental biological phenomenon that is prevalent in living organisms. The term was introduced in the 1970s to describe the controlled demise of individual cells in specific physiological or pathological circumstances [10], The majority of cell death mechanisms rely on the via caspase-dependent apoptotic pathway, which plays a crucial role in programmed cell death [11]. The morphological characteristics of apoptotic chondrocytes obtained from human osteoarthritic cartilage consist of nuclear condensation, DNA fragmentation, chromosome condensation, cytosolic vesiculation, cellular crumpling, and fragmentation into small membrane-enclosed vesicles [12]. These characteristics are commonly observed in the apoptotic process. Apoptosis is a phenomenon that is typically observed infrequently in normal chondrocytes.
Apoptosis, the initial recognized mechanism of programmed cell death, is orchestrated by a group of intracellular caspases that target various intracellular substrates, leading to cellular contraction, chromatin fragmentation, membrane vesicle formation, and breakdown into membrane-enclosed vesicles known as apoptotic bodies. Specifically, caspase-3 and caspase-8 target DNA fragmentation factor (DFF or inhibitor of cystatin-activated DNase[ICAD]), initiating the liberation and activation of its dimerization chaperone, DFF40, which subsequently cleaves chromatin DNA into nucleosome fragments [13]. Cysteine proteases (caspases) are a class of enzymes that play a key role in apoptosis, regulating not only apoptosis, but also non-apoptotic functions, including cell proliferation, differentiation, and migration [14]. Effector cysteine aspartic proteases are activated to cleave a diverse range of cellular substrates, such as cytokeratins, poly(ADP-ribose) polymerase (PARP), plasma membrane cytoskeletal protein α-fodrin, and nuclear mitotic apparatus protein (NuMA). Caspase-3 is essential in the process of apoptosis through its ability to cleave inhibitors of cysteine-activated nuclease (ICAD) and prevent cysteine-activated nuclease (CAD) from acting as an endonuclease, thereby facilitating the characteristic apoptotic features observed in various tissues, such as cartilage, including DNA fragmentation and cellular demise [15, 16]. Caspase-1 is activated by the inflammasome, which is formed by nod-like receptors (NLR) or members of the pyrin and HIN structural domain-containing protein (PYHIN) family, leading to the cleavage and activation of pro-IL-1β and pro-IL-18 cytokines, thereby inducing pro-inflammatory responses [17].In addition, alterations in mitochondrial membrane potential affect the activity of some enzymes, such as the release of cytochrome C and the activation of apoptosis-inducing factors. In the process of chondrocyte apoptosis, dysregulation of gene expression occurs, notably with the imbalance of Bcl-2 family genes leading to programmed cell death. The anti-apoptotic members of the Bcl-2 family exhibit hydrophobic helices on the cell surface, necessary for binding to their pro-apoptotic counterparts and facilitating pro-survival functions [18]. Whereas mutations in the p53 gene are closely related to the development of OA, some growth factors, cytokines and hormones also affect the gene expression of chondrocytes, which in turn affects the apoptotic process. During the process of chondrocyte apoptosis, alterations in the permeability of the cell membrane result in modifications to the flow of various molecules, including Ca2+ and Na+ influx and cytochrome C efflux. These changes impact cellular metabolism and function, ultimately contributing to the progression of chondrocyte apoptosis. Additionally, significant modifications to the cytoskeleton occur during chondrocyte apoptosis, such as destabilization of microtubules and microfilaments, leading to potential alterations in cell morphology and motility. The modification of the cytoskeleton has a significant impact on cell signaling, material transport processes, and ultimately promotes chondrocyte apoptosis. This process also results in alterations to the functions of various organelles such as mitochondria and endoplasmic reticulum. For instance, the disruption of mitochondrial membrane potential leads to decreased ATP synthesis, affecting cellular energy metabolism, and the dysfunction of the endoplasmic reticulum results in aberrant protein synthesis and folding, ultimately exacerbating cellular damage and promoting apoptosis (Fig. 1).

Figure 1
In conclusion, our findings indicate that chondrocyte apoptosis is a complex process involving morphological changes and multiple levels of regulation. In OA, changes in genes like caspase and Bcl-2 play a role in regulating cell death and inflammation. When the rate of chondrocyte apoptosis surpasses a specific threshold, a reduction in chondrocyte population occurs. This decline not only hinders the synthesis and secretion of cartilage matrix, but also disrupts the equilibrium of enzyme systems responsible for maintaining cartilage matrix synthesis and degradation. Consequently, the imbalance ultimately results in qualitative and quantitative alterations in the cartilage matrix, further exacerbating chondrocyte apoptosis and perpetuating a cycle of cartilage degeneration. Therefore, the inhibition of chondrocyte apoptosis represents an effective strategy for prevention and treatment OA.
Therapeutic targets related to apoptosis for OA
Apoptosis presents numerous potential targets for the development of therapeutic drugs, and recent progress in understanding the underlying mechanisms of apoptosis has reignited interest in this field. Apoptosis presents numerous potential targets for the development of therapeutic drugs, and recent progress in understanding the underlying mechanisms of apoptosis has reignited interest in this field. Caspase-3 gene silencing has been reported to downregulate the expression of TNF-α-mediated inflammatory genes TNFR1, FADD, and IL-1β, thereby attenuating the apoptotic pathway in vitro. Furthermore, the silencing of the Caspase-3 gene resulted in a significant decrease in OARSI scores and the expression of key genes involved in OA, including Caspase-3, Caspase-9, MMP13, and TNF-α, in a rat model of surgically induced OA. These findings suggest that targeting Caspase-3 gene expression may hold promise as a novel therapeutic approach for the treatment of cartilage injury and OA [19]. Furthermore, the protective effects induced by ubiquitin-specific protease 13 (USP13) were found to be correlated with the improvement of nuclear factor erythroid 2-related factor 2 (Nrf-2) and the inhibition of Caspase-3 [20]. In the rabbit anterior cruciate ligament transection model, intra-articular administration of hyaluronic acid (HA) mitigates the severity of OA and decreases the quantity of apoptotic chondrocytes [21].Through analysis of web-based pharmacological data, the researchers identified that the β-sitosterol component of OD targeted key factors Bax, Bcl2, and JUN, resulting in the inhibition of chondrocyte apoptosis. This intervention also led to a significant reduction in inflammation and cartilage degeneration in OA. As a result, the study suggests that targeting apoptosis key factors Bax, Bcl2, and JUN may hold promise as therapeutic targets for OA [22].
Moreover, numerous non-coding RNAs, such as long-stranded non-coding RNAs and cyclic RNAs, have been identified as regulators of chondrocyte apoptosis through diverse mechanisms. For instance, in OA cartilage, the expression of miR-146a is induced by various microbes and pro-inflammatory cytokines, including TNF-α and IL-1β [23].On the contrary, the upregulation of miR-146a-5p in human OA tissues resulted in a notable increase in Caspase-3 activation, PARP degradation, and Bax expression, while concurrently suppressing Bcl-2 expression, ultimately promoting chondrocyte apoptosis [24].MiR-146a upregulates apoptosis in human chondrocytes by selectively suppressing Smad4 expression in mechanically damaged cartilage [25]. Furthermore, the study found that miR-1236 expression was significantly increased in OA knee cartilage compared to normal cartilage. This upregulation of miR-1236 expression was shown to suppress cell proliferation and promote apoptosis. Interestingly, co-transfection of miR-1236 with PIK3R3 was able to reverse the apoptotic effects induced by miR-1236 mimics. Based on these findings, Wang et al. suggest that miR-1236 may serve as a promising biomarker or therapeutic target for OA [26]. Mitochondria are essential for cell function and survival, as oxidative stress and disruptions in mitochondrial respiratory function have been linked to cell death and degeneration. A proteomics analysis revealed significant alterations in mitochondrial proteins related to energy production, maintenance of mitochondrial membrane integrity, and detoxification of free radicals during apoptosis, including reduced levels of mitochondrial superoxide dismutase (SOD) and increased intracellular production of reactive oxygen species (ROS) in chondrocytes affected by OA [27]. The depolarization of mitochondria results in the release of apoptotic factors, including cytochrome c, apoptosis-inducing factor, and caspase-9, from the intermembrane space [28].
In conclusion, the involvement of apoptosis in OA is intricately linked to members of the Caspase family, including Caspase-3, Caspase-9, as well as other targets such as USP13, Bax, Bcl2, and JUN. Further exploration of these targets may offer novel insights for the treatment of apoptosis in OA. Additionally, the regulation of mitochondria and alterations in non-coding RNA, such as miR-146a-5p and miR-1236, could serve as potential therapeutic targets for modulating chondrocyte apoptosis in OA. These factors interact synergistically to facilitate the apoptotic process in chondrocytes. The primary therapeutic strategies for addressing chondrocyte apoptosis in OA encompass the inhibition of apoptosis, stimulation of cell regeneration, and anti-inflammatory interventions. Novel therapeutic modalities like stem cell therapy and gene therapy are currently under investigation and hold promise for enhancing treatment efficacy in OA patients.
Pyroptosis and OA
Cellular pyroptosis exhibits distinct morphological features and characteristics, including cell swelling and lysis, cell membrane rupture leading to the release of cytoplasmic contents, and chromosomal DNA fragmentation, representing a novel form of cell death. Pyroptosis is characterized as a type of regulated cell death (RCD) that relies significantly on gasdermin family proteins, which facilitate the formation of pores in the plasma membrane. This process is typically triggered by inflammatory cysteine aspartic proteases and represents a more intricate mode of cell demise compared to apoptosis [29]. In recent years, a growing body of research has demonstrated the occurrence of cellular pyroptosis in various cell types beyond mononuclear phagocyte lineages, including vascular endothelial cells and neuroglia. This finding offers a solid theoretical foundation for exploring the potential implications of pyroptosis in non-immune diseases, such as cardiovascular and neurodegenerative disorders.
Gasdermin is a recently identified group of proteins that possess the capability to form membrane pores, with gasdermin D (GSDMD) believed to be the primary mediator of pyroptosis [30]. This family contains six homologous proteins in humans, gasdermin A-E (GSDMDA-GSDDE) and DFNB59 [31]。When the GSDMD pore formation is disrupted, it hinders the activation of inflammatory vesicles and cysteinyl aspartic enzymes, thereby impeding their biomolecular signaling. This inhibition can mitigate pyroptosis by preventing the excessive activation of GSDMD and indirectly interfering with the subsequent inflammatory cascade response [32].The disruption of the cell membrane during pyroptosis results in the extracellular release of intracellular inflammatory mediators, such as IL-1β [33]. The multiprotein complex responsible for the recognition of pathogen-associated molecular patterns (PAMP) and damage-associated molecular patterns (DAMP) subsequently triggers the activation of caspase-1.
Nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) is a crucial component of the NLR family of inflammasomes, serving as a well-defined inflammasome. The NLRP3 inflammasome is composed of the NLRP3 protein, apoptosis-associated speck-like protein ASC, and caspase-1 precursor. Upon receiving activation signals, ASC facilitates the recruitment of NLRP3 and caspase-1 precursors through interactions with their structural domains, leading to the activation of caspase-1 and the subsequent release of pro-inflammatory factors, including IL-1β and IL-18, which trigger caspase-1-dependent classical cell death, known as pyroptosis [34]. The NLRP3 inflammasome facilitates Caspase-1-dependent classical cellular pyroptosis, leading to the processing and cleavage of precursor forms of IL-1β and IL-18 into their active mature forms. In contrast to apoptosis and simple cell necrosis, cellular pyroptosis is characterized by its pro-inflammatory nature. Research has demonstrated that chondrocyte pyroptosis-induced cartilage inflammation and matrix degradation are potential features of OA chondrocytes, and are closely linked to the manifestation of joint pain and localized joint dysfunction [35, 36]. Numerous risk factors, including obesity, basic calcium phosphate (BCP), and aging, have been shown to trigger the assembly of the NLRP3 inflammasome, consequently leading to the development of OA [35]. In an OA model, the expression of NLRP3 was found to be elevated in the medial cartilage compared to the lateral cartilage. Additionally, the inhibition of the MAPK/NF-κB/NLRP3 pathway was shown to mitigate chondrocyte pyroptosis, proteoglycan loss, collagen degradation, articular cartilage degeneration, and subchondral bone destruction [37]. Activator-induced pyroptosis worsens diseases by activating inflammasome in chondrocytes through the MAPK/NF-κB pathway, leading to the release of inflammatory factors like IL-1β, IL-18, TNF-α, and MMP [1, 38,39,40]. , This increases the level of inflammation in the joint fluid and surrounding tissues. Facilitates the upregulation of catabolic enzymes and pro-inflammatory factors, resulting in the degradation of cartilage and exacerbation of the inflammatory response. The protein NLRP3, associated with OA, has been shown to inhibit the activation of the NLRP3 inflammasome and impede the growth of osteoarthritic synoviocytes by the introduction of exogenous stromal cell-derived factor-1 (SDF-1) through the stimulation of the AMPK signaling pathway [41] (Figure 2).

Figure 2
In conclusion, cellular pyroptosis significantly contributes to the pathological advancement of OA by influencing cartilage degradation, inflammatory response, and synovial destruction. The differential expression of key proteins and cytokines, including NLRs, caspases, interleukins, and GSDMD, is closely associated with the progression of OA. By enhancing our comprehension of the correlation between cellular pyroptosis and OA and investigating efficient regulatory strategies, it is anticipated that novel opportunities will emerge for the prevention and treatment of OA.
Therapeutic targets related to pyroptosis for OA
Gasdermin D (GSDMD) initiates pyroptosis by integrating numerous endogenous and exogenous signals from inflammasome. In the event that the GSDMD pore formation is disrupted, the activation of inflammasome and cysteine-aspartic enzymes, along with their associated biomolecular signals, is largely impeded. This intervention has the potential to pyroptosis by inhibiting the excessive activation of GSDMD, thereby preventing the subsequent impairment of the inflammatory cascade response. Therefore, the targeting of GSDMD blockade demonstrates greater selectivity in comparison to specific forms of inflammasome inhibition. So [42,43,44], In theory targeting GSDMD blockade may represent the most promising intervention strategy currently available. A study investigated the impact of GSDMD on post-traumatic OA. Suppression of GSDMD mitigated alterations in cartilage, synovitis, and subchondral osteosclerosis. The study showed that GSDMD deficiency did not prevent rheumatoid arthritis or gouty arthritis, indicating its unique role in OA [45]. The NF-κB pathway plays a crucial role in the initiation of NLRP3 inflammasome, making it a key target for inhibiting pyroptosis. A study on knee OA (KOA) demonstrated a strong correlation between NLRP1 and NLRP3 inflammasome and the inflammatory response and pyroptosis in fibroblast-like synoviocytes (FLS). The suppression of these inflammasome results in a notable decrease in pyroptosis-induced cytoplasmic cleavage [5]. Zhang et al. showed that synovial macrophages undergo pyroptosis in a rat model of KOA, and that inhibiting cystatinase-1 and GSDMD with siRNAs can reduce synovitis and fibrosis in KOA [46].Bostin [47] developed an in vitro model of synovial fibroblast pyroptosis induced by LPS/ATP, and found that NLRP3 inflammasome were highly expressed in synovial tissues of OA patients. Knocking out NLRP3 gene can significantly reduce the expression level of downstream signal molecules ASC, Caspase-1, GSDMD gene and protein, and can slow down the occurrence of focal death of synovial fibroblasts. Hence, the modulation of NLRP3 inflammasome to mediate Caspase-1-dependent cellular pyroptosis is crucial in determining the level of inflammatory response and prognosis in OA. In addition, research has demonstrated there are many pharmaceutical preparations such as inhibitor CY-09 [48], chrysin [49], and baicalein [50](via intra-articular injection), and Physical therapy procedures, as well as physiotherapeutic procedures [51], in reducing the production of NLRP3 inflammatory vesicles, inhibiting pyroptosis, catabolism, and inflammatory response of chondrocytes, and enhancing cartilage integrity.
In addition, Chen [52] identified CASP6, NOD1, and PYCARD as prognostic factors by exploring the role of cellular pyroptosis-related genes in OA and their expression in different chondrocyte subtypes at the level of individual cells, utilizing a dataset related to OA for single-cell RNA sequencing and RNA-seq. However, these studies did not explore the molecular mechanisms in animal models, and related studies need to be further considered and improved in the future. In addition, some many non-coding RNAs also play a role in the process of juxtaposition in OA, such as miR-223, as a key miRNA that can bind to the 3UTR of NLRP3 mRNA, which provides a favorable effect on the inhibition of the activation of the NLRP3 inflammasome and the treatment of OA [53]. MiR-203a-3 inhibited cartilage matrix degradation, oxidative stress, and chondrocyte pyroptosis by regulating the MYD88/NF-κB pathway [54].The aforementioned targets underscore the potential importance of key regulators influencing pyroptosis in OA.
In conclusion, there exist specific associations between cellular pyroptosis and OA. Cellular pyroptosis can result in the demise of articular chondrocytes, consequently facilitating the degeneration and deterioration of articular cartilage. Furthermore, caspase-1-dependent cellular pyroptosis can trigger an inflammatory reaction, intensifying joint inflammation and pain symptoms. Moreover, various research studies have demonstrated the significance of GSDMD and the NLRP3 inflammasome in OA by instigating cellular death and inflammation, thereby exacerbating the progression of the disease. Many targets, including ASC, Caspase-1, GSDMD genes, and proteins, play a significant role in regulating the process of OA by influencing key genes such as IL-1β and MMP. Additionally, non-coding RNAs like miR-223 and MiR-203a-3 have emerged as important areas of research in the context of cell death in OA. This evidence underscores the close relationship between cellular pyroptosis and the development of OA. Exploring the targets of cell death in OA offers a novel theoretical framework and potential therapeutic approach for treating this condition.
Autophagy and OA
Cellular autophagy is a crucial mechanism through which cytoplasmic proteins and organelles are degraded via the lysosomal pathway to meet metabolic demands and facilitate organelle turnover. This dynamic and evolutionarily conserved process is integral to maintaining intracellular homeostasis by encapsulating cytoplasmic constituents within double-membrane vesicles (autophagosomes) for subsequent degradation in the lysosome [55]. Initially believed to be a non-selective degradation mechanism activated in response to starvation, autophagy has since been redefined as a targeted process that selectively degrades specific cellular components, thereby safeguarding the cell against various stressors and pathogens [56]. The regulation of autophagy primarily occurs through the activation of adenosine monophosphate-activated protein kinase (AMPK) and the suppression of the target of rapamycin (mTOR) signaling pathway [57].
Autophagy markers are expressed in healthy human chondrocytes and at reduced levels in OA tissues, which has stimulated great interest in this field [58]. As aging issues rise, more and more attention has been paid to cellular autophagy, especially in relation to chronic, degenerative diseases, and attempts have been made to elucidate the pathogenesis of OA with the perspective of cellular autophagy. During the initial stages of OA, chondrocytes rely on autophagy to safeguard against maladaptive responses to varying environmental stimuli. Subsequently, as cartilage undergoes progressive degeneration, autophagic diminishes, leading to cellular demise [59]. Autophagy in OA is modulated by AMPK signaling, with liver protein kinase B1 (LKB1) serving as the principal upstream kinase of AMPK. AMPK activation subsequently enhances chondrocyte autophagy through SIRT1, while concurrently suppressing apoptosis to mitigate OA. However, in the advanced stages of OA, chondrocyte autophagy diminishes while apoptosis increased. The process of apoptosis is initiated by excessive autophagic degradation of intracellular proteins and organelles, resulting in cellular demise. A common characteristic of the co-occurrence of apoptosis and autophagy in chondrocytes is the diminished presence of crucial autophagic regulators [60]. The regulation of autophagy in chondrocytes plays a crucial role in preserving cellular function and retarding the process of cartilage degeneration. Research indicates that [61], The promotion of autophagy has the potential to suppress apoptosis and inflammatory reactions in chondrocytes, thus safeguarding cartilage tissue. It has been noted that autophagy dysfunction in chondrocytes is a common occurrence in the advancement of OA. Carames et al. illustrated a notable decrease in the quantity of autophagic vesicles within rat articular chondrocytes when compared to younger rats. Additionally, the researchers observed a progressive down-regulation of autophagy-related genes ATG-5 and LC3 with advancing age, alongside an elevation in the levels of markers associated with chondrocyte apoptosis [62].Furthermore, autophagy inhibits the expression of chondrocyte catabolic genes and promotes the secretion of chondrogenic anabolic genes [63]. Activation of autophagy attenuates cartilage destruction by inhibiting the NF-kB signaling pathway in chondrocytes to down-regulate inflammatory catabolic genes such as MMP3 and − 9, ADAMTS5 and CCL-1 [64]. Research has indicated that the Wnt/β-catenin and NF-κB signaling pathways are significant contributors to the regulation of autophagy [64, 65].The studies have indicated that meniscal autophagy is a significant factor in the development of OA, with evidence suggesting that meniscal injury precedes articular cartilage degeneration following anterior cruciate ligament transection (ACLT) in rat models. Furthermore, the secretion of meniscal cells has been shown to decrease the levels of MMP13 and ADAMTS5 in chondrocytes treated with IL-1β, indicating that the activation of autophagy in meniscal cells may potentially impede the advancement of OA [66] (in Fig. 3).

Figure 3
In conclusion, our research highlights the significant role of autophagy in the pathogenesis of OA, demonstrating its therapeutic potential through the inhibition of chondrocyte apoptosis, preservation of the extracellular matrix of cartilage, attenuation of the inflammatory response, and mitigation of oxidative stress damage. Specifically, autophagy activation in chondrocytes serves as a protective mechanism in the early stages of degenerative cartilage disease, safeguarding cells against environmental stressors. However, as cartilage degeneration progresses, the inability of chondrocytes to sustain autophagy leads to cellular damage and potential cell death.
Therapeutic targets related to autophagy for OA
Autophagy has a significant impact on chondrocyte viability and plays a key role in regulating the expression of genes related to OA within the extracellular matrix (ECM). Activation of autophagy can exhibit dual effects, serving as both a cytoprotective and cytotoxic mechanism, with outcomes on cellular function that are contingent upon the stage of the disease. Its protective mechanism mainly includes that AdipoRon can promote autophagy by activating AMPK/mTOR signaling significantly attenuating calcification in OA chondrocytes [67], Sucrose-induced autophagy in vitro is dependent on the activation of the AKT/mTOR/P70S6K signaling pathway and blocks IL-1β-induced apoptosis as well as the expression of related OA genes [68]. Sirt I (Sirtuins) a class of deacetylases, can regulate autophagy by interacting with Atg7, and autophagy in OA was reduced by overexpression of Sirt I. Autophagic cell death in OA chondrocytes can be inhibited by increasing Sirt I activity [69]. In addition, hydroxytyrosol (HT) protects chondrocytes from oxidative stress-induced DNA damage and cell death via the deacetylase SIRT-1, and silencing of this enzyme prevents HT-promoted autophagy processes and cell survival [70]. Furthermore, certain studies have documented contrasting cytotoxic impacts of autophagy-related genes. Plant homologous structural domain finger protein 23 (PHF23) has emerged as a novel autophagy-inhibiting gene, with research indicating elevated expression of PHF23 in human OA cartilage and synovium. Knockdown of PHF23 has been shown to elevate levels of autophagy-associated proteins and reduce levels of apoptosis-associated proteins in IL-1β-induced OA-like chondrocytes [71]. P63 is a member of the P53 family, and Yufan et al. [72] demonstrated that up-regulation of P63 in cartilage tissues of OA patients inhibited chondrocyte autophagy, which led to the malignant progression of OA. It has also been shown that decreased expression of FOXO transcription factors in chondrocytes is associated with a decrease in antioxidant proteins and autophagy-related proteins, as well as, increased susceptibility to oxidative stress-induced cell death [73]。.
Several drugs and natural products have been found to promote autophagic activity in chondrocytes, such as rapamycin and allicin. These substances promote cellular autophagy by inhibiting the mTOR signaling pathway or activating the AMPK signaling pathway. In addition, a number of cytokines and growth factors can affect chondrocyte survival and function by regulating autophagy [74]. Autophagy has garnered heightened interest as a critical process in maintaining cellular equilibrium. Research has indicated that mitochondrial autophagy, a novel form of autophagy, is intricately linked to the advancement of OA [75]. Mitochondrial autophagy is commonly acknowledged as the principal mechanism for maintaining mitochondrial quality control, as the preservation of intact mitochondrial structure is essential for the survival of chondrocytes [76]. Mitochondrial dysfunction can lead to metabolic disorders and inflammatory responses in chondrocytes, resulting in the progression of OA. Zhang et al. showed that mitochondrial autophagy and mitochondrial dynamics are involved in the regulation of biological stress in chondrocytes [77] Moreover, Tang et al. demonstrated in a study of AGE-induced chondrocyte injury that bone marrow MSC-Exos suppressed chondrocyte apoptosis and cartilage matrix degradation by facilitating drp1-mediated mitochondrial autophagy [78]. Although many strategies have been developed to assess autophagy, most of the existing methods are static assessments, making accurate dynamic monitoring difficult to achieve [79]. mRFP-GFP-LC3 is an optional method for dynamic detection of autophagy, but imprecise fluorescence quantification remains a problem and needs further improvement and optimization. In addition, current drugs that activate or inhibit autophagy lack specificity, which may increase the risk of side effects during treatment [80].
In conclusion, autophagy serves as a crucial mechanism in the maintenance of chondrocyte homeostasis and presents a potential target for therapeutic intervention in OA. The signaling pathways, including AMPK/mTOR, AKT/mTOR/P70S6K, Sirtuins, PHF23, FOXO, and transcription factors, have been identified as effective targets for intervention in OA by modulating cell death, anabolism, catabolism, and inflammation. Especially in the early stages of OA pathogenesis, the increased level of autophagy in chondrocytes plays a role in protecting articular cartilage, which in turn slows down OA degeneration. Therefore, we need to conduct more in-depth studies on the autophagy regulatory mechanisms to develop more specific autophagy-targeting agents and ultimately provide a more effective approach for the treatment of OA in the clinic.
OA and Ferroptosis
Ferroptosis is an iron-dependent, oxidative mechanism of programmed cell death that is distinct from other forms of cell death. It is metabolically regulated in relation to amino acid and iron uptake, storage, utilization, efflux, and phospholipid synthesis. This mode of cell death is distinct from apoptosis and is induced by the ferroptosis-dependent peroxidation of membrane phospholipids, ultimately leading to the disassembly of all plasma membranes and termination of cellular life [81]. f Ferroptosis is characterized by mitochondrial contraction, heightened membrane density, diminished or absent cristae, and disruption of the outer membrane [82]. Ferroptosis is a form of iron-dependent programmed cell death distinguished by the buildup of lipid peroxides, which ultimately results in the disruption of the plasma membrane and organelle membranes, culminating in cellular demise. Redox-active iron is a critical factor in the acceleration of lipid peroxidation reactions. The glutathione-dependent antioxidant defense system is primarily responsible for suppressing lipid peroxidation, with a key role being played by glutathione peroxidase 4 (GPX4) within this system [83]. Reduced glutathione (GSH) serves as a crucial cofactor for GPX4, and its inhibition significantly hinders the enzymatic activity of GPX4. GSH is biosynthesized from the amino acid cysteine through the coordinated function of solute transporter family SLC3A2 and solute transporter family SLC7A11 proteins [84]. The Cystine/GSH/GPX4 system serves as a traditional mechanism for iron removal inhibition. System Xc, composed of the transmembrane protein complex SLC7A11 and the SLC3A2 subunit, functions as an amino acid transport protein with high specificity for cystine and glutamate, essential for glutathione (GSH) synthesis. The GSH pathway is recognized as a critical antioxidant defense pathway. Therefore, a potential method for suppressing ferroptosis involves the direct utilization of complex proteins that hinder the function of System Xc (Fig. 4).

Figure 4
Ferroptosis is implicated in the decreased activity of chondrocytes in OA, and this review examines recent research on the pathological mechanisms of Ferroptosis that result in chondrocyte death in this condition. It has been proposed that the tumor suppressor gene P53 suppresses the expression of SLC7A11, leading to diminished GSH synthesis and inhibition of GPX4, ultimately triggering Ferroptosis in various cancer cell lines [85]。it has been reported, IL-1β and TNF-α induce transferrin receptor expression, increasing iron uptake and accumulation, promoting ferroptosis under inflammatory conditions. Disruption of mitochondrial membranes increases cytoplasmic iron levels and disrupts oxidative phosphorylation, generating ROS and further promoting iron-mediated cell death [86]. In contrast to apoptosis, Ferroptosis lacks specific markers for its identification or selective staining of Ferroptosis cells [87]. The initiation and progression of Ferroptosis are predominantly regulated by three biochemical mechanisms: lipid ROS, ferrous iron, and GPX4 activity. Elevated levels of ferrous iron coupled with diminished GPX4 activity are indicative of the onset of Ferroptosis [88]. Erastin-treated polyunsaturated fatty acids (PUFA) undergo a Fenton reaction with reduced Fe2+ in the presence of large amounts of ROS to produce lipid peroxides (LPO) [89]. Ferrostatin-1 is an iron death-specific inhibitor that attenuates IL-1β and FAC-induced changes in Ferroptosis-associated ROS and protein expression and promotes activation of the Nrf2 antioxidant system. Yao et al. discovered that IL-1β and ferric ammonium citrate (FAC) both elicited alterations in the expression of reactive oxygen species (ROS) and iron-death-related proteins in chondrocytes, while the iron-death inducer erastin upregulated MMP-13 expression and downregulated collagen II expression in chondrocytes. Conversely, treatment with ferrostatin-1, a specific inhibitor of Ferroptosis, mitigated IL-1β- and FAC-induced cytotoxicity, ROS accumulation, and changes in the expression of ferroptosis-associated proteins [90]. This research represents the initial evidence of Ferroptosis taking place in chondrocytes. Additionally, Binetal’s recent discovery suggests that IL-6 triggers Ferroptosis in chondrocytes through the elevation of intracellular oxidative stress and iron buildup [91]. Ferroptosis has been linked to the death of chondrocytes, and the prevention of chondrocyte ferroptosis has been shown to mitigate cartilage degeneration. In a study by Ansari et al., human chondrocytes were induced with IL-1β to replicate the pathological environment of OA, resulting in a notable rise in reactive oxygen species (ROS) production and the buildup of mitochondrial impairment [92]. The similarities between OA and Ferroptosis, such as disrupted iron homeostasis, impaired mitochondrial function, and accumulation of lipid peroxidation, suggest a potential relationship between Ferroptosis and the advancement of OA. A study by Yao et al. revealed that the removal of iron from chondrocytes accelerates the progression of OA, highlighting the role of iron in the disease. Recent research has further supported the involvement of chondrocyte iron-induced cell death in the development of OA, showing that treatments with the antioxidant ferrostatin-1 or the iron chelator deferoxamine (DFX) can mitigate the progression of OA [93]. Hypoxia-inducible factor 2α (HIF-2α) has been documented to have a significant impact on cartilage development, progression of OA, and cellular resistance to ferritin [94]. There are in vitro studies using IL-1β to treat chondrocytes in the OA microenvironment. The results showed that HIF-1α and LPO levels were up-regulated and GSH levels were down-regulated in chondrocytes under IL-1β stimulation. During OA, synovium undergoes interstitial vascularization, fibrosis, and hyperplasia, leading to synovitis [95]. This is closely associated with joint dysfunction, injury and pain. These factors also contribute to cartilage degeneration in OA. Studies have shown that patients with OA have higher iron concentrations in synovial tissue compared to healthy individuals of the same age [96]. Furthermore, the dysfunction of iron homeostasis-regulating genes (HFE) results in cellular iron overload. Elevated levels of iron ions in HFE knockout chondrocytes lead to increased production of reactive oxygen species (ROS), as well as collagen II and MMP13, which are recognized markers of OA [97] .The above demonstrates that Ferroptosis is associated with the pathogenesis of OA. In animal models of iron overload, large amounts of intracellular iron can be seen to accumulate in the femoral joint cartilage and the infrapatellar fat pad of the knee. This leads to changes in iron transport proteins, cytokines, and cartilage structure, which are important factors in the early development of OA [98]. The transporter protein DMT1 transports iron into cells and is an important cause of iron overload. The use of DMT1 inhibitors reduces inflammation and ECM degradation and slows the progression of OA [99]. A two-year longitudinal study revealed that individuals with OA exhibited elevated serum ferritin levels compared to healthy controls, with the extent of elevation showing a positive association with joint stenosis [100]. The aforementioned studies indicate that disruption of iron homeostasis contributes to cartilage damage and extracellular matrix degradation, playing a significant role in the development of OA.
In conclusion, our study revealed that Ferroptosis plays a crucial role in chondrocytes and is particularly vulnerable to cellular and tissue damage with increased accumulation. This susceptibility is associated with inflammatory factors such as IL-1β and IL-6, which have been shown in previous research to induce Ferroptosis in OA. Additionally, iron inhibitors Fer-1 and DFO have shown promise as therapeutic agents for bone-related diseases, The subsequent discussion will delve into the precise mechanism of action of Ferroptosis in OA, along with the identification of potential targets within the Ferroptosis pathway for the purpose of enhancing therapeutic interventions for individuals afflicted with OA.
Therapeutic targets related to Ferroptosis for OA
Recent research has demonstrated the significant regulatory impact of Ferroptosis on autoimmune diseases and its involvement in the pathogenesis of OA, particularly in chondrocytes. Consequently, the development of pharmaceutical interventions targeting Ferroptosis may offer novel therapeutic avenues for the management of OA. It has been determined that enhancing the sensitivity of chondrocytes to Ferroptosis through intervention can enhance endochondral homeostasis. The P21 gene, a marker of senescence, has been shown to inhibit Ferroptosis. Zheng et al. conducted a study which revealed that P21 knockdown exacerbated the decrease in Col2a1, reduced GPX4 expression in vivo, and increased MMP13 expression in osteoarthritic chondrocytes, ultimately worsening OA [101]. Research conducted by Zhao et al. utilizing whole genome RNA sequencing, genomics, and proteomics has revealed that the expression of G protein-coupled receptor 30 (GPR30) in OA cartilage tissues is decreased compared to normal tissues. Furthermore, their findings suggest that the activation of GPR30 can suppress Ferroptosis of chondrocytes by inhibiting YAP1 phosphorylation. These results present a novel avenue for potential therapeutic interventions targeting GPR30 in arthritis [102]. Furthermore, it has been discovered that heat shock protein A family member 5 (HSPA5) acts as an inhibitor of Ferroptosis. Overexpression of HSPA5 has been shown to inhibit IL-1β-induced chondrocyte Ferroptosis. Conversely, knockdown of the RNA-binding protein SND1 results in the up-regulation of HSPA5 and GPX4 expression in rat cartilage. This up-regulation leads to the inhibition of inflammatory injury and Ferroptosis, ultimately slowing down the progression of OA [103]. On the other hand, upregulation of the membrane protein excitatory amino acid transport protein 1 (EAAT1) results in elevated intracellular glutamate (Glu) concentrations and induces the activation of the glutathione system, thereby suppressing Ferroptosis and consequently retarding the progression of OA [104], treatment with D-mannose, a C-2 epimer of glucose, reversed alterations in levels of lipid peroxidation (LPO) and reduced glutathione (GSH). This treatment also led to a decrease in malondialdehyde (MDA) levels, a byproduct of LPO, while simultaneously increasing RNA and protein levels of the desmoplastic inhibitors GPX4 and SLC7A11. Thus, it reduces the sensitivity of chondrocytes to Ferroptosis [105].
Traditional Chinese medicines have been found to inhibit iron-induced cell death in bone spur chondrocytes. For example, Icariside II has been shown to decrease the expression of iron transporter proteins, activate the GSH/GPX4 axis, and suppress the expression of genes SLC7A11 and SLC3A2L, which are involved in Ferroptosis. This ultimately leads to a significant reduction in chondrocyte death [106]. Theaflavin-3,3’-Digallate (TF3) is a polyphenolic compound derived from black tea. Research has demonstrated that TF3 has the ability to counteract Erastin-induced iron dysregulation in human cultured chondrocytes, lipid ROS accumulation, and elevated Fe2 + levels in mitochondria by activating the Nrf2/Gpx4 signaling pathway. These findings suggest that TF3 may hold promise as a therapeutic adjunct for the treatment of OA [107]. Furthermore, Astragalus extract has the ability to decrease intracellular iron levels through the inhibition of TfR1 and the promotion of FPN, thereby modulating the Nrf2/system xc-/GPX4 signaling pathway, scavenging free radicals, and inhibiting lipid peroxidation [108].
During the progression of OA, joint chondrocytes generate significant quantities of reactive oxygen species (ROS) due to inflammatory reactions, mechanical strain, and various stimuli, resulting in heightened oxidative stress. The progression of Ferroptosis intensifies oxidative stress levels, culminating in chondrocyte impairment and programmed cell death, ultimately contributing to articular cartilage deterioration. This process exacerbates joint dysfunction, impacting the patient’s daily life and working ability. Targeted inhibition of chondrocyte Ferroptosis, particularly through modulation of the GSH/GPX4 axis, represents a promising therapeutic avenue for OA. As research on Ferroptosis and its role in OA advances, it is increasingly evident that pharmacological agents targeting Ferroptosis hold potential for novel treatment strategies. However, further research and development efforts are necessary to fully realize the therapeutic benefits. The profound impact of Ferroptosis on the pathogenesis of OA underscores the importance of investigating the underlying mechanisms, offering valuable insights and approaches for the management of this debilitating condition.
Cuproptosis and OA
Copper, a crucial element/ion in living organisms, plays a significant role in various human physiological processes, such as the mitochondrial respiratory system and antioxidant defense system. In physiological settings, maintaining low intracellular levels of copper ions is advantageous for sustaining cellular functions and meeting metabolic demands. When the body is unable to regulate copper metabolism or experiences an increase in intracellular copper ions for various reasons, it can result in significant disruptions to bodily functions and potentially irreversible damage.
Recent research has elucidated the mechanism of copper-induced cell death, a form of programmed cell death triggered by copper ions that may be linked to apoptosis, cysteine proteases, or the accumulation of reactive oxygen species. This process is initiated by the disruption of cell membrane regulation of excessive extracellular copper ions, leading to the influx of high levels of copper ions into the cell. These ions then interact with mitochondrial lipoic acid-associated protein fractions to generate compounds that accumulate in the mitochondria, ultimately resulting in the depletion of Fe-S clusters dependent on FDX-1. This disruption not only hinders the energy supply pathway of the tricarboxylic acid cycle, but also triggers stress responses related to proteotoxicity, resulting in damage to mitochondrial membranes and impaired function of enzymes associated with the tricarboxylic acid cycle, ultimately leading to irreversible cell death. In normal circumstances, divalent copper ions ingested are absorbed as monovalent copper ions by SLC31A1, a solute carrier family member located on the membrane of intestinal epithelial cells, and are then transported to the bloodstream as copper cyanoproteins through the reduction of duodenal six-transmembrane proteins [109], Interacts with COX17, CCS, ATOX-1, and other molecular chaperone proteins with varied functions, facilitating its transportation to different bodily locations for the manifestation of biological effects or elimination through feces, urine, bile, etc. Interacts with COX17, CCS, ATOX-1, and other molecular chaperone proteins with varied functions, facilitating its transportation to different bodily locations for the manifestation of biological effects or elimination through feces, urine, bile, etc [110]. Likewise, deletion of the murine intestinal SLC31A1/2 gene led to a deficiency in copper levels in peripheral tissues and diminished functionality of copper ion-dependent enzymes [111]. SOD1, a protein closely linked to oxidative stress within the cytoplasm and mitochondrial intermembrane space (IMS), acquires copper ions from the molecular chaperone CCS to regulate redox balance, mitigate reactive oxygen species (ROS) generation, and uphold copper ion homeostasis [112]. It has been shown that overexpression of CCS leads to affect copper ion homeostasis and ultimately triggers Cuproptosis. The binding of copper ions by the CuA and CuB sites in the two core subunits of cytochrome oxidase COX is facilitated by from the cytoplasm to the mitochondria for incorporation into COX [113]. The aforementioned processes encompass the absorption and transportation of copper ions, as well as the associated mechanisms and regulatory functions. as shown in Fig. 5.

Figure 5
Research has demonstrated an intricate correlation between copper metabolism and arthritis progression, which will be thoroughly examined in this study with a focus on the impact of copper ion metabolism on cartilage in both physiological and arthritic conditions. In the physiological state, copper ions serve as crucial cofactors for enzymes involved in cartilage metabolism, notably lysyl oxidase (LOX), responsible for collagen cross-linking in connective tissue [114], Superoxide dismutase (SOD), which is responsible for the scavenging of ROS [115] and cytochrome c oxidase (Cox), which is involved in the mitochondrial respiratory chain [116]. Furthermore, copper ions play a crucial role in maintaining bone homeostasis through their influence on bone metabolism, density, and strength [117], and excess Cu can lead to ROS production, lipid peroxidation and inflammation, which can be harmful to bones [118]. The hypothesized mechanism underlying copper-induced cell death involves the binding of copper ions to thioctylated proteins within the tricarboxylic acid cycle (TCA), leading to aberrant oligomerization of these proteins and a reduction in Fe-S cluster protein levels. This cascade of events triggers a proteotoxic stress response culminating in cellular demise [119]. A cross-sectional study indicates that serum copper levels are elevated in patients with OA and rheumatoid arthritis (RA) compared to non-arthritic individuals when observed at a macroscopic level. Since 1990, the potential involvement of copper ions in the regulation of cytokine secretion has been suggested, indicating a potential significant role in inflammatory processes. This was demonstrated by Vinci C. et al., who showed that copper can inhibit interleukin 1 (IL-1) activity. Current evidence suggests that copper ions may play a role in the pathogenesis of arthritic diseases through inflammatory mechanisms [120], it is possible that the activation of the HIF signaling pathway contributes to its anti-inflammatory effects by promoting macrophage polarization towards the M2 anti-inflammatory phenotype, downregulating the expression of pro-inflammatory cytokines such as TNF-α and IL-18, and upregulating the expression of the anti-inflammatory cytokine IL-10 [121]. Research has demonstrated that copper possesses anti-inflammatory, angiogenic, and osteogenic properties that can enhance the process of bone healing [122]. The underlying cause is that controlled release promotes polarization of macrophages from pro-inflammatory M1 macrophages to anti-inflammatory M2 macrophages. In addition, copper deficiency may impair monoamine oxidase and LOX activity, decreasing collagen stability and the strength of bone structure, and LOX, of which copper is a major component, plays a key role in the formation of proteoglycans and collagen, which are components of the cartilage matrix [123], This predicts that impaired copper-related LOX function may exacerbate the development of OA [124].
In conclusion, a potential association exists between copper-induced cell death, a newly identified form of programmed cell death, and OA. This linkage is mediated not only through the tricarboxylic acid cycle (TCA), but also involves inflammatory factors like TNF-α and IL-18. The utilization of genes associated with copper-induced cell death in constructing disease prediction models demonstrates improved accuracy in forecasting the likelihood of developing OA. These findings suggest the potential utility of copper-induced cell death in the diagnosis and prevention of OA.
Therapeutic targets related to Cuproptosis for OA
As our understanding of the involvement of copper ions in arthritis has advanced, numerous therapeutic strategies have been developed. A recent study conducted both in vitro and in vivo has demonstrated the potential efficacy of nanoparticles functionalized with copper sulfate and anti-beta-2-microglobulin antibodies (B2M-CuS NPs) in the management of OA. It is important to note, however, that excessive copper levels can interfere with normal copper metabolism. The influx of surplus copper ions into the cell results in their interaction with mitochondrial lipoic acid-related protein components, leading to the formation of aggregates within the mitochondria. This process ultimately causes the FDX-1-dependent depletion of Fe-S clusters, resulting in mitochondrial dysfunction and damage to enzymes involved in the tricarboxylic acid cycle. Consequently, the accumulation of lipid peroxides and other compounds ensues, culminating in copper-induced cell death in oxidative stress conditions. Cuproptosis mainly involves the abnormal accumulation of copper ions in cells and its toxic effects, but at present, due to its relatively little research, Cuproptosis target therapy for OA is not a widely accepted or recognized therapeutic method. Currently, there are several main areas of target therapy. Copper-related compounds are used in the treatment of OA. When the corresponding copper compounds are formed, they induce apoptosis through the toxicity response ROS, selectively targeting and eliminating senescent chondrocytes, which can effectively promote the formation of cartilage for the treatment of OA [125]. In addition, endoplasmic reticulum stress, interferes with chondrocyte homeostasis and initiates the unfolded protein response (UPR), and chronic or irreversible endoplasmic reticulum stress triggers UPR-initiated cell death, so targeting endoplasmic reticulum stress per se or interfering with the UPR signaling to reduce chondrocyte death may be a promising future treatment for OA [126]. Research has demonstrated that ammonium tetrathiomolybdate (TM) has the ability to form complexes with intracellular copper, thereby impeding the advancement of OA with minimal adverse reactions, and additionally enhancing the well-being of individuals afflicted with OA [127]. Nanoparticles are commonly integrated into modern cartilage tissue engineering methodologies to augment chondrocyte functionality and exhibit significant promise in the management of degenerative joint pathologies [128]. The multifunctional composite heat-sensitive hydrogel (HPP@Cu gel) effectively mitigates inflammation by scavenging reactive oxygen and nitrogen species, modulating macrophage polarization from M1 to M2 phenotype, and suppressing the production of inflammatory mediators. These activities have been shown to attenuate cartilage degradation and reduce inflammatory factor production in OA rat models [129]. In addition, the natural compound curcumin has been used as a copper transporter protein to kill cancer cells through intracellular copper delivery [130]. Researchers have predominantly concentrated on the catalytic properties of copper, with limited exploration into the therapeutic implications of varying copper concentrations. Consequently, further investigation is warranted in this domain.
In conclusion, the bidirectional regulatory impact of copper ions on the pathological processes of OA suggests that strategically enhancing copper ion metabolism may hold promise for the treatment of this condition. Moreover, a more comprehensive understanding of the relationship between copper metabolism and cell death mechanisms is essential to optimize the protective effects of copper on joint and cartilage health while mitigating chondrocyte cell death. Further scientific investigations and clinical trials are warranted to validate the efficacy and safety of copper supplementation in OA management.。.
Conclusion
Cell death is a crucial process in biological systems that plays a vital role in maintaining homeostasis within the body and ensuring the proper functioning of tissues and organs. Various types of cell death exist, with apoptosis and necrosis being the most prevalent. Apoptosis, a programmed cell death mechanism, involves orchestrated changes in cellular structure and function, culminating in autolysis and subsequent phagocytosis by neighboring cells. Cellular necrosis is characterized by cellular injury caused by physical, chemical, or biological agents, leading to cell swelling, rupture, and release of cellular contents. In contrast, ferroptosis and copper death are ion-dependent mechanisms of cell death triggered by lipid peroxidation and oxidative stress, contributing to inflammation and exacerbation of OA with advancing age. A comprehensive examination of the mechanisms of cell death in OA is essential for elucidating the pathogenesis and progression of the disease. Contemporary research indicates that various factors, such as mechanical stress, oxidative stress, inflammatory response, autophagy, apoptosis, and the modulation of signaling pathways associated with Ferroptosis, may play a role in the initiation and control of cell death in OA.
The pathogenesis of OA is intricate, and its clinical management is significantly constrained. Nevertheless, as knowledge of the etiology and pathogenesis of OA advances, an increasing number of potential targets are being explored for the prevention and mitigation of the disease. Future research may focus on investigating the relationship between the pathogenesis of OA and various types of cell death. Hence, in the management of OA, it is imperative to address not only treatments aimed at mitigating the inflammatory response and other pathological alterations, but also to focus on the clearance of necrotic cells and facilitation of chondrocyte regeneration and repair. By effectively addressing these processes, symptoms of OA can be ameliorated and disease progression can be attenuated.
Data availability
No datasets were generated or analysed during the current study.
References
Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet 2019, 393(10182):1745–1759.
Abramoff B, Caldera FE. Osteoarthritis: Pathology, diagnosis, and Treatment options. Med Clin North Am. 2020;104(2):293–311.
Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12(7):412–20.
Yao Q, Wu X, Tao C, Gong W, Chen M, Qu M, Zhong Y, He T, Chen S, Xiao G. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Signal Transduct Target Ther. 2023;8(1):56.
Zhao LR, Xing RL, Wang PM, Zhang NS, Yin SJ, Li XC, Zhang L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATP–induced pyroptosis in knee osteoarthritis. Mol Med Rep. 2018;17(4):5463–9.
Xie J, Wang Y, Lu L, Liu L, Yu X, Pei F. Cellular senescence in knee osteoarthritis: molecular mechanisms and therapeutic implications. Ageing Res Rev. 2021;70:101413.
Hubert V, Weiss S, Rees AJ, Kain R. Modulating chaperone-mediated autophagy and its clinical applications in Cancer. Cells 2022, 11(16).
Motuhi SE, Mehiri M, Payri CE, La Barre S, Bach S. Marine Natural products from New Caledonia–A Review. Mar Drugs 2016, 14(3).
Hollville E, Romero SE, Deshmukh M. Apoptotic cell death regulation in neurons. FEBS J. 2019;286(17):3276–98.
Liu X, Gu Y, Bian Y, Cai D, Li Y, Zhao Y, Zhang Z, Xue M, Zhang L. Honokiol induces paraptosis-like cell death of acute promyelocytic leukemia via mTOR & MAPK signaling pathways activation. Apoptosis. 2021;26(3–4):195–208.
Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284(33):21777–81.
Bennell KL, Bowles KA, Payne C, Cicuttini F, Williamson E, Forbes A, Hanna F, Davies-Tuck M, Harris A, Hinman RS. Lateral wedge insoles for medial knee osteoarthritis: 12 month randomised controlled trial. BMJ. 2011;342:d2912.
Liu X, Zou H, Widlak P, Garrard W, Wang X. Activation of the apoptotic endonuclease DFF40 (caspase-activated DNase or nuclease). Oligomerization and direct interaction with histone H1. J Biol Chem. 1999;274(20):13836–40.
Wang XR, Cull B. Apoptosis and autophagy: current understanding in Tick-Pathogen interactions. Front Cell Infect Microbiol. 2022;12:784430.
Howley B, Fearnhead HO. Caspases as therapeutic targets. J Cell Mol Med. 2008;12(5A):1502–16.
Labbe K, Saleh M. Cell death in the host response to infection. Cell Death Differ. 2008;15(9):1339–49.
Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275(5302):983–6.
Liu X, Dai S, Zhu Y, Marrack P, Kappler JW. The structure of a Bcl-xL/Bim fragment complex: implications for bim function. Immunity. 2003;19(3):341–52.
Zhu W, Yang X, Liu S, Wang Y, Li W, Zhong Q, Zhang L, Xu J. Lentivirus-based shRNA of Caspase-3 gene silencing inhibits chondrocyte apoptosis and delays the progression of surgically induced osteoarthritis. Biotechnol J. 2024;19(1):e2300031.
Huang J, Ye Z, Wang J, Chen Q, Huang D, Liu H. USP13 mediates PTEN to ameliorate osteoarthritis by restraining oxidative stress, apoptosis and inflammation via AKT-dependent manner. Biomed Pharmacother. 2021;133:111089.
Pelletier JP, Mineau F, Boileau C, Martel-Pelletier J. Diacerein reduces the level of cartilage chondrocyte DNA fragmentation and death in experimental dog osteoarthritic cartilage at the same time that it inhibits caspase-3 and inducible nitric oxide synthase. Clin Exp Rheumatol. 2003;21(2):171–7.
Lou C, Lin C, Wang W, Jiang H, Cai T, Lin S, Xue X, Lin J, Pan X. Extracts of Oldenlandia diffusa protects chondrocytes via inhibiting apoptosis and associated inflammatory response in osteoarthritis. J Ethnopharmacol. 2023;316:116744.
Yamasaki K, Nakasa T, Miyaki S, Ishikawa M, Deie M, Adachi N, Yasunaga Y, Asahara H, Ochi M. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009;60(4):1035–41.
Zhang H, Zheng W, Li D, Zheng J. miR-146a-5p promotes Chondrocyte apoptosis and inhibits autophagy of Osteoarthritis by Targeting NUMB. Cartilage. 2021;13(2suppl):S1467–77.
Jin L, Zhao J, Jing W, Yan S, Wang X, Xiao C, Ma B. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int J Mol Med. 2014;34(2):451–63.
Wang WT, Huang ZP, Sui S, Liu JH, Yu DM, Wang WB. microRNA-1236 promotes chondrocyte apoptosis in osteoarthritis via direct suppression of PIK3R3. Life Sci. 2020;253:117694.
Zhang Y, Yan M, Shan W, Zhang T, Shen Y, Zhu R, Fang J, Mao H. Bisphenol A induces pyroptotic cell death via ROS/NLRP3/Caspase-1 pathway in osteocytes MLO-Y4. Food Chem Toxicol. 2022;159:112772.
Shoshan-Barmatz V, Keinan N, Abu-Hamad S, Tyomkin D, Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta. 2010;1797(6–7):1281–91.
Chang X, Kang Y, Yang Y, Chen Y, Shen Y, Jiang C, Shen Y. Pyroptosis: a novel intervention target in the progression of Osteoarthritis. J Inflamm Res. 2022;15:3859–71.
He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25(12):1285–98.
Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol. 2020;20(3):143–57.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547(7661):99–103.
Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. 2000;38(1):31–40.
Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol. 2018;9:2379.
An S, Hu H, Li Y, Hu Y. Pyroptosis plays a role in Osteoarthritis. Aging Dis. 2020;11(5):1146–57.
Donate R, Tamaddon M, Ribeiro V, Monzon M, Oliveira JM, Liu C. Translation through collaboration: practice applied in BAMOS project in in vivo testing of innovative osteochondral scaffolds. Biomater Transl. 2022;3(2):102–4.
Jiang T, Gong Y, Zhang W, Qiu J, Zheng X, Li Z, Yang G, Hong Z. PD0325901, an ERK inhibitor, attenuates RANKL-induced osteoclast formation and mitigates cartilage inflammation by inhibiting the NF-kappaB and MAPK pathways. Bioorg Chem. 2023;132:106321.
Hu J, Zhou J, Wu J, Chen Q, Du W, Fu F, Yu H, Yao S, Jin H, Tong P, et al. Loganin ameliorates cartilage degeneration and osteoarthritis development in an osteoarthritis mouse model through inhibition of NF-kappaB activity and pyroptosis in chondrocytes. J Ethnopharmacol. 2020;247:112261.
Ortega N, Behonick DJ, Werb Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004;14(2):86–93.
Hu Q, Ecker M. Overview of MMP-13 as a Promising Target for the treatment of Osteoarthritis. Int J Mol Sci 2021, 22(4).
Wang S, Mobasheri A, Zhang Y, Wang Y, Dai T, Zhang Z. Exogenous stromal cell-derived factor-1 (SDF-1) suppresses the NLRP3 inflammasome and inhibits pyroptosis in synoviocytes from osteoarthritic joints via activation of the AMPK signaling pathway. Inflammopharmacology. 2021;29(3):695–704.
Kanneganti A, Malireddi RKS, Saavedra PHV, Vande Walle L, Van Gorp H, Kambara H, Tillman H, Vogel P, Luo HR, Xavier RJ, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of familial Mediterranean Fever. J Exp Med. 2018;215(6):1519–29.
Rashidi M, Simpson DS, Hempel A, Frank D, Petrie E, Vince A, Feltham R, Murphy J, Chatfield SM, Salvesen GS, et al. The pyroptotic cell death Effector Gasdermin D is activated by gout-Associated Uric acid crystals but is dispensable for cell death and IL-1beta release. J Immunol. 2019;203(3):736–48.
Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, Civitelli R, Abu-Amer Y, Kanneganti TD, Link DC, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. 2018;16(11):e3000047.
Yang T, Sun K, Wang C, Swarnkar G, Quan S, Kress D, Xiao J, Alippe Y, Zheng H, Brophy RH, et al. Gasdermin D deficiency attenuates arthritis induced by traumatic injury but not autoantibody-assembled immune complexes. Arthritis Res Ther. 2021;23(1):286.
Zhang L, Xing R, Huang Z, Zhang N, Zhang L, Li X, Wang P. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediators Inflamm 2019, 2019:2165918.
Bostan E, Gokoz O, Atakan N. The role of NLRP1 and NLRP3 inflammasomes in the etiopathogeneses of pityriasis lichenoides chronica and mycosis fungoides: an immunohistochemical study. Arch Dermatol Res. 2023;315(2):231–9.
Zhang Y, Lin Z, Chen D, He Y. CY-09 attenuates the progression of osteoarthritis via inhibiting NLRP3 inflammasome-mediated pyroptosis. Biochem Biophys Res Commun. 2021;553:119–25.
Liao T, Ding L, Wu P, Zhang L, Li X, Xu B, Zhang H, Ma Z, Xiao Y, Wang P. Chrysin attenuates the NLRP3 Inflammasome Cascade to Reduce Synovitis and Pain in KOA rats. Drug Des Devel Ther. 2020;14:3015–27.
Bai H, Yuan R, Zhang Z, Liu L, Wang X, Song X, Ma T, Tang J, Liu C, Gao L. Intra-articular Injection of Baicalein Inhibits Cartilage Catabolism and NLRP3 Inflammasome Signaling in a Posttraumatic OA Model. Oxid Med Cell Longev 2021, 2021:6116890.
He Z, Nie P, Lu J, Ling Y, Guo J, Zhang B, Hu J, Liao J, Gu J, Dai B, et al. Less mechanical loading attenuates osteoarthritis by reducing cartilage degeneration, subchondral bone remodelling, secondary inflammation, and activation of NLRP3 inflammasome. Bone Joint Res. 2020;9(10):731–41.
Chen Y, Zhang Y, Ge Y, Ren H. Integrated single-cell and bulk RNA sequencing analysis identified pyroptosis-related signature for diagnosis and prognosis in osteoarthritis. Sci Rep. 2023;13(1):17757.
Liu W, Liu A, Li X, Sun Z, Sun Z, Liu Y, Wang G, Huang D, Xiong H, Yu S, et al. Dual-engineered cartilage-targeting extracellular vesicles derived from mesenchymal stem cells enhance osteoarthritis treatment via miR-223/NLRP3/pyroptosis axis: toward a precision therapy. Bioact Mater. 2023;30:169–83.
Chen J, Liu Z, Sun H, Liu M, Wang J, Zheng C, Cao X. MiR-203a-3p attenuates apoptosis and pyroptosis of chondrocytes by regulating the MYD88/NF-kappaB pathway to alleviate osteoarthritis progression. Aging. 2023;15(23):14457–72.
Wang L, Ye X, Zhao T. The physiological roles of autophagy in the mammalian life cycle. Biol Rev Camb Philos Soc. 2019;94(2):503–16.
Kawabata T, Yoshimori T. Beyond starvation: an update on the autophagic machinery and its functions. J Mol Cell Cardiol. 2016;95:2–10.
Corona Velazquez AF, Jackson WT. So many roads: the multifaceted regulation of Autophagy induction. Mol Cell Biol 2018, 38(21).
Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 2010;62(3):791–801.
Li YS, Zhang FJ, Zeng C, Luo W, Xiao WF, Gao SG, Lei GH. Autophagy in osteoarthritis. Joint Bone Spine. 2016;83(2):143–8.
Almonte-Becerril M, Navarro-Garcia F, Gonzalez-Robles A, Vega-Lopez MA, Lavalle C, Kouri JB. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of osteoarthritis within an experimental model. Apoptosis. 2010;15(5):631–8.
Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11(1):35–44.
Carames B, Olmer M, Kiosses WB, Lotz MK. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 2015;67(6):1568–76.
Sasaki H, Takayama K, Matsushita T, Ishida K, Kubo S, Matsumoto T, Fujita N, Oka S, Kurosaka M, Kuroda R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheum. 2012;64(6):1920–8.
Liu Y, Li X, Jin A. Rapamycin inhibits Nf-KappaB activation by Autophagy to reduce catabolism in human chondrocytes. J Invest Surg. 2020;33(9):861–73.
Ma L, Liu Y, Zhao X, Li P, Jin Q. Rapamycin attenuates articular cartilage degeneration by inhibiting beta-catenin in a murine model of osteoarthritis. Connect Tissue Res. 2019;60(5):452–62.
Zhu H, Liu H, Chen X, Xu X, Zhang S, Xie D. Enhancing autophagy and energy metabolism in the meniscus can delay the occurrence of PTOA in ACLT rat. Front Cell Dev Biol. 2022;10:971736.
Gomez-Virgilio L, Silva-Lucero MD, Flores-Morelos DS, Gallardo-Nieto J, Lopez-Toledo G, Abarca-Fernandez AM, Zacapala-Gomez AE, Luna-Munoz J, Montiel-Sosa F, Soto-Rojas LO et al. Autophagy: A Key Regulator of Homeostasis and Disease: An Overview of Molecular Mechanisms and Modulators. Cells 2022, 11(15).
Khan NM, Ansari MY, Haqqi TM. Sucrose, but not glucose, blocks IL1-beta-Induced Inflammatory response in human chondrocytes by inducing autophagy via AKT/mTOR pathway. J Cell Biochem. 2017;118(3):629–39.
Le Clanche S, Bonnefont-Rousselot D, Sari-Ali E, Rannou F, Borderie D. Inter-relations between osteoarthritis and metabolic syndrome: a common link? Biochimie. 2016;121:238–52.
Li X, Yang X, Maimaitijuma T, Cao XY, Jiao Y, Wu H, Meng ZC, Liu H, Guan ZP, Cao YP. Plant homeodomain finger protein 23 inhibits autophagy and promotes apoptosis of chondrocytes in osteoarthritis. Chin Med J (Engl). 2019;132(21):2581–7.
Liao FX, Huang F, Ma WG, Qin KP, Xu PF, Wu YF, Wang H, Chang J, Yin ZS. The New Role of Sirtuin1 in human osteoarthritis chondrocytes by regulating Autophagy. Cartilage. 2021;13(2suppl):S1237–48.
Wangyang Y, Zheng X, Liu GW, Li DY, Feng YB, Guo TY, Ma C, Wang T. Upregulation of P63 inhibits chondrocyte autophagy thereby enhancing the malignant progression of osteoarthritis. Pharmazie. 2017;72(6):361–4.
Akasaki Y, Alvarez-Garcia O, Saito M, Carames B, Iwamoto Y, Lotz MK. FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol. 2014;66(12):3349–58.
Chen H, Tan XN, Hu S, Liu RQ, Peng LH, Li YM, Wu P. Molecular mechanisms of chondrocyte proliferation and differentiation. Front Cell Dev Biol. 2021;9:664168.
Sun K, Jing X, Guo J, Yao X, Guo F. Mitophagy in degenerative joint diseases. Autophagy. 2021;17(9):2082–92.
Kan S, Duan M, Liu Y, Wang C, Xie J. Role of Mitochondria in Physiology of chondrocytes and diseases of Osteoarthritis and Rheumatoid Arthritis. Cartilage. 2021;13(2suppl):S1102–21.
Zhang J, Hao X, Chi R, Qi J, Xu T. Moderate mechanical stress suppresses the IL-1beta-induced chondrocyte apoptosis by regulating mitochondrial dynamics. J Cell Physiol. 2021;236(11):7504–15.
Tang S, Tang T, Gao G, Wei Q, Sun K, Huang W. Bone marrow mesenchymal stem cell-derived exosomes inhibit chondrocyte apoptosis and the expression of MMPs by regulating Drp1-mediated mitophagy. Acta Histochem. 2021;123(8):151796.
Mizushima N, White E, Rubinsztein DC. Breakthroughs and bottlenecks in autophagy research. Trends Mol Med. 2021;27(9):835–8.
Raudenska M, Balvan J, Masarik M. Crosstalk between autophagy inhibitors and endosome-related secretory pathways: a challenge for autophagy-based treatment of solid cancers. Mol Cancer. 2021;20(1):140.
Djulbegovic MB, Uversky VN. Ferroptosis - an iron- and disorder-dependent programmed cell death. Int J Biol Macromol. 2019;135:1052–69.
Zerbes RM, Bohnert M, Stroud DA, von der Malsburg K, Kram A, Oeljeklaus S, Warscheid B, Becker T, Wiedemann N, Veenhuis M, et al. Role of MINOS in mitochondrial membrane architecture: cristae morphology and outer membrane interactions differentially depend on mitofilin domains. J Mol Biol. 2012;422(2):183–91.
Dar NJ, John U, Bano N, Khan S, Bhat SA. Oxytosis/Ferroptosis in Neurodegeneration: the underlying role of Master Regulator glutathione peroxidase 4 (GPX4). Mol Neurobiol. 2024;61(3):1507–26.
Xia C, Xing X, Zhang W, Wang Y, Jin X, Wang Y, Tian M, Ba X, Hao F. Cysteine and homocysteine can be exploited by GPX4 in ferroptosis inhibition independent of GSH synthesis. Redox Biol. 2024;69:102999.
Shao M, Jiang Q, Shen C, Liu Z, Qiu L. Sinapine induced ferroptosis in non-small cell lung cancer cells by upregulating transferrin/transferrin receptor and downregulating SLC7A11. Gene. 2022;827:146460.
Yang F, Jiang X, Cao L, Gu Q, Teng X, He L. Diverse sesquiterpenoids from the roots of Croton crassifolius and their Inhibitory effects on Ferroptosis. Chem Biodivers. 2022;19(4):e202101028.
Turchi R, Faraonio R, Lettieri-Barbato D, Aquilano K. An Overview of the Ferroptosis Hallmarks in Friedreich’s Ataxia. Biomolecules 2020, 10(11).
Jafari-Gharabaghlou D, Pilehvar-Soltanahmadi Y, Dadashpour M, Mota A, Vafajouy-Jamshidi S, Faramarzi L, Rasouli S, Zarghami N. Combination of metformin and phenformin synergistically inhibits proliferation and hTERT expression in human breast cancer cells. Iran J Basic Med Sci. 2018;21(11):1167–73.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, Wang G, Guo Z, Ye Y, Guo F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. 2021;27:33–43.
Bin S, Xin L, Lin Z, Jinhua Z, Rui G, Xiang Z. Targeting miR-10a-5p/IL-6R axis for reducing IL-6-induced cartilage cell ferroptosis. Exp Mol Pathol. 2021;118:104570.
Ansari MY, Haqqi TM. Interleukin-1beta induced stress granules sequester COX-2 mRNA and regulates its Stability and translation in Human OA Chondrocytes. Sci Rep. 2016;6:27611.
Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G, Hou L, Xu J, Guo J, Guo F. Deferoxamine alleviates osteoarthritis by inhibiting Chondrocyte Ferroptosis and activating the Nrf2 pathway. Front Pharmacol. 2022;13:791376.
Zhang ZJ, Hou YK, Chen MW, Yu XZ, Chen SY, Yue YR, Guo XT, Chen JX, Zhou Q. A pH-responsive metal-organic framework for the co-delivery of HIF-2alpha siRNA and curcumin for enhanced therapy of osteoarthritis. J Nanobiotechnol. 2023;21(1):18.
Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51(2):249–57.
Yazar M, Sarban S, Kocyigit A, Isikan UE. Synovial fluid and plasma selenium, copper, zinc, and iron concentrations in patients with rheumatoid arthritis and osteoarthritis. Biol Trace Elem Res. 2005;106(2):123–32.
Simao M, Gavaia PJ, Camacho A, Porto G, Pinto IJ, Ea HK, Cancela ML. Intracellular iron uptake is favored in Hfe-KO mouse primary chondrocytes mimicking an osteoarthritis-related phenotype. BioFactors. 2019;45(4):583–97.
Ni Z, Li Y, Song D, Ding J, Mei S, Sun S, Cheng W, Yu J, Zhou L, Kuang Y, et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. 2022;13(7):579.
Yanatori I, Kishi F. DMT1 and iron transport. Free Radic Biol Med. 2019;133:55–63.
Yoshiiwa T, Miyazaki M, Notani N, Ishihara T, Kawano M, Tsumura H. Analysis of the relationship between Ligamentum Flavum thickening and lumbar segmental instability, disc degeneration, and Facet Joint Osteoarthritis in lumbar spinal stenosis. Asian Spine J. 2016;10(6):1132–40.
Zheng Z, Shang X, Sun K, Hou Y, Zhang X, Xu J, Liu H, Ruan Z, Hou L, Guo Z, et al. P21 resists ferroptosis in osteoarthritic chondrocytes by regulating GPX4 protein stability. Free Radic Biol Med. 2024;212:336–48.
Zhao Z, Niu S, Chen J, Zhang H, Liang L, Xu K, Dong C, Su C, Yan T, Zhang Y, et al. G protein-coupled receptor 30 activation inhibits ferroptosis and protects chondrocytes against osteoarthritis. J Orthop Translat. 2024;44:125–38.
Lv M, Cai Y, Hou W, Peng K, Xu K, Lu C, Yu W, Zhang W, Liu L. The RNA-binding protein SND1 promotes the degradation of GPX4 by destabilizing the HSPA5 mRNA and suppressing HSPA5 expression, promoting ferroptosis in osteoarthritis chondrocytes. Inflamm Res. 2022;71(4):461–72.
Wen Z, Xia G, Liang C, Wang X, Huang J, Zhang L, Shan D, Wu S, Cao X. Selective clearance of senescent chondrocytes in osteoarthritis by targeting excitatory amino acid transporter protein 1 to induce ferroptosis. Antioxid Redox Signal. 2023;39(4–6):262–77.
Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W, Yuan W, Yi Y, Wang J, Liu J. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a HIF-2alpha-dependent manner. Cell Prolif. 2021;54(11):e13134.
Yu R, Zhou Y, Shi S, Wang X, Huang S, Ren Y. Icariside II induces ferroptosis in renal cell carcinoma cells by regulating the miR-324-3p/GPX4 axis. Phytomedicine. 2022;102:154182.
Xu C, Ni S, Xu N, Yin G, Yu Y, Zhou B, Zhao G, Wang L, Zhu R, Jiang S et al. Theaflavin-3,3’-Digallate Inhibits Erastin-Induced Chondrocytes Ferroptosis via the Nrf2/GPX4 Signaling Pathway in Osteoarthritis. Oxid Med Cell Longev 2022, 2022:3531995.
He Q, Yang J, Pan Z, Zhang G, Chen B, Li S, Xiao J, Tan F, Wang Z, Chen P, et al. Biochanin A protects against iron overload associated knee osteoarthritis via regulating iron levels and NRF2/System xc-/GPX4 axis. Biomed Pharmacother. 2023;157:113915.
Dame C, Horn D, Schomburg L, Grunhagen J, Chillon TS, Tietze A, Vogt A, Buhrer C. Fatal congenital copper transport defect caused by a homozygous likely pathogenic variant of SLC31A1. Clin Genet. 2023;103(5):585–9.
Araya M, Andrews M, Pizarro F, Arredondo M. Chaperones CCS, ATOX and COXIV responses to copper supplementation in healthy adults. Biometals. 2012;25(2):383–91.
Qiu Z, Liu Q, Wang L, Xiong Y, Wu J, Wang M, Yan X, Deng H. The copper transporter, SLC31A1, transcriptionally activated by ELF3, imbalances copper homeostasis to exacerbate cisplatin-induced acute kidney injury through mitochondrial dysfunction. Chem Biol Interact. 2024;393:110943.
Skopp A, Boyd SD, Ullrich MS, Liu L, Winkler DD. Copper-zinc superoxide dismutase (Sod1) activation terminates interaction between its copper chaperone (Ccs) and the cytosolic metal-binding domain of the copper importer Ctr1. Biometals 2019, 32(4):695–705.
Li L, Guo W, Wu K, Zhao Y, Luo Q, Zhang Q, Liu J, Xiong S, Wang F. Identification of binding sites of cisplatin to human copper chaperone protein Cox17 by high-resolution FT-ICR-MS. Rapid Commun Mass Spectrom. 2016;30(Suppl 1):168–72.
Vallet SD, Ricard-Blum S. Lysyl oxidases: from enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019;63(3):349–64.
Trist BG, Hilton JB, Hare DJ, Crouch PJ, Double KL. Superoxide dismutase 1 in Health and Disease: how a Frontline antioxidant becomes neurotoxic. Angew Chem Int Ed Engl. 2021;60(17):9215–46.
Swaminathan AB, Gohil VM. The role of COA6 in the mitochondrial copper delivery pathway to cytochrome c oxidase. Biomolecules 2022, 12(1).
Gaffney-Stomberg E. The Impact of Trace Minerals on Bone Metabolism. Biol Trace Elem Res. 2019;188(1):26–34.
Qu X, He Z, Qiao H, Zhai Z, Mao Z, Yu Z, Dai K. Serum copper levels are associated with bone mineral density and total fracture. J Orthop Translat. 2018;14:34–44.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler RD, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61.
Wang H, Zhang R, Shen J, Jin Y, Chang C, Hong M, Guo S, He D. Circulating level of Blood Iron and Copper Associated with inflammation and Disease Activity of Rheumatoid Arthritis. Biol Trace Elem Res. 2023;201(1):90–7.
Li G, Cheng T, Yu X. The impact of Trace Elements on Osteoarthritis. Front Med (Lausanne). 2021;8:771297.
Li S, Zhang L, Liu C, Kim J, Su K, Chen T, Zhao L, Lu X, Zhang H, Cui Y, et al. Spontaneous immunomodulation and regulation of angiogenesis and osteogenesis by Sr/Cu-borosilicate glass (BSG) bone cement to repair critical bone defects. Bioact Mater. 2023;23:101–17.
Lin W, Xu L, Li G. Molecular insights into Lysyl oxidases in Cartilage Regeneration and Rejuvenation. Front Bioeng Biotechnol. 2020;8:359.
Rucker RB, Kosonen T, Clegg MS, Mitchell AE, Rucker BR, Uriu-Hare JY, Keen CL. Copper, lysyl oxidase, and extracellular matrix protein cross-linking. Am J Clin Nutr. 1998;67(5 Suppl):S996–1002.
Zhou J, Liu C, Sun Y, Francis M, Ryu MS, Grider A, Ye K. Genetically predicted circulating levels of copper and zinc are associated with osteoarthritis but not with rheumatoid arthritis. Osteoarthritis Cartilage. 2021;29(7):1029–35.
Rellmann Y, Eidhof E, Dreier R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cell Signal. 2021;78:109880.
Qi H, Shi H, Yan M, Zhao L, Yin Y, Tan X, Qi H, Li H, Weng K, Tang Y, et al. Ammonium tetrathiomolybdate relieves oxidative stress in cisplatin-induced acute kidney injury via NRF2 signaling pathway. Cell Death Discov. 2023;9(1):259.
Sang Y, Zhang J, Liu C, Liu K, Yao H, Zhao H, Xu W, Xu Y, Hou G. Ameliorating osteoarthritis in mice using silver nanoparticles. J Vis Exp 2023(196).
Zhu C, Han S, Zeng X, Zhu C, Pu Y, Sun Y. Multifunctional thermo-sensitive hydrogel for modulating the microenvironment in Osteoarthritis by polarizing macrophages and scavenging RONS. J Nanobiotechnol. 2022;20(1):221.
Yang Y, Liang S, Geng H, Xiong M, Li M, Su Q, Jia F, Zhao Y, Wang K, Jiang J, et al. Proteomics revealed the crosstalk between copper stress and cuproptosis, and explored the feasibility of curcumin as anticancer copper ionophore. Free Radic Biol Med. 2022;193(Pt 2):638–47.
Funding
This work was supported by supported by the Jilin Scientific and Technological Development Program [Grant number: 20210101205JC], and Natural Science Foundation of Jilin provincial science and Technology Department [Grant number: YDZJ202301ZYTS040].
Author information
Authors and Affiliations
Contributions
M.G. and Z.L. wrote the main manuscript text and Q.Y. involvedin design , Y.W. and W.Y conduct of study, J.Y and M.G approval of the manuscript.All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Informed consent
Not applicable.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guan, M., Yu, Q., Zhou, G. et al. Mechanisms of chondrocyte cell death in osteoarthritis: implications for disease progression and treatment. J Orthop Surg Res 19, 550 (2024). https://doi.org/10.1186/s13018-024-05055-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-024-05055-6