
Abstract
Background
Lennert lymphoma (LL) is a variant of peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS), also known as a lymphoepithelioid variant of PTCL. Because of the rarity and lack of clear-cut diagnostic criteria, LL is susceptible tomisdiagnosis. Although previously diagnosed with LL might be reclassified and evaluated with the advent of of molecular and/or genetic findings, cytomorphology and immunohistochemistry are still the key to give rise to correct diagnosis.
Case presentation
We report a case of a patient who was diagnosed as LL based on cytomorphology and immunohistochemistry. Routine stain (Hematoxlin and Eosin-H&E) revealed tumor cells were mainly small to medium-sized CD4(+) T cells, the CD8 +/TIA-1 + cytotoxic cells were less minority, no expressions of follicle helper T cell markers (CD10, BCL6, PD1, CXCL13, ICOS) or CD21(+) hyperplastic FDC network, or proliferation of high edndothelial venules were noted; however, numerous epithelioid histiocytes are noted in the background and scattered EBV(+) cells were also present. The patient was achieved complete remission after six courses of chemotherapy with cyclophosphamide, epirubicin, vincristine, etoposide, and prednisone regimen. She was followed for 5 years without recurrence or progression.
Conclusions
Classic LL is not difficult to diagnose by cytomorphology and immunohistochemistry, and the mutation profiles can be helpful to distinguish LL from other lymphomas.
Similar content being viewed by others


Background
Karl Lennert in 1968 suggested that lymphoepithelioid lymphoma being a subtype of Hodgkin’s disease [1]. In 1976, Burke and Butler defined the morphologic diagnostic criteria for LL [2]. By 1988, it was recognized as T-cell lymphoma [3,4,5,6]. In the 1992 (Kiel) classification, LL was classified as a T-cell-derived low-grade malignant lymphoma. Recently, Etebari et al. [7] supported the classification of LL as a distinct entity by gene expression profiling and microRNAs detection studies. As LL is rare and barely studied, the 4th and 5th editions of the WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues classified it as a variant of PTCL-NOS.
Background cells EBER + LL received even less attention, however, the pathogenic role of epstein-barr virus in LL cases are still unclear. Here, We will report a few background cells EBER + LL, focus on LL’s tumor cells and background cells about morphology and immunohistochemical, and PCR showed a polyclonal pattern for IgH and a monoclonal TCRβ-chain rearrangement, which can be distinguished from the other lymphomas. In addition, with the rapid development of molecular pathology in recent years, new findings on the molecular pathological features of LL have been reported. This case was briefly analyzed and the literature was reviewed in order to deepen the understanding of LL.
Case presentation
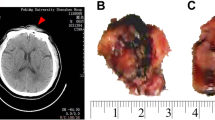
The patient, a 58-year-old female, presented with a painful mass in the left inguinal region for 10 days in our emergency department in April 2018 with no apparent cause. At the time of hospitalization, the inguinal mass was about 6 cm × 4 cm in size. She was unable to return to the back-up position when lying down flat and had no abdominal pain, abdominal distension, urinary frequency, urinary urgency, no fever, night sweats, weight loss, and other symptoms. She was physically healthy before and did not have a history of dermatological disease, autoimmune disease, lymphoma, etc. Laboratory tests results were slightly abnormal and routine blood neutrophil percentage was 85.0%, lymphocyte and platelet counts were normal, lactate dehydrogenase 327 U/L, glutamate transaminase 37 U/L, and β2 microglobulin 5.3 mg/L. Whole abdominal enhanced computed tomography (CT) scan showed multiple significantly enlarged lymph nodes in the cardiophrenic angle, retroperitoneum, abdominal cavity (prominently in the anterior pancreas and the ileocecal region), and left inguinal region and so lymphoma was considered. Two inguinal lymph nodes, with sizes of 60 mm × 50 mm × 40 mm and 15 mm × 12 mm × 10 mm, respectively with grayish, solid, and medium texture on the section were sent for examination (Fig. 1A). The specimens were fixed with 4% neutral formaldehyde, fully extracted, paraffin-embedded, and the tissues were sectioned at a thickness of 4 μm and stained with hematoxylin and eosin (H&E) for microscopic observation. The diaminobenzidine method was used for immunohistochemistry, and the in situ VENTANA ISH iVIEW Blue Detection Kit was used for Epstein–Barr virus (EBV)-encoded RNA (EBER) detection. Immunohistochemistry and special staining reagents were purchased from Roche and staining was carried out according to the manufacturer’s instructions. Bone marrow was collected for T-cell clonality analysis by polymerase chain reaction for the TCR gene as well as for the mature T/NK lymphoma gene mutation in detection panel.

Gross morphology (A). H&E shows structural disruption of the lymph nodes with a diffuse growth pattern (B: digital scan, H&E × 1). Epithelioid histiocytes (C: digital scan, H&E × 100) in small clusters (red circles, magnified on the right) or scattered distribution and multinucleated giant cells (yellow arrows). Epithelioid histocytes morphology (D: H&E × 400): cucumber-like nuclei (yellow arrows), straw-shoe-like nuclei (red arrows, magnified on the right). Abnormal small to medium-sized T lymphocytes (E: H&E × 400) and individual large cells (yellow arrows), scattered individual RS-like cells (F: H&E × 400, yellow arrows)
Both lymph nodes showed complete structural disruption on H&E microscopy (Fig. 1B). Epithelioid histiocytes were abundant, localized in small clusters(Fig. 1C), with abundant lightly stained cytoplasm, indistinct borders, round or ovoid nuclei or with “cucumber-like” presentation (Fig. 1D), thin nuclear membrane, little chromatin, even vacuolated, 1–2 small nucleoli in the nucleus, no nuclear grooves or obvious nucleoli. When examined with H&E microscopically, the tumor cells were predominantly small to medium-sized, with thickened and unevenly distributed chromatin, a few cells had clear cytoplasm (Fig. 1E), and karyokinesis was rare; a few Reed–Sternberg (RS)-like cells (Fig. 1F), neutrophils, a few eosinophils, and plasma cells, and no high endothelial venule hyperplasdia were seen.
Tumor cells expressed complete T-cell antigens (CD2, CD3, CD5, CD7, CD45RO), BCL2, and vimentin. A few scattered RS-like cells and anaplastic RS-like cells expressed CD15 and CD30 (with varying cytoplasmic granular staining), did not express CD20,PAX5 and ALK (Fig. 2A–D). A few background cells were EBER-positive (Fig. 2E). The pattern of tumor cells expressed CD4 which was consitent with CD3(Fig. 2F, G). A few T-cells expressed CD8 and TIA-1 in a uniform and scattered distribution (Fig. 2H, I). Ki67 index was 30%(Fig. 2J) .Epithelioid histiocytes express CD68 (KP-1). CD21 showed a residual follicular dendritic cell (FDC) network and no proliferating FDC network. There were no abnormal vessels in CD31. Tumor cells did not express follicle helper T-cell (TFH) markers (CD10, BCL-6, CXCL13, PD-1, ICOS) (Fig. 2K–O), CD23, CD56, GrB, CD1a, Langerin, S-100, and GATA-3. Acid-fast staining was negative. PCR showed a polyclonal pattern for IgH and a monoclonal TCRβ-chain rearrangement, and no mutations were detected in the mature T/NK lymphoma gene mutation detection panel. The final diagnosis was Lennert lymphoma (LL).

Immunohistochemical staining results showed A-E (IHC × 400): RS-like cells were positive for CD15 and CD30, while PAX-5 and ALK were negative, and a few cells were EBER+. F-J (IHC ×400): CD3 and CD4 positive, reactive T-cells showed diffuse CD8 and TIA-1 positivity, Ki67 index were about 30%. K–O (K, M IHC × 200, L, N, O IHC × 400): negative for TFH markers CD10, Bcl6, PD-1, CXCL13, and ICOS, with a little granular staining of PD-1 histocytes cytoplasm (top right magnification) and CXCL13 noted as an internal control (top right magnification)
Clinical assessment was Ann Arbor stage IV. Six-course cyclophosphamide, epirubicin, vincristine, etoposide, and prednisone (CHOEP) regimen chemotherapy was administered. In June 2018, according to the 2014 edition of Lugano criteria, combined with positron emission tomography/computed tomography (PET/CT) results, the patient was evaluated as in complete remission. Moreover, She was followed for 5years without recurrence or progression.
Discussion and conclusions
There are no clinically significant gender differences in LL, and this is mainly seen in middle-aged and elderly patients. Some patients have B symptoms and rash, hepatosplenomegaly, and generalized lymph node enlargement. Extra-nodal involvement is uncommon (bone marrow is the most common site of extra-nodal involvement). Some patients have elevated serum lactate dehydrogenase, and most patients have Ann Arbor stage III or IV [8, 9].
The morphology of LL shows a large number of epithelioid histiocytes in typical small clusters with scattered distribution, usually without high endothelial venule hyperplasia [6]. Heterotypic cells are predominantly small to medium-sized T lymphocytes, where large cells are seen, occasionally RS-like cells, and commonly eosinophils and plasma cells are seen; the mitotic figures are uncommon, and extra-nodal involvement is rare [1, 2, 6, 8].
LL expresses complete T-cell markers (CD2, CD3, CD5, CD7) and can also be deficient in one or above markers. The expression of CD4 helper T-cells [4, 6], CD8, and cytotoxic molecules (e.g., TIA-1, GrB, etc.) [9, 10], involving the origin of LL, is still controversial. However, LL is mostly CD4/CD8 single positive, predominantly CD4 positive, and rarely CD4/CD8 dual positive. LL background components are relatively complex, and attention should be paid to IHC-positive localization [4, 10]. In this case, the combined CD3CD4 and Ki67 index results showed that the tumor cells were centered on small to medium-sized CD4 + T lymphocytes. CD8 + TIA1 + cells were less than 10%, the uniform distribution of CD8 + and TIA1 + cytotoxic T cells suggests EB virus infection or tumor immune response.
Although RS like cells and EB virus infection without literature confirmed has inevitable connection, but EB virus infection can cause the cell changes, such as infectious monocyte hyperplasia and some Hodgkin’s lymphoma, so there is reason to believe that the correlation between the two; and small clusters epithelioid histiocytes reaction are the same, the cases of background cells EBER-positive PTCL NOS and angioimmunoblastic T-cell lymphoma(AITL) had a high probability of this phenomenon [11], but the tumor microenvironment may play an important role in it [7]. In addition, attention should be paid to the difference between the pseudoaggregation of epithelioid tissue cells and granuloma, as detailed in Table 1. Although EBER + LL only accounts for about 31% [9, 15], the significance of EB virus infection in LL is still unclear and may be one of the pathogenic factors of LL. The heterogeneity of this tumor, increases the difficulty of differentiation from Hodgkin’s lymphoma, and highlights the diagnostic defect of LL.
TFH markers (CD10, BCL6, CXCL13, PD-1, ICOS) were not expressed [11], but Kurita et al. [9] reported 57.7% of 26 LL patients were positive for at least one marker and mainly PD-1 and CXCL13, 3.8% were positive for CD10, and all patients were negative for BCL6, the study reveal TFH markers negative LL patients compared with positive patients tend to good prognosis. The absence of FDC network proliferation in most cases is also beneficial to distinguish AITL. CD56 expression is seen in rare cases, and does not express ALK. The ki67 index is usually not high, and a study by Hartmann et al. [11] of eight LL cases showed a median Ki67 index of 15% (range 5–40%). The immunohistochemical expressions and TCR gene rearrangement of the above-mentioned literatures are summarized in Table 2.
- unsupported, + supportive. Pathogenic factors include bacteria, fungi, viruses, parasites and other microbial infection and foreign body.
Recent studies have shown that PTCL-NOS exhibits abnormal GATA-3 and TBX21 gene expression profiles (GEP). PTCL-GATA3 subtype had a poor prognosis, with a 5-year OS of 19% and the most frequent genomic abnormalities. The reduction of copy number in PTCL-GATA3 was often accompanied by frequent deletion of tumor suppressor genes related to the CDKA2A/B-TP53 axis and PTEN-PI3K pathway, while the increase of copy number was often accompanied by amplification of cell cycle regulatory genes such as MYC and STAT3. Copy number alterations (CNAs) events such as CDKNA2 have independent prognostic significance in PTCL-NOS and PTCL-GATA3 subtypes. PTCL-TBX21 subtype has a better prognosis, with a 5-year OS rate of 38%. Compared with PTCL-GATA3, PTCL-TBX21 subtype has fewer genomic abnormalities. CNAs are often related to T cell differentiation and cytotoxicity, and the mutated genes are more genes related to epigenetic regulation. Genes regulating NF-kB signaling (ZC3H12DC/p3450 and TNFAIP3) and tumor suppressor kinase (LATS1) were frequently deleted in the PTCL-TBX21 subtype [12, 13], and some authors have classified PTCL-NOS into four morphological patterns, corresponding to the two GEP molecular types described above, with LL classified as pattern four and this pattern is present only in the TBX21 subtype. Moreover, immunohistochemistry (four antibodies: GATA-3, TBX 21, CCR 4 and CXCR 3) was applied to reproduce the two GEP molecular types with 85% agreement [14]. Therefore, the application of GEP and the corresponding four immunohistochemical antibodies is beneficial to identify other PTCL-NOS and predict the prognosis of patients. In this case, immunohistochemistry was GATA-3 negative, which was not sufficient to rule out GATA-3 molecular typing due to the absence of CCR4 immunohistochemistry for the GATA-3 corresponding receptor.
Etebari et al. [7] performed a gene expression profiling analysis of 12 LL and 68 other PTCL-NOS cases, and 455 genes were differentially expressed between the two groups, of which 385 were upregulated in LL. Analysis of upregulated genes in LL or PTCL-NOS using the molecular signature database revealed that genes related to the regulation of, for example, cell differentiation upregulation, apoptosis, and immune response were significantly enriched in LL. In contrast, genes related to cell growth/maintenance and cell proliferation were downregulated in LL or upregulated in PTCL-NOS. Moreover, microRNAs expression profiling of three LL cases and 20 PTCL-NOS cases showed that nine cellular miRNAs were differentially expressed in LL. According to the GEP and microRNAs signatures, it was revealed that the PI3K/Akt/mTOR signaling axis plays a central role in the molecular pathology of LL through STAT5. Thus, the use of GEP and microRNAs assays can distinguish LL from other PTCL-NOS and support the classification of LL as a distinct entity and not just a morphological variant of PTCL-NOS. In our patient, only bone marrow TCR gene rearrangement was performed, suggesting tumor involvement of the bone marrow, and clonal proliferation of T-cells was confirmed. The histopathology of this case showed typical morphological and immunohistochemical staining images of LL. Unfortunately, microRNAs and epigenetic testing were not performed due to the patient’s wishes. At present, more studies are needed on the genetic alterations of LL molecules.
LL needs to be differentiated from lymphomas such as Hodgkin’s lymphoma (HL), anaplastic large cell lymphoma (ALCL), TFH lymphoma, and adult T-cell lymphoma/leukemia (ATLL) [6,7,8,9]; the combination of clinical manifestations, H&E morphology, immunohistochemical staining, and TCR gene rearrangement is less difficult to identify. The number and expression patterns of CD30-expressing cells, and the marker of ALK, especially for identifying HL and ALCL (as: lymphohistiocytic pattern, small cell pattern, or Hodgkin-like pattern). It is necessary to identify follicular helper T cell lymphoma in lymph nodes. The latter TFH immunohistochemical markers (bcl 6, CD10, PD1, CXCL13, ICOS) should express at least two or more positive markers, obvious FDC network and high endothelial vein hyperplasia, etc., which alse can be identified by genetic testing. The latest study shows that the AITL genome has low abnormal complexity, and the most common CNAs are the Chr 5 increase, often accompanied by an increase in Chr 21, which is significantly associated with the IDH2 R172 mutation. In IDH 2 wild-type cases, there is often loss of PI3K-AKT-mTOR activation and up-regulated and pathway negative regulators, in addition to many gene abnormalities related to DNA repair and cellular metabolism pathways; PTCL-TFH has similar genetic basis to AITL, but lacks IDH2 R172 mutation [12, 13]. The development of immunohistochemical techniques and molecular genetic testing has facilitated the correct categorization and further research on LL.
The overall prognosis of LL is poor [15]. In particular, EBER + LL with more clinical complications predicts a worse prognosis, thus making clinical treatment challenging. The current first-line treatments for LL are cyclophosphamide, vincristine, doxorubicin, and prednisone(CHOP); CHOEP; adjusted doses of CHOEP, and high-dose therapy and autologous stem cell salvage therapy as consolidation therapy [16].
This case was treated with CHOEP chemotherapy and followed up for 5 years without recurrence and progression, and we will continue regular follow-up.
Materials
Immunohistochemistry
The antibodies provided by Fuzhou Maixin Biotechnology Development Co.,Ltd.: CD3(rat.no.MAB-0740),CD10(rat.no.MAB-0668),CD15(rat.no.MAB-0779),CD20(rat.no.Kit-0001),CD21(rabbit.no.RMA-0811),CD23(rabbit.no.RMA-0504),CD30(rat.no.MAB-0023),CD31(rat.no.MAB-0720),CD45RO(rat.no.MAB-0039),CD56(rat.no.MAB-0743),CD68(KP-1)(rat.no.Kit-0026),BCL6(rat.no.MAB-0746),PD1(rat.no.MAB-0734),CXCL13(sheep.no.GAB-0616),TIA-1(rat.no.MAB-0798),GrB(rat.no.MAB-0352),Bcl2(rat.no.MAB-0711),PAX5(rat.no.MAB-0706),ALK(rat.no.MAB-0281),CD1a(rat.no.MAB-0336),Langerin(rat.no.MAB-0633),S100(rat.no.Kit-0007),GATA3(rat.no.MAB-0695),Vimentin(rat.no.MAB-0735).
The antibodies provided by ZSGB-BIO: CD2(rat.no.ZM-0278),CD4(rabbit.no.ZA-0519),CD5(rat.no.ZM-0280),CD7(rabbit.no.ZA-0589),CD8(rabbit.no.ZA-0508),ICOS(rabbit.no.ZA-0690).
All antibodies were strictly operated according to the reagent instructions and were done with negative and positive controls.
In situ hybridization for Epstein-Barr virus-encoded RNA
The EBER probe kit was provided by Roche Diagnostics GmbH, operated according to the kit instructions and performed strictly negative and positive controls.
T-cell clonality analysis
T-cell clonality was evaluated using T-cell receptor(TCR).
Data availability
All data generated or analysed during this study are included in this article. Further enquiries should be directed to the corresponding author.
Abbreviations
- CHOEP:
-
Cyclophosphamide, epirubicin, vincristine, etoposide, and prednisone
- CT:
-
Computed tomography
- EBER:
-
Epstein–Barr virus-encoded RNA
- EBV:
-
Epstein–Barr virus
- FDC:
-
Follicular dendritic cell
- GEP:
-
Gene expression profiling
- H&E:
-
Hematoxylin and eosin
- HL:
-
Hodgkin’s lymphoma
- LL:
-
Lennert lymphoma
- TFH:
-
Follicle helper T-cell
References
Lennert K, Mestdagh J. Lymphogranulomatosen Mit Konstant Hohem Epitheloidzellgehalt. Virchows Arch Pathol Anat. 1968;344:1–20.
Burke JS, Butler JJ. Malignant lymphoma with a high content of epitheloid histiocytes (Lennert’s lymphoma). Am J Clin Pathol. 1976;66:1–9.
Gödde-Salz E, Feller AC, Lennert K. Cytogenetic and immunohistochemical analysis of lymphoepithelioid cell lymphoma (Lennert’s lymphoma): further substantiation of its T-cell nature. Leuk Res. 1986;10:313–23.
Feller AC, Griesser GH, Mak TW, Lennert K. Lymphoepithelioid lymphoma (Lennert’s lymphoma) is a monoclonal proliferation of helper/inducer T cells. Blood. 1986;68:663–7.
O’Connor NT, Feller AC, Wainscoat JS, et al. T-cell origin of Lennert’s lymphoma. Br J Haematol. 1986;64:521–8.
Patsouris E, Noël H, Lennert K. Histological and immunohistological findings in lymphoepithelioid cell lymphoma (Lennert’s lymphoma). Am J Surg Pathol. 1988;12:341–50.
Etebari M, Navari M, Agostinelli C, et al. Transcriptional analysis of Lennert lymphoma reveals a unique profile and identifies novel therapeutic targets. Front Genet. 2019;10:780.
Patsouris E, Engelhard M, Zwingers T, Lennert K. Lymphoepithelioid cell lymphoma (Lennert’s lymphoma): clinical features derived from analysis of 108 cases. Br J Haematol. 1993;84:346–8.
Kurita D, Miyoshi H, Yoshida N, et al. A clinicopathologic study of Lennert lymphoma and possible prognostic factors: the importance of follicular helper T-cell markers and the association with angioimmunoblastic T-cell lymphoma. Am J Surg Pathol. 2016;40:1249–60.
Geissinger E, Odenwald T, Lee SS, et al. Nodal peripheral T-cell lymphomas and, in particular, their lymphoepithelioid (Lennert’s) variant are often derived from CD8(+) cytotoxic T-cells. Virchows Arch. 2004;445:334–43.
Hartmann S, Agostinelli C, Klapper W, et al. Revising the historical collection of epithelioid cell-rich lymphomas of the Kiel Lymph Node Registry: what is Lennert’s lymphoma. Nowadays? Histopathology. 2011;59:1173–82.
Marchi E, O’Connor OA. The rapidly changing landscape in mature T-cell lymphoma (MTCL) biology and management. CA Cancer J Clin. 2020;70:47–70.
Iqbal J, Wright G, Wang C, et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014;123:2915–23.
Amador C, Greiner TC, Heavican TB, et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood. 2019;134:2159–70.
Weisenburger DD, Savage KJ, Harris NL, et al. Peripheral T-cell lymphoma, not otherwise specified: a report of 340 cases from the international peripheral T-cell lymphoma project. Blood. 2011;117:3402–8.
Horwitz SM, Ansell S, Ai WZ, Lymphomas T-C, et al. Version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw Version 2 2022. 2022;20:285–308.
Acknowledgements
The authors thank this patient for her cooperation in this study.
Funding
There was no financial support for this study.
Author information
Authors and Affiliations
Contributions
Shun Ding and Jiao Chen was responsible for project progress, collection of data and writing of the manuscript. Fengjie Qi was responsible for project design, overall planning. Jiajun Su, Jiewen Liu and Weihua Yin were responsible for project progress, collection of data. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics approval
This study protocol was reviewed and approved by the committee name and affiliation of Shenzhen Luohu Hospital Group, Luohu People’s Hospital.
Consent
The patient provided written informed consent for the publication of this case, including the publication of images.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, S., Chen, J., Su, J. et al. Clinicopathological diagnosis of Lennert lymphoma: a case report and review of the literature. Diagn Pathol 19, 111 (2024). https://doi.org/10.1186/s13000-024-01533-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13000-024-01533-x